

Abstract
Protein homeostasis and cellular fitness in the presence of proteotoxic stress is promoted by heat shock factor 1 (Hsf1), which controls basal and stress-induced expression of molecular chaperones and other targets. The major heat shock proteins and molecular chaperones Hsp70 and Hsp90, in turn, participate in a negative feedback loop that ensures appropriate coordination of the heat shock response with environmental conditions. Features of this regulatory circuit in the budding yeast Saccharomyces cerevisiae have been recently defined, most notably regarding direct interaction between Hsf1 and the constitutively expressed Hsp70 protein Ssa1. Here, we sought to further examine the Ssa1/Hsf1 regulation. We found that Ssa1 interacts independently with both the previously defined CE2 site in the Hsf1 C-terminal transcriptional activation domain and with an additional site that we identified within the N-terminal activation domain. Consistent with both sites bearing a recognition signature for Hsp70, we demonstrate that Ssa1 contacts Hsf1 via its substrate-binding domain and that abolishing either regulatory site results in loss of Ssa1 interaction. Removing Hsp70 regulation of Hsf1 globally dysregulated Hsf1 transcriptional activity, with synergistic effects on both gene expression and cellular fitness when both sites are disrupted together. Finally, we report that Hsp70 interacts with both transcriptional activation domains of Hsf1 in the related yeast Lachancea kluyveri. Our findings indicate that Hsf1 transcriptional activity is tightly regulated to ensure cellular fitness and that a general and conserved Hsp70–HSF1 feedback loop regulates cellular proteostasis in yeast.
Keywords: Heat shock factor protein 1 (HSF1), heat shock protein (HSP), proteostasis, Saccharomyces cerevisiae, stress response, cell stress, gene regulation, heat shock response, Hsp70, protein chaperone, transcription factor
Introduction
Cells respond to proteotoxic insults such as heat or oxidative stress by activating a highly conserved transcriptional program known as the heat shock response (HSR).2 A key feature of the HSR is a rapid increase in expression of protein molecular chaperones and other heat shock genes that play roles in safeguarding cellular physiology and restoring and maintaining protein homeostasis (proteostasis) (1). The master regulator of the HSR in eukaryotes from yeast to humans is the transcription factor heat shock factor 1 (HSF1), a multidomain protein with a highly conserved DNA-binding domain (DBD) and trimerization domain, as well as a less well-conserved C-terminal transcriptional activation domain (C-AD). In some fungal species, a second transcriptional activation domain is present N-terminal to the DBD (N-AD) (Fig. 1A and Fig. S5) (2). HSF1 recognizes heat shock elements (HSEs) with the preferred binding sequences nGAAn, which are localized in distinct iterative repeat motifs in promoters of target genes: three or more continuous, inverted repeats (perfect), noncontinuous repeats with a 5-bp skip between two of the HSEs (gap), and an architecture with 5-bp skips between each nGAAn repeat (step) (3, 4). In Saccharomyces cerevisiae, Hsf1 is encoded by a single essential gene, although recent work demonstrated that the HSF1 gene is dispensable if the two core molecular chaperones Hsp70 and Hsp90 are expressed under a heterologous promoter (5). In contrast to yeast, human cells encode several HSF proteins with roles in stress response and development, dysregulation of which is implicated in several human diseases (6). Specifically, some cancer cell lines have been shown to be dependent on overactivation of HSF1, and an increase in HSF1 activity has been linked to poor prognosis in breast and colon cancers (7). In several neurodegenerative disorders, including Parkinson's disease and ALS, a decrease in HSF levels and activity is linked to reduction in cellular survival and stress tolerance (6).
Figure 1.

The yeast Hsp70, but not Hsp90, interacts with Hsf1 during heat shock and attenuation. A, schematic of the yeast Hsf1 domain structure with C-terminal FLAG tag used for immunoprecipitation (IP). 3X, leucine zipper trimerization domain. CE2 is shown in dark blue. B, real-time luciferase reporter assay of Hsf1 activity over a 90-min heat shock at 37 °C. Error bars, S.D. of three biological replicates. C, Ssa1, but not Hsp90, co-immunoprecipitates with Hsf1-FLAG at the time points indicated in B with block arrows. GFP-FLAG is used as a negative control for nonspecific Ssa1 interaction. Immunoblots are representative of three independent experiments. AU, arbitrary units.
In both yeast and humans, the prevailing model for HSF regulation involves repression of transcriptional activity by molecular chaperones during optimal conditions, titration of those chaperones during a proteotoxic stress, and the resulting production of molecular chaperones, leading to eventual HSR attenuation. Studies in metazoan systems demonstrated association between Hsps, most notably Hsp90, and HSF1 both in vivo and in vitro (8–10). A regulatory chaperone–Hsf1 interaction in yeast was long supported by genetic studies in which the loss of constitutive Hsp70 or loss of a functional Hsp90 led to derepression of Hsf1 activity (11–13). Additionally, the constitutively expressed Hsp70 Ssa1 was found to utilize two surface-exposed cysteine residues to regulate Hsf1 as a sensor for thiol-reactive stress (14). Recent work by Pincus and co-workers (15) has provided detailed and definitive support for the existence of a carefully tuned Hsp70–Hsf1 regulatory circuit in S. cerevisiae. Ssa1 was demonstrated to interact with Hsf1 under basal growth conditions and to dissociate within 5 min of heat stress, coincident with HSR induction. Binding was re-established within 15 min in a manner consistent with transcriptional attenuation. Whereas earlier regulatory models implicated the abundant phosphorylation of Hsf1 during HSR induction as a primary mediator of activation, it is now understood that this post-translational modification modulates the magnitude of the response (15–17). Further support for the notion that Hsp70 binding and release is the primary activation switch was provided by experiments showing that titration of the chaperone via overexpression of a transcriptionally inert Hsf1 protein fusion resulted in inappropriate Hsf1 induction. Consistent with this model, a binding site for Ssa1 in the C-terminal domain was defined that fell within a previously documented Hsf1 regulatory site (CE2) (18) and was shown to be required for maintaining Hsf1 in an inactive state (19). Despite these many recent advances, our understanding of Hsf1 control by molecular chaperones remains incomplete. Is there a role for Hsp90, as suggested by genetic studies? How is the N-AD, previously shown to be competent for a subset of HSR activation, regulated (20, 21)? Additionally, is the concept of an Hsp70-specific chaperone switch extrapolatable to other organisms?
In this study, we further explored the nature of the Hsp70–Hsf1 regulatory interaction in yeast. We determined that whereas yeast Hsp70 associates with Hsf1 before and during an extended heat shock, Hsp90 fails to do so, reinforcing the model that this chaperone does not play a direct role in HSR regulation. Hsp70 was found to associate independently with both the previously defined CE2 site in the Hsf1 C-AD and a novel site that we identify in the N-AD. Site-directed substitutions in either Hsp70-binding site eliminated chaperone interaction and resulted in moderate derepression of the HSR, whereas an Hsf1 mutant lacking both sites exhibited pronounced up-regulation of Hsf1 target genes as well as a marked growth inhibition. We exploited these three HSF1 alleles to probe differential AD control over the Hsf1-dependent transcriptome. Contrary to previous studies, we observed no AD specificity for target genes and no clear relationship between AD regulation and HSE architecture. Finally, we demonstrate that both Hsp70-binding sites are conserved in the related yeast Lachancea kluyveri. Together, these results provide further evidence for a general and conserved multisite Hsp70–HSF1 feedback loop required for optimal cellular proteostasis.
Results
The molecular chaperone Hsp70, but not Hsp90, interacts with Hsf1 during heat shock and attenuation
Hsp90 is encoded by the constitutively expressed HSC82 and stress-inducible HSP82 genes in S. cerevisiae (22). Despite previous genetic evidence for a role for Hsp90 in HSR regulation, MS experiments failed to identify either gene product in affinity purifications of a dual epitope-tagged version of Hsf1 (12, 15). We therefore set out to ask whether Hsp90 may be involved in attenuation of Hsf1 activity during a prolonged thermal stress. A single FLAG epitope tag was appended to the C terminus of Hsf1, and the modified protein was expressed as the only copy of Hsf1 under the control of a CYC promoter from a plasmid in hsf1Δ knockout cells (Fig. 1A). To accurately follow the transcriptional dynamics of the HSR, we took advantage of a previously published destabilized reporter system. This reporter is based on a firefly luciferase variant fused to CL1 and PEST protein degradation sequences that also includes an ARE mRNA degradation element to ensure that both the mRNA and protein products are rapidly targeted for degradation (lucCP+) (23). We generated an HSE-lucCP+ construct and performed a real-time luciferase assay to assess HSR activation state. In keeping with previous mRNA-based approaches, upon shift from 30 to 37 °C, luciferase activity was detectable within 12 min and peaked at ∼36 min, and attenuation to nearly basal levels was complete by 80 min (Fig. 1B). This response was specific to control by the HSE, as luciferase production under the control of the unrelated promoters GRE2 and CYC1 remained unchanged (Fig. S1). We subsequently monitored the interaction of Hsp90 and Hsp70 with Hsf1 throughout the course of a 90-min heat shock via co-immunoprecipitation, taking samples at distinct time points corresponding to induction, peak, and attenuation phases of the HSR as indicated in Fig. 1B. Ssa1 associated with Hsf1 throughout the experiment, consistent with rapid release and rebinding as reported previously and extending well into the late attenuation phase (Fig. 1C). However, no association of Hsf1 with Hsp90 was observed using antiserum previously shown to recognize both Hsc82 and Hsp82, indicating that neither the constitutively expressed nor the heat-inducible Hsp90 isoform appears to form a stable repressing complex during the HSR in yeast (24). No binding of Ssa1 or Hsc/p82 was observed to a control GFP-FLAG protein fusion, eliminating the possibility that we were observing nonspecific binding (Fig. 1C). These results suggest that the influence of the Hsp90 chaperone system on the HSR may be indirect, in a manner distinct from the direct binding observed to HSF1 in metazoans (25).
Interaction between Hsp70 and Hsf1 depends on the Hsp70 substrate-binding domain and independent binding sites within the activation domains of Hsf1
Hsp70 chaperone proteins are composed of an N-terminal nucleotide-binding domain (NBD) connected by a flexible linker to a substrate-binding domain (SBD). Cycles of binding and release of substrate proteins to the SBD are controlled by ATPase activity within the NBD, further influenced by partner proteins including Hsp40 chaperones and nucleotide exchange factors that bind the NBD (26). Ssa1 has been shown to directly bind Hsf1 within the CE2 motif in the C-AD in a manner that can be competed by decoy constructs that contain one or more iterations of this motif (19). We additionally identified a second Ssa1-interacting region localized to the N-AD that resides between residues 50 and 100 using Hsf1 fragments fused to GFP-FLAG (Fig. S2). These findings are consistent with a model whereby Hsp70 interacts with two distinct regions of Hsf1, presumably via direct binding by the SBD. To test this hypothesis, we generated plasmids expressing each Hsp70 functional domain independently (Fig. 2A). The isolated Hsp70 SBD is capable of recognizing and stably binding substrate, and the NBD is also an independently folding unit (27, 28). A portion of the Hsf1 C-AD containing the CE2 Ssa1 binding element fused to GFP-FLAG (520–568HGF) and the aforementioned N-AD fusion (50–100HGF) were co-expressed in cells with either the Ssa1-NBD or Ssa1-SBD, and co-immunoprecipitations were performed. Both Hsf1 constructs exhibited binding to endogenous Ssa1 as well as the isolated SBD, but not the NBD (Fig. 2, C and D). Neither full-length Ssa1 nor the isolated domains associated with the control GFP-FLAG protein. Furthermore, the fusion proteins were found to be diffuse throughout the cells, indicating that the interaction with Ssa1 is not the result of aggregation (Fig. 2B and Fig. S3). These data indicate that Ssa1 is likely recognizing motifs within Hsf1 as substrates via the peptide-binding cleft within the SBD.
Figure 2.

Ssa1 recognizes Hsf1 via the substrate binding domain. A, the SBD and NBD were expressed as independent domains in WT cells along with the N-AD (50–100 Hsf1-GFP-FLAG (HGF)) or C-AD (520–568HGF) constructs bearing Ssa1-binding sites. B, the GFP-FLAG and HGF fusion proteins exhibit diffuse localization throughout the cell. C, Ssa1-NBD does not co-immunoprecipitate with either the N-AD or C-AD. D, Ssa1-SBD is sufficient to interact with both the N-AD and C-AD through co-immunoprecipitation. In both experiments, the endogenous Ssa1 protein is also present and exhibits binding to the Hsf1 fragments. IP, immunoprecipitation.
To further investigate how Ssa1 recognizes Hsf1, we identified potential Hsp70-binding sites through computational analysis of the Hsf1 amino acid sequence using the LIMBO algorithm, previously trained on known Hsp70 substrate peptide sequences (29). Using prediction parameters for high sensitivity, a single potential site was identified within residues 50–100 of the N-AD (residues 90–97), and between residues 520 and 568, two overlapping sites were found and encompass amino acids 533–543, the latter matching analysis done by Pincus and co-workers (19). To assess the relevance of these predictions, three core amino acids within each site were substituted with residues expected to disrupt Hsp70 binding (L92S,V93A,R94H and Y537S,L538S,L539S) in the context of the GFP-FLAG fusions (Fig. 3A). As expected, Hsp70 co-immunoprecipitated with the WT N- and C-terminal Hsf1-GFP-FLAG (HGF) constructs. In contrast to the binding observed with both WT Hsf1 sequences, the substitution constructs showed no association with Ssa1, validating the in silico predictions (Fig. 3B). Additionally, we noted that production of minor degradation products observed in both of the Hsf1 fusion constructs was nearly eliminated in the substitution mutants, suggesting that Ssa1 binding to these regions may induce conformational changes in both ADs that promote proteolysis in vivo. Furthermore, both the N-AD– and CE2–binding sites are present within regions of predicted disorder (Fig. 3C). Together, these data indicate that Ssa1 recognizes classic Hsp70 substrate-like peptide sequences found in both Hsf1 activation domains to promote stable complex formation in the absence of cellular stress.
Figure 3.

Ssa1-binding sites are required for Ssa1 to interact with the N-AD and C-AD. A, mutations designed to disrupt the LIMBO algorithm-predicted potential Hsp70-binding site in the N-AD and the known CE2 site (dark blue) in the C-AD are highlighted in red. B, Ssa1 requires intact Hsp70-binding sites to associate with the indicated N-AD or C-AD constructs described previously in the legend to Fig. 2, as demonstrated by failure to bind fusions bearing the indicated mutations (designated m50–100HGF and m520–568HGF). Immunoblots are representative of three independent experiments. C, both the N-AD and C-AD are predicted to be intrinsically disordered, with confidence scores for disorder above 0.5, including the Hsp70-binding sites highlighted in light red. Disorder was predicted by the DISOPRED3 algorithm (35).
Loss of Ssa1-binding sites leads to synergistic dysregulation of Hsf1 transcriptional activity and growth perturbation
Whereas the loss of the CE2 Ssa1-binding site has been shown to result in increased Hsf1 activity, no such information is available for the N-AD–binding site identified in this study (15, 21). Loss of the majority of the N-AD results in significantly increased Hsf1 activity and increased DNA binding in the absence of stress, indicating that a repressive element is present within residues 1–146, consistent with our identified Ssa1-binding domain (19, 21). To investigate the role of Hsp70 binding in regulating Hsf1 transcriptional activity at both activation domains, we moved the amino acid substitutions that eliminated Ssa1 binding to either the N-AD or CE2 sites or both together into full-length Hsf1 and expressed these mutants as the sole HSF1 allele. We first verified the expression level of each construct through cellular lysis and immunoblotting. The allele containing the N-AD mutation, hsf1-mN, and the CE2 mutation, hsf1-mC, were both expressed at a level similar to WT Hsf1, whereas expression of hsf1-mNmC was consistently slightly decreased (Fig. 4A). The reduced expression of hsf1-mNmC, however, contrasts with the increased levels of Ssa1 (4-fold) and Hsp90 (3.4-fold) proteins observed. We detected little to no significant increase in steady-state levels of these chaperones in cells expressing either single-domain HSF1 mutant alone. Interestingly, the growth of cells expressing the mutant constructs was slower than that of cells expressing WT HSF1 at 30 °C, with hsf1-mNmC being particularly hindered (Fig. 4B). These growth phenotypes were further exacerbated at 37 °C. Therefore, Ssa1 association with Hsf1 appears to be essential for optimal growth and heat stress tolerance, with apparent synergy between the two distinct chaperone-binding modules.
Figure 4.
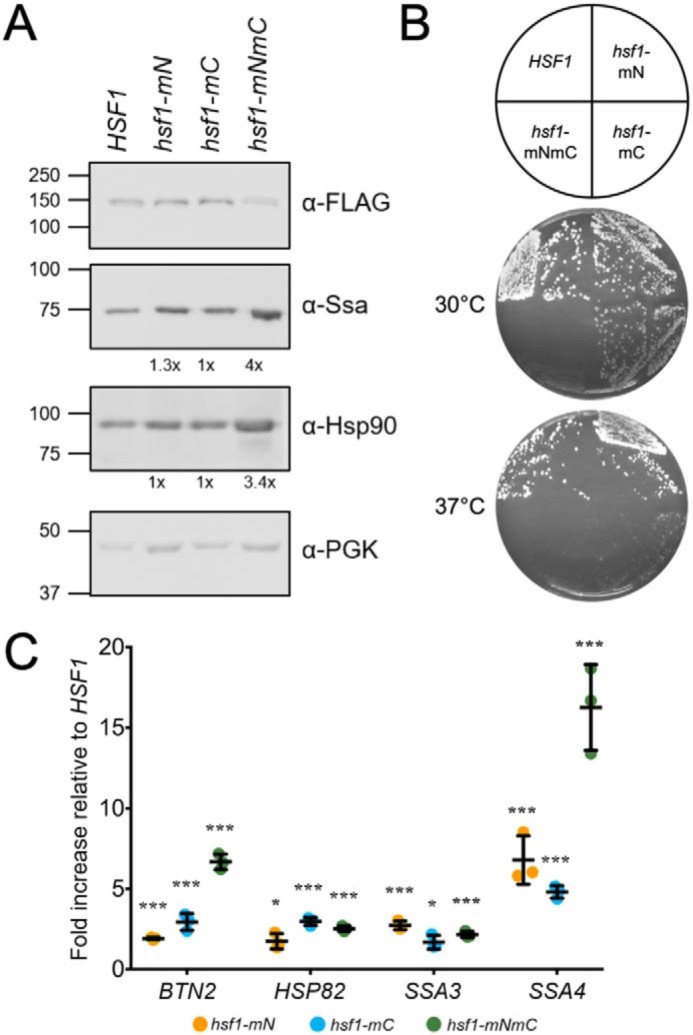
Disruption of Ssa1-binding sites in full-length Hsf1 results in constitutive activation and severe slow growth phenotypes. A, immunoblotting of cell lysates from the indicated strains expressing plasmid-based HSF1 alleles with a C-terminal FLAG tag, lacking one (hsf1-mN, hsf1-mC) or both (hsf1-mNmC) of the Ssa1-binding sites. Steady-state levels of Hsp70 (Ssa1) and Hsp90 relative to WT cells and normalized to the load control, PGK, were determined by averaging densitometric quantitation of three independent experiments and are shown below the respective panels. B, the same strains were plated onto YPD solid medium as indicated in the key and grown at 30 or 37 °C for 2 days. C, quantitative RT-PCR of the Hsf1 target genes BTN2, HSP82, SSA3, and SSA4 is shown as -fold increase in gene expression relative to the WT strain. Results from three independent experiments are shown. *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
To more directly probe the consequences of eliminating Ssa1 regulation of Hsf1 transcriptional activity, we assessed the steady-state levels of the highly heat shock–induced Hsf1-specific genes SSA3, HSP82, SSA4, and BTN2 using quantitative RT-PCR (qRT-PCR). The transcript levels induced in hsf1-mN, hsf1-mC, and hsf1-mNmC cells were significantly greater than those produced by WT for all four targets, ranging from a 1.5-fold increase of BTN2 transcripts to a 16-fold increase in expression of SSA4 (Fig. 4C). However, loss of regulation of the N-AD or C-AD did not induce the same level of derepression in all four target genes. For BTN2 and SSA4, loss of regulation at both termini appeared to act synergistically, whereas for HSP82 and SSA3, loss of regulation at either terminus was sufficient to drive transcription at the same level as observed in hsf1-mNmC cells. For all four genes tested, the levels of derepression observed were still substantially below those observed with heat shock, consistent with the absence of potentiating phosphorylation in the ADs as reported (Fig. S4) (15). These results indicated that Ssa1-mediated regulation of either AD may differentially affect transcriptional activity and that with at least some target genes, both ADs could contribute to total transcriptional output. We envisioned multiple scenarios in which this could occur. In the first model, the N-AD masks the activity of the C-AD and plays little to no role in activating transcription alone. In another model, each activation domain is capable of directing transcription of a specific subset of Hsf1 targets, perhaps dictated by promoter context and HSE architecture (30–32). In a third scenario, both ADs are capable of inducing transcription, and the presence of multiple Ssa1 regulatory sites provides a range of activation potential.
To attempt to distinguish between these possibilities, we expanded the gene expression analysis to include the entire transcriptome using RNA-Seq to compare all three HSF1 mutants at 30 °C with both WT cells and WT cells heat-shocked at 37 °C (Table S1). Consistent with the qRT-PCR results, whereas elimination of Ssa1-binding sites within the ADs leads to increases in Hsf1 transcriptional activity, the canonical HSR program is not fully engaged. During heat shock, concurrent with enhanced transcription of heat-induced genes, some gene classes are repressed, notably ribosomal protein genes (33, 34). This phenomenon is clearly observed upon examining the RPS8B and SSA4 loci, adjacently located on chromosome V. RNA-Seq reads for SSA4 mirror the results seen in our qRT-PCR data in which at 30 °C, hsf1-mN and hsf1-mC mutants displayed modest derepression that was further increased in the hsf1-mNmC strain to levels near those observed with heat shock in WT cells (Fig. 5A). Conversely, the ribosomal protein gene RPS8B maintained nearly unchanged levels of expression in the hsf1-mN, hsf1-mC, and hsf1-mNmC samples as compared with WT at 30 °C.
Figure 5.

Global dysregulation of the Hsf1 transcriptional program in the absence of regulation by Ssa1. A, mapped reads of RNA abundance from the indicated strains and for WT cells (HSF1) heat-shocked at 37 °C for 15 min for the neighboring genes RPS8B and SSA4 displayed using IGV version 2.4.15. Vertical scale is 0–25,000 for all tracks. B, radar plot of the -fold change in FPKM for the set of target genes that require Hsf1 for basal and induced gene expression, normalized to WT at 30 °C and plotted with a logarithmic radial scale. C, radar plot of the -fold change in FPKM values for the set of target genes that require Hsf1 only for heat shock induction, normalized to WT at 30 °C and plotted with a logarithmic radial scale. D, radar plot of the -fold change in FPKM values for genes with defined HSE architecture. Step, gap, and perfect HSE arrangements are as described under “Introduction.” Genes with perfect HSE architecture were further divided into those with three inverted HSE repeats or with greater than three inverted repeats.
When the FPKM (fragments per kilobase of transcript per million reads mapped) values of 18 Hsf1 target genes previously shown to be dependent on Hsf1 for basal levels of expression are normalized to transcript reads from WT cells at 30 °C, a general pattern emerges in which transcripts from hsf1-mN, hsf1-mC, and hsf1-mNmC cells exhibit up-regulation (Table S2 and Fig. 5B) (5). For example, the genes HSP42, AHA1, and HSP78 display increasing levels of induction in hsf1-mC cells compared with hsf1-mN cells, with the highest levels observed in the double hsf1-mNmC mutant. In some cases, genes in this class are even further induced upon heat stress in WT cells, due either to the activity of the general stress response transcription factors Msn2/4 or to potentiated Hsf1 activity in response to phosphorylation (33, 34). Hsf1 is the primary, if not sole, stress regulator of some genes, such as STI1, HSP82, and BTN2 (3). Transcription in the hsf1-mNmC double mutant is equal to, or even greater than, the induction that these genes undergo in cells experiencing a 37 °C heat shock. Furthermore, when we evaluated the transcript levels of genes dependent on Hsf1 for heat shock induction but not basal expression, similar trends were observed (5) (Table S2 and Fig. 5C). In the majority of these 42 previously identified transcripts, each single activation domain mutant, as well as the double mutant, demonstrated increased transcription compared with WT Hsf1. The lack of observed specificity for one AD versus the other in the surveyed Hsf1 target genes suggests that each AD has the potential to promote transcription to some degree. Not all genes follow the same trends, however, likely because of the involvement of other regulatory factors including other transcription factors, such as Msn2/4, which is also responsible for the regulation of HSP26 and GPH1 (36).
Previous investigations revealed a possible role for HSE promoter architecture in dictating Hsf1 transcriptional activity. Specifically, genes with “perfect” HSE positioning and spacing, such as the SSA gene family, maintained heat inducibility in HSF truncation mutants lacking the C-AD, whereas those with “step,” or “gap,” architecture (e.g. CUP1, HSP82, and HSC82) were reliant on this domain (20, 32, 37). Because these previous assessments all utilized Hsf1 loss-of-function mutants, our differential gain-of-function mutants provided a unique opportunity to ask whether we would observe the same dependence when AD repression was constitutively relieved via loss of the Ssa1-binding site. However, when we grouped Hsf1-dependent genes based on their promoter HSE architecture, no clear correlation emerged (Table S3 and Fig. 5D). Interestingly, genes containing perfect HSEs with greater than three repeats exhibited stronger synergistic transcriptional activation in the hsf1-mNmC mutant when compared with the other classes. Because these HSEs are thought to engage multiple Hsf1 trimers simultaneously, these results could suggest that loss of Hsp70 regulation at both sites within an Hsf1 monomer may result in increased cooperativity between trimers. Together, our gene expression analysis experiments demonstrate clear and specific chronic derepression of Hsf1 by abolishing Ssa1 binding within either AD and synergistic effects of simultaneously disrupting both Hsp70-binding sites.
The Hsp70–Hsf1 chaperone switch may be conserved in ancestrally related yeast
The presence of additional protein sequence N-terminal to the DBD is a unique feature of some fungal species, including the yeast L. kluyveri, a yeast related to the ancestor of S. cerevisiae (Fig. S5) (38). We therefore speculated that if this region likewise functions as a transcriptional AD, it may also be regulated by Hsp70. To test this theory, and to further validate the chaperone switch model, we aligned the L. kluyveri and S. cerevisiae Hsf1 amino acid sequences using the LIMBO algorithm to identify potential Hsp70-binding sites within the N-AD and C-AD regions (Fig. 6A). Protein fusions were generated between the entirety of the L. kluyveri N-AD or C-AD and the GFP-FLAG moiety. Initially, we expressed the L. kluyveri constructs in S. cerevisiae and successfully co-immunoprecipitated S. cerevisiae Hsp70, but not Hsp90, with both fusions but not the GFP-FLAG control (Fig. 6B). We then moved the same plasmids to the L. kluyveri ura3 strain and verified that our anti-Ssa antiserum recognized the endogenous Ssa protein (39). Upon FLAG immunoprecipitation, the L. kluyveri Hsp70 specifically associated with both protein fusions (Fig. 6C). These results provide additional evidence that Hsp70 interacts with fungal Hsf1 transcription factors through independent binding sites located within conserved domains, suggesting conservation of the chaperone switch.
Figure 6.

Ancestrally related L. kluyveri yeast demonstrate potential for Hsp70-mediated regulation of Hsf1 transcriptional activity. A, schematic of S. cerevisiae (S.c.) and L. kluyveri (L.k.) Hsf1 proteins with sequence alignments of LIMBO-identified potential Hsp70 recognition sites within both ADs highlighted in the bottom half of the panel. B, Ssa1, but not Hsp90, co-immunoprecipitates with L. kluyveri N-AD (Lk-N-HGF) and C-AD (Lk-C-HGF) protein fusions when expressed in S. cerevisiae. C, the presumptive L. kluyveri Ssa homolog, but not Hsp90, co-immunoprecipitates with L. kluyveri N-AD (Lk-N-HGF) and C-AD (Lk-C-HGF) protein fusions when expressed in L. kluyveri. IP, immunoprecipitation.
Discussion
Repression of yeast Hsf1 transcriptional activity during optimal conditions and attenuation following a thermal stress has been recently demonstrated to depend exclusively on direct binding by Hsp70 (15, 19). The mechanism that determines how this interaction between Hsp70 and Hsf1 is capable of preventing transcriptional activity is still incompletely understood. In this study, we confirm the Hsp70-binding site (CE2) located within the C-AD and further define a second site located within the N-AD. Introducing substitutions that eliminate Hsp70 binding within each site independently, and to a greater extent simultaneously, creates gain-of-function HSF1 alleles that are constitutively activated in the absence of stress. This dysregulation of Hsf1-dependent transcription results in compromised growth and, contrary to expectations, does not provide enhanced protection from elevated temperatures but rather hypersensitivity. Together, these results suggest that Hsf1 transcriptional competence must be tightly regulated to maximize cellular fitness.
Previous studies in metazoans supported a model wherein Hsp90 interacts with monomeric Hsf1 to prevent trimerization and translocation into the nucleus (25). Additionally, studies in Candida albicans have demonstrated that the loss of Hsp90 results in derepression of the heat shock response and a reduced capacity for macrophage infection, suggesting that Hsp90 may play a role in Hsf1 regulation in that organism (40, 41). However, in budding yeast, Hsf1 is constitutively trimerized and localized to the nucleus, where it can bind DNA and drive basal transcription of several key proteins, including Hsp70 and Hsp90 (5, 18). In this scenario, regulation by Hsp90 at the level of nuclear translocation and trimerization is unnecessary, consistent with our in vivo experiments that do not support a stable interaction between Hsp90 and Hsf1. It is possible that chronic loss of Hsp90 may contribute to overall proteotoxic stress due to accumulation of misfolded proteins, rather than direct regulation of Hsf1. This model then requires a sensor for misfolded proteins that can signal Hsf1 to activate the HSR, a role that Hsp70 is well-suited to play. Hsp70 appears to be capable of distinguishing between different types of stress, as two cysteines in Ssa1 were shown to be required for Hsf1 activation by thiol-reactive compounds but not for activation by heat shock (14).
Expression of Hsf1-dependent HSR genes is partially dependent on the architecture of the HSEs within the promoter region. Sequences with high affinity are prebound by Hsf1 prior to proteotoxic stress, with Hsf1 binding of low-affinity sequences occurring during derepression of Hsf1 activity (18, 42, 43). Whereas the different promoter affinities are likely driven via variable and/or cooperative interactions with individual DBDs in each Hsf1 monomer, the activation potential of HSR genes may be additionally dependent on the individual ADs. In this model, genes dependent on Hsf1 for either basal or induced expression would segregate into several groups: those that required derepression of the N-AD, the C-AD, or both. When we analyzed the behavior of these genes via RNA-Seq, the predominant pattern we observed was that the hsf1-mN and hsf1-mC mutants each drove a higher level of transcription compared with WT at 30 °C and that the double hsf1-mNmC mutant was even further activated. These data are consistent with the ADs operating in a nonspecific fashion, at least in the absence of heat shock, which also induces abundant phosphorylation of Hsf1 and significant enhancement of transcriptional potency. The observation that the hsf1-mNmC mutant exhibited the greatest synergistic transcriptional activation in Hsf1-dependent genes with three or more HSE units arranged in the perfect orientation suggests that cooperativity between trimers of Hsf1 may also be impacted by the dysregulation of the activation domains. In addition to cooperativity between trimers within a promoter region, Hsf1 also has the potential to interact with other promoter-bound Hsf1 trimers during the chromatin remodeling undertaken during activation of the HSR (44). These constitutively active Hsf1 mutants may be promoting chromatin remodeling even under nonstress conditions, although the presence of at least one Hsp70-binding site is capable of maintaining some repression of Hsf1 transcriptional activity. Work from Gross and co-workers (44, 45) recently identified the intriguing phenomenon of transcriptional clustering, wherein spatially distant heat shock loci coalesce in three-dimensional space upon Hsf1 binding to potentiate gene activation. It is tempting to speculate that Ssa1 may regulate this step as well, possibly inhibiting clustering through steric hindrance or obfuscation of Hsf1 regions promoting association. Recent work by Pincus and colleagues (19) suggests that the N-terminal repressive domain is linked to Hsf1 DNA binding. Our findings do not rule out the possibility that elimination of Hsp70 binding in the N-AD results in increased DNA binding, but they are most consistent with this regulatory site playing the same role for the N-AD as the CE2 site plays for the C-AD.
The potential for Hsp70-mediated regulation of Hsf1 activity is not limited to S. cerevisiae and L. kluyveri. Although the presence of an extended N-terminal activation domain appears to be limited to some fungal species, all known HSFs possess a C-AD (Fig. S5). However, whereas the DNA-binding domain and trimerization domain are well-conserved at the primary sequence level between species, the AD(s) exhibits little to no conservation. The identification of Hsp70-binding sites in the fungal Hsf1 ADs is consistent with the previous demonstration of a similar interaction in human HSF1, although no binding site was defined in this study (8). It is therefore likely that regulation of HSF by both Hsp70 and Hsp90 could occur in some species. Although HSFs in metazoans and some fungi are differentially localized during nonstress conditions, the trimerization and translocation of HSF1 into the nucleus of human cells does not always result in transcriptional activity (46). This suggests that whereas Hsp90 may repress HSF1 trimerization and translocation, Hsp70 may prevent transcriptional activity even while HSF1 is DNA-bound in both yeast and human cells. Furthermore, in budding yeast, where Hsf1 is essential due to basal transcription of HSP70 and HSP90 genes, the presence of the extended N-terminal activation domain may provide another regulatory step as well as additional transcriptional activation potential (5). Whether a correlation exists between organisms in which HSF is constitutively nuclear and the presence of an extended N terminus with an Hsp70-binding site remains to be determined. Clearly, despite over 30 years of investigation, our knowledge of the intricate mechanisms of HSF regulation remains incomplete. Given the growing appreciation of the importance of the HSF-controlled proteostasis network in human health and disease, the challenge of fully understanding this enigmatic transcription factor remains a priority.
Experimental procedures
Strains and plasmids
The Saccharomyces cerevisiae strains used in this study are BY4741 (MATα his3Δ0 leu2Δ0 met15Δ0 ura3Δ0) and the isogenic strain DNY248 (hsf1Δ::KanMX, pRS316-yHSF1), kindly provided by Dennis Thiele (Duke University, Durham, NC) (47). The L. kluyveri ura3 FM628 strain was a kind gift from Ambro van Hoof (McGovern Medical School, Houston, TX) (39). All plasmids were transformed into BY4741, DNY248, and FM628 using the rapid yeast transformation protocol (48).
To construct the C-terminal FLAG-tagged HSF1, HSF1 was amplified from BY4741 genomic DNA with a 3′ primer containing a triple FLAG tag repeat and XhoI restriction site and a 5′ primer containing an XbaI site. Restriction digestion and ligation was used to insert the HSF1-FLAG amplicon into the pRS413-TEF expression vector, and XmaI was used instead of XhoI in a separate 3′ primer to create the pRS413-CYC expression vector (49). Construction of pRS416-TEF-Lk-N-HGF and pRS416-TEF-Lk-C-HGF required amplification of HSF1 nucleotides 1–498 and 1108–1671 from L. kluyveri and followed the same XbaI, XhoI cloning scheme. To construct the Hsp70-binding site mutants, primers were designed with nucleotide mismatches at the identified sites and used to amplify the N- or C-terminal constructs. PCR overlap was then used to create the full-length HSF1 amplicons, and insert and vector were subject to restriction digestion and ligation. The control plasmid pRS413-TEF-GFP-FLAG was constructed by amplification of GFP with 5′-XhoI/5′-XmaI and 3′-XbaI-Flag primers and insertion into the pRS413-TEF/pRS413-CYC expression vectors, respectively. All fusions were screened for aggregation via microscopy of the GFP moiety (Fig. S3). Plasmids were rescued from Escherichia coli and verified by sequencing. Generation of full-length WT and mutant HSF1 expression plasmids in DNY248 was conducted using a standard plasmid shuffle technique with counterselection of the WT HSF1 expression plasmid by 5-fluoroorotic acid. To measure the real-time transcriptional dynamics of Hsf1, plasmid pAG413-GRE2-lucCP+ (a kind gift from M. Proft, Institute for Plant Molecular and Cellular Biology, Valencia, Spain) was modified as follows (23). A portion of the SSA3 promoter, from −236 to +14, was amplified from BY4741 using oligonucleotides with SacI or XmaI sites at the 5′ and 3′ ends, respectively. Restriction digestion using SacI and XmaI was used to liberate the GRE2 promoter, and ligation was used to insert the HSE-containing promoter. To switch the HIS3 cassette in the original reporter plasmid, the URA3 gene was amplified from pRS416 using oligonucleotides containing homologous 5′ and 3′ regions of the HIS3 gene and pAG413. The resulting URA3 amplicon and pAG413-HSE-lucCP+ were co-transformed into BY4741 cells, selecting for Ura+ His− transformants arising through homologous recombination. The modified plasmid was rescued into E. coli. The Ssa1 nucleotide-binding domain and substrate-binding domain were liberated from plasmids pRS416-GPD-ssa1-ATPase and pRS416GPD-ssa1-SBD using SpeI and XhoI and ligated into plasmid pRS425TEF. Plasmids were transformed into E. coli, rescued, and verified by restriction digestion and sequencing.
Cellular culture and growth analysis
S. cerevisiae and L. kluyveri strains were cultured in yeast extract, peptone, dextrose medium (YPD) or on synthetic complete (SC) medium (Sunrise Science, San Diego, CA) lacking the nutrient for appropriate plasmid selection, as indicated. All cells were grown at 30 °C unless otherwise noted. To analyze growth of DNY248 hsf1Δ::kanMX-expressing WT and mutant Hsf1 proteins, cells were grown in selective media to mid-log phase and washed, and optical density was equalized. Wooden sticks were used to spread cultures on YPD solid medium agar plates. The cells were allowed to grow at the temperatures and amount of time indicated.
Cellular lysis and immunoprecipitation
Strains were grown to mid-log phase, and proteins were isolated following glass bead lysis as described previously (50). For FLAG immunoprecipitation, 30 ml of mid-log phase cells were lysed, and the total cell lysate was combined with anti-FLAG M2 Affinity gel (Sigma) and rocked for 2 h at 4 °C in a total volume of 700 μl of TEGN (20 mm Tris-HCl, pH 7.9, 0.5 mm EDTA, 10% glycerol (v/v), 50 mm NaCl) plus protease inhibitors (50). Beads were washed eight times with 750 μl of TEGN + protease inhibitors, and proteins were eluted at room temperature for 25 min in the presence of 40 μl of FLAG peptide (200 μg/ml). 6× SDS sample buffer (350 mm Tris-HCl, pH 6.8, 36% glycerol (v/v), 10% SDS (w/v), 5% β-mercaptoethanol (w/v), and 0.012% bromphenol blue (w/v)) was added to cell lysates and immunoprecipitated samples before boiling at 65 °C for 10 min.
Immunoblot analysis
Proteins were separated with SDS-PAGE and transferred to polyvinylidene difluoride membrane (EMD Millipore). Immunoblot analysis was performed using anti-Ssa1/2 polyclonal antibody at a 1:10,000 dilution (generous gift from M. Ptashne, Sloan Kettering Institute), anti-FLAG mAb at a 1:4,000 dilution (Sigma), anti-Hsp90 polyclonal at a 1:4,000 dilution (kindly provided by Dr. Avrom Caplan, CUNY, NY), using a procedure described previously (50). Blots were coated with Hy-Glo ECL spray briefly before exposure and image capture by the ImageQuant LAS 4000 mini (GE Healthcare) product and software. Protein bands were quantitated using Image Studio Lite (LI-COR Biosciences).
Fluorescence microscopy
For all images, live cells were wet-mounted onto glass slides and immediately imaged using a ×100 objective with an FITC filter to detect GFP on an Olympus IX81 microscope (Waltham, MA), captured with a Hamamatsu (Bridgewater, NJ) ORCA camera.
Real-time luciferase activity assay
Cells expressing the pHSE-lucCP+ plasmid were grown to mid-log phase at 30 °C. Activity of Hsf1 was determined by adding luciferin (final concentration 0.5 mm) and distributing 150-μl aliquots of the cultures into a white 96-well plate (Lumitrac 200, Greiner). Cells were incubated in a Synergy MX Microplate reader (Biotek Instruments, Winooski, VT) at 37 °C for 90 min, and luminescence was read every 3 min (23). Graph was prepared using GraphPad Prism 7.
RNA isolation and qRT-PCR
Cells were grown in 20 ml of YPD medium at 30 °C or heat-shocked at 37 °C for 15 min, harvested at A600 = 0.7, and centrifuged, and the pellet was immediately frozen in liquid nitrogen. Total RNA was isolated by the hot phenol method as described previously (51). For qRT-PCR assays, 1 μg of RNA was converted to cDNA using the iScript cDNA synthesis kit (Bio-Rad). Relative expression of the genes HSP82, BTN2, SSA3, and SSA4 was measured by qRT-PCR using iTaq Universal SYBR Green Supermix (Bio-Rad) and calculated using standard methods (52). Due to variance in ACT1 gene expression in response to heat shock, TAF10 was used as the normalization control gene. All experiments were conducted with three biological replicates. Significance was calculated using GraphPad QuickCalcs Welch's unpaired t test calculator.
RNA-Seq
RNA was isolated as above with three independent biological replicates per sample; sequencing, quality control, and alignment to the S. cerevisiae genome were performed by Novogene (Chula Vista, CA). RNA was sequenced by paired-end 150-bp Illumina sequencing, and ∼20 million reads passed quality control with over 85% uniquely mapped to S. cerevisiae ORFs through Tophat (version 2.0.12). Gene expression was analyzed on the HTSeq platform (version 0.6.1), with analysis and normalization of differential expression by DESeq (version 1.12.0). Statistical validation by DESeq (version 1.12.0) and one-way analysis of variance is provided in Table S4. Transcript read counts were visualized in the Integrative Genomics Viewer (IGV) software available from the Broad Institute (53). Normalized and corrected FPKM values, as supplied by Novogene, were analyzed in Microsoft Excel to produce radar plots demonstrating gene expression.
Author contributions
S. P. and K. A. M. conceptualization; S. P. and K. A. M. data curation; S. P., D. G., and K. A. M. formal analysis; S. P., D. G., and K. A. M. investigation; S. P. and D. G. methodology; S. P. writing-original draft; S. P., D. G., and K. A. M. writing-review and editing; K. A. M. supervision; K. A. M. funding acquisition; K. A. M. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Allan Drummond and Dr. David Pincus (University of Chicago) for helpful discussions and Amoldeep Kainth and Dr. David Gross (Louisiana State University, Shreveport, LA) for advice and support. We thank Dr. Dennis Thiele (Duke University) for providing the hsf1Δ knockout strain and Dr. Markus Proft (Institute for Plant Molecular and Cellular Biology, Valencia, Spain) for sharing the lucCP+ system. Dr. Ambro van Hoof (McGovern Medical School, Houston, TX) is acknowledged for assistance with RNA-Seq.
This work was supported by National Institutes of Health Grant R01-GM127287. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Raw and processed data are available from the Gene Expression Omnibus (GEO) archive, record GSE129832.
This article contains Tables S1–S4 and Figs. S1–S5.
- HSR
- heat shock response
- HSF
- heat shock factor
- AD
- activation domain
- C-AD
- C-terminal transcriptional activation domain
- N-AD
- N-terminal transcriptional activation domain
- DBD
- DNA-binding domain
- Hsp
- heat shock protein
- CE2
- control element 2
- HSE
- heat shock element
- NBD
- nucleotide-binding domain
- SBD
- substrate-binding domain
- HGF
- Hsf1-GFP-FLAG
- qRT-PCR
- quantitative RT-PCR
- FPKM
- fragments per kilobase of transcript per million reads mapped.
References
- 1. Morano K. A., Grant C. M., and Moye-Rowley W. S. (2012) The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics 190, 1157–1195 10.1534/genetics.111.128033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akerfelt M., Morimoto R. I., and Sistonen L. (2010) Heat shock factors: integrators of cell stress, development, and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555 10.1038/nrm2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamamoto A., Mizukami Y., and Sakurai H. (2005) Identification of a novel class of target genes and a novel type of binding sequence of heat shock transcription factor in Saccharomyces cerevisiae. J. Biol. Chem. 280, 11911–11919 10.1074/jbc.M411256200 [DOI] [PubMed] [Google Scholar]
- 4. Hashikawa N., Yamamoto N., and Sakurai H. (2007) Different mechanisms are involved in the transcriptional activation by yeast heat shock transcription factor through two different types of heat shock elements. J. Biol. Chem. 282, 10333–10340 10.1074/jbc.M609708200 [DOI] [PubMed] [Google Scholar]
- 5. Solís E. J., Pandey J. P., Zheng X., Jin D. X., Gupta P. B., Airoldi E. M., Pincus D., and Denic V. (2016) Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol. Cell 63, 60–71 10.1016/j.molcel.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gomez-Pastor R., Burchfiel E. T., and Thiele D. J. (2018) Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell. Biol. 19, 4–19 10.1038/nrm.2017.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mendillo M. L., Santagata S., Koeva M., Bell G. W., Hu R., Tamimi R. M., Fraenkel E., Ince T. A., Whitesell L., and Lindquist S. (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562 10.1016/j.cell.2012.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abravaya K., Myers M. P., Murphy S. P., and Morimoto R. I. (1992) The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 6, 1153–1164 10.1101/gad.6.7.1153 [DOI] [PubMed] [Google Scholar]
- 9. Baler R., Zou J., and Voellmy R. (1996) Evidence for a role of Hsp70 in the regulation of the heat shock response in mammalian cells. Cell Stress Chaperones 1, 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neef D. W., Jaeger A. M., Gomez-Pastor R., Willmund F., Frydman J., and Thiele D. J. (2014) A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 9, 955–966 10.1016/j.celrep.2014.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Craig E. A., and Jacobsen K. (1984) Mutations of the heat inducible 70 kilodalton genes of yeast confer temperature sensitive growth. Cell 38, 841–849 10.1016/0092-8674(84)90279-4 [DOI] [PubMed] [Google Scholar]
- 12. Duina A. A., Kalton H. M., and Gaber R. F. (1998) Requirement for Hsp90 and a CyP-40-type cyclophilin in negative regulation of the heat shock response. J. Biol. Chem. 273, 18974–18978 10.1074/jbc.273.30.18974 [DOI] [PubMed] [Google Scholar]
- 13. Harris N., MacLean M., Hatzianthis K., Panaretou B., and Piper P. W. (2001) Increasing Saccharomyces cerevisiae stress resistance, through the overactivation of the heat shock response resulting from defects in the Hsp90 chaperone, does not extend replicative lifespan but can be associated with slower chronological ageing of nondividing cells. Mol. Genet. Genomics 265, 258–263 10.1007/s004380000409 [DOI] [PubMed] [Google Scholar]
- 14. Wang Y., Gibney P. A., West J. D., and Morano K. A. (2012) The yeast Hsp70 Ssa1 is a sensor for activation of the heat shock response by thiol-reactive compounds. Mol. Biol. Cell 23, 3290–3298 10.1091/mbc.e12-06-0447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng X., Krakowiak J., Patel N., Beyzavi A., Ezike J., Khalil A. S., and Pincus D. (2016) Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. eLife 5, e18638 10.7554/eLife.18638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sorger P. K., Lewis M. J., and Pelham H. R. B. (1987) Heat shock factor is regulated differently in yeast and HeLa cells. Nature 329, 81–84 10.1038/329081a0 [DOI] [PubMed] [Google Scholar]
- 17. Budzyński M. A., Puustinen M. C., Joutsen J., and Sistonen L. (2015) Uncoupling stress-inducible phosphorylation of heat shock factor 1 from its activation. Mol. Cell. Biol. 35, 2530–2540 10.1128/MCB.00816-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jakobsen B. K., and Pelham H. R. (1988) Constitutive binding of yeast heat shock factor to DNA in vivo. Mol. Cell. Biol. 8, 5040–5042 10.1128/MCB.8.11.5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krakowiak J., Zheng X., Patel N., Feder Z. A., Anandhakumar J., Valerius K., Gross D. S., Khalil A. S., and Pincus D. (2018) Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. eLife 7, e31668 10.7554/eLife.31668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Santoro N., Johansson N., and Thiele D. J. (1998) Heat shock element architecture is an important determinant in the temperature and transactivation domain requirements for heat shock transcription factor. Mol. Cell. Biol. 18, 6340–6352 10.1128/MCB.18.11.6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sorger P. K. (1990) Yeast heat shock factor contains separable transient and sustained response transcriptional activators. Cell 62, 793–805 10.1016/0092-8674(90)90123-V [DOI] [PubMed] [Google Scholar]
- 22. Borkovich K. A., Farrelly F. W., Finkelstein D. B., Taulien J., and Lindquist S. (1989) Hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol. Cell. Biol. 9, 3919–3930 10.1128/MCB.9.9.3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rienzo A., Pascual-Ahuir A., and Proft M. (2012) The use of a real-time luciferase assay to quantify gene expression dynamics in the living yeast cell. Yeast 29, 219–231 10.1002/yea.2905 [DOI] [PubMed] [Google Scholar]
- 24. Morano K. A., Santoro N., Koch K. A., and Thiele D. J. (1999) A trans-activation domain in yeast heat shock transcription factor is essential for cell cycle progression during stress. Mol. Cell. Biol. 19, 402–411 10.1128/MCB.19.1.402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zou J., Guo Y., Guettouche T., Smith D. F., and Voellmy R. (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 Complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480 10.1016/S0092-8674(00)81588-3 [DOI] [PubMed] [Google Scholar]
- 26. Zuiderweg E. R., Hightower L. E., and Gestwicki J. E. (2017) The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 22, 173–189 10.1007/s12192-017-0776-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Flaherty K. M., DeLuca-Flaherty C., and McKay D. B. (1990) Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 346, 623–628 10.1038/346623a0 [DOI] [PubMed] [Google Scholar]
- 28. Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. E., and Hendrickson W. A. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614 10.1126/science.272.5268.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Durme J., Maurer-Stroh S., Gallardo R., Wilkinson H., Rousseau F., and Schymkowitz J. (2009) Accurate prediction of DnaK-peptide binding via homology modelling and experimental data. PLoS Comput. Biol. 5, e1000475 10.1371/journal.pcbi.1000475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hashikawa N., and Sakurai H. (2004) Phosphorylation of the yeast heat shock transcription factor is implicated in gene-specific activation dependent on the architecture of the heat shock element. Mol. Cell. Biol. 24, 3648–3659 10.1128/MCB.24.9.3648-3659.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nieto-Sotelo J., Wiederrecht G., Okuda A., and Parker C. S. (1990) The yeast heat shock transcription factor contains a transcriptional activation domain whose activity is repressed under nonshock conditions. Cell 62, 807–817 10.1016/0092-8674(90)90124-W [DOI] [PubMed] [Google Scholar]
- 32. Tamai K. T., Liu X., Silar P., Sosinowski T., and Thiele D. J. (1994) Heat shock transcription factor activates yeast metallothionein gene expression in response to heat and glucose starvation via distinct signalling pathways. Mol. Cell. Biol. 14, 8155–8165 10.1128/MCB.14.12.8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Causton H. C., Ren B., Koh S. S., Harbison C. T., Kanin E., Jennings E. G., Lee T. I., True H. L., Lander E. S., and Young R. A. (2001) Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12, 323–337 10.1091/mbc.12.2.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gasch A. P., Spellman P. T., Kao C. M., Carmel-Harel O., Eisen M. B., Storz G., Botstein D., and Brown P. O. (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11, 4241–4257 10.1091/mbc.11.12.4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jones D. T., and Cozzetto D. (2015) DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863 10.1093/bioinformatics/btu744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moskvina E., Schüller C., Maurer C. T. C., Mager W. H., and Ruis H. (1998) A search in the genome of Saccharomyces cerevisiae for genes regulated via stress response elements. Yeast 14, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 37. Sakurai H., and Takemori Y. (2007) Interaction between heat shock transcription factors (HSFs) and divergent binding sequences: binding specificities of yeast HSFs and human HSF1. J. Biol. Chem. 282, 13334–13341 10.1074/jbc.M611801200 [DOI] [PubMed] [Google Scholar]
- 38. Kellis M., Birren B. W., and Lander E. S. (2004) Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428, 617–624 10.1038/nature02424 [DOI] [PubMed] [Google Scholar]
- 39. Marshall A. N., Montealegre M. C., Jiménez-López C., Lorenz M. C., and van Hoof A. (2013) Alternative splicing and subfunctionalization generates functional diversity in fungal proteomes. PLoS Genet. 9, e1003376 10.1371/journal.pgen.1003376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leach M. D., Tyc K. M., Brown A. J., and Klipp E. (2012) Modeling the regulation of thermal adaptation in Candida albicans, a major fungal pathogen of humans. PLoS One 7, e32467 10.1371/journal.pone.0032467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leach M. D., Farrer R. A., Tan K., Miao Z., Walker L. A., Cuomo C. A., Wheeler R. T., Brown A. J. P., Wong K. H., and Cowen L. E. (2016) Hsf1 and Hsp90 orchestrate temperature-dependent global transcriptional remodelling and chromatin architecture in Candida albicans. Nat Commun. 7, 11704 10.1038/ncomms11704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Erkine A. M., Magrogan S. F., Sekinger E. A., and Gross D. S. (1999) Cooperative binding of heat shock factor to the yeast HSP82 promoter in vivo and in vitro. Mol. Cell. Biol. 19, 1627–1639 10.1128/MCB.19.3.1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pincus D., Anandhakumar J., Thiru P., Guertin M. J., Erkine A. M., and Gross D. S. (2018) Genetic and epigenetic determinants establish a continuum of Hsf1 occupancy and activity across the yeast genome. Mol. Biol. Cell 29, 3168–3182 10.1091/mbc.E18-06-0353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chowdhary S., Kainth A. S., and Gross D. S. (2017) Heat shock protein genes undergo dynamic alteration in their three-dimensional structure and genome organization in response to thermal stress. Mol. Cell. Biol. 37, e00292–17 10.1128/MCB.00292-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chowdhary S., Kainth A. S., Pincus D., and Gross D. S. (2019) Heat shock factor 1 drives intergenic association of its target gene loci upon heat shock. Cell Rep. 26, 18–28.e5 10.1016/j.celrep.2018.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cotto J. J., Kline M., and Morimoto R. I. (1996) Activation of heat shock factor 1 DNA binding precedes stress-induced serine phosphorylation: evidence for a multistep pathway of regulation. J. Biol. Chem. 271, 3355–3358 10.1074/jbc.271.7.3355 [DOI] [PubMed] [Google Scholar]
- 47. Liu X. D., Liu P. C., Santoro N., and Thiele D. J. (1997) Conservation of a stress response: human heat shock transcription factors functionally substitute for yeast HSF. EMBO J. 16, 6466–6477 10.1093/emboj/16.21.6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gietz D., St Jean A., Woods R. A., and Schiestl R. H. (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20, 1425 10.1093/nar/20.6.1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mumberg D., Müller R., and Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 10.1016/0378-1119(95)00037-7 [DOI] [PubMed] [Google Scholar]
- 50. Verghese J., and Morano K. A. (2012) A lysine-rich region within fungal BAG domain-containing proteins mediates a novel association with ribosomes. Eukaryot. Cell 11, 1003–1011 10.1128/EC.00146-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., and Struhl K. (2003) Current Protocols in Molecular Biology, Ringbou Ed, John Wiley & Sons, New York [Google Scholar]
- 52. Nolan T., Hands R. E., and Bustin S. A. (2006) Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559–1582 10.1038/nprot.2006.236 [DOI] [PubMed] [Google Scholar]
- 53. Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., and Mesirov J. P. (2011) Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.