

Abstract
Degradation of polysaccharides is central to numerous biological and industrial processes. Starch-active polysaccharide monooxygenases (AA13 PMOs) oxidatively degrade starch and can potentially be used with industrial amylases to convert starch into a fermentable carbohydrate. The oxidative activities of the starch-active PMOs from the fungi Neurospora crassa and Myceliophthora thermophila, NcAA13 and MtAA13, respectively, on three different starch substrates are reported here. Using high-performance anion-exchange chromatography coupled with pulsed amperometry detection, we observed that both enzymes have significantly higher oxidative activity on amylose than on amylopectin and cornstarch. Analysis of the product distribution revealed that NcAA13 and MtAA13 more frequently oxidize glycosidic linkages separated by multiples of a helical turn consisting of six glucose units on the same amylose helix. Docking studies identified important residues that are involved in amylose binding and suggest that the shallow groove that spans the active-site surface of AA13 PMOs favors the binding of helical amylose substrates over nonhelical substrates. Truncations of NcAA13 that removed its native carbohydrate-binding module resulted in diminished binding to amylose, but truncated NcAA13 still favored amylose oxidation over other starch substrates. These findings establish that AA13 PMOs preferentially bind and oxidize the helical starch substrate amylose. Moreover, the product distributions of these two enzymes suggest a unique interaction with starch substrates.
Keywords: carbohydrate-binding protein, polysaccharide, metalloprotein, copper, copper monooxygenase, amylose, auxiliary activity (AA) enzyme, oxygenase, polysaccharide monooxygenase, starch
Introduction
Polysaccharide monooxygenases (PMOs)3 are copper-dependent enzymes that use an oxidative mechanism to hydroxylate the glycosidic bond of polysaccharides, including cellulose (1, 2), hemicellulose (3–6), chitin (7, 8), xylan (9, 10), and starch (11, 12). They are commonly referred to as lytic polysaccharide monooxygenases and are classified by the Carbohydrate Active Enzyme database as auxiliary activity (AA) enzymes (13). The Type-2 mononuclear copper active site in PMOs is surface exposed, allowing the enzyme access to the glycosidic bonds on the surface of insoluble crystalline substrates. It is unlikely that PMOs separate single polysaccharide chains from the substrates, an energy-intensive step required by glycoside hydrolases (GHs). PMOs regioselectively hydroxylate either the C1-H or C4-H through co-substrate (O2 or H2O2)–dependent mechanisms (14–16), leading to glycosidic bond cleavage. The new chain ends generated can then be hydrolyzed by GHs. The synergy between PMOs and GHs increases the overall conversion of recalcitrant polysaccharides to fermentable sugars and thus has significant potential in the biofuels industry (17–19).
Starch is an α(1→4) linked glucose biopolymer that serves as a form of energy storage in plants. The main components of plant-based starch are amylose (∼15%–35%) and amylopectin (∼65%–85%) (20, 21). Amylose chains typically contain several hundred to thousands of α(1→4)-linked D-glucose units that form both single and double helices (Fig. 1). These double helices align in a parallel fashion to form crystalline structures (22–24). In this most common form, A-type, each amylose helix turn consists of six glucose units. Amylopectin chains contain several thousands to hundreds of thousands of glucose units with α(1→6)-branched linkages approximately every 30 glucose units along the main chain. Past research showed that the crystalline nature of amylose leads to a dense, insoluble, and hydrolytically resistant biopolymer (21, 25–27). In contrast, amylopectin has a lower density and is more accessible for amylase hydrolysis.
Figure 1.
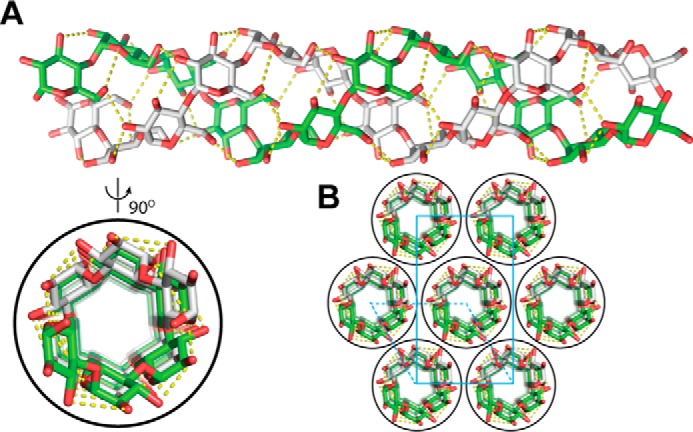
Structure of A-amylose. A and B, double helix of A-type amylose (A, AmyA_double.pdb) and typical packing of crystal unit of amylose based on the file “A-amylose_2009-popov_expanded.pdb” (B) obtained from http://polysac3db.cermav.cnrs.fr.4
Starch-active PMOs (AA13 PMOs) are one of seven families in the PMO superfamily (28–30) and represent a small fraction (∼1%) of the total known PMOs. Although these enzymes were discovered in fungi through the starch-specific carbohydrate binding module 20 (CBM20), a significant number of AA13 PMOs have now been identified that lack this domain. Carbohydrate binding modules (CBM) are noncatalytic domains that increase enzyme affinity for specific polysaccharides (31, 32). PMOs are able to naturally function without a CBM, as only about 30% of the more than 15,000 putative PMOs are associated with a CBM. On the other hand, 60% of putative AA13 PMOs contain the starch-specific CBM20 domain, suggesting that CBMs play a significant role in this family compared with other classes of PMOs. Nonetheless, AA13 PMOs generate malto-aldonic acids via oxidation of the C1 position of α(1→4) glycosidic linkages (Fig. 2, top reaction) (11, 12), which resembles the oxidative cleavage of β(1→4) and β(1→3) glycans performed by other PMO families (2, 3, 7–9, 33). AA13 PMOs are currently the only PMOs that hydroxylate α(1→4) glycosidic linkages. It is unknown whether AA13 PMOs cleave the branching α(1→6) linkages (Fig. 2, bottom reaction). Additionally, AA13 PMOs differ from other PMO families in that they possess a shallow groove found on the substrate-binding surface, which likely accommodates helical starch substrates (12).
Figure 2.
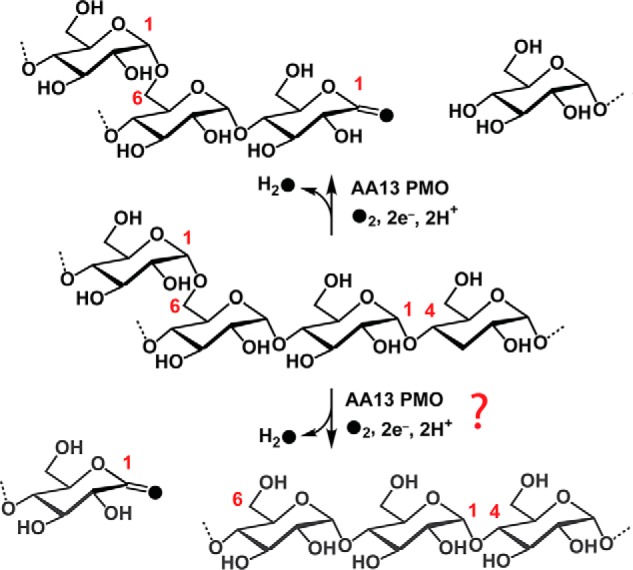
Oxidative cleavage of starch by AA13 PMOs. AA13 PMOs oxidize the C1 position of α(1→4) linkage (top arrows). It is unknown whether AA13 PMOs oxidize the α(1→6) linkage of starch substrates (bottom arrows).
Despite the low occurrence of AA13 PMOs, their unique ability to oxidize α(1→4) glycosidic linkages places them in an important biological niche. Consistent with activity on this substrate, AA13 PMO expression is up-regulated when fungi are grown on starch (34, 35). Understanding AA13 activity is an important step toward understanding how organisms utilize this polysaccharide. In this study, AA13 PMO activity on starch substrates with varying extents of α(1→6) branching was investigated. Two AA13 PMOs, one from Neurospora crassa (NcAA13) and the other from Myceliophthora thermophila (MtAA13), were characterized using activity assays, pulldown assays, and docking models. The data establish that AA13 PMOs preferentially oxidize the nonbranched helical polysaccharide amylose over α(1→6)-branched starch polysaccharides and identify residues within the catalytic domain involved in substrate binding that are conserved among AA13 PMOs. In addition, AA13 PMOs generate major malto-aldonic acid products with degrees of polymerization of 6n (n = 1, 2, 3, etc.) via the oxidation of glycosidic linkages separated by multiples of a helical turn on the same helix of amylose substrate, which is suggestive of the AA13 mode of action on the crystalline surface of amylose. These insights contribute to elucidating the mechanism of AA13 PMOs in particular and the PMO superfamily in general.
Results
Sequence similarity networks of the Pfam PF03067 indicated that all AA13 PMOs (165 sequences) are closely related (Fig. 3A). The tight clustering of these PMOs suggests that all AA13 likely act on the same type of starch polysaccharide. The sequences in this cluster are derived from 122 organisms, with no obvious distinction between the presence or absence of the starch-specific CBM20 domain (Table S1). Moreover, ∼40% of AA13 PMOs do not contain a CBM20 domain, suggesting that the presence of the CBM is not necessarily associated with physiological function in all cases. NcAA13, which naturally contains a CBM20 domain, and MtAA13, which lacks a CBM20 domain, were selected as representative examples of the two domain architectures associated with AA13 PMOs. To make direct biochemical comparisons between these two PMOs, truncations removing the linker and CBM20 of NcAA13, as well as a chimeric protein, MtAA13+CBM, adding the CBM20 of NcAA13 to MtAA13, were recombinantly expressed and purified (Fig. 3B and supporting information). Following copper reconstitution, each enzyme bound 0.87–0.93 equivalents of copper.
Figure 3.
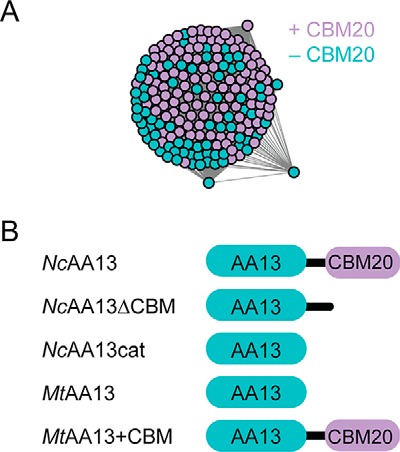
Overview of AA13 PMOs. A, SSN for AA13 PMOs generated using an E value of 10−80. Nodes are color-coded based on the native AA13 PMO sequence containing a CBM20 (purple) or only an AA13 catalytic domain (cyan). B, domain architectures of the AA13 PMO constructs used in this study.
Substrate preference of AA13 PMOs
The activity of NcAA13 and MtAA13 on amylose, amylopectin, and cornstarch was assessed to investigate specificity with various starch polysaccharides. HPAEC-PAD was used to analyze the soluble oxidized products (Fig. 4, A and B). These chromatograms exhibit peaks corresponding to malto-aldonic acids with various degrees of polymerization (DPs), as shown previously (11). Without malto-aldonic acid standards with DP > 7, exact product quantification was not possible. Therefore, activity was calculated as the sum of the areas of all product peaks. Both AA13 PMOs were significantly more active on amylose than on the other two starch substrates. NcAA13 produced ∼25 and 125 times more soluble oxidized products from amylose than from amylopectin and cornstarch, respectively (Fig. 4A and Fig. S1). Similarly, MtAA13 produced ∼20 and 90 times more soluble oxidized products from amylose than from amylopectin and cornstarch, respectively (Fig. 4B and Fig. S1).
Figure 4.

HPAEC-PAD chromatograms of NcAA13 and MtAA13. A and B, activity assays of NcAA13 (A) and MtAA13 (B) with amylose (blue lines), amylopectin (red lines), and cornstarch (green lines) substrates. A chromatogram of malto-aldonic acid standards (yellow lines) has been included for reference. For clarity, the chromatograms for the amylose assays were scaled down by 10-fold. A1–A30 indicate the malto-aldonic acid products with degrees of polymerization of 1–30. Chromatograms for NcAA13ΔCBM, NcAA13cat, and MtAA13+CBM can be found in the supporting information.
Truncations of NcAA13 that lacked the CBM20 domain were assayed to determine the effect of the CBM on oxidative activity. NcAA13ΔCBM and NcAA13cat produced a larger number of distinct oxidized products from amylose than amylopectin and cornstarch, but the overall product yield was lower (Figs. S1–S4). NcAA13ΔCBM displayed a similar product profile as NcAA13, generating oxidized malto-aldonic acid products (A) with a DP from A3 to over A36 (Fig. S2). Smaller soluble oxidized products (A1–A3) were observed for NcAA13cat, suggesting that it is not able to access the more crystalline regions of amylose.
Closer examination of the HPAEC-PAD chromatograms revealed that all constructs exhibited a unique product distribution pattern with amylose. The peak corresponding to A6 had the highest intensity. The peak intensity then gradually decreased, with the lowest intensity for A8 and A9, and then increased in intensity up to A12. This pattern is repeated for every pair of A6n and A6(n+1) products, where n is an integer from 1 to at least 5, as detected within the observable range of the HPAEC-PAD method. Among these A6n peaks, the intensity decreases with increasing values of n. The peak intensity progressively decreased from A6 to A5 and A4. Although A1–A3 products were not observed in the WT chromatograms, smaller oxidized products were observed for NcAA13cat (Fig. S3). The product distribution pattern from assays on amylose indicated that A6n is the major product from both NcAA13 and MtAA13. This unique pattern was not observed in assays with amylopectin and cornstarch, which contain shorter and less ordered helices. Instead, the product peaks from these substrates increased up to A6 and then gradually decreased.
CBM20 contribution to oxidative activity
NcAA13 and MtAA13 are ideal PMOs to explore the role of CBM20 domains in starch oxidation. NcAA13 contains both an AA13 catalytic domain and a CBM20 domain, whereas MtAA13 consists of only a catalytic AA13 domain. The sequence identity between the two catalytic domains is 77%. In addition to the full-length enzymes, two truncations of NcAA13 (NcAA13ΔCBM and NcAA13cat) and a chimeric MtAA13 PMO (MtAA13+CBM) were further characterized for their ability to degrade starch. Because the above results indicated that amylose was the preferred polysaccharide substrate for both NcAA13 and MtAA13, assays were performed using this substrate. Low enzyme concentrations were used to increase enzyme-substrate loading, thereby minimizing off-pathway oxidations. The reaction progress of each construct was monitored over 2 h.
Initial reactions were performed using 25 mg/ml amylose (Fig. 5A). Both NcAA13 and MtAA13 generated soluble oxidized products, with activity beginning to plateau after approximately 1 h. Both enzymes appeared to retain activity after 2 h, albeit with significantly lower turnover. Both truncations of NcAA13 formed nominal amounts of soluble oxidized products despite an O2 reduction activity similar to that of the WT (Fig. S5). The chimeric MtAA13+CBM construct was the only enzyme to display linear rates of oxidized product formation over the entire time course. In addition, the concentration of soluble oxidized products was higher compared with all other constructs, suggesting that the chimeric enzyme can more efficiently utilize amylose than WT MtAA13.
Figure 5.

Formation of oxidized products from amylose. A, product formation with 25 mg/ml amylose. B, product formation with 50 mg/ml amylose. All assays contained PMO (1 μm), ascorbic acid (2 mm), and amylose (25 or 50 mg/ml) in 50 mm sodium acetate buffer (pH 5.0). Error bars represent one standard deviation of the mean (n = 3). Some error bars are smaller than the size of the data point.
Substrate occupancy at the active site is an important criterion to minimize off-pathway oxidations that lead to enzyme inactivation (36, 37). In line with this, the observed loss in activity with 25 mg/ml amylose may have been a result of a subsaturating amylose concentration. Therefore, the reaction progress was then monitored using 50 mg/ml amylose (Fig. 5B). MtAA13+CBM showed a linear formation of oxidized products with similar relative activity to that observed at 25 mg/ml. Both WT PMOs displayed linear rates of soluble product formation, suggesting that there was no enzyme inactivation via auto-oxidation. The progress curves of the WT enzymes were ∼1.5-fold lower than that of the chimeric enzyme, indicating that the chimeric enzyme can more efficiently utilize amylose. Increasing the substrate concentration had little effect on the oxidative active activity of NcAA13.
Starch binding by AA13 PMOs
To assess the ability of AA13 PMOs to bind starch polysaccharides and quantify the contribution of the CBM20 domain to substrate binding, pulldown assays were performed using amylose, amylopectin, and cornstarch (Fig. 6). More than 50% of all constructs were pulled down by cornstarch and amylopectin except for MtAA13 by amylopectin. Conversely, amylose binding was strongly influenced by the presence of the CBM20 domain. Compared with WT NcAA13, approximately 35% and 50% reductions in amylose binding were observed for NcAA13ΔCBM and NcAA13cat, respectively. Although MtAA13 exhibited poor binding to amylose (∼30%), addition of NcAA13's CBM20 domain to MtAA13 (MtAA13+CBM) resulted in a more than 50% increase in enzyme binding. Together, these results show that the CBM20 domain has the greatest influence over amylose binding affinity. The favorable binding of cornstarch and amylopectin, regardless of domain architecture, may be a result of their more “porous” structure compared with amylose as well as α(1→6) branch points, which could provide additional, nonspecific interactions with the enzyme surface. However, less than 5% of the total enzyme concentration was found in the supernatant following resuspension of the polysaccharide pellet.
Figure 6.
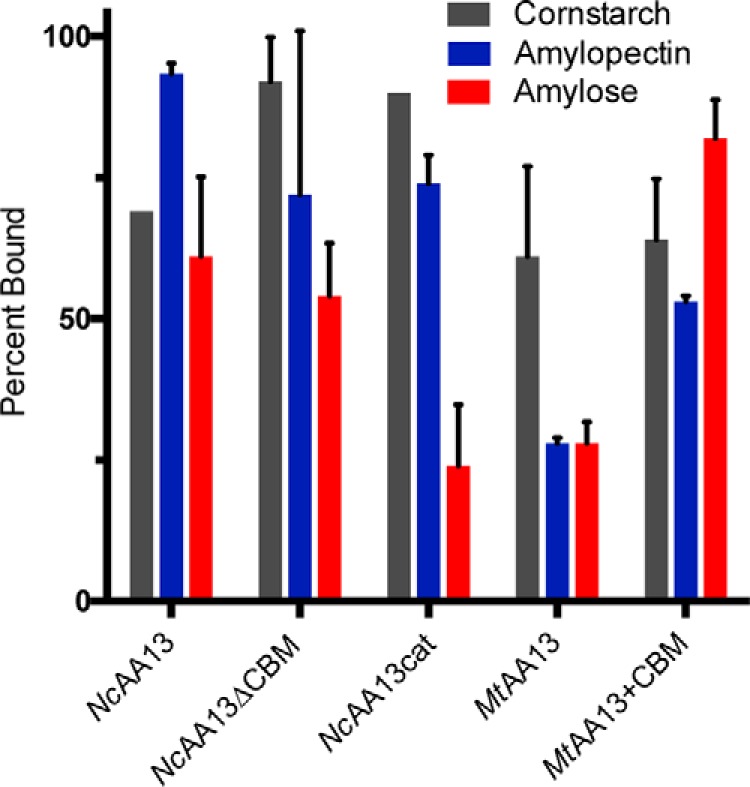
Starch binding to AA13 PMOs. The ability of each AA13 PMO to bind cornstarch, amylopectin, and amylose was assessed through pulldown assays. Assays containing PMO (10 μm) and polysaccharide substrate (25 mg/ml) in 50 mm sodium acetate buffer (pH 5.0) were shaken for 2 h. The starch substrate was pelleted, and the supernatant containing the unbound protein was analyzed by SDS-PAGE. Error bars represent one standard deviation of the mean (n = 3).
Amylose docking model
To better understand the substrate preference of AA13 PMOs, the interaction of AoAA13 from Aspergillus oryzae was examined with structurally defined substrates. AoAA13 is currently the only AA13 PMO for which a structure is available (PDB code 4OPB) (12). AoAA13 shares 72% and 76% sequence identity with NcAA13 and MtAA13, respectively, thus serving as a reasonable structural model for these two PMOs. The copper active site of AoAA13 is located midway through a shallow groove spanning the active-site surface (12). This shallow groove was proposed to have a role in the interaction of AA13 PMOs with branched-type substrates (12, 28). To test this hypothesis, molecular docking between the active-site groove and an amylose random coil, A-type single helix, and A-type double helix was performed. The docked conformations of the three types of substrates fit relatively well into the active-site groove of AoAA13. The optimal binding interactions of AoAA13 with these substrates are shown in Fig. 7 and Figs. S6 and S7.
Figure 7.

Docking model of an amylose double helix bound to AoAA13. A, illustration of the amylose double helix bound to the active-site surface. B, residues identified in the model to make hydrogen bonds (yellow dashed lines) with amylose. This model was generated from a docking experiment using PDB code 4OPB. C, close-up of the active site. The blue dashed lines indicate the distance from the copper center (brown) to the C1 (5.1 Å) and C4 (5.8 Å) positions of the glycosidic linkage positioned over the active site. D, sequence logo for the catalytic domain of the putative AA13 PMO family, showing conserved contact sites for modeled substrates: random (blue dots), single helix (red dots), or double helix (orange dots) amylose. All binding sites predicted in the docking model involve highly conserved residues in the AA13 family, with the exception of the Glu167/Pro172/Lys173 contact sites, which are found in a highly variable region of the sequence. Gaps in the logo caused by nonconserved insertions were manually removed for clarity. Numbers do not exactly match the sequence numbering of NcAA13, as many other sequences in the family have relatively conserved insertions.
In all three models, each substrate was found to dock along the active-site surface groove, positioning a glycosidic linkage in proximity (5.1–5.8 Å) to the copper active site (Fig. 7B and Figs. S6 and S7). As a result, equatorial coordination of the co-substrate would be favored, but axial coordination cannot be ruled out. Because different structural analysis tools give different results in H-bond determination, a range in the number of H-bonds formed between the AA13 PMO and each substrate is reported, representing the H-bonds identified using a variety of analyses. A random amylose coil forms 16–19 H-bonds with 11 residues of AA13 PMOs (Cys25, Ile27, Glu29, Arg44, Ala52, Arg53, Asn88, Asp90, Pro172, Lys173, and Gln219) (Fig. S6). Single-helix A-amylose forms six to 12 H-bonds with six residues of AA13 PMO (Glu29, Arg53, Asn88, Glu167, Gln219, and Gln222) (Fig. S7). Double-helix A-amylose forms seven to 13 H-bonds with six residues of AA13 PMOs (Glu29, Arg53, Asn88, Glu167, Lys173, and Gln219) (Fig. 7B). The four residues Glu29, Arg53, Asn88, and Gln219 are involved in H-bonds with all three substrates, whereas Glu167 only forms an H-bond with helical substrates. These H-bonding residues identified in the docking models are highly conserved (Fig. 7C), suggesting that these models are likely applicable to all AA13 PMOs.
The Gibbs free energies for binding of substrates to the active-site groove were calculated using Autodoc Vina as described previously (38). The binding energy to the active-site groove for the amylose double helix (−11.8 kcal/mol) is slightly higher than that of the single helix (−10.5 kcal/mol) and almost 2-fold greater than that of the random coil (−6.2 kcal/mol). The differences in affinities between the amylose random coil and the amylose helices can be explained through the differences in their degrees of freedom. The number of degrees of freedom of the coil conformation is much higher than that of the helical conformations. It is thus more energetically difficult to adopt the optimized binding position between the amylose random coil and the active-site groove. In other words, the conformational entropy would significantly reduce the binding affinity of the amylose random coil to the AA13 PMO. Therefore, although the amylose random coil forms more noncovalent bond contacts, it a has much lower affinity to the groove compared with that of the helical substrates. This result suggests that the AA13 PMO surface groove could play a role in the preference of amylose helical type substrates over random coil types.
Discussion
AA13 PMOs degrade insoluble starch polysaccharides and, in the process, generate a fairly wide distribution of products that can be derived from long soluble fragments or from the crystalline lattice. The observed substrate selectivity and product distributions indicate that AA13 PMOs prefer to act on the nonbranched α-helical substrate amylose. The lack of A1 and A2 products and the small amount of A3 and A4 products suggest that AA13 PMOs require at least four glucose units on either side of the cleavage position, consistent with the docking models (Fig. 7 and Figs. S6 and S7). Accessibility of the glycosidic linkages in the amylose helix is directly responsible for the unique product distribution observed for AA13 PMOs. Amylose helices, which have pseudo C6 symmetry, consist of six glucose units per turn. The packing of amylose helices into sheets and stacks of sheets (Fig. 1) would make the 6n sites (corresponding to n helical turns) the most accessible linkages on the crystalline surface to the PMO active site. This packing only allows the PMO access to one face of the amylose helix; the oxidized products corresponding to A6n positions should be the most abundant, and, indeed, that is the case. Conversely, the PMO is not restricted to one face of amylopectin because its shorter, less ordered helices are not as tightly packed. Thus, the unique pattern is not observed.
A mechanism for AA13 degradation of crystalline amylose can be proposed based on the product distribution observed (Fig. 8). AA13 PMOs oxidize the target linkage at the first position encountered on the amylose helix (Fig. 8, step 1). This will release An fragments when this cleavage is near the end of the chain. Subsequent cleavage at the next accessible glycosidic linkage along the same helix results in A6 products (Fig. 8, step 2). All additional turnovers of the PMO can then oxidize all available sites along the crystal face to generate A6 fragments (Fig. 8, step 3). Cleavage reactions that occur when the PMO has skipped an available site either through processivity or substrate dissociation and reassociation (Fig. 8, step 4) would yield A6n products (Fig. 8, step 5); these smaller fragments can be further processed to generate A6 fragments. The three-dimensional orientation of α(1→4) glycosidic linkages between A6n and A12n of the helical substrate are not accessible unless the enzyme migrates around the helix or the helix unravels to a random coil.
Figure 8.
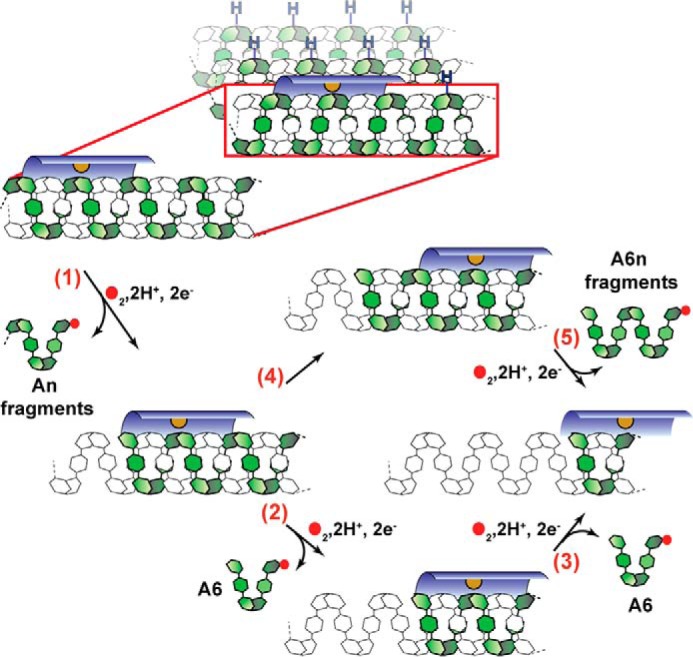
Proposed mechanism for AA13 PMO action on amylose. The AA13 PMO binds to the amylose double helix in the active-site surface groove (dark blue), with the active site positioned above the α(1→4) linkage. Accessible glycosidic C-H bonds on the amylose sheet are designated with a blue H. The copper active site is shown in orange. Glucose units are shown as hexagons. The amylose chain undergoing oxidation is shown in green. Filled red circles indicate oxygen atoms. H2O release resulting from each cleavage event is omitted for clarity. Oxidation of the glycosidic linkage results in the release of an An fragment (n = degree of polymerization) (step 1). The PMO can then oxidize and cleave the next available bond to generate an A6 fragment (step 2) and subsequently move along the same helix to generate additional A6 fragments (step 3). Alternatively, following the first oxidation event, the PMO can bind a new portion of the amylose helix to generate A6n fragments (step 5).
The accessibility of every sixth glycosidic bond in the amylose helix and the observation that the A6 products are largest in peak height and area suggest processive action of AA13 PMOs. Single-molecule imaging of AA9 PMOs on cellulosic substrate indicates that these enzymes are not processive (39), but the mode of action for AA13 PMOs may differ. Similar product distributions are observed for only the catalytic domains (NcAA13cat and MtAA13), ruling out CBM-mediated processive action of AA13 PMOs. However, processive hydrolases do not require a CBM (40), and the tunnel-shaped active site is a key structural feature promoting processivity (41, 42). AA13 PMOs have a groove along the substrate-binding surface that is not observed in other AA PMO families, which could allow the PMO to move along nonbranched amylose helices; α(1→6) branch points would impede PMO action. A metric commonly used to measure processivity of glycoside hydrolases is the molar ratio of the major product (disaccharide) to that of odd-numbered oligosaccharides (monosaccharides and trisaccharides) (43). The larger the ratio, the higher the degree of processivity, with ratios for processive hydrolases typically greater than 10 (44–46). To investigate the possibility of processivity in AA13 PMOs, peak areas of soluble product fragments can be used as a proxy for concentrations to generate a similar ratiometric comparison. Peak area is derived from the electrochemical detector response and is not a strict quantitative measure of concentration. With that proviso, regardless of AA13 PMO and the time point chosen (15, 30, or 60 min), the sum of A6 and A12 peak areas compared with the products in between (A5–A11) resulted in a ratio of <2. These product ratios are significantly lower than that of processive glycosyl hydrolases and are consistent with nonprocessive action by AA13 PMOs.
The significantly higher activity of NcAA13 and MtAA13 on amylose over cornstarch and amylopectin has several implications. Previous studies have shown that the oxidative function of AA13 PMOs has been shown to confer more efficient hydrolase action on cornstarch (12). Because cornstarch is made up of both amylose and amylopectin, our results are consistent with AA13 action on the amylose portion of this substrate. The tight packing of starch in amylose helices naturally makes this form of starch more resistant to hydrolytic action compared with amylopectin.
AA13 PMOs acting on crystalline, recalcitrant substrates is consistent with other AA PMO families; PMOs disrupt the crystalline lattice to facilitate access of substrate for GHs. AA13 PMOs appear to favor extended amylose chains lacking α(1→6) branch points. The lower activity on cornstarch and amylopectin likely arises from the less favorable binding affinity of nonhelical substrates in combination with α(1→6) branch points. Although AA13 PMOs can bind starch substrates with α(1→6) branch points (Fig. 6), these branch points likely prevent productive substrate binding and point to the inability of AA13 PMOs to oxidize these linkages and release soluble products. In the same way that PMOs increase accessibility of crystalline substrates to hydrolases, amylases or debranching enzymes that hydrolyze α(1→6) branch points (47) may also provide synergy for PMO-mediated starch degradation. The helical amylose chains can then bind favorably in the unique substrate-binding groove found in AA13 PMOs, positioning the glycosidic bond near the active-site copper. Starches with high amylose content are very resistant to hydrolytic amylase action, which could in part be due to the crystallinity of the polysaccharide (48). Indeed, AA13 PMO oxidative activity is necessary to create new chain ends for hydrolases; the AA13 PMO from Aspergillus nidulans, AnAA13, and β-amylase enhance the degradation of retrograde starch (12).
Our data support previous findings that CBMs have important roles in polysaccharide binding, which leads to protection from enzyme inactivation (37, 49–52). Previous work has also investigated the binding properties of PMOs appended with a CBM20 domain (53). Our work shows that the CBM20 domain clearly confers an advantage to binding helical starch substrates (Fig. 6), although it is not an absolute requirement for catalytic activity. NcAA13 produced higher quantities of longer oxidized products compared with MtAA13 (Fig. 4), and removal of the CBM20 domain from NcAA13 (NcAA13cat) resulted in shorter oxidized products. MtAA13 appears to have evolved without a CBM20 to productively oxidize amylose with similar product distributions to that of NcAA13. Sequence alignments of the two PMOs point to no obvious residues in the catalytic domain (e.g. additional aromatic or H-bonding residues) that would confer such an advantage. Although the NcAA13 truncations are able to bind starch substrates, the relatively low activity suggests that the glycosidic linkage cannot be correctly positioned in the active site, likely leading to oxidative inactivation. The CBM20 domain may have a role in allowing the PMO to access crystalline portions of the biomass; such a role has been proposed for cellulose-active PMOs (50).
Taken together, AA13 PMOs serve an important role oxidizing α(1→4) glycosidic bonds in the degradation of starch. Although the exact physiological role of AA13 PMOs is not clear, our studies show that AA13 PMOs highly prefer extended helical amylose chains over other types of starch substrates. Moreover, AA13 PMOs generate A6n products through the cleavage of glycosidic linkages separated by multiples of a six-glucose unit helical turn on the same helix in amylose. Because plant starch is primarily comprised of α(1→6)-branched amylopectin, the full mechanism of action between starch-active PMOs and hydrolases active on starch may be more complicated. However, the tight packing of amylose prevents hydrolytic action, and thus the oxidative activity of AA13 PMOs likely plays a central role in amylose degradation. The residues identified to be involved in amylose binding are highly conserved, suggesting that the substrate preference and product distribution observed here for NcAA13 and MtAA13 constructs are common features of all AA13 PMOs. This work has provided a clearer picture of starch degradation and provides useful insight for future studies of AA13 PMOs. Rigorous single-molecule imaging techniques and molecular simulations will help to resolve the molecular mechanism of AA13 PMOs and their ability to generate the unique product profile observed here.
Experimental procedures
Cloning, expression, and purification
NcAA13 (NCU08746; UniProt KB Q7SCE9) and MtAA13 (Mycth_2313229; UniProt KB G2QP40) were cloned from conidia of N. crassa and M. thermophila, respectively. Overlapping PCR was used to attach the linker and CBM20 domain of NcAA13 to MtAA13. All constructs were inserted into the pCSR1 vector, and the sequences were verified to be correct (University of California Berkeley Sequencing Facility). The plasmids were then linearized for transformation via electroporation into N. crassa as described previously (54). Transformants were screened for protein expression, and the positive transformant producing the highest level of recombinant protein was selected for large-scale growths.
AA13 PMOs were expressed in N. crassa following methods reported previously (11). Cultures (10 liters in 250-ml flasks) of N. crassa were grown at 30 °C for 3 days. The secretome was separated from the fungal biomass and then concentrated 100-fold. All constructs were purified to homogeneity following methods described previously (11). AA13 PMOs with a CBM20 were purified using an amylose resin column (New England Biolabs), followed by size exclusion chromatography (Superdex 75 16/600 pg, GE Healthcare). All other constructs were purified using anion-exchange chromatography (MonoQ 10/100 GL, GE Healthcare) and size exclusion chromatography (Superdex 75 16/600 pg, GE Healthcare). Following purification, enzymes were treated with EDTA (1 mm) to remove all metals. Dialysis against buffer A (50 mm sodium acetate (pH 5.0)) containing CuSO4 (20 μm) was used to reconstitute each PMO. A PD-10 desalting column equilibrated with buffer A was used to remove excess CuSO4. Reconstitution of each enzyme was confirmed using the copper-specific chelator bathocuproine disulfonate (BCS). PMO (50 μm), BCS (1 mm), and ascorbic acid (1 mm) in 50 mm sodium acetate (pH 5.0) were incubated at room temperature for 30 min. Spectrophotometric determination of [Cu(BCS)2]3− (ϵ483 = 12,500 M−1 cm−1) was used to determine the copper content of each PMO (55).
Polysaccharide oxidation assay
Activity assays monitoring oxidized products were carried out as described previously with slight modifications (11, 56). Cornstarch (Sigma-Aldrich, S4126), amylopectin (Sigma-Aldrich, 10120), and potato amylose (Sigma-Aldrich, A0512) were dispersed in buffer A at 100 mg/ml. Substrates were pelleted and resuspended in buffer A three times to thoroughly wash and remove contaminants. Assays contained Cu(II)PMO (5 μm), ascorbic acid (2 mm), and polysaccharide substrate (50 mg/ml) in buffer A. The assay mixtures were incubated at 42 °C with shaking at 1100 rpm for 2 h. In negative controls, Cu(II)PMO was replaced with CuSO4 (1 or 5 μm). The reactions were quenched by addition of an equal volume of 0.2 m NaOH. Time point assays were performed to quantify the products formed using Cu(II)PMO (1 μm), ascorbic acid (2 mm), and amylose (25 mg/ml or 50 mg/ml) in buffer A. Aliquots were taken from the reaction and quenched with an equal volume of NaOH (0.2 m) after 10, 30, 60, and 120 min. Product analysis was carried out with HPAEC-PAD using a Dionex ICS-5000 system (Thermo Fisher) as described previously (56). Activity was calculated as the sum of the areas of all peaks corresponding to malto-aldonic acids.
Sequence similarity network (SSN) generation
SSN creation has been described previously (57). Pfam PF03067 was used as the input for the SSN. An alignment score of 80 (E value = 10−80) was used to generate the SSN, which was visualized using Cytoscape 3.3.0 (58). The network was created by selecting the only cluster (171 sequences) that contained known starch-active PMOs. Six sequences, which did not contain a predicted signal peptide that would be cleaved before a histidine residue, were left out of the analysis. 100 sequences contained a C-terminal CBM20 domain and were identified and colored manually.
Sequence logo
Sequences were curated from the 165 sequences identified using the SSN. Signal peptides were trimmed from sequences, and a multiple sequence alignment was made using ClustalOmega (59). The multiple sequence alignment was then used as an input to make a sequence logo using WebLogo (60). Gaps in the logo because of low conservation of insertions were manually removed for clarity. Sequences corresponding to the CBM20 domain were removed so that the AA13 domain was depicted in the sequence logo.
Rates of O2 reduction
O2 reduction for each AA13 PMO construct was determined by visualizing the rate of hydrogen peroxide formation in the absence of polysaccharide substrate (61). The PMO (1 μm), HRP 1.3 μm), and Amplex Red (100 μm) were added to 50 mm MOPS (pH 7.0) in the presence or absence of superoxide dismutase (250 units/ml) in a 96-well plate. Reactions were initiated by addition of ascorbic acid (2 mm). Formation of resorufin was then monitored at 560 nm at 30-s intervals for 30 min using a SpectraMax340 spectrophotometer (Molecular Devices). Control reactions (without PMO) were carried out in parallel, and the resulting background rates (0.6–0.9 units s−1 μm−1) were subtracted from the reported experimental values. All assays were performed in quadruplicate. The activity of each AA13 PMO was derived from the linear region of resorufin formation, typically corresponding to the 0- to 10-min time range.
Polysaccharide binding assays
Each PMO construct's binding affinity for amylose, amylopectin, or cornstarch (25 or 50 mg/ml) was tested. Binding assays (100 μl) were carried out in 500-μl EppendorfTM microcentrifuge tubes with 5 μm enzyme in buffer A and shaken at 1100 rpm for 120 min at 40 °C. The reaction mixture was then centrifuged at 13,000 rpm for 5 min to pellet the insoluble substrate. The supernatant was removed, and the pellet was resuspended in buffer A (100 μl). The resuspended pellet was shaken for an additional hour to remove nonspecifically bound PMO prior to centrifugation. Subsequently, an aliquot (20 μl) of each sample's supernatant (containing unbound PMO) as well as an aliquot containing nonspecifically bound PMO was mixed with SDS loading dye, boiled, and then resolved on a stain-free SDS-PAGE gel using a Bio-Rad ChemiDoc MP Imager system. Band intensities were quantified using Image Lab software (Bio-Rad). All binding assays were performed in three independent replicates.
Docking studies
Autodock Vina (62) was employed to model the binding of a flexible starch coil, an amylose single helix, and an amylose double helix to the active-site groove of the AA13 PMO from A. oryzae AoAA13 (PBD code 4OPB (12)). Atomic coordinates for a random starch coil, an amylose single helix, and an amylose double helix were obtained from the Database of Polysaccharide 3D structures (http://polysac3db.cermav.cnrs.fr)4 (63). The substrates were randomly docked to the active-site groove of AA13 PMO using the Broyden–Fletcher–Goldfarb–Shanno method (64) for optimization with a 26 × 26 × 60 Å3 grid covering the entire active-site groove and centered on the copper active site. During the computation, the amylose single helix, double helix, and receptor were set to be fully rigid, whereas the random starch coil was fully flexible. Atom types were treated through the all-atom Chemistry at Harvard Macromolecular Mechanics (CHARMM) force field (65, 66). The charge of Cu(II) ions was determined from restrained electrostatic potential analysis with molecular orbital quantum chemical calculation using the B3LYP functional and 6–31G(d) basis set. Residues Nϵ-Me-His1, His91, and Tyr224, which coordinate the Cu(II) ion, were also included in the calculation. These residues were capped by -CO-CH3 and -NH-CH3. The parameter exhaustiveness, which is associated with the accuracy of the computation, was set at 400. The noncovalent bond interactions between substrates and AoAA13 were evaluated using multiple approaches, including Protein-Ligand Interaction Profiler (67), and manual examination using PyMOL (68).
Author contributions
V. V. V., J. A. H., T. C. D., S. T. N., E. A. S., and M. A. M. conceptualization; V. V. V., J. A. H., and T. C. D. formal analysis; V. V. V., J. A. H., T. C. D., S. T. N., E. A. S., and M. A. M. methodology; V. V. V., J. A. H., T. C. D., S. J. W. H., E. A. S., and M. A. M. writing-review and editing; J. A. H., T. C. D., S. J. W. H., S. T. N., and E. A. S. investigation; J. A. H. and T. C. D. writing-original draft; M. A. M. supervision; M. A. M. funding acquisition; M. A. M. project administration.
Supplementary Material
Acknowledgments
We thank the members of the M. A. M. laboratory for critical reading of the manuscript.
This research was funded by National Science Foundation Grant 1565770 and Vietnam National Foundation for Science and Technology Development Grant 106-NN.02-2016.33. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7 and Table S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- PMO
- polysaccharide monooxygenase
- AA
- auxiliary activity
- GH
- glycoside hydrolase
- CBM
- carbohydrate binding module(s)
- HPAEC
- high-performance anion-exchange chromatography
- PAD
- pulsed amperometry detection
- DP
- degree of polymerization
- BCS
- bathocuproine disulfonate
- SSN
- sequence similarity network.
References
- 1. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J-.C. N., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., and Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. 108, 15079–15084 10.1073/pnas.1105776108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 10.1021/ja210657t [DOI] [PubMed] [Google Scholar]
- 3. Agger J. W., Isaksen T., Várnai A., Vidal-Melgosa S., Willats W. G., Ludwig R., Horn S. J., Eijsink V. G., and Westereng B. (2014) Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. 111, 6287–6292 10.1073/pnas.1323629111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simmons T. J., Frandsen K. E. H., Ciano L., Tryfona T., Lenfant N., Poulsen J. C., Wilson L. F. L., Tandrup T., Tovborg M., Schnorr K., Johansen K. S., Henrissat B., Walton P. H., Lo Leggio L., and Dupree P. (2017) Structural and electronic determinants of lytic polysaccharide monooxygenase reactivity on polysaccharide substrates. Nat. Commun. 8, 1064 10.1038/s41467-017-01247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bennati-Granier C., Garajova S., Champion C., Grisel S., Haon M., Zhou S., Fanuel M., Ropartz D., Rogniaux H., Gimbert I., Record E., and Berrin J.-G. (2015) Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol. Biofuels 8, 90 10.1186/s13068-015-0274-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu B., Olson Å., Wu M., Broberg A., and Sandgren M. (2017) Biochemical studies of two lytic polysaccharide monooxygenases from the white-rot fungus Heterobasidion irregulare and their roles in lignocellulose degradation. PLoS ONE 12, e0189479 10.1371/journal.pone.0189479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., and Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 10.1126/science.1192231 [DOI] [PubMed] [Google Scholar]
- 8. Hemsworth G. R., Henrissat B., Davies G. J., and Walton P. H. (2014) Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat. Chem. Biol. 10, 122–126 10.1038/nchembio.1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Couturier M., Ladevèze S., Sulzenbacher G., Ciano L., Fanuel M., Moreau C., Villares A., Cathala B., Chaspoul F., Frandsen K. E., Labourel A., Herpoël-Gimbert I., Grisel S., Haon M., Lenfant N., et al. (2018) Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat. Chem. Biol. 14, 306–310 10.1038/nchembio.2558 [DOI] [PubMed] [Google Scholar]
- 10. Frommhagen M., Sforza S., Westphal A. H., Visser J., Hinz S. W., Koetsier M. J., van Berkel W. J., Gruppen H., and Kabel M. A. (2015) Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol. Biofuels 8, 101 10.1186/s13068-015-0284-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vu V. V., Beeson W. T., Span E. A., Farquhar E. R., and Marletta M. A. (2014) A family of starch-active polysaccharide monooxygenases. Proc. Natl. Acad. Sci. 111, 13822–13827 10.1073/pnas.1408090111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lo Leggio L., Simmons T. J., Poulsen J.-C., Frandsen K. E., Hemsworth G. R., Stringer M. A., von Freiesleben P., Tovborg M., Johansen K. S., De Maria L., Harris P. V., Soong C.-L., Dupree P., Tryfona T., Lenfant N., et al. (2015) Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat. Commun. 6, 5961 10.1038/ncomms6961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bissaro B., Røhr Å. K., Müller G., Chylenski P., Skaugen M., Forsberg Z., Horn S. J., Vaaje-Kolstad G., and Eijsink V. G. H. (2017) Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 13, 1123–1128 10.1038/nchembio.2470 [DOI] [PubMed] [Google Scholar]
- 15. Hangasky J., Detomasi T., and Marletta M. (2019) Glycosidic bond hydroxylation by polysaccharide monooxygenases. Trends Chem. 1, 198–209 10.1016/j.trechm.2019.01.007 [DOI] [Google Scholar]
- 16. Hangasky J. A., Iavarone A. T., and Marletta M. A. (2018) Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc. Natl. Acad. Sci. 115, 4915–4920 10.1073/pnas.1801153115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hemsworth G. R., Johnston E. M., Davies G. J., and Walton P. H. (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol. 33, 747–761 10.1016/j.tibtech.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 18. Eibinger M., Ganner T., Bubner P., Rošker S., Kracher D., Haltrich D., Ludwig R., Plank H., and Nidetzky B. (2014) Cellulose surface degradation by a lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J. Biol. Chem. 289, 35929–35938 10.1074/jbc.M114.602227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu J., Arantes V., Pribowo A., Gourlay K., and Saddler J. N. (2014) Substrate factors that influence the synergistic interaction of AA9 and cellulases during the enzymatic hydrolysis of biomass. Energy Environ. Sci. 7, 2308–2315 10.1039/C4EE00891J [DOI] [Google Scholar]
- 20. Vamadevan V., and Bertoft E. (2015) Structure-function relationships of starch components. Starch 67, 55–68 10.1002/star.201400188 [DOI] [Google Scholar]
- 21. Pérez S., and Bertoft E. (2010) The molecular structures of starch components and their contribution to the architecture of starch granules: A comprehensive review. Starch 62, 389–420 10.1002/star.201000013 [DOI] [Google Scholar]
- 22. Sarko A., and Wu H.-C. H. (1978) The crystal structures of A-, B- and C-polymorphs of amylose and starch. Starch 30, 73–78 10.1002/star.19780300302 [DOI] [Google Scholar]
- 23. Hsien-Chih H. W., and Sarko A. (1978) The double-helical molecular structure of crystalline a-amylose. Carbohydr. Res. 61, 27–40 10.1016/S0008-6215(00)84464-X [DOI] [Google Scholar]
- 24. Hsein-Chih H. W., and Sarko A. (1978) The double-helical molecular structure of crystalline b-amylose. Carbohydr. Res. 61, 7–25 10.1016/S0008-6215(00)84463-8 [DOI] [Google Scholar]
- 25. Wang S., Li C., Copeland L., Niu Q., and Wang S. (2015) Starch retrogradation: a comprehensive review. Compr. Rev. Food Sci. Food Saf. 14, 568–585 10.1111/1541-4337.12143 [DOI] [Google Scholar]
- 26. Smith A. M., Zeeman S. C., and Smith S. M. (2005) Starch degradation. Annu. Rev. Plant Biol. 56, 73–98 10.1146/annurev.arplant.56.032604.144257 [DOI] [PubMed] [Google Scholar]
- 27. Berry C. S. (1986) Resistant starch: formation and measurement of starch that survives exhaustive digestion with amylolytic enzymes during the determination of dietary fibre. J. Cereal Sci. 4, 301–314 10.1016/S0733-5210(86)80034-0 [DOI] [Google Scholar]
- 28. Vu V. V., and Marletta M. A. (2016) Starch-degrading polysaccharide monooxygenases. Cell Mol. Life Sci. 73, 2809–2819 10.1007/s00018-016-2251-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Filiatrault-Chastel C., Navarro D., Haon M., Grisel S., Herpoël-Gimbert I., Chevret D., Fanuel M., Henrissat B., Heiss-Blanquet S., Margeot A., and Berrin J.-G. (2019) AA16, a new lytic polysaccharide monooxygenase family identified in fungal secretomes. Biotechnol. Biofuels. 12, 55 10.1186/s13068-019-1394-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tandrup T., Frandsen K. E. H., Johansen K. S., Berrin J.-G., and Lo Leggio L. (2018) Recent insights into lytic polysaccharide monooxygenases (LPMOs). Biochem. Soc. Trans. 46, 1431–1447 10.1042/BST20170549 [DOI] [PubMed] [Google Scholar]
- 31. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 10.1042/BJ20040892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guillén D., Sánchez S., and Rodríguez-Sanoja R. (2010) Carbohydrate-binding domains: multiplicity of biological roles. Appl. Microbiol. Biotechnol. 85, 1241–1249 10.1007/s00253-009-2331-y [DOI] [PubMed] [Google Scholar]
- 33. Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G., and Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 10.1074/jbc.M113.530196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu G., Li J., Liu Q., Sun W., Jiang M., and Tian C. (2018) Transcriptional analysis of Myceliophthora thermophila on soluble starch and role of regulator AmyR on polysaccharide degradation. Bioresour. Technol. 265, 558–562 10.1016/j.biortech.2018.05.086 [DOI] [PubMed] [Google Scholar]
- 35. Nekiunaite L., Arntzen M. Ø., Svensson B., Vaaje-Kolstad G., and Abou Hachem M. (2016) Lytic polysaccharide monooxygenases and other oxidative enzymes are abundantly secreted by Aspergillus nidulans grown on different starches. Biotechnol. Biofuels 9, 187–203 10.1186/s13068-016-0604-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loose J. S. M., Arntzen M. Ø., Bissaro B., Ludwig R., Eijsink V. G. H., and Vaaje-Kolstad G. (2018) Multipoint precision binding of substrate protects lytic polysaccharide monooxygenases from self-destructive off-pathway processes. Biochemistry 57, 4114–4124 10.1021/acs.biochem.8b00484 [DOI] [PubMed] [Google Scholar]
- 37. Mutahir Z., Mekasha S., Loose J. S. M., Abbas F., Vaaje-Kolstad G., Eijsink V. G. H., and Forsberg Z. (2018) Characterization and synergistic action of a tetra-modular lytic polysaccharide monooxygenase from Bacillus cereus. FEBS Lett. 592, 2562–2571 10.1002/1873-3468.13189 [DOI] [PubMed] [Google Scholar]
- 38. Caffalette C. A., Corey R. A., Sansom M. S. P., Stansfeld P. J., and Zimmer J. (2019) A lipid gating mechanism for the channel-forming O antigen ABC transporter. Nat. Commun. 10, 824 10.1038/s41467-019-08646-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eibinger M., Sattelkow J., Ganner T., Plank H., and Nidetzky B. (2017) Single-molecule study of oxidative enzymatic deconstruction of cellulose. Nat. Commun. 8, 894 10.1038/s41467-017-01028-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Igarashi K., Koivula A., Wada M., Kimura S., Penttilä M., and Samejima M. (2009) High speed atomic force microscopy visualizes processive movement of Trichoderma reesei cellobiohydrolase I on crystalline cellulose. J. Biol. Chem. 284, 36186–36190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Varrot A., Frandsen T. P., von Ossowski I., Boyer V., Cottaz S., Driguez H., Schülein M., and Davies G. J. (2003) Structural basis for ligand binding and processivity in cellobiohydrolase Cel6A from Humicola insolens. Structure 11, 855–864 [DOI] [PubMed] [Google Scholar]
- 42. Davies G., and Henrissat B. (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3, 853–859 10.1016/S0969-2126(01)00220-9 [DOI] [PubMed] [Google Scholar]
- 43. Horn S. J., Sørlie M., Vårum K. M., Väljamäe P., and Eijsink V. G. (2012) Measuring processivity. Methods Enzymol. 510, 69–95 10.1016/B978-0-12-415931-0.00005-7 [DOI] [PubMed] [Google Scholar]
- 44. Hamre A. G., Lorentzen S. B., Väljamäe P., and Sørlie M. (2014) Enzyme processivity changes with the extent of recalcitrant polysaccharide degradation. FEBS Lett. 588, 4620–4624 10.1016/j.febslet.2014.10.034 [DOI] [PubMed] [Google Scholar]
- 45. Fox J. M., Levine S. E., Clark D. S., and Blanch H. W. (2012) Initial- and processive-cut products reveal cellobiohydrolase rate limitations and the role of companion enzymes. Biochemistry 51, 442–452 10.1021/bi2011543 [DOI] [PubMed] [Google Scholar]
- 46. von Ossowski I., Ståhlberg J., Koivula A., Piens K., Becker D., Boer H., Harle R., Harris M., Divne C., Mahdi S., Zhao Y., Driguez H., Claeyssens M., Sinnott M. L., and Teeri T. T. (2003) Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Cel7A: a comparison with Phanerochaete chrysosporium Cel7D. J. Mol. Biol. 333, 817–829 10.1016/S0022-2836(03)00881-7 [DOI] [PubMed] [Google Scholar]
- 47. Møller M. S., Henriksen A., and Svensson B. (2016) Structure and function of α-glucan debranching enzymes. Cell. Mol. Life Sci. 73, 2619–2641 10.1007/s00018-016-2241-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buléon A., Pontoire B., Riekel C., Chanzy H., Helbert W., and Vuong R. (1997) Crystalline ultrastructure of starch granules revealed by synchrotron radiation microdiffraction mapping. Macromolecules 30, 3952–3954 10.1021/ma970136q [DOI] [Google Scholar]
- 49. Kruer-Zerhusen N., Alahuhta M., Lunin V. V., Himmel M. E., Bomble Y. J., and Wilson D. B. (2017) Structure of a Thermobifida fusca lytic polysaccharide monooxygenase and mutagenesis of key residues. Biotechnol. Biofuels 10, 243 10.1186/s13068-017-0925-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Courtade G., Forsberg Z., Heggset E. B., Eijsink V. G. H., and Aachmann F. L. (2018) The carbohydrate-binding module and linker of a modular lytic polysaccharide monooxygenase promote localized cellulose oxidation. J. Biol. Chem. 293, 13006–13015 10.1074/jbc.RA118.004269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crouch L. I., Labourel A., Walton P. H., Davies G. J., and Gilbert H. J. (2016) The contribution of non-catalytic carbohydrate binding modules to the activity lytic polysaccharide monooxygenases. J. Biol. Chem. 291, 7439–7449 10.1074/jbc.M115.702365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Forsberg Z., Nelson C. E., Dalhus B., Mekasha S., Loose J. S., Crouch L. I., Røhr Å. K., Gardner J. G., Eijsink V. G., and Vaaje-Kolstad G. (2016) Structural and functional analysis of a lytic polysaccharide monooxygenase important for efficient utilization of chitin in Cellvibrio japonicus. J. Biol. Chem. 291, 7300–7312 10.1074/jbc.M115.700161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nekiunaite L., Isaksen T., Vaaje-Kolstad G., and Abou Hachem M. (2016) Fungal lytic polysaccharide monooxygenases bind starch and β-cyclodextrin similarly to amylolytic hydrolases. FEBS Lett. 590, 2737–2747 10.1002/1873-3468.12293 [DOI] [PubMed] [Google Scholar]
- 54. Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 10.1021/ja409384b [DOI] [PubMed] [Google Scholar]
- 55. Badarau A., and Dennison C. (2011) Copper trafficking mechanism of CXXC-containing domains: Insight from the pH-dependence of their Cu(I) affinities. J. Am. Chem. Soc. 133, 2983–2988 10.1021/ja1091547 [DOI] [PubMed] [Google Scholar]
- 56. Phillips C. M., Beeson W. T., Cate J. H., and Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 10.1021/cb200351y [DOI] [PubMed] [Google Scholar]
- 57. Gerlt J. A., Bouvier J. T., Davidson D. B., Imker H. J., Sadkhin B., Slater D. R., and Whalen K. L. (2015) Enzyme function initiative-enzyme similarity tool (EFI-EST): a web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 1854, 1019–1037 10.1016/j.bbapap.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kittl R., Kracher D., Burgstaller D., Haltrich D., and Ludwig R. (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5, 79–92 10.1186/1754-6834-5-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trott O., and Olson A. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sarkar A., and Pérez S. (2012) PolySac3DB: an annotated data base of 3 dimensional structures of polysaccharides. BMC Bioinformatics 13, 302 10.1186/1471-2105-13-302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shanno D. F. (1970) Conditioning of quasi-Newton methods for function minimization. Math. Comput. 24, 647–656 10.1090/S0025-5718-1970-0274029-X [DOI] [Google Scholar]
- 65. Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., and Olson A. J. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B [DOI] [Google Scholar]
- 66. Morris G. M., Goodsell D. S., Huey R., and Olson A. J. (1996) Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J. Comput. Aided. Mol. Des. 10, 293–304 10.1007/BF00124499 [DOI] [PubMed] [Google Scholar]
- 67. Salentin S., Schreiber S., Haupt V. J., Adasme M. F., and Schroeder M. (2015) PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 43, W443–W447 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.8, Schroedinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.