

Abstract
Cholera toxin (CT) travels by vesicle carriers from the cell surface to the endoplasmic reticulum (ER) where the catalytic A1 subunit of CT (CTA1) dissociates from the rest of the toxin, unfolds, and moves through a membrane-spanning translocon pore to reach the cytosol. Heat shock protein 90 (HSP90) binds to the N-terminal region of CTA1 and facilitates its ER-to-cytosol export by refolding the toxin as it emerges at the cytosolic face of the ER membrane. HSP90 also refolds some endogenous cytosolic proteins as part of a foldosome complex containing heat shock cognate 71-kDa protein (HSC70) and the HSC70/HSP90-organizing protein (HOP) linker that anchors HSP90 to HSC70. We accordingly predicted that HSC70 and HOP also function in CTA1 translocation. Inactivation of HSC70 by drug treatment disrupted CTA1 translocation to the cytosol and generated a toxin-resistant phenotype. In contrast, the depletion of HOP did not disrupt CT activity against cultured cells. HSC70 and HSP90 could bind independently to disordered CTA1, even in the absence of HOP. This indicated HSP90 and HSC70 recognize distinct regions of CTA1, which was confirmed by the identification of a YYIYVI-binding motif for HSC70 that spans residues 83–88 of the 192-amino acid CTA1 polypeptide. Refolding of disordered CTA1 occurred in the presence of HSC70 alone, indicating that HSC70 and HSP90 can each independently refold CTA1. Our work suggests a novel translocation mechanism in which sequential interactions with HSP90 and HSC70 drive the N- to C-terminal extraction of CTA1 from the ER.
Keywords: bacterial toxin, heat shock protein (HSP), heat shock protein 90 (Hsp90), translocation, endoplasmic reticulum-associated protein degradation (ERAD), bacterial pathogenesis
Introduction
Cholera toxin (CT)2 is an AB5 toxin that is endocytosed after binding to GM1 gangliosides on the plasma membrane of a host cell (1). Most internalized toxin is routed to the lysosomes for degradation, but 5–10% of the cell-associated toxin travels to the endoplasmic reticulum (ER) by retrograde vesicular transport (2–4). In the ER, the catalytically active A1 subunit is separated from the holotoxin through the action of protein-disulfide isomerase (5, 6). The dissociated CTA1 subunit spontaneously unfolds due to its intrinsic thermal instability (7) and is processed by the ER-associated degradation (ERAD) system for export to the cytosol (8, 9). CTA1 evades the ubiquitin-dependent proteasomal degradation that is often coupled to ERAD translocation (7, 10, 11) and modifies its G protein target to elicit a cytopathic effect via elevated levels of intracellular cAMP (12). Because CTA1 is an unstable protein, it must be refolded and stabilized by host factors in the cytosol (13).
ERAD-mediated export usually involves the action of p97, an oligomeric AAA ATPase that uses the energy from ATP hydrolysis to pull ERAD substrates through a membrane-spanning translocon pore (14). CTA1 translocation does not require p97 (15, 16) but instead relies upon heat shock protein 90 (HSP90), a cytosolic chaperone (17, 18). We have proposed a ratchet mechanism for CTA1 export in which HSP90 provides directionality to the translocation event by refolding the toxin as it emerges at the cytosolic face of the translocon pore (18). The refolded toxin could not slide back into the narrow translocon pore, thus ensuring efficient, unidirectional movement from the ER to the cytosol. HSP90 only recognizes the disordered conformation of CTA1, and ATP hydrolysis by HSP90 is required for both CTA1 refolding and CTA1 translocation to the cytosol (18). This novel, HSP90-driven translocation mechanism could also apply to endogenous host proteins that move from the ER to the cytosol in a p97-independent process (19, 20) and to AB toxins such as diphtheria toxin (DT) that move from the endosomes to the cytosol (21–26).
HSP90 refolds cytosolic proteins as part of a foldosome complex involving heat shock cognate 71-kDa protein (HSC70), HSC70/HSP90-organizing protein (HOP), and heat shock protein 40 (HSP40) (27–29). Substrate refolding occurs via an initial interaction with HSP40, followed by transfer to HSC70 and subsequent complex formation with HOP and HSP90 (30–34). HOP is a noncatalytic linker that recruits HSP90 to the HSC70-bound client protein and is often required for HSP90 binding to the client protein (35, 36). Heat shock protein 70 (HSP70) is a stress-inducible variant of HSC70 that is highly expressed in cancer cells. HSP70 and HSC70 share 86% amino acid sequence identity, and they perform redundant functions in protein quality control (37–39).
HSP90 can bind directly to CTA1 in an unusual interaction that does not require co-chaperones (17, 18), but we still predicted that HSC70 and HOP would also be involved with the ER-to-cytosol export of CTA1. HSC70/HSP70 inactivation disrupted CTA1 passage into the cytosol and generated a toxin-resistant phenotype. HOP, in contrast, was not required for CT intoxication. HSC70 could directly bind to either folded or disordered CTA1, whereas HSP90 only interacts with unfolded conformations of CTA1 (18). Like HSP90 (18), HSC70 generated a gain-of-structure in unfolded CTA1. However, HSC70 recognized an internal YYIYVI sequence in CTA1 as opposed to the previously reported interaction between HSP90 and the N-terminal region of CTA1 (40). Our work thus demonstrates atypical client-chaperone interactions, independent of the foldosome complex, drive CTA1 export from the ER to the cytosol.
Results
CT intoxication involves HSC70 and HSP70 but not HOP
RNAi was initially used to examine the functional roles of HSC70, HSP70, and HOP in CT intoxication. Western blot analysis confirmed the specific depletion of each targeted protein in siRNA-transfected HeLa cells (Figs. 1, A and B). After a 2-h exposure to CT, each of these cell populations exhibited a cAMP response comparable with the mock transfected cells and cells transfected with the control siRNA (Fig. 1C). The individual HSC70, HSP70, and HOP proteins were therefore dispensable for CT activity against cultured cells.
Figure 1.

Inactivation of both HSC70 and HSP70, but not HOP, blocks the cytopathic effect of CT. HeLa cells were mock-transfected or transfected with siRNA targeting HOP, HSC70, HSP70, or both HSC70 and HSP70. A and B, cell extracts were probed by Western blot analysis with antibodies against (A) HOP or (B) HSC70 and HSP70, as indicated. In all panels, blotting against actin was used as a loading control. One of 4 representative experiments is shown for each panel. C, cells were exposed to 100 ng/ml of CT for 2 h before intracellular cAMP levels were determined. Results from siRNA-transfected cells or PES-treated cells were expressed as percentages of the values obtained from mock-transfected or mock-treated cells, respectively. Data from at least 4 independent experiments per condition are presented as box-and-whisker plots. The asterisk indicates a statistically significant difference from the control (p < 0.0001, Student's t test).
HSC70 and HSP70 could perform redundant functions in CT intoxication. To examine this possibility, we first attempted a co-transfection with HSC70 and HSP70 siRNA. This strategy apparently generated a stress condition that resulted in enhanced HSP70 expression, to the point of overcoming the RNAi effect (Fig. 1B). Toxin-challenged cells were therefore exposed to 2-phenylethynesulfonamide (PES), an inhibitor of both HSP70 and HSC70 (38, 41), as an alternative approach. Cells treated with PES exhibited substantially lower levels of toxin-induced cAMP compared with the untreated control cells (Fig. 1C). PES did not affect the basal level of cAMP from control cells, but cells treated with PES and the adenylate cyclase agonist forskolin exhibited a 2.0-fold (0.5 range, n = 2) increase in cAMP production when compared with cells incubated with forskolin alone. Our intoxication data did not compensate for this stimulatory effect, which highlights the potent inhibition of CT activity by PES. From these collective observations, HSC70 and HSP70 appear to play redundant roles in the CT intoxication process.
HSC70/HSP70 is required for CTA1 translocation to the cytosol
Like HeLa cells, PES-treated CHO cells exhibited a 3.0-fold (0.25 range, n = 2) increase in forskolin-driven cAMP production, which was not corrected for in the corresponding intoxication assays. Despite this stimulatory effect, PES completely protected CHO cells from the cytopathic activity of CT (Fig. 2A).
Figure 2.
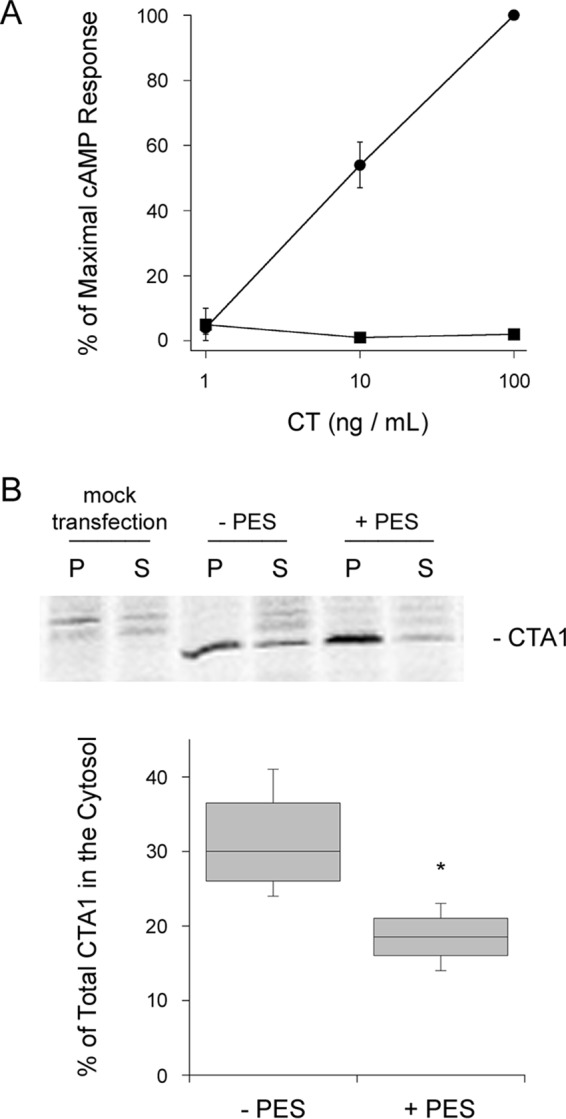
HSC70/HSP70 inactivation blocks CTA1 translocation to the cytosol. A, CHO cells were incubated with varying concentrations of CT for 2 h in the absence (circles) or presence (squares) of 20 μm PES before intracellular cAMP levels were determined. Data are presented as mean ± S.E. from 4 independent experiments with triplicate samples. B, CHO cells were mock transfected or transfected with the ssCTA1/pcDNA3.1 expression plasmid that directs CTA1 for co-translational insertion into the ER. After a 1-h incubation with [35S]methionine, the ER-to-cytosol export of CTA1 was monitored through immunoprecipitation of organelle pellets (P) and cytosolic supernatant (S) fractions generated from untreated (−PES) or PES-treated cells. The percentages of radiolabeled CTA1 detected in the cytosolic fractions are presented as box-and-whisker plots (n = 4). The asterisk indicates a statistically significant difference from the control (p = 0.02, Student's t test).
Loss of HSP90 function has been shown to protect cultured cells from CT by inhibiting the ER-to-cytosol export of CTA1 (17). We predicted the loss of HSC70/HSP70 activity would also reduce the efficiency of CTA1 translocation to the cytosol. To test this prediction, CHO cells were transfected with the ssCTA1/pcDNA3.1 expression plasmid that targets CTA1 for co-translational insertion into the ER via an N-terminal signal sequence. Proteolytic processing removes the signal sequence from CTA1 in the ER lumen, and the toxin is then dislocated back into the cytosol. Rapid removal of the CTA1 signal sequence in the ER results in exclusive detection of the processed, mature form of CTA1 (42, 43). This strategy bypasses the trafficking events required for CT delivery to the ER and thus allows direct detection of drug-induced disruptions to CTA1 translocation.
PES-treated cells did not efficiently export ssCTA1 from the ER to the cytosol (Fig. 2B). CHO cells expressing ssCTA1 were radiolabeled for 1 h before separate organelle and cytosolic fractions were generated by digitonin permeabilization and centrifugation. Triton X-100 was added to both fractions, followed by CTA1 immunoprecipitation. Quantification of the immunoisolated material by PhosphorImager analysis of SDS-PAGE gels found untreated cells exported 31% of radiolabeled CTA1 from the ER to the cytosol, whereas PES-treated cells had only transferred 19% of radiolabeled CTA1 to the cytosol. PES treatment thus reduced CTA1 translocation to the cytosol by 40%.
Cyclophilin A is not required for CT intoxication
The A chains of some ADP-ribosylating toxins move from the endosomes to the cytosol in a process involving HSP90, HSP70, and cyclophilin A (44). CTA1 translocation from the ER involves HSP90 and HSC70/HSP70, and cyclophilins can act as co-chaperones in the foldosome complex (45). We therefore examined the possible role of cyclophilin A in the CT intoxication process. Cells treated with the cyclophilin inhibitor cyclosporin A (CysA) (46) were challenged with either DT or CT. DT is an ADP-ribosylating toxin that inhibits protein synthesis, and its A chain moves from the endosomes to the cytosol in an HSP90-dependent manner (21, 22, 47). Consistent with previous reports (22, 48), we found CysA-treated cells were resistant to the cytotoxic action of DT (Fig. 3A). In contrast, treatment with CysA did not protect cells from CT (Fig. 3B). The endosome-to-cytosol translocation of DT A chain and the ER-to-cytosol translocation of CTA1 thus share some common host factors but are distinct molecular events.
Figure 3.

DT and CT exhibit distinct chaperone-toxin interactions. Untreated CHO cells and CHO cells treated with 10 μm CysA were challenged with the stated concentrations of (A) DT or (B) CT for 4 and 2 h, respectively. The mean ± S.E. from four independent experiments with triplicate samples are shown.
HSC70 binds directly to CTA1
HSP90 exhibits a direct, high-affinity interaction with disordered CTA1 that facilitates the translocation event (17). A direct interaction between HSC70 and CTA1 could likewise contribute to toxin translocation. To determine whether HSC70 binds directly to CTA1, we perfused HSC70 over a CTA1-coated surface plasmon resonance (SPR) sensor at 15 or 37 °C (Fig. 4A). Previous studies have shown by CD, tryptophan fluorescence, and FTIR spectroscopy that CTA1 maintains a folded conformation at low temperature but transitions to a partially unfolded state at the physiological temperature of 37 °C (7, 49). HSC70 could bind to both the folded conformation of CTA1 present at 15 °C and the disordered conformation of CTA1 present at 37 °C. Binding did not require HSP40, which is often necessary to deliver client proteins to HSC70 (30–32). Likewise, the binding of HSP90 to CTA1 does not depend upon previous association with HSP40, HSC70, or HOP (17). Both HSC70 and HSP90 form tight complexes with CTA1, as little dissociation is seen after removal of the chaperone from the perfusion buffer (Fig. 4A) (17, 18). We previously calculated a 7 nm KD affinity between HSP90 and CTA1 at 37 °C (17). HSC70 exhibited a similar 5 nm KD affinity for CTA1 at 37 °C (Fig. 4B). However, host-toxin interactions are not identical for the two chaperones: HSC70 will bind to either folded or disordered CTA1, whereas HSP90 only recognizes disordered CTA1 (18).
Figure 4.

HSC70 directly binds to both folded and disordered CTA1. A, HSC70 was perfused over a CTA1-coated SPR sensor at 15 or 37 °C. HSC70 was removed from the perfusion buffer 300 s after injection. B, five concentrations of HSC70 (1600, 800, 400, 200, and 100 ng/ml) were perfused over a CTA1-coated SPR sensor at 37 °C. HSC70 was removed from the perfusion buffer after 300 s. Best fit curves (orange lines) derived from five independent experiments with each protein concentration were calculated using a 1:1 Langmuir fit. A KD of 4.5 ± 3.6 nm was calculated from all five data sets, which produced an average ka of 6.2 × 105 ± 2.2 × 105 m−1 s−1 and a kd of 1.7 × 10−3 ± 0.7 × 10−3 s−1. Errors represent S.D.
HSC70 and HSP90 recognize distinct regions of CTA1
Additional SPR experiments were performed with various combinations of CTA1, HSC70, and HSP90. In Fig. 5A, HSP90 was perfused over a CTA1-coated SPR sensor at 37 °C. HSP90 did not appreciably dissociate from CTA1 after its removal from the perfusion buffer, which was consistent with our published reports (17, 18). Addition of HSC70 to the sensor-bound CTA1/HSP90 complex produced a further increase in the refractive index unit (RIU), which did not appreciably drop after removal of HSC70 from the perfusion buffer. This demonstrated HSC70 and HSP90 can both bind to disordered CTA1 at the same time. In a related experiment, CTA1 was perfused over an HSC70-coated SPR sensor at 37 °C (Fig. 5B). CTA1 did not appreciably dissociate from HSC70 after its removal from the perfusion buffer. Addition of HSP90 to the sensor-bound HSC70/CTA1 complex produced a further increase in the RIU, which did not decrease after removal of HSP90 from the perfusion buffer. HSP90 could thus bind to an existing complex of CTA1 and HSC70, just as HSC70 could bind to an existing complex of CTA1 and HSP90. Any CTA1 captured on the HSC70-coated slide would, by necessity, have its HSC70-binding site occupied before the addition of HSP90 to the perfusion buffer. Consistent with previous reports (50), HSP90 only exhibited minimal direct binding to HSC70 (Fig. 5C). HSC70 and HSP90 therefore recognize distinct regions of CTA1 and do not compete for a single binding site.
Figure 5.

HSC70 and HSP90 can bind simultaneously to CTA1. A, HSP90 with 1 mm ATP was perfused over a CTA1-coated SPR sensor at 37 °C. After removing HSP90 from the perfusion buffer (first empty triangle), HSC70 was perfused over the CTA1/HSP90 complex (solid triangle). Removal of HSC70 from the perfusion buffer is indicated by the second empty triangle. Antibody controls confirmed both HSP90 and HSC70 remained associated with CTA1 at the end of the experiment (data not shown). B, CTA1 was perfused over an HSC70-coated SPR sensor at 37 °C. After removing CTA1 from the perfusion buffer (first empty triangle), HSP90 with 1 mm ATP was perfused over the HSC70/CTA1 complex (solid triangle). Removal of HSP90 from the perfusion buffer is denoted by the second empty triangle. Antibody controls confirmed both CTA1 and HSP90 remained associated with the HSC70 sensor at the end of the experiment (data not shown). C, HSP90 with 1 mm ATP was perfused over an HSC70-coated SPR sensor at 37 °C in the absence of CTA1.
HSC70 recognizes an internal YYIYVI sequence in CTA1
To identify the CTA1-binding site(s) for HSC70, we generated a series of peptides that contained overlapping amino acid sequences from the entire length of the CTA1 subunit. As summarized in Table 1, HSC70 bound two consecutive peptides spanning CTA1 amino acid residues 73–98. These peptides contained a shared YYIYVI sequence that likely represented the core HSC70-binding site. The YYIYVI sequence alone could not be generated as a soluble hexapeptide, but we found a truncated GQTILSGHST peptide that lacked the YYIYVI motif and a mutant GQTILSGHSTYYLYVL peptide with leucine for isoleucine substitutions in the YYIYVI motif could not bind to HSC70 (Table 1). These results indicated the YYIYVI motif was recognized by HSC70.
Table 1.
HSC70 recognizes a YYIYVI sequence in CTA1
Peptides containing overlapping sequences of amino acids from CTA1 were appended to SPR sensor slides. HSC70 was then perfused over each slide; binding to the peptide-coated sensor is indicated by a + sign. A mutant (m) CTA1 sequence is denoted in bold.
| CTA1 residues | Peptide sequence | HSC70 interaction |
|---|---|---|
| 1–16 | NDDKLYRADSRPPDEI | − |
| 11–26 | RPPDEIKQSGGLMPRG | − |
| 21–36 | GLMPRGQSEYFDRGTQ | − |
| 31–46 | FDRGTQMNINLYDHAR | − |
| 41–56 | LYDHARGTQTGFVRHD | − |
| 51–66 | GFVRHDDGYVSTSISL | − |
| 63–78 | SISLRSAHLVGQTILS | − |
| 73–88 | GQTILSGHSTYYIYVI | + |
| 83–98 | YYIYVIATAPNMFNVN | + |
| 93–108 | NMFNVNDVLGAYSPHP | − |
| 103–118 | AYSPHPDEQEVSALGG | − |
| 113–128 | VSALGGIPYSQIYGWY | − |
| 123–138 | QIYGWYRVHFGVLDEQ | − |
| 133–148 | GVLDEQLHRNRGYRDR | − |
| 143–158 | RGYRDRYYSNLDIAPA | − |
| 153–168 | LDIAPAADGYGLAGFP | − |
| 163–177 | GLAGFPPEHRAWREE | − |
| 173–188 | AWREEPWIHHAPPGCG | − |
| 179–192 | WIHHAPPGCGNAPR | − |
| 73–82 | GQTILSGHST | − |
| 73–88m | GQTILSGHSTYYLYVL | − |
A competition assay further established the specificity of HSC70 binding to the YYIYVI sequence (Fig. 6). For this experiment, CTA1 was appended to an SPR sensor slide and exposed to perfusion buffer containing either HSC70 alone or HSC70 in combination with a CTA1-related peptide. HSC70 could bind to CTA1 in the absence of peptide and in the presence of the mutant GQTILSGHSTYYLYVL peptide. However, HSC70 could not bind to CTA1 in the presence of the GQTILSGHSTYYIYVI peptide. These observations confirmed HSC70 specifically recognizes the YYIYVI sequence in CTA1.
Figure 6.
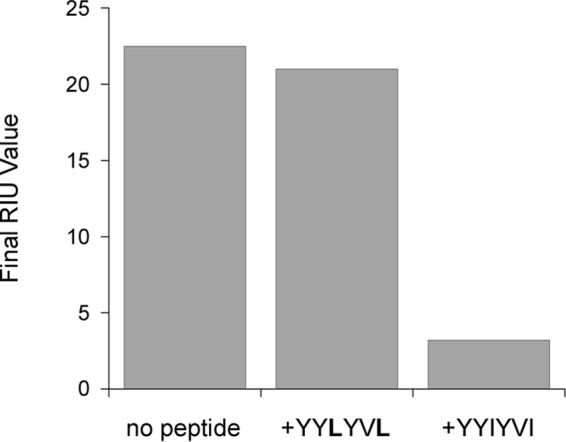
The YYIYVI sequence competitively inhibits HSC70 binding to CTA1. HSC70 was perfused over a CTA1-coated SPR sensor in the absence of peptide, in the presence of mutant GQTILSGHSTYYLYVL peptide, or in the presence of the WT GQTILSGHSTYYIYVI peptide. Ligands were removed from the perfusion buffer after 200 s. Following an additional 100 s of dissociation, final RIU values were recorded.
HSC70 refolds disordered CTA1
HSP90 couples CTA1 refolding with CTA1 extraction from the ER (18). To determine whether HSC70 could also refold CTA1, HSC70 was combined with uniformly 13C-labeled CTA1 for structural measurements involving isotope-edited FTIR spectroscopy. 13C labeling does not affect protein structure but causes a 45–50 cm−1 spectral downshift, which allows its amide I spectrum to be resolved from the spectrum of an unlabeled protein in the same sample (49, 51). Measurements of 13C-labeled CTA1 alone at 10 and 37 °C were taken as reference points for the folded and disordered conformations of CTA1, respectively. The folded conformation of CTA1 present at 10 °C contained 46% α-helical and 38% β-sheet content (Fig. 7A, Table 2). Due to its thermal instability (7), CTA1 lost substantial secondary structure when it was warmed to 37 °C: compared with the 10 °C measurement, at 37 °C there was a 15% drop in α-helix content with a corresponding increase in irregular (i.e. disordered) structure from 15 to 32% (Fig. 7B, Table 2). Addition of HSC70 to the disordered, 37 °C conformation of CTA1 produced a gain-of-structure resulting in a toxin conformation with 38% α-helical, 37% β-sheet, and 17% irregular structure (Fig. 7C, Table 2). These observations demonstrated an interaction between HSC70 and disordered CTA1 leads to the refolding of CTA1.
Figure 7.

HSC70 induces a gain-of-structure in disordered CTA1. The FTIR spectrum of uniformly 13C-labeled CTA1 was recorded in the absence or presence of HSC70. In the curve fitting panels of the left column, the dashed line represents the sum of all deconvoluted components from the measured spectrum (overlapping gray line). Deconvoluted components are presented in dark blue (α-helix), light blue (β-sheet), red (irregular), gray (other/turns), and black (side chains and, in C, the unlabeled protein). The right column presents the respective second derivatives. A, 13C-labeled CTA1 structure at 10 °C. B, 13C-labeled CTA1 structure after 30 min at 37 °C. C, 13C-labeled CTA1 structure in the presence of unlabeled HSC70 at 37 °C. CTA1 was heated to 37 °C for 30 min before the addition of HSC70.
Table 2.
Refolding of CTA1 by HSC70
| Condition | % of CTA1 structure |
|||
|---|---|---|---|---|
| α-Helix | β-Sheet | Irregular | Other | |
| 10 °C | 46 ± 1 | 38 ± 3 | 15 ± 2 | 3 ± 0 |
| 37 °C | 31 ± 1 | 36 ± 1 | 32 ± 1 | 1 ± 1 |
| 37 °C + HSC70 | 38 ± 0 | 37 ± 2 | 17 ± 1 | 9 ± 1 |
Deconvolution of amide I bands from the FTIR spectroscopy data of Fig. 7 was used to calculate the percentages of CTA1 structure under the stated conditions. The averages ± ranges from two curve fittings are shown.
Discussion
Unfolded CTA1 passes through an ER translocon pore to reach the cytosol and its G protein target. Translocation does not require p97 (15, 16) but instead involves an HSP90-dependent ratchet mechanism that couples CTA1 refolding with CTA1 extraction from the ER: co-translational refolding ensures unidirectional toxin export, as the refolded toxin could not slide back into the translocation pore and could therefore only move out of the ER (18). In this work, we provide evidence that HSC70/HSP70 also plays a role in CTA1 extraction to the cytosol.
HSC70 and HSP90 perform complementary but distinct functions in CTA1 translocation. Both HSC70 and HSP90 are required for toxin export to the cytosol and productive intoxication. Both chaperones also independently refold CTA1. However, there are also differences in these toxin-chaperone interactions. HSC70 recognizes both folded and disordered CTA1, whereas HSP90 only interacts with disordered CTA1 (18). HSC70 and HSP90 thus appear to recognize distinct motifs in CTA1 and, as such, do not compete for a single binding site in the toxin. We indeed found that HSC70 recognizes a YYIYVI motif spanning residues 83–88 of the 192-amino acid CTA1 polypeptide, whereas HSP90 binds to the N-terminal region of CTA1 (40).
Several chaperones in the ER lumen interact with CTA1 either before or after its release from the CT holotoxin (49, 52–55). Chaperone binding to the free, disordered CTA1 subunit prevents toxin aggregation. It also directs the toxin to the Hrd1 translocon pore for export to the cytosol. HSP90 and HSC70 then recognize CTA1 at the cytosolic face of the ER membrane and continue the translocation event. A continuum of chaperone interactions thus guides CTA1 from the ER lumen to the cytosol.
Our collective data support a model of CTA1 translocation that involves (i) extraction of CTA1 from the ER in an N-terminal to C-terminal direction; (ii) initial contact of HSP90 with the N terminus of CTA1 as the toxin emerges from the translocon pore; and (iii) subsequent interaction between HSC70 and an internal region of CTA1. The ability of HSC70 to bind both folded and disordered conformations of CTA1 may facilitate its interaction with CTA1 after HSP90 has initiated the refolding process that is coupled with toxin extraction from the ER. Although HSC70 alone can refold CTA1 in vitro, the internal location of its binding site in CTA1 suggests HSP90 must initiate the coupled refolding/translocation process at the N terminus of CTA1 before HSC70 can contribute to CTA1 refolding in vivo. The combined action of HSP90 and HSC70 thereby delivers CTA1 to the cytosol in a folded conformation. Demonstration of a functional role for HSC70 in CTA1 translocation and identification of the HSC70-binding site in CTA1 has thus provided additional molecular detail regarding the process of toxin translocation from the ER to the cytosol.
The 5 nm affinity between HSC70 and CTA1 is unusual but not unprecedented for protein-protein interactions involving HSP70/HSC70 (56, 57). This tight binding suggests the two proteins remain in a complex after toxin delivery to the cytosol. The free CTA1 subunit is in a disordered state at physiological temperature (7), so continued association with HSC70 could allow CTA1 to maintain a folded, functional conformation for the ADP-ribosylation of Gsα. HSP90 appears to play a similar role in facilitating CTA1 activity at 37 °C (18). Future work using recombinant CTA1 with a mutant YYLYVL motif that cannot bind HSC70 will examine the possible additional role of HSC70 in promoting the cytosolic activity of CTA1. Bacterial pathogenesis often involves the manipulation and modification of host factors by toxins and other virulence factors. The interplay between CTA1 and HSC70/HSP90 represents another example of this phenomenon.
HSP90 is thought to recognize a feature of ADP-ribosylating toxins that allows it to facilitate the translocation of CTA1 and the A chains from several AB toxins that enter the cytosol from acidified endosomes (40, 44). HSC70 may also recognize a general feature of ADP-ribosylating toxins, as this chaperone works with HSP90 to deliver CTA1 and several endosome-translocating toxins to the cytosol (22, 58, 59). Peptidyl-prolyl cis/trans isomerases such as cyclophilin A also function in the endosome-to-cytosol export of ADP-ribosylating toxins (44), yet we found cyclophilin A was not required for the cytopathic activity of CT. A general HSP90/HSC70-driven translocation mechanism in which toxin folding is coupled with toxin extraction to the cytosol may therefore be active at both the ER and endosome membrane, but the contributing co-chaperones apparently differ for the two translocation sites and possibly for different toxins. Moreover, the HSP90-driven mechanism appears to be specific for ADP-ribosylating toxins: HSP90 and HSC70 are not required for the ER-to-cytosol export of ricin A chain (an RNA N-glycosidase) (60); HSP90 is not required for the endosome-to-cytosol export of anthrax toxin lethal factor (an A chain metalloprotease) (48); and HSC70 is not required for the endosome-to-cytosol export of botulinum toxin light chain (another A chain metalloprotease) (61).
HSP90 and HSC70 are core components of the foldosome, a multimeric cytosolic machine active in protein renaturation. Typical foldosome activity begins with substrate binding to HSP40, transfer to HSC70, and further complex formation with HOP and HSP90. Our studies have recorded unique toxin-chaperone interactions in which HSP90 and HSC70 can each bind independently to CTA1 in the absence of HSP40 or HOP. The work presented here documents a role for HSC70/HSP70 in the ER-to-cytosol export of a soluble ERAD substrate and establishes a noncanonical pattern of chaperone-mediated protein folding that drives toxin extraction from the ER.
Experimental procedures
Materials
CysA and PES were purchased from Sigma. DT and CT were purchased from List Biological Laboratories (Campbell, CA). His-tagged CTA1 and 13C-labeled CTA1 were purified in the lab as previously described (49, 62). HSC70 was purchased from Enzo Life Sciences (Farmingdale, NY) or Abcam (Cambridge, United Kingdom), and HSP90 was purchased from Biovision (Milpitas, CA). Antibodies against HSP90 were purchased from Calbiochem (Darmstadt, Germany). Tissue culture reagents, Lipofectamine, and Lipofectamine 2000 were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). Tablets of PBS with 0.05% Tween 20 (PBST) were purchased from Medicago (Research Triangle Park, NC). [35S]Methionine was purchased from PerkinElmer Life Sciences (Waltham, MA). HSP70 siRNA (sc-29352), STI1/HOP siRNA (9sc-106905), HSC70 siRNA (sc-29349), and Control siRNA-A (sc-37007) were purchased from Santa Cruz Biotechnology (Dallas, TX). Peptides containing amino acid sequences from CTA1 were purchased from Peptide 2.0 (Chantilly, VA).
siRNA transfections
HeLa cells grown to 60–80% confluence in 24-well-plates were transfected with the indicated siRNAs (1 μm final concentrations) using Lipofectamine 2000 and Opti-MEM according to the manufacturer's instructions. After an overnight incubation, the medium was replaced with Dulbecco's modified Eagle's medium containing 10% FBS. Western blot analysis and intoxication assays were performed 48 h post-transfection.
Western blot analysis
For siRNA experiments, cell extracts were resolved by SDS-PAGE with 15% polyacrylamide gels and probed using the following antibodies: 1:5,000 rabbit anti-HOP (STI1) (Abcam); 1:5,000 rat anti-HSC70 (Abcam); 1:5,000 mouse anti-HSP70 (StressMarq Biosciences, Victoria, BC, Canada); 1:10,000 rabbit anti-βactin (Abcam); and 1:10,000 horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA), 1:10,000 HRP-conjugated goat anti-mouse IgG (Jackson ImmunoResearch), or 1:10,000 HRP-conjugated rabbit anti-rat IgG (Abcam). Incubations with primary antibodies were overnight at 4 °C, whereas incubations with secondary antibodies were 30 min at room temperature.
Intoxication assays
HeLa or CHO cells grown to ∼80% confluence in 24-well-plates were challenged with varying concentrations of DT or CT in serum-free medium. Where indicated, cells were also exposed to 20 μm PES for 1–2 h or 10 μm CysA for 30 min before toxin addition. Drugs were also present during the toxin incubations.
After a 2-h incubation with CT, the cells were washed and lysed at 4 °C with acidic ethanol (1 m HCl in ethanol at a 1:100 dilution). Cell extracts were dried, resuspended in assay buffer, and processed for cAMP content using a commercial competition assay according to the manufacturer's instructions (GE Healthcare). The basal levels of cAMP from unintoxicated cells were background subtracted from all experimental values, which were then expressed as percentages of the cAMP value obtained from cells exposed to 100 ng/ml of CT in the absence of drug or siRNA treatment (i.e. the maximal control cAMP response). Triplicate wells were used for each condition.
After a 4-h incubation with DT, cells were bathed in methionine-free medium supplemented with 10 μCi/ml of [35S]methionine for 30 min. Radiolabeled cells were washed with PBS and placed in PBS containing 10% trichloroacetic acid (TCA) for 30 min at 4 °C. After a second 10-min 4 °C wash with PBS containing 10% TCA, the cells were solubilized in 0.2 n NaOH at room temperature. A scintillation counter was then used to determine the NaOH-solubilized, TCA-precipitated cpm from proteins containing [35S]methionine. Results from drug- and/or toxin-treated cells were expressed as percentages of the value obtained from matched, unintoxicated control cells. Triplicate wells were used for each condition.
CTA1 transfection/translocation assay
CHO cells grown to 80% confluence in 6-well-plates were transfected with the pcDNA3.1/ssCTA1 plasmid encoding a CTA1 construct with the N-terminal CTA signal sequence (ssCTA1) using Lipofectamine as the transfection reagent (17). At 24 h post-transfection, the cells were placed in methionine-free medium for 1 h, incubated in the same medium supplemented with 150 μCi/ml of [35S]methionine for 1 h, and then lifted from the plate with PBS/EDTA. The collected cells were exposed to 0.04% digitonin in HCN buffer (50 mm Hepes, pH 7.5, 150 mm NaCl, 2 mm CaCl2, 10 mm N-ethylmaleimide, and protease inhibitor mixture) for 10 min at 4 °C before a 10-min centrifugal spin at 16,000 × g generated separate cytosol (supernatant) and organelle (pellet) fractions. The cytosol-containing supernatant was placed in a 1-ml final volume of PBS containing 1% Triton X-100. The pellet was reconstituted in 1 ml of PBS with 1% Triton X-100. An anti-CT antibody (Sigma) coupled to protein A-conjugated Sepharose beads was then added to both fractions for an overnight incubation at 4 °C. Immunoisolated samples were resolved by SDS-PAGE using 15% polyacrylamide gels and visualized with PhosphorImager analysis. To determine the extent of CTA1 export from the ER to the cytosol, background-subtracted band intensities were quantified for the ER pellet and cytosolic supernatant fractions. Export efficiency was calculated by dividing the intensity of the supernatant band by the sum intensities of the pellet and supernatant bands. When indicated, 20 μm PES was present during the methionine starvation and radiolabeling steps.
SPR
As previously described (63), experiments were performed with a Reichert (Depew, NY) SR7000 SPR refractometer using a flow rate of 41 μl/min for all steps. His-tagged CTA1 was appended to a nickel-nitrilotriacetic acid sensor slide (52), and amide coupling was used to append HSC70 or CTA1-derived peptides to a Reichert SPR sensor slide with a mixed self-assembled monolayer (63). Our SPR experiments used a dual-chamber SPR instrument. After activation of both channels with NHS/EDC, ligand was appended to only one channel. Both channels were then blocked with ethanolamine and subjected to a 10-min perfusion with PBST buffer to establish the 0 RIU baseline that corresponded the mass of the sensor-bound protein/peptide. Both channels were subsequently exposed to the analyte(s) of interest (CTA1, HSP90 with 1 mm ATP, and/or HSC70). The second channel without immobilized ligand was used as a reference to account for nonspecific binding to the sensor. Analyte concentration was 1600 ng/ml in PBST unless otherwise noted. A 5-min perfusion of ligand was followed by a 5-min PBST wash. Reichert Labview software was used for data collection. The BioLogic (Campbell, Australia) Scrubber 2 software and WaveMetrics (Lake Oswego, OR) Igor Pro software were used to analyze the data and generate figures. Because the SPR measurements to measure KD did not reach steady state, affinity calculations were derived from kinetic rather than equilibrium analysis. The Scrubber 2 software generated ka and kd values for each of our replicate experiments, which were then used to calculate KD.
For competition assays, heat-denatured CTA1 was appended to a Biacore (GE Healthcare) Dock CM5 SPR sensor chip by amide coupling. HSC70 was mixed in PBST with a 10-fold molar excess of the indicated peptide for 10 min at room temperature before injection into the Biacore X100 instrument at a flow rate of 10 μl/min. Data were collected with Biacore X100 Control Software version 2.0.1 and analyzed with Scrubber 2 and Igor Pro software.
Isotope-edited FTIR spectroscopy
Samples for FTIR spectroscopy (25 μg of 13C-labeled CTA1-His6 and 83 μg of HSC70) were prepared in D2O-based 20 mm sodium borate buffer (pD 7.0) as previously described (49). All measurements were performed on a Jasco 4200 FTIR spectrometer (Easton, MD) with Peltier temperature controller (Pike Technologies, Madison, WI). When indicated, measurements were taken 10 min after chaperone addition. All traces were background-subtracted using a buffer alone control. Analysis of secondary structure was performed as described in detail in Ref. 49, using the following wavenumber assignments for 13C-labeled CTA1 secondary structures: 1568–1596 cm−1 for the main β-sheet component, 1596–1604 for irregular structure, 1605–1628 for α-helix, 1629–1635 for the higher-frequency and low extinction coefficient component of antiparallel β-sheet, and 1635–1648 cm−1 for various turns tabulated as “other” structures.
Author contributions
H. B. and K. T. conceptualization; H. B., A. K., J. G., S. A. T., and K. T. formal analysis; H. B., A. K., and J. G. investigation; H. B., A. K., and J. G. methodology; H. B. and K. T. writing-original draft; H. B., A. K., J. G., S. A. T., and K. T. writing-review and editing; S. A. T. and K. T. supervision; K. T. funding acquisition.
This work was supported by National Institutes of Health NIAID Grant R01AI099493 (to K. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CT
- cholera toxin
- CysA
- cyclosporin A
- DT
- diphtheria toxin
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- FBS
- fetal bovine serum
- HRP
- horseradish peroxidase
- HSC70
- heat shock cognate 71-kDa protein
- HSP40
- heat shock protein 40
- HSP70
- heat shock protein 70
- HSP90
- heat shock protein 90
- HOP
- HSC70/HSP90-organizing protein
- PES
- 2-phenylethynesulfonamide
- PBST
- phosphate-buffered saline with 0.05% Tween 20
- RIU
- refractive index unit
- SPR
- surface plasmon resonance.
References
- 1. Heggelund J. E., Bjornestad V. A., and Krengel U. (2015) Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond. in The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th edition (Alouf J. E., Ladant D., and Popoff M. R., eds), pp. 195–229, Elsevier, Waltham, MA [Google Scholar]
- 2. Guimaraes C. P., Carette J. E., Varadarajan M., Antos J., Popp M. W., Spooner E., Brummelkamp T. R., and Ploegh H. L. (2011) Identification of host cell factors required for intoxication through use of modified cholera toxin. J. Cell Biol. 195, 751–764 10.1083/jcb.201108103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lencer W. I., de Almeida J. B., Moe S., Stow J. L., Ausiello D. A., and Madara J. L. (1993) Entry of cholera toxin into polarized human intestinal epithelial cells: identification of an early brefeldin A-sensitive event required for A1-peptide generation. J. Clin. Invest. 92, 2941–2951 10.1172/JCI116917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Orlandi P. A., Curran P. K., and Fishman P. H. (1993) Brefeldin A blocks the response of cultured cells to cholera toxin. Implications for intracellular trafficking in toxin action. J. Biol. Chem. 268, 12010–12016 [PubMed] [Google Scholar]
- 5. Taylor M., Burress H., Banerjee T., Ray S., Curtis D., Tatulian S. A., and Teter K. (2014) Substrate-induced unfolding of protein-disulfide isomerase displaces the cholera toxin A1 subunit from its holotoxin. PLoS Pathog. 10, e1003925 10.1371/journal.ppat.1003925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsai B., Rodighiero C., Lencer W. I., and Rapoport T. A. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948 10.1016/S0092-8674(01)00289-6 [DOI] [PubMed] [Google Scholar]
- 7. Pande A. H., Scaglione P., Taylor M., Nemec K. N., Tuthill S., Moe D., Holmes R. K., Tatulian S. A., and Teter K. (2007) Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 374, 1114–1128 10.1016/j.jmb.2007.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Teter K., and Holmes R. K. (2002) Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect. Immun. 70, 6172–6179 10.1128/IAI.70.11.6172-6179.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teter K. (2013) Cholera toxin interactions with host cell stress proteins. in Moonlighting Cell Stress Proteins in Microbial Infections (Henderson B., ed), pp. 323–338, Springer, New York [Google Scholar]
- 10. Hazes B., and Read R. J. (1997) Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36, 11051–11054 10.1021/bi971383p [DOI] [PubMed] [Google Scholar]
- 11. Rodighiero C., Tsai B., Rapoport T. A., and Lencer W. I. (2002) Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 3, 1222–1227 10.1093/embo-reports/kvf239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muanprasat C., and Chatsudthipong V. (2013) Cholera: pathophysiology and emerging therapeutic targets. Future Med. Chem. 5, 781–798 10.4155/fmc.13.42 [DOI] [PubMed] [Google Scholar]
- 13. Teter K. (2013) Toxin instability and its role in toxin translocation from the endoplasmic reticulum to the cytosol. Biomolecules 3, 997–1029 10.3390/biom3040997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolf D. H., and Stolz A. (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys Acta 1823, 117–124 10.1016/j.bbamcr.2011.09.002 [DOI] [PubMed] [Google Scholar]
- 15. Kothe M., Ye Y., Wagner J. S., De Luca H. E., Kern E., Rapoport T. A., and Lencer W. I. (2005) Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J. Biol. Chem. 280, 28127–28132 10.1074/jbc.M503138200 [DOI] [PubMed] [Google Scholar]
- 16. McConnell E., Lass A., and Wójcik C. (2007) Ufd1-Npl4 is a negative regulator of cholera toxin retrotranslocation. Biochem. Biophys. Res. Commun. 355, 1087–1090 10.1016/j.bbrc.2007.02.077 [DOI] [PubMed] [Google Scholar]
- 17. Taylor M., Navarro-Garcia F., Huerta J., Burress H., Massey S., Ireton K., and Teter K. (2010) Hsp90 is required for transfer of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 285, 31261–31267 10.1074/jbc.M110.148981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burress H., Taylor M., Banerjee T., Tatulian S. A., and Teter K. (2014) Co- and post-translocation roles for Hsp90 in cholera intoxication. J. Biol. Chem. 289, 33644–33654 10.1074/jbc.M114.609800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carlson E. J., Pitonzo D., and Skach W. R. (2006) p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 25, 4557–4566 10.1038/sj.emboj.7601307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wójcik C., Rowicka M., Kudlicki A., Nowis D., McConnell E., Kujawa M., and DeMartino G. N. (2006) Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 17, 4606–4618 10.1091/mbc.e06-05-0432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ratts R., Zeng H., Berg E. A., Blue C., McComb M. E., Costello C. E., vanderSpek J. C., and Murphy J. R. (2003) The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J. Cell Biol. 160, 1139–1150 10.1083/jcb.200210028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schuster M., Schnell L., Feigl P., Birkhofer C., Mohr K., Roeder M., Carle S., Langer S., Tippel F., Buchner J., Fischer G., Hausch F., Frick M., Schwan C., Aktories K., Schiene-Fischer C., and Barth H. (2017) The Hsp90 machinery facilitates the transport of diphtheria toxin into human cells. Sci. Rep. 7, 613 10.1038/s41598-017-00780-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haug G., Aktories K., and Barth H. (2004) The host cell chaperone Hsp90 is necessary for cytotoxic action of the binary ι-like toxins. Infect. Immun. 72, 3066–3068 10.1128/IAI.72.5.3066-3068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haug G., Leemhuis J., Tiemann D., Meyer D. K., Aktories K., and Barth H. (2003) The host cell chaperone Hsp90 is essential for translocation of the binary Clostridium botulinum C2 toxin into the cytosol. J. Biol. Chem. 278, 32266–32274 10.1074/jbc.M303980200 [DOI] [PubMed] [Google Scholar]
- 25. Kaiser E., Kroll C., Ernst K., Schwan C., Popoff M., Fischer G., Buchner J., Aktories K., and Barth H. (2011) Membrane translocation of binary actin-ADP-ribosylating toxins from Clostridium difficile and Clostridium perfringens is facilitated by cyclophilin A and Hsp90. Infect. Immun. 79, 3913–3921 10.1128/IAI.05372-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lang A. E., Ernst K., Lee H., Papatheodorou P., Schwan C., Barth H., and Aktories K. (2014) The chaperone Hsp90 and PPIases of the cyclophilin and FKBP families facilitate membrane translocation of Photorhabdus luminescens ADP-ribosyltransferases. Cell Microbiol. 16, 490–503 10.1111/cmi.12228 [DOI] [PubMed] [Google Scholar]
- 27. Li J., and Buchner J. (2013) Structure, function, and regulation of the Hsp90 machinery. Biomed. J. 36, 106–117 10.4103/2319-4170.113230 [DOI] [PubMed] [Google Scholar]
- 28. Cano L. Q., Lavery D. N., and Bevan C. L. (2013) Foldosome regulation of androgen receptor action in prostate cancer. Mol. Cell Endocrinol. 369, 52–62 10.1016/j.mce.2013.01.023 [DOI] [PubMed] [Google Scholar]
- 29. Zuehlke A., and Johnson J. L. (2010) Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 93, 211–217 10.1002/bip.21292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Minami Y., and Minami M. (1999) Hsc70/Hsp40 chaperone system mediates the Hsp90-dependent refolding of firefly luciferase. Genes Cells 4, 721–729 10.1046/j.1365-2443.1999.00299.x [DOI] [PubMed] [Google Scholar]
- 31. Fan C.-Y., Lee S., and Cyr D. M. (2003) Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones 8, 309–316 10.1379/1466-1268(2003)008<0309:MFROHF>2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hernández M. P., Sullivan W. P., and Toft D. O. (2002) The assembly and intermolecular properties of the hsp70-Hop-hsp90 molecular chaperone complex. J. Biol. Chem. 277, 38294–38304 10.1074/jbc.M206566200 [DOI] [PubMed] [Google Scholar]
- 33. Young J. C., Moarefi I., and Hartl F. U. (2001) Hsp90: a specialized but essential protein-folding tool. J. Cell Biol. 154, 267–273 10.1083/jcb.200104079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hernández M. P., Chadli A., and Toft D. O. (2002) HSP40 binding is the first step in the HSP90 chaperoning pathway for the progesterone receptor. J. Biol. Chem. 277, 11873–11881 10.1074/jbc.M111445200 [DOI] [PubMed] [Google Scholar]
- 35. Chen S., and Smith D. F. (1998) Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J. Biol. Chem. 273, 35194–35200 10.1074/jbc.273.52.35194 [DOI] [PubMed] [Google Scholar]
- 36. Johnson B. D., Schumacher R. J., Ross E. D., and Toft D. O. (1998) Hop modulates Hsp70/Hsp90 interactions in protein folding. J. Biol. Chem. 273, 3679–3686 10.1074/jbc.273.6.3679 [DOI] [PubMed] [Google Scholar]
- 37. Daugaard M., Rohde M., and Jaattela M. (2007) The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 581, 3702–3710 10.1016/j.febslet.2007.05.039 [DOI] [PubMed] [Google Scholar]
- 38. Leu J. I., Pimkina J., Pandey P., Murphy M. E., and George D. L. (2011) HSP70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol. Cancer Res. 9, 936–947 10.1158/1541-7786.MCR-11-0019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu T., Daniels C. K., and Cao S. (2012) Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 136, 354–374 10.1016/j.pharmthera.2012.08.014 [DOI] [PubMed] [Google Scholar]
- 40. Taylor M., Britt C. B. T., Fundora J., and Teter K. (2015) Modulation of cholera toxin structure/function by Hsp90 in Proceedings from the Physical Biology of Proteins and Peptides Conference (Olivares-Quiroz L., Guzman-Lopez O., and Jardón-Valadez E., eds), pp. 67–79, Springer, New York [Google Scholar]
- 41. Leu J. I., Pimkina J., Frank A., Murphy M. E., and George D. L. (2009) A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 36, 15–27 10.1016/j.molcel.2009.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Teter K., Allyn R. L., Jobling M. G., and Holmes R. K. (2002) Transfer of the cholera toxin A1 polypeptide from the endoplasmic reticulum to the cytosol is a rapid process facilitated by the endoplasmic reticulum-associated degradation pathway. Infect. Immun. 70, 6166–6171 10.1128/IAI.70.11.6166-6171.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Teter K., Jobling M. G., Sentz D., and Holmes R. K. (2006) The cholera toxin A13 subdomain is essential for interaction with ADP-ribosylation factor 6 and full toxic activity but is not required for translocation from the endoplasmic reticulum to the cytosol. Infect. Immun. 74, 2259–2267 10.1128/IAI.74.4.2259-2267.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ernst K., Schnell L., and Barth H. (2017) Host cell chaperones Hsp70/Hsp90 and peptidyl-prolyl cis/trans isomerases are required for the membrane translocation of bacterial ADP-ribosylating toxins. Curr. Top. Microbiol. Immunol. 406, 163–198 [DOI] [PubMed] [Google Scholar]
- 45. Riggs D. L., Cox M. B., Cheung-Flynn J., Prapapanich V., Carrigan P. E., and Smith D. F. (2004) Functional specificity of co-chaperone interactions with Hsp90 client proteins. Crit. Rev. Biochem. Mol. Biol. 39, 279–295 10.1080/10409230490892513 [DOI] [PubMed] [Google Scholar]
- 46. Handschumacher R. E., Harding M. W., Rice J., Drugge R. J., and Speicher D. W. (1984) Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226, 544–547 10.1126/science.6238408 [DOI] [PubMed] [Google Scholar]
- 47. Murphy J. R. (2011) Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins (Basel) 3, 294–308 10.3390/toxins3030294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dmochewitz L., Lillich M., Kaiser E., Jennings L. D., Lang A. E., Buchner J., Fischer G., Aktories K., Collier R. J., and Barth H. (2011) Role of CypA and Hsp90 in membrane translocation mediated by anthrax protective antigen. Cell Microbiol. 13, 359–373 10.1111/j.1462-5822.2010.01539.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taylor M., Banerjee T., Ray S., Tatulian S. A., and Teter K. (2011) Protein-disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J. Biol. Chem. 286, 22090–22100 10.1074/jbc.M111.237966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Czar M. J., Owens-Grillo J. K., Dittmar K. D., Hutchison K. A., Zacharek A. M., Leach K. L., Deibel M. R. Jr., and Pratt W. B. (1994) Characterization of the protein-protein interactions determining the heat shock protein (hsp90·hsp70·hsp56) heterocomplex. J. Biol. Chem. 269, 11155–11161 [PubMed] [Google Scholar]
- 51. Tatulian S. A. (2013) Structural characterization of membrane proteins and peptides by FTIR and ATR-FTIR spectroscopy. in Lipid-Protein Interactions: Methods and Protocols (Kleinschmidt J. H., ed), pp. 177–218, Humana Press, New York: [DOI] [PubMed] [Google Scholar]
- 52. Massey S., Burress H., Taylor M., Nemec K. N., Ray S., Haslam D. B., and Teter K. (2011) Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect. Immun. 79, 4739–4747 10.1128/IAI.05503-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Orlandi P. A. (1997) Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J. Biol. Chem. 272, 4591–4599 [PubMed] [Google Scholar]
- 54. Winkeler A., Gödderz D., Herzog V., and Schmitz A. (2003) BiP-dependent export of cholera toxin from endoplasmic reticulum-derived microsomes. FEBS Lett. 554, 439–442 10.1016/S0014-5793(03)01217-1 [DOI] [PubMed] [Google Scholar]
- 55. Williams J. M., Inoue T., Banks L., and Tsai B. (2013) The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol. Biol. Cell 24, 785–795 10.1091/mbc.e12-07-0522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Takayama S., Xie Z., and Reed J. C. (1999) An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 274, 781–786 10.1074/jbc.274.2.781 [DOI] [PubMed] [Google Scholar]
- 57. Kundel F., De S., Flagmeier P., Horrocks M. H., Kjaergaard M., Shammas S. L., Jackson S. E., Dobson C. M., and Klenerman D. (2018) Hsp70 inhibits the nucleation and elongation of tau and sequesters tau aggregates with high affinity. ACS Chem. Biol. 13, 636–646 10.1021/acschembio.7b01039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ernst K., Liebscher M., Mathea S., Granzhan A., Schmid J., Popoff M. R., Ihmels H., Barth H., and Schiene-Fischer C. (2016) A novel Hsp70 inhibitor prevents cell intoxication with the actin ADP-ribosylating Clostridium perfringens ι toxin. Sci. Rep. 6, 20301 10.1038/srep20301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ernst K., Schmid J., Beck M., Hägele M., Hohwieler M., Hauff P., Ückert A. K., Anastasia A., Fauler M., Jank T., Aktories K., Popoff M. R., Schiene-Fischer C., Kleger A., Müller M., Frick M., and Barth H. (2017) Hsp70 facilitates trans-membrane transport of bacterial ADP-ribosylating toxins into the cytosol of mammalian cells. Sci. Rep. 7, 2724 10.1038/s41598-017-02882-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spooner R. A., Hart P. J., Cook J. P., Pietroni P., Rogon C., Höhfeld J., Roberts L. M., and Lord J. M. (2008) Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 105, 17408–17413 10.1073/pnas.0809013105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tehran D. A., Pirazzini M., Leka O., Mattarel A., Lista F., Binz T., Rossetto O., and Montecucco C. (2017) Hsp90 is involved in the entry of clostridial neurotoxins into the cytosol of nerve terminals. Cell Microbiol. 19, 12647 [DOI] [PubMed] [Google Scholar]
- 62. Banerjee T., Pande A., Jobling M. G., Taylor M., Massey S., Holmes R. K., Tatulian S. A., and Teter K. (2010) Contribution of subdomain structure to the thermal stability of the cholera toxin A1 subunit. Biochemistry 49, 8839–8846 10.1021/bi101201c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Taylor M., Banerjee T., VanBennekom N., and Teter K. (2012) Detection of toxin translocation into the host cytosol by surface plasmon resonance. J. Visual. Exp. 59, e3686 10.3791/3686 [DOI] [PMC free article] [PubMed] [Google Scholar]