

Abstract
Apolipoprotein B mRNA editing enzyme catalytic subunit-like protein 3B (APOBEC3B or A3B), as other APOBEC3 members, is a single-stranded (ss)DNA cytosine deaminase with antiviral activity. A3B is also overexpressed in multiple tumor types, such as carcinomas of the bladder, cervix, lung, head/neck, and breast. A3B generates both dispersed and clustered C-to-T and C-to-G mutations in intrinsically preferred trinucleotide motifs (TCA/TCG/TCT). A3B-catalyzed mutations are likely to promote tumor evolution and cancer progression and, as such, are associated with poor clinical outcomes. However, little is known about cellular processes that regulate A3B. Here, we used a proteomics approach involving affinity purification coupled to MS with human 293T cells to identify cellular proteins that interact with A3B. This approach revealed a specific interaction with cyclin-dependent kinase 4 (CDK4). We validated and mapped this interaction by co-immunoprecipitation experiments. Functional studies and immunofluorescence microscopy experiments in multiple cell lines revealed that A3B is not a substrate for CDK4–Cyclin D1 phosphorylation nor is its deaminase activity modulated. Instead, we found that A3B is capable of disrupting the CDK4-dependent nuclear import of Cyclin D1. We propose that this interaction may favor a more potent antiviral response and simultaneously facilitate cancer mutagenesis.
Keywords: DNA viruses, cell cycle, virus, proteomics, retrovirus, APOBEC3B, cancer mutagenesis, CDK4, innate antiviral immunity
Introduction
Human apolipoprotein B mRNA editing enzyme catalytic subunit-like protein 3B (APOBEC3B or A3B)3 is a member of a larger family of zinc-dependent cytosine deaminases that convert cytosine bases to uracils in single-stranded (ss)DNA (1–6). APOBEC3 family members have overlapping roles in restricting the replication of a variety of DNA-based viruses. For instance, several APOBEC3 family members including A3B inflict DNA C-to-U lesions in reverse-transcribing retroviruses and retrotransposons (6–9). In addition, A3B has elicited restriction activity against EBV, HBV, papillomaviruses, and polyomaviruses (9–13).
Recently, A3B has also been implicated as a source of mutation in various cancer types, with major influences in primary breast tumors (comprising 20% of base substitutions) and in metastatic disease (mutational contribution greater than 50%) (2, 4, 14–23). Additionally, A3B has been correlated with poor clinical outcomes including drug resistance (24–32). Efforts are being focused on developing therapies to inhibit A3B-mediated mutagenesis in cancer as an adjuvant to current treatment strategies (33). However, post-translational regulation of A3B enzymatic activity, the molecular mechanisms of how it gains access to ssDNA, and how it enters the nuclear compartment have remained elusive “black boxes.”
Interestingly, A3B is the only family member shown to be constitutively nuclear (34–37). After mitosis and nuclear membrane reconstitution, A3B is rapidly reimported into the nuclear compartment where it remains constitutively present until the next mitotic cell division (34, 35, 38). Although the mechanism of nuclear import has yet to be fully elucidated, the identification of A3B-interacting factors may shed light in this process.
The cell cycle is comprised of a complex set of interacting proteins that tightly regulate progression through each phase. A key component of cell-cycle regulation is the family of serine/threonine cyclin-dependent kinases (CDK) (39–43). CDKs cooperate with various Cyclin proteins to regulate cell-cycle checkpoints (41). Cells must progress through four phases of the cell cycle to divide and replicate: G1, S phase (DNA synthesis), G2, and M phase (mitosis). The key regulator of the G1/S transition is CDK4, which forms a complex with Cyclin D1 (encoded by CCND1) and inactivates the retinoblastoma protein RB1 through phosphorylation of residue Ser780 (44, 45). This relieves RB1-mediated inhibition of the transcription factor E2F, which commits the cell to progression through the cell cycle (43, 46–48). Although there are many CDK-dependent points of cell-cycle regulation, the regulation of the G1/S transition via CDK4–Cyclin D1 is perturbed in a large proportion of human cancers (49–52).
Our studies here began with the goal of identifying direct protein–protein interactors that post-translationally regulate A3B. We used affinity purification coupled to MS (AP-MS) to identify CDK4 as a high-confidence A3B interactor. AP-MS results were validated by showing that A3B interacts directly with CDK4 but not with the closely related family members CDK2 or CDK6. Additionally, we use structure-guided mutagenesis to define the regions of both A3B and CDK4 required for interaction. We could also engraft key residues from CDK4 into CDK2 and endow it with the ability to interact with A3B. Finally, we demonstrated that both endogenous and exogenous expression of A3B could disrupt CDK4-dependent import of Cyclin D1, thereby providing a novel post-translational function for A3B.
Results
AP-MS revealed a specific interaction between A3B and the cell-cycle regulator CDK4
To identify direct cellular regulators of A3B, we utilized AP-MS to identify A3B-interacting proteins. We expressed an A3B-2×Strep-3×Flag-tagged (hereafter referred to as A3B-SF) construct in 293T cells followed by affinity purification via anti-Strep resin and stringent high salt washes to enrich for tightly bound proteins. Immunoblot analysis, Coomassie gels, and activity assays using ssDNA oligonucleotide substrates were used to validate the presence, enrichment, and activity of affinity-purified A3B (schematic in Fig. 1A and data not shown). As controls, eGFP-SF, A3A-SF, and A3G-SF constructs were expressed in 293T cells and affinity purified in parallel. Co-immunoprecipitated samples were digested with trypsin and subjected to analysis by MS (Fig. 1A). To our surprise, we identified CDK4 as a putative A3B-interacting protein based on the recovery of multiple unique peptides across six independent AP-MS experiments (cumulatively spanning 24.7% of CDK4 amino acid sequence; Fig. 1, B and C). Additionally, CDK4 was not identified in the eGFP-SF, A3A-SF, or A3G-SF control experiments. Interestingly, even though CDK4 shares high homology with closely related family members (70% identity with CDK6 and 42% with CDK2), it was the only CDK and the only cell-cycle protein identified in our AP-MS datasets. Furthermore, Cyclin D1, the canonical binding partner of CDK4, was not present in any of the A3B-SF AP-MS datasets.
Figure 1.

AP-MS reveals an interaction between A3B and CDK4. A, schematic of the AP-MS workflow used to identify A3B-interacting proteins. B, summary of the total spectral counts (SC) and percent coverage for A3B and CDK4. C, amino acid sequence of CDK4 with AP-MS peptides highlighted in red.
Experimental validation of A3B–CDK4 interaction
To validate our AP-MS results implicating an A3B–CDK4 interaction, we performed an immunoprecipitation (IP) of eGFP-SF and A3B-SF in 293T cells. Immunoblot analysis confirmed that endogenous CDK4 could IP with A3B-SF but not with eGFP-SF (Fig. 2A). A similar approach was taken to IP endogenous A3B; however, despite multiple attempts in various cell lines using several commercial and custom in-house antibodies, immunoprecipitation of endogenous A3B was unsuccessful due to an overt failure to bind A3B, cross-reactivity to related APOBEC3s, and/or inferred low affinity (i.e. all were non-IP grade reagents; data not shown).
Figure 2.

Confirmation of the A3B–CDK4 interaction through IP experiments. A, co-IP of endogenous CDK4 in 293T cells transfected with A3B-SF. Parallel reactions with eGFP-SF serve as a negative control. Upper two immunoblots show the indicated proteins in whole cell lysates (input), and the bottom two immunoblots show the immunoprecipitated samples (elution). kDa markers are shown to the left of each blot and the primary antibody used for detection is shown to the right. B, co-IP results for Strep- and Flag-tagged CDK4 (upper blots) and Strep- and Flag-tagged A3B (bottom blots) from 293T cells. The asterisk denotes residual signal from the IP antibody due to insufficient stripping. C, immunoblots showing an interaction between E. coli expressed His-tagged A3BE255A and untagged CDK4. Immunoblot labels are as follows: insoluble pellet (P), lysate (L), and His purification (H). D, co-IP of the indicated Flag-tagged CDK4 constructs with Strep-tagged A3 constructs in 293T cells. E, fluorescence microscopy images of PLA results showing the interaction between A3B and CDK4 as well as CDK4 and Cyclin D1 in MCF10A cells treated with PMA to induce A3B expression (scale bar, 50 μm).
To overcome these technical limitations, we further verified the interaction in co-IP experiments using two different sets of epitope-tagged A3B and CDK4 constructs. We expressed either Strep- or Flag-tagged A3B and CDK4 constructs in 293T cells and performed co-IPs probing for A3B first, CDK4s, and vice versa. Immunoblot analysis of both Strep and Flag co-IPs revealed that, regardless of resin or epitope tag, these two proteins reproducibly immunoprecipitated CDK4 (Fig. 2B).
Although our AP-MS approach was performed in the presence of both DNA and RNA nucleases, we sought to determine whether the A3B–CDK4 interaction could occur in a heterologous system, which would indicate a direct protein–protein interaction. Utilizing a His-tagged full-length A3B catalytically dead construct (A3BE255A) expressed in Escherichia coli, we were able to successfully purify co-expressed CDK4 (Fig. 2C). These results showed that no other human protein or specific RNA/DNA intermediate is required for the A3B–CDK4 interaction, such as the canonical CDK4 binding partner Cyclin D1.
Although CDK4 was not identified in our A3A or A3G AP-MS data sets, sequence homology between A3B and these APOBEC3 family members is high (89% homology between A3A and A3B C-terminal domain and 57% homology between A3G and A3B). Therefore, we sought to reconfirm our AP-MS results by directly examining the ability of A3A and A3G to interact with CDK4. As shown in Fig. 2D, A3B is the only deaminase able to co-IP CDK4.
A3B is comprised of two deaminase domains, an N-terminal domain (NTD) and a C-terminal domain (CTD). The CTD is catalytically active and responsible for deaminating cytosines in ssDNA. The NTD is catalytically inactive and implicated in protein oligomerization and mediating interactions with nucleic acids (53, 54). To begin to determine the binding interface for CDK4, we performed comparative co-IPs with full-length Strep-tagged A3B (A3BFL), A3BNTD, and A3BCTD constructs. These experiments revealed that CDK4 could IP with A3BFL and A3BNTD, but not with A3BCTD (Fig. 2D).
To address whether this interaction was occurring endogenously within a cell, a proximity ligation assay (PLA) was performed using A3B, CDK4, and Cyclin D1 antibodies to visualize these interactions in MCF10A cells treated with 25 ng/ml of phorbol 12-myristate 13-acetate (PMA) to induce A3B expression. Notably, A3B and CDK4 showed PLA foci in the nuclear compartment, whereas CDK4 and Cyclin D1 PLA foci were cell-wide (Fig. 2E). In further support of our proteomics results, A3B and Cyclin D1 did not form PLA foci.
CDK4 does not affect A3B phosphorylation, localization, or DNA deaminase activity
Interestingly, we noticed that A3BFL and A3BNTD displayed altered mobilities in co-IP experiments with overexpressed CDK4 (seen throughout Fig. 2, A–D). It is well-known that altered mobility of protein species in SDS-PAGE can be attributed to post-translational modifications such as phosphorylation (55, 56). This raised the possibility that A3B may be phosphorylated by CDK4.
We used kinase prediction algorithms (GPS 2.0 and Scansite 2.0 (57, 58)) to identify putative phosphorylation sites in A3BNTD that could be targeted by CDK4. These programs identified a 95TPXP98 motif within A3BNTD that closely resembles the (S/T)PXK consensus motif recognized by CDK4 (59–63). To determine whether CDK4 was capable of phosphorylating A3B, we performed an in vitro kinase assay by incubating purified RB1–3×HA (positive control), A3G-3×HA (negative control), A3B-3×HA, and two phospho-mutants at amino acid Thr95 (phospho-null A3BT95A and phosphomimetic A3BT95D) with active recombinant CDK4-Cyclin D1 in the presence of [γ-32P]ATP. Autoradiograph and immunoblot analysis revealed [γ-32P] incorporation into RB1 but not into A3G or A3B, suggesting that the altered migration of A3B observed in Fig. 2 is not due to CDK4-mediated phosphorylation (Fig. 3A).
Figure 3.

Multiple A3B properties are not altered by CDK4. A, in vitro kinase assay of the indicated HA-tagged proteins in the presence of recombinant CDK4-Cyclin D1 and [γ-32P]ATP. The top two panels show immunoblots of the indicated proteins, and the bottom panel shows a representative autoradiograph of the same reaction following separation by SDS-PAGE. B, fluorescence microscopy of 293T cells stably expressing A3B–eGFP following transfection with siCtrl or siCDK4 RNAs (scale bar, 20 μm). Histogram showing CDK4 mRNA levels from RT-qPCR relative to TBP mRNA (TATA-binding protein). C, PAGE analysis of DNA deamination reaction products following incubation of ssDNA substrates with U2OS whole cell extracts ± A3B and CDK4 depletion as indicated (see text and “Experimental procedures” for details). Recombinant A3A and no APOBEC3 provide positive and negative controls, respectively. Immunoblot controls are shown below.
Several studies have demonstrated that A3BNTD contains cis elements that are required for active nuclear import (35–38). Because CDK4 is also actively imported into the nuclear compartment (45), we were curious if it is required for nuclear import of A3B. To address this idea, we challenged A3B nuclear import with knockdown of CDK4. Despite binding the localization domain of A3B, CDK4 depletion did not seem to alter the steady-state accumulation of A3B within the nuclear compartment (Fig. 3B).
Next, because DNA deamination is the canonical activity of APOBEC3 enzymes, we asked whether CDK4 alters A3B deaminase activity. U2OS cells were engineered to stably express either an A3B-specific short hairpin (sh)RNA construct (shA3B) or a nonspecific shRNA construct as a control (shCTRL), and they were then transfected with either a CDK4-specific small interfering RNA (siRNA) or scrambled siRNA as a control. Using an established ssDNA deamination assay and immunoblots as additional controls, we observed that CDK4 depletion does not cause a major alteration in the enzymatic activity of endogenous A3B (Fig. 3C).
A3BNTD residues required for CDK4 binding
We next sought to map the surface(s) of the NTD that mediate the interaction with CDK4. As illustrated in Fig. 4, A and B, structural knowledge of A3BNTD and A3A was used to inform the construction of a series of chimeric proteins to map the CDK4 interaction (64–66). A3A was chosen as a chimeric partner because it displays cell-wide localization in many cell types, it is represented by multiple high-resolution crystal structures, and it does not interact with CDK4 (35, 64, 65, 67) (Fig. 2D). A comparison between A3A and A3BNTD structures (PDB 4XXO and 5TKM, respectively) led us to focus on four surface-exposed regions with major differences in amino acid composition (sections 1–4 in Fig. 4A). Co-IPs and immunoblot analysis were performed with cells co-expressing CDK4 and A3A/B1–4 chimeric constructs. Interestingly, A3A/B1–3 proteins showed WT binding to CDK4, whereas A3A/B4 had a compromised capacity to bind (Fig. 4C). These results suggested that amino acid residues 80–85 may be required for mediating the interaction with CDK4 (highlighted in blue in Fig. 4B). Because the A3BNTD nuclear localization surfaces defined by residues Asp19, Glu24, and Thr95–Ala102 do not overlap with these CDK4-binding residues, our results implicate a distinct A3B protein surface in the interaction with CDK4.
Figure 4.

CDK4 interacts with a N-terminal region of A3B. A, amino acid alignment of A3BNTD and A3A with chimeric junctions indicated in red and structural elements shown above the alignment (α helices, β strands, and loop regions). B, ribbon schematic overlay of the crystal structures of A3BNTD (red, PDB 5TKM) and A3A (gray, PDB 4XXO). A3BNTD residues 80–85 are highlighted in blue (see text for details). C, anti-Flag (CDK4) co-IP of indicated Strep-tagged A3BNTD constructs from 293T cells. Parallel reactions were done with empty vector and A3A-S as negative controls. See Fig. 2A legend for a description of the IP labeling scheme.
N-terminal region of CDK4 is required for A3B binding
As described above, CDK4 is unlikely to be using obvious mechanisms to regulate A3B activities. We therefore hypothesized instead that A3B could be regulating CDK4. As CDK4 family members serve different functions, we initially wanted to establish which family members bind to A3B. Similar to APOBEC3s, the CDK family shares high homology in protein sequence and function (e.g. CDK4 and CDK2 alignment in Fig. 5A). CDK4 is one of four classical cell-cycle CDKs, sharing the most sequence homology with CDK6 and CDK2 (70 and 42% identity, respectively), both of which regulate cell-cycle progression (41, 42). Co-IP of CDK4, -2, and -6 was performed in the presence of A3B followed by immunoblot analysis. As shown in Fig. 5B, A3B bound to CDK4, but not to CDK6 or CDK2, further indicating a specific interaction.
Figure 5.

N-terminal region of CDK4 is required for A3B binding. A, amino acid alignment of CDK4 and CDK2 with chimeric junctions indicated in red and structural elements shown above the alignment (α helices, β strands, and loop regions). B–D, anti-Strep (A3B) co-IPs of the indicated Flag-tagged CDK constructs from 293T cells. See Fig. 2A legend for a description of the IP labeling scheme.
Next, we wanted to delineate the CDK4 interaction surface to better understand what biological role the complex might play. We generated several reciprocal chimeras that exchanged surface-exposed regions of CDK4 with CDK2. CDK2 was used as a chimeric partner for several reasons. First, CDK2 is negative for A3B binding. Second, available high-resolution crystal structures allow for structure-guided mutagenesis (39, 68). Third, CDK2 binds an alternative cyclin (Cyclin E) (42) thereby providing clarity for lack of Cyclin D1 in proteomics and A3B–CDK4 binding. Chimeric junctions were guided by available crystal structures as well as by amino acid conservation between the protein coding sequences (Fig. 5A). Co-IPs and immunoblot analysis were performed on cells co-expressing A3B and CDK4/2A-C chimeras. As described above, CDK2 serves as a negative control and does not bind A3B. Interestingly, all CDK4/2A-C chimeric constructs had the ability to bind A3B, indicating that region “A” (aa 1–96) is sufficient for binding (Fig. 5C). To further support this finding, a reciprocal CDK2/4A chimera was constructed and used for co-IP. This construct lost the ability to bind A3B, confirming that region A of CDK4 is necessary for binding to A3B (Fig. 5D). Having identified a variant of CDK4 (CDK2/4A) defective for binding A3B, we also sought to reconfirm that the ssDNA deaminase activity of A3B is not altered by this interaction. HeLa cells were transfected with either WT A3B-HA or the catalytic mutant (A3BCM-HA) in the presence of either empty vector or Flag-tagged CDK constructs. As above, DNA deaminase activity assays and control immunoblots combined to show that neither CDK4 nor the A3B-binding mutant (CDK2/4A) alters A3B deaminase activity (data not shown).
A3B disrupts CDK4-dependent nuclear import of Cyclin D1
Interestingly, the N-terminal region of CDK4 identified for A3B binding also contains the known CDK4–Cyclin D1 binding interface (44), which together with the aforementioned data suggests that these interactions may be mutually exclusive. This observation may also explain why Cyclin D1 was not identified in our proteomics analysis, why no exogenous Cyclin D1 is needed for co-IP validations, and why CDK4A/CDK2 (68% CDK2 residues) retains the ability to bind to A3B (even though CDK2 normally binds a different Cyclin (59)). These insights, along with domain mapping of the A3B–CDK4 interaction above, support a model in which A3B and Cyclin D1 share an overlapping binding interface on CDK4.
To ask whether A3B binding to CDK4 could disrupt the nuclear import of Cyclin D1, A3B–eGFP was expressed in HeLa cells and immunofluorescence microscopy was used to examine Cyclin D1 localization. We observed that CDK4 and Cyclin D1 are both predominantly nuclear in the absence of exogenous A3B, as anticipated from prior studies (45) (see untransfected GFP-negative cells in Fig. 6A and quantification in Fig. 6B). However, in the presence of A3B–eGFP, A3B and CDK4 were found to co-localize in the nuclear compartment, whereas Cyclin D1 became more cell-wide (see transfected A3B–eGFP-positive cells in Fig. 6A, and quantification in Fig. 6B). Additionally, A3B–eGFP expression caused significant accumulation of Cyclin D1 in foci that appeared to form within the nuclear compartment (Fig. 6C). In support of these observations, endogenous A3B depletion in U2OS cells caused significant Cyclin D1 relocalization (increased nuclear to cytoplasmic expression ratios) and a trend toward fewer Cyclin D1 foci, compared with control short-hairpin RNA-transduced cells (shA3B versus shCTRL in Fig. 6, D–G).
Figure 6.

A3B disrupts CDK4-dependent nuclear import of Cyclin D1. A, three sets of representative immunofluorescence microscopy images of HeLa cells transfected with A3B–eGFP and stained for endogenous Cyclin D1, endogenous CDK4, and DAPI (×60 magnification image scale bar, 20 μm). A ×100 magnification of the boxed regions is also provided for Cyclin D1-stained, A3B–eGFP expressing cells for better visualization of Cyclin D1 redistribution and foci (white arrows). B and C, whiskered dot plots for quantification of nuclear to cytoplasmic ratio of Cyclin D1 as well as number of Cyclin D1 foci, respectively (n = 20 cells; p values calculated using an unpaired Student's t test). D, immunoblots showing A3B levels in U2OS cells stably expressing an A3B-specific short hairpin RNA construct (shA3B) or a nonspecific shRNA construct as a control (shCTRL). E, two sets of representative immunofluorescence microscopy images of U2OS cells stably expressing shCTRL or shA3B constructs and stained for endogenous Cyclin D1 and 4′,6-diamidino-2-phenylindole (DAPI) (scale bar, 10 μm). F and G, whiskered dot plots for quantification of nuclear to cytoplasmic ratio of Cyclin D1 as well as number of Cyclin D1 foci, respectively (n = 20 cells; p values calculated using an unpaired Student's t test).
Discussion
A3B is unique among the human APOBEC3 family of DNA cytosine deaminase enzymes in that it constitutively localizes to the nucleus and is up-regulated in several different cancer types. Here, we use a combination of AP-MS, structural-guided mutagenesis, genetic, and immunofluorescence microscopy approaches to demonstrate that CDK4 is a cellular A3B-interacting protein. We show that A3B, and not homologous family members A3A and A3G, can specifically co-IP with CDK4. In addition, we demonstrate that A3B does not co-IP with other closely related CDK family members such as CDK2 and CDK6. Using structure-guided analysis of recently solved crystal structures and co-IP techniques we show that the interaction between A3B and CDK4 is mediated through the N-terminal domain of A3B (utilizing but not limited to aa 80–85) and the N-terminal region of CDK4 (aa 1–96). Additionally, we conclude that CDK4 is not likely to be regulating A3B via established mechanisms (CDK4–Cyclin D1 phosphorylation or CDK4-dependent nuclear import) or a potentially novel mechanism (DNA deamination inhibition). Last, PLA and immunofluorescence microscopy studies show A3B and CDK4 co-localization in the nuclear compartment, as well as aberrant Cyclin D1 localization.
Our results favor a model in which A3B may regulate CDK4–Cyclin D1 by sequestering CDK4 in the nuclear compartment, which in turn disrupts Cyclin D1 localization, likely via the secondary consequence of less available CDK4 for Cyclin D1 interaction (Fig. 7). A3B binds the N-terminal region of CDK4 and thereby disrupts CDK4-dependent import of Cyclin D1, likely due to overlapping and therefore competing binding surfaces. This is supported by studies that mapped the CDK4–Cyclin D1-binding interface and identified the N-terminal domain, specifically CDK4 residues 50–56 (PISTVRE), as the Cyclin D1-binding surface (44). Additional evidence supporting a mutually exclusive interaction for A3B–CDK4 is as follows: Cyclin D1 was not identified in proteomics analysis; exogenous Cyclin D1 is not needed for co-IP validations; and CDK4A/CDK2 (68% CDK2 residues) retained the ability to bind to A3B, even though CDK2 normally binds to a different Cyclin (59). Additionally, aberrant localization of Cyclin D1 is only observed in cells expressing A3B–eGFP as neighboring untransfected cells show normal nuclear enrichment for Cyclin D1. Our studies thereby demonstrate that A3B disrupts CDK4-dependent import of Cyclin D1, which may perturb cell-cycle regulation and stall cells in G1/S.
Figure 7.
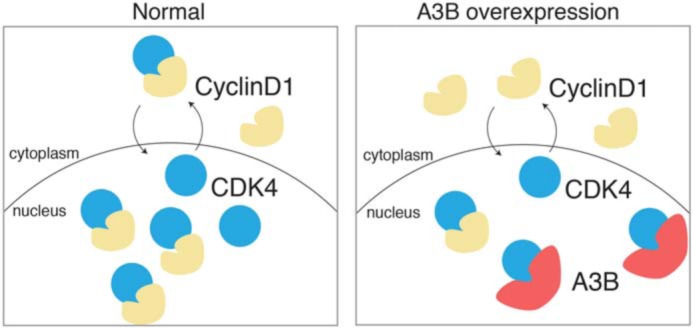
Working model for A3B–CDK4 interaction. A cartoon depicting the normal nucleo-cytoplasmic shuttling of CDK4 and CDK4–Cyclin D1 complexes (left panel). In comparison, A3B-mediated sequestration of CDK4 in the nuclear compartment causes a cell-wide distribution of Cyclin D1 (right panel).
It is possible that the A3B–CDK4 interaction benefits the innate antiviral response of cells. The APOBEC3s can restrict a wide variety of DNA-based pathogens and have been shown to be induced by viral infection (10, 69–75). Divergent viral families manipulate the cell cycle in an attempt to optimize viral growth conditions and/or viral fitness. Manipulation of the cell cycle for optimized viral fitness may aid in overcoming mechanisms of APOBEC3-mediated restriction. For example, the EBV protein BORF2 causes cell-cycle arrest of host cells undergoing lytic viral DNA replication, and it also binds and inhibits A3B to prevent hypermutation of the EBV genome (10, 76). As a second example, the HIV-1 Vif protein has been reported to cause host cells to arrest in the G2 phase of the cell cycle, and it too binds APOBEC3 proteins and targets them for proteasomal degradation to escape restriction (77, 78). Additionally, irregular cell-cycle phenotypes, such as cell-cycle arrest, cell-cycle delay, and aborted cell division (multi-nucleation), have been reported in the literature for A3B but remain poorly understood (35, 79–81).
Interestingly, many viruses that are susceptible to DNA deamination by A3B, including EBV and HPV, also subvert the cell cycle to their advantage. For instance, EBNA3C (encoded by EBV) and E7 (encoded by HPV) are able to perturb the control of G1/S phase transition to replicate their genomes at the same time as cellular DNA replication (82–84). Other viruses can induce a G2/M arrest to provide an optimized cellular environment for maximal levels of viral replication (85–87). One speculation for how this might facilitate an antiviral response is by altering the cell cycle to expose more viral ssDNA replication intermediates to A3B mutagenesis. One example for this is the recent study on A3B and the herpesvirus EBV (and KSHV) where hypermutation of the viral genome is driven by A3B during lytic replication (10). In addition to EBV, studies on BK and JC polyomaviruses have indicated that the preferred trinucleotide motifs of A3B are depleted from viral genomes, specifically on the DNA strand that is most likely to be single-stranded (i.e. the nontranscribed strand or the DNA replication lagging strand template (11)). Despite these and other possible models, full elucidation of the molecular mechanism and potential benefits of the A3B–CDK4 interaction to the cell will require systematic studies with different viruses in pathologically relevant systems.
It is also possible that the A3B–CDK4 interaction plays a role in healthy, uninfected cells. However, A3B expression appears to be kept at very low levels in normal cell types, and it is more likely that the cell-cycle phenotype described here could become exacerbated upon A3B overexpression in cancer (potentially benefitting the evolvability of different tumor cell types). One canonical role of CDK4 is to control the nuclear import of Cyclin D1, which facilitates the downstream progression of the G1/S phase transition (45, 89–94). Several different cancer types have been shown to up-regulate A3B, which may lead to G1/S phase stalling via its interaction with CDK4. As the G1/S phase transition involves numerous points of exposed ssDNA (DNA replication, transcription, and DNA repair), this may result in increased substrate availability and could lead to increased genomic DNA deamination, nonclustered and clustered mutation, and overall genomic instability. Although a role for A3B in cancer mutagenesis and evolvability has been well-documented, universally accepted genomic substrates have yet to be described. Conflicting studies show A3B can target transcription-associated substrates as well as replication and recombination-associated substrates (18, 54, 74, 95, 96). Therefore, it is plausible that A3B-mediated mutagenesis can be exacerbated during G1/S stalling, which may create more exposed ssDNA substrates.
Experimental procedures
Constructs
Strep and Flag epitope-tagged constructs were cloned into pcDNA4/TO using standard PCR cloning techniques. C-terminal 2×Strep and/or 3×Flag tags were cloned into pcDNA4/TO using XhoI–ApaI. N-terminal 3×Flag tag was cloned into pcDNA4/TO using HindIII–NotI. The full set of pcDNA3.1(+) human APOBEC-HA expression constructs have been described (71) (A3A (GenBankTM accession NM_145699), A3B (NM_004900), and A3G (NM_021822)]. CDK2 (BC003065.2), CDK4(CR542247.1), and CDK6 (BC052264) were cloned into pcDNA4/TO-2×Strep using HindIII–NotI and N terminally tagged pcDNA4/TO-3×Flag using NotI–XbaI. RB1 (NM_000321.2) was cloned in the pcDNA3.1(+)–3×HA using HindIII–KpnI. A3B phosphorylation mutants were generated using standard PCR-based site-directed mutagenesis techniques. The A3Bi–GFP construct has been described (35). A3B–eGFP–pQCXIH was made by cloning A3B-eGFP into pQCXIH using NotI–AgeI. Chimeric A3A/B1–4, CDK4/2A-C, and CDK2/4A constructs were cloned using standard overlap PCR methods. Bacterial expression constructs were cloned into pRSF-Duet using SbfI and HindIII for A3B and NaeI and PacI for CDK4. A3B knockdown and control shRNA constructs were validated previously (2, 97).
Cells
293T and HeLa cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum and penicillin-streptomycin. 293T cells stably expressing A3B–eGFP were made by transducing 293T cells with A3B–eGFP–pQCXIH virus following an established protocol (98). MCF10A cells were cultured in Dulbecco's modified Eagle's medium/F-12 supplemented with 5% equine serum, EGF (20 ng/ml), insulin (10 μg/ml), hydrocortisone (0.5 mg/ml), cholera toxin (100 ng/ml), and penicillin-streptomycin. U2OS cells were cultured in McCoy's 5A medium supplements with 10% fetal bovine serum and penicillin-streptomycin.
AP-MS
293T cells were transfected with pcDNA4/TO-A3B-2×Strep-3×Flag, A3A-2×Strep-3×Flag, A3G-2×Strep-3×Flag, or eGFP-2×Strep-3×Flag using TransIT-LT1 (Mirus). Cells were harvested in 1× PBS 48 h post-transfection. Cells were washed two times in 1× PBS followed by lysis (50 mm Tris-HCl, pH 8.0, 1% Tergitol, 150 mm NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm DTT, 1× Protease Inhibitor (Roche), RNase and DNase). Lysates were subjected to sonication prior to clearing by centrifugation. Cleared lysates were then added to Strep-Tactin Superflow resin (IBA) followed by end-over-end rotation for 2 h at 4 °C. Following IP, the anti-Strep resin was washed three times in high salt wash buffer (20 mm Tris-HCl, pH 8.0, 1.5 mm MgCl2, 1 m NaCl, 0.2% Tergitol, 0.5 mm DTT, and 5% glycerol) followed by three washes in low salt wash buffer (same as high salt but with 150 mm NaCl). To remove detergents for proteomics submission, samples were subjected to three washes of no-detergent wash buffer (20 mm Tris-HCl, pH 8.0, 1.5 mm MgCl2, 150 mm NaCl, 0.5 mm DTT, and 5% glycerol). Protein was eluted from the resin in elution buffer (100 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 2.5 mm desthiobiotin). Samples were validated using immunoblotting, DNA deaminase activity assays (discussed below), and Coomassie staining. In-solution samples were analyzed by LC-MS/MS at the Harvard Proteomic Core.
Co-IP experiments and immunoblotting
Semi-confluent 293T cells were transfected with plasmids using TransIT-LT1 (Mirus) per manufacturer's protocol. Cells were harvested in 1× PBS 48 h post-transfection. Cells were washed two times in 1× PBS followed by lysis (150 mm NaCl, 50 mm Tris-HCl, pH 8.0, 0.5% Tergitol, 1× Protease inhibitor (Roche), RNase, and DNase). Cells were vortexed vigorously and incubated at 4 °C for 30 min prior to clearing by centrifugation. Cleared lysates were then added to Strep-Tactin Superflow resin (IBA) or anti-Flag M2 Magnetic Beads (Sigma M8823) followed by end-over-end rotation overnight at 4 °C. Beads were then washed three times in lysis buffer followed by elution in elution buffer (lysis buffer + 0.15 mg/ml of Flag Peptide (Sigma) or 2.5 mm desthiobiotin). Protein was analyzed by immunoblot. Antibodies used include mouse anti-Flag, 1:10,000 (Sigma F1804), rabbit anti-HA, 1:5000 (Cell Signaling C29F4), rabbit anti-APOBEC3B, 1:2000 (5210-87-13 (99)), rabbit anti-Strep Tag II, 1:5000 (Abcam, ab76949), and mouse anti-CDK4, 1:2000 (Santa Cruz, sc-23896).
DNA deaminase activity assays
Deamination reactions were performed at 37 °C for 2 h using purified protein, 4 pmol of oligonucleotide (5′-ATTATTATTATTCAAATGGATTTATTTATTTATTTATTTATTT-fluorescein), 0.025 units of uracil DNA glycosylase (UDG), 1× UDG buffer (New England Biolabs), and 1.75 units of RNase A, as described in Ref. 24. Reaction mixtures were treated with 100 mm NaOH at 95 °C for 10 min to achieve complete backbone breakage. Reaction mixtures were separated on 15% Tris borate/EDTA/urea gels to separate substrate from product. Gels were scanned using a Fujifilm FLA-7000 image reader.
Protein purification from E. coli
pRSF-Duet-A3BE255A and pRSF-Duet-A3BE255A-CDK4 were transformed into BL21 CaCl2 E. coli and colonies were used to inoculate 1 liter of 2× YT media. Cultures were grown to an OD of 0.5 and 50 μm zinc sulfate was added 30 min prior to induction with isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 0.5 mm. Induced cultures were grown overnight at 16 °C and pelleted. Pellets were resuspended in lysis buffer (1% Triton X-100, 50 mm HEPES, pH 7.4, 150 mm NaCl, 1× Protease Inhibitor (Roche Applied Science), 0.1 mg/ml of RNase A, 0.1 mg/ml of DNase, 100 μg/ml of lysozyme, 5 mm imidazole, and 1.5 mm MgCl2) incubated on ice for 10 min and sonicated (Branson Sonifier). Lysate was cleared with centrifugation and insoluble pellet and input lysate were collected and boiled for immunoblot. Cleared lysate was subjected to protein purification using nickel-nitrilotriacetic acid-agarose (Qiagen). Bound protein was washed in lysis buffer (without RNase A, DNase, and lysozyme) and purified protein was eluted by boiling. Samples were run for immunoblot analysis for confirmation of expression and purification.
mRNA quantification
CDK4 mRNA was quantified by RT-qPCR relative to the stable housekeeping transcript, TBP, using specific primers and probed (Roche Lightcycler). RNA isolation and RT-qPCR methods have been described (100).
siRNA knockdown
siRNA targeting CDK4 (Dharmacon, L-003238-00) and control siRNA (Dharmacon, D-001810-10-05) were purchased and diluted to a working concentration of 20 μm. A final concentration of 20 nm was used in 293T cells stably expressing A3B–eGFP. siRNAs were delivered to cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific) per the manufacturer's protocol.
PLA
MCF10A cells were treated with 25 ng/ml of PMA for 18 h. Cells were fixed with 4% paraformaldehyde in PBS (PFA in PBS) for 15 min at room temperature. Cells were permeabilized using 0.5% Triton X-100 for 10 min at 4 °C. The remainder of proximity ligation assay was performed using Duolink In Situ Red Starter Kit Mouse/Rabbit (Sigma, DUO92101). Antibodies were mouse anti-CDK4, 1:50 (Santa Cruz, sc-23896), rabbit anti-Cyclin D1, 1:100 (Abcam, ab134175), rabbit anti-APOBEC3B, 1:50 (5210-87-13 (99)), and mouse anti-Cyclin D1, 1:100 (Santa Cruz, sc-20044).
In vitro kinase assay
Purified RB1–3×HA and A3–3×HA proteins were produced in 293T cells, purified using anti-HA magnetic beads (Thermo Fisher, 88836), and validated using immunoblot techniques. Recombinant CDK4–Cyclin D1 proteins were produced in Baculovirus-infected Sf9 cells (Abcam, ab55695). Purified proteins were incubated with 0.1 μg of recombinant CDK4–Cyclin D1 in kinase reaction buffer (50 mm HEPES, pH 7.5, 10 mm MgCl2, 2.5 mm EGTA, 20 μm DTT, 0.1 mm ATP, and 5.4 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) at 30 °C for 2 h. Reactants were subjected to SDS-PAGE and proteins were detected by immunoblot, whereas phosphorylated proteins were detected by autoradiography.
Immunofluorescence microscopy
Microscopy procedures have been described (88). HeLa cells were seeded on glass coverslips in 6-well plates and were transiently transfected with 500 ng each of A3B–eGFP or empty vector (Mirus, TransIT-LT1) and incubated for 48 h. The cells were fixed with 4% PFA in PBS for 15 min at room temperature. Cells were permeabilized using 0.5% Triton X-100 for 10 min at 4 °C. Slides were blocked in 5% normal goat serum, 4% BSA in PBS for 1 h at room temperature. Primary antibodies were diluted in blocking buffer and slides were incubated in primary antibody overnight at 4 °C. Antibodies were mouse anti-CDK4, 1:50 (Santa Cruz, sc-23896), rabbit anti-Cyclin D1, 1:100 (Abcam, ab134175), anti-rabbit Cy5 (Abcam, ab6564), and anti-mouse Alexa Fluor 594 (Invitrogen, A11032). All slides were treated with 0.1% Hoechst dye to stain nuclei. The slides were mounted with 50% glycerol and imaged using a Nikon inverted Ti-E deconvolution microscope. All images were analyzed for foci and nuclear-to-cytoplasmic ratio using ImageJ-Fiji Software and p values were calculated using an unpaired Student's t test.
Author contributions
J. M. M., D. J. S., and R. S. H. conceptualization; J. M. M., M. M. K., E. M. L., and D. J. S. data curation; J. M. M., D. J. S., and R. S. H. formal analysis; J. M. M., D. J. S., and R. S. H. supervision; J. M. M., E. K. L., W. L. B., and D. J. S. validation; J. M. M., M. M. K., E. M. L., and E. K. L. investigation; J. M. M. methodology; J. M. M. and R. S. H. writing-original draft; J. M. M., D. J. S., and R. S. H. writing-review and editing; E. K. L., W. L. B., D. J. S., and R. S. H. project administration; R. S. H. funding acquisition.
Acknowledgments
We thank Chuanmao Zhang for CDK4 constructs and Ivan D'Orso for pcDNA4/TO Strep- and Flag-tagged base vectors. We thank the University of Minnesota Microbiology, Immunology and Cancer Biology Graduate Program for academic support. A subset of the imaging studies was done with a Nikon inverted Ti-E deconvolution microscope provided by the University of Minnesota-University Imaging Centers.
This work was supported by the University of Minnesota Masonic Cancer Center, Academic Health Center, and College of Biological Sciences and in part by National Science Foundation Graduate Research Fellowship Grant 00039202 (to J. L. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- APOBEC3B
- apolipoprotein B mRNA editing enzyme catalytic subunit-like protein 3B
- AP-MS
- affinity purification mass spectrometry
- CDK
- cyclin-dependent kinase
- co-IP
- co-immunoprecipitation
- PLA
- proximity ligation assay
- RB
- retinoblastoma protein
- shRNA
- short hairpin RNA
- ssDNA
- single-stranded DNA
- SF
- 2×Strep-3×Flag
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- PMA
- phorbol 12-myristate 13-acetate
- HA
- hemagglutinin
- eGFP
- enhanced green fluorescent protein
- qPCR
- quantitative PCR
- aa
- amino acid(s)
- EBV
- Epstein-Barr virus
- HBV
- hepatitis B virus.
References
- 1. Conticello S. G. (2008) The AID/APOBEC family of nucleic acid mutators. Genome Biol. 9, 229 10.1186/gb-2008-9-6-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burns M. B., Lackey L., Carpenter M. A., Rathore A., Land A. M., Leonard B., Refsland E. W., Kotandeniya D., Tretyakova N., Nikas J. B., Yee D., Temiz N. A., Donohue D. E., McDougle R. M., Brown W. L., et al. (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 10.1038/nature11881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harris R. S. (2015) Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Res. 17, 8 10.1186/s13058-014-0498-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts S. A., and Gordenin D. A. (2014) Hypermutation in human cancer genomes: footprints and mechanisms. Nat. Rev. Cancer 14, 786–800 10.1038/nrc3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuong K. J., and Loeb L. A. (2013) APOBEC3B mutagenesis in cancer. Nat. Genet. 45, 964–965 10.1038/ng.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Refsland E. W., and Harris R. S. (2013) The APOBEC3 family of retroelement restriction factors. in Intrinsic Immunity (Cullen B. R., ed) pp. 1–27, Springer-Verlag, Berlin: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stavrou S., and Ross S. R. (2015) APOBEC3 proteins in viral immunity. J. Immunol. 195, 4565–4570 10.4049/jimmunol.1501504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stenglein M. D., Burns M. B., Li M., Lengyel J., and Harris R. S. (2010) APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 17, 222–229 10.1038/nsmb.1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ooms M., Krikoni A., Kress A. K., Simon V., and Münk C. (2012) APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 86, 6097–6108 10.1128/JVI.06570-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng A. Z., Yockteng-Melgar J., Jarvis M. C., Malik-Soni N., Borozan I., Carpenter M. A., McCann J. L., Ebrahimi D., Shaban N. M., Marcon E., Greenblatt J., Brown W. L., Frappier L., and Harris R. S. (2019) Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat. Microbiol. 4, 78–88 10.1038/s41564-018-0284-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verhalen B., Starrett G. J., Harris R. S., and Jiang M. (2016) Functional upregulation of the DNA cytosine deaminase APOBEC3B by polyomaviruses. J. Virol. 90, 6379–6386 10.1128/JVI.00771-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vieira V. C., Leonard B., White E. A., Starrett G. J., Temiz N. A., Lorenz L. D., Lee D., Soares M. A., Lambert P. F., Howley P. M., and Harris R. S. (2014) Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. mBio 5, e02234–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Warren C. J., Westrich J. A., Doorslaer K. V., and Pyeon D. (2017) Roles of APOBEC3A and APOBEC3B in human papillomavirus infection and disease progression. Viruses 9, 233 10.3390/v9080233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burns M. B., Temiz N. A., and Harris R. S. (2013) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983 10.1038/ng.2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roberts S. A., Lawrence M. S., Klimczak L. J., Grimm S. A., Fargo D., Stojanov P., Kiezun A., Kryukov G. V., Carter S. L., Saksena G., Harris S., Shah R. R., Resnick M. A., Getz G., and Gordenin D. A. (2013) An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45, 970–976 10.1038/ng.2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lefebvre C., Bachelot T., Filleron T., Pedrero M., Campone M., Soria J. C., Massard C., Lévy C., Arnedos M., Lacroix-Triki M., Garrabey J., Boursin Y., Deloger M., Fu Y., Commo F., Scott V., et al. (2016) Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Med. 13, e1002201 10.1371/journal.pmed.1002201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexandrov L. B., Nik-Zainal S., Wedge D. C., Aparicio S. A., Behjati S., Biankin A. V., Bignell G. R., Bolli N., Borg A., Børresen-Dale A. L., Boyault S., Burkhardt B., Butler A. P., Caldas C., Davies H. R., et al. (2013) Signatures of mutational processes in human cancer. Nature 500, 415–421 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nik-Zainal S., Alexandrov L. B., Wedge D. C., Van Loo P., Greenman C. D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L. A., Menzies A., Martin S., Leung K., Chen L., Leroy C., et al. (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 10.1016/j.cell.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swanton C., McGranahan N., Starrett G. J., and Harris R. S. (2015) APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 5, 704–712 10.1158/2159-8290.CD-15-0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Helleday T., Eshtad S., and Nik-Zainal S. (2014) Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 15, 585–598 10.1038/nrg3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Venkatesan S., Rosenthal R., Kanu N., McGranahan N., Bartek J., Quezada S. A., Hare J., Harris R. S., and Swanton C. (2018) Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann. Oncol. 29, 563–572 10.1093/annonc/mdy003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ullah I., Karthik G. M., Alkodsi A., Kjällquist U., Stalhåmmar G., Lövrot J., Martinez N. F., Lagergren J., Hautaniemi S., Hartman J., and Bergh J. (2018) Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J. Clin. Invest. 128, 1355–1370 10.1172/JCI96149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bertucci F., Ng C. K. Y., Patsouris A., Droin N., Piscuoglio S., Carbuccia N., Soria J. C., Dien A. T., Adnani Y., Kamal M., Garnier S., Meurice G., Jimenez M., Dogan S., Verret B., et al. (2019) Genomic characterization of metastatic breast cancers. Nature 569, 560–564 10.1038/s41586-019-1056-z [DOI] [PubMed] [Google Scholar]
- 24. Law E. K., Sieuwerts A. M., LaPara K., Leonard B., Starrett G. J., Molan A. M., Temiz N. A., Vogel R. I., Meijer-van Gelder M. E., Sweep F. C., Span P. N., Foekens J. A., Martens J. W., Yee D., and Harris R. S. (2016) The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Science Adv. 2, e1601737 10.1126/sciadv.1601737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sieuwerts A. M., Lyng M. B., Meijer-van Gelder M. E., de Weerd V., Sweep F. C., Foekens J. A., Span P. N., Martens J. W., and Ditzel H. J. (2014) Evaluation of the ability of adjuvant tamoxifen-benefit gene signatures to predict outcome of hormone-naive estrogen receptor-positive breast cancer patients treated with tamoxifen in the advanced setting. Mol. Oncol. 8, 1679–1689 10.1016/j.molonc.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsuboi M., Yamane A., Horiguchi J., Yokobori T., Kawabata-Iwakawa R., Yoshiyama S., Rokudai S., Odawara H., Tokiniwa H., Oyama T., Takeyoshi I., and Nishiyama M. (2016) APOBEC3B high expression status is associated with aggressive phenotype in Japanese breast cancers. Breast Cancer 23, 780–788 10.1007/s12282-015-0641-8 [DOI] [PubMed] [Google Scholar]
- 27. Cescon D. W., Haibe-Kains B., and Mak T. W. (2015) APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc. Natl. Acad. Sci. U.S.A. 112, 2841–2846 10.1073/pnas.1424869112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walker B. A., Wardell C. P., Murison A., Boyle E. M., Begum D. B., Dahir N. M., Proszek P. Z., Melchor L., Pawlyn C., Kaiser M. F., Johnson D. C., Qiang Y. W., Jones J. R., Cairns D. A., Gregory W. M., et al. (2015) APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 6, 6997 10.1038/ncomms7997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nik-Zainal S., Wedge D. C., Alexandrov L. B., Petljak M., Butler A. P., Bolli N., Davies H. R., Knappskog S., Martin S., Papaemmanuil E., Ramakrishna M., Shlien A., Simonic I., Xue Y., Tyler-Smith C., Campbell P. J., and Stratton M. R. (2014) Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 46, 487–491 10.1038/ng.2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tokunaga E., Yamashita N., Tanaka K., Inoue Y., Akiyoshi S., Saeki H., Oki E., Kitao H., and Maehara Y. (2016) Expression of APOBEC3B mRNA in primary breast cancer of Japanese women. PLoS ONE 11, e0168090 10.1371/journal.pone.0168090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fanourakis G., Tosios K., Papanikolaou N., Chatzistamou I., Xydous M., Tseleni-Balafouta S., Sklavounou A., Voutsinas G. E., and Vastardis H. (2016) Evidence for APOBEC3B mRNA and protein expression in oral squamous cell carcinomas. Exp. Mol. Pathol. 101, 314–319 10.1016/j.yexmp.2016.11.001 [DOI] [PubMed] [Google Scholar]
- 32. Middlebrooks C. D., Banday A. R., Matsuda K., Udquim K. I., Onabajo O. O., Paquin A., Figueroa J. D., Zhu B., Koutros S., Kubo M., Shuin T., Freedman N. D., Kogevinas M., Malats N., Chanock S. J., et al. (2016) Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat. Genet. 48, 1330–1338 10.1038/ng.3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wagner J. R., Demir Ö., Carpenter M. A., Aihara H., Harki D. A., Harris R. S., and Amaro R. E. (2019) Determinants of oligonucleotide selectivity of APOBEC3B. J. Chem. Inf. Model. 59, 2264–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lackey L., Demorest Z. L., Land A. M., Hultquist J. F., Brown W. L., and Harris R. S. (2012) APOBEC3B and AID have similar nuclear import mechanisms. J. Mol. Biol. 419, 301–314 10.1016/j.jmb.2012.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lackey L., Law E. K., Brown W. L., and Harris R. S. (2013) Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 12, 762–772 10.4161/cc.23713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pak V., Heidecker G., Pathak V. K., and Derse D. (2011) The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J. Virol. 85, 8538–8547 10.1128/JVI.02645-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stenglein M. D., Matsuo H., and Harris R. S. (2008) Two regions within the amino-terminal half of APOBEC3G cooperate to determine cytoplasmic localization. J. Virol. 82, 9591–9599 10.1128/JVI.02471-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salamango D. J., McCann J. L., Demir Ö., Brown W. L., Amaro R. E., and Harris R. S. (2018) APOBEC3B Nuclear localization requires two distinct N-terminal domain surfaces. J. Mol. Biol. 430, 2695–2708 10.1016/j.jmb.2018.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jeffrey P. D., Russo A. A., Polyak K., Gibbs E., Hurwitz J., Massagué J., and Pavletich N. P. (1995) Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320 10.1038/376313a0 [DOI] [PubMed] [Google Scholar]
- 40. Jeffrey P. D., Tong L., and Pavletich N. P. (2000) Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 14, 3115–3125 10.1101/gad.851100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malumbres M., and Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 10.1038/nrc2602 [DOI] [PubMed] [Google Scholar]
- 42. Malumbres M. (2014) Cyclin-dependent kinases. Genome Biol. 15, 122 10.1186/gb4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murray A. W. (2004) Recycling the cell cycle: cyclins revisited. Cell 116, 221–234 10.1016/S0092-8674(03)01080-8 [DOI] [PubMed] [Google Scholar]
- 44. Day P. J., Cleasby A., Tickle I. J., O'Reilly M., Coyle J. E., Holding F. P., McMenamin R. L., Yon J., Chopra R., Lengauer C., and Jhoti H. (2009) Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. U.S.A. 106, 4166–4170 10.1073/pnas.0809645106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen H. B., Xu X. W., Wang G. P., Zhang B. Y., Wang G., Xin G. W., Liu J. J., Jiang Q., Zhang H. Y., and Zhang C. M. (2017) CDK4 protein is degraded by anaphase-promoting complex/cyclosome in mitosis and reaccumulates in early G1 phase to initiate a new cell cycle in HeLa cells. J. Biol. Chem. 292, 10131–10141 10.1074/jbc.M116.773226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nevins J. R. (2001) The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10, 699–703 10.1093/hmg/10.7.699 [DOI] [PubMed] [Google Scholar]
- 47. Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 10.1101/gad.12.15.2245 [DOI] [PubMed] [Google Scholar]
- 48. Massagué J. (2004) G1 cell-cycle control and cancer. Nature 432, 298–306 10.1038/nature03094 [DOI] [PubMed] [Google Scholar]
- 49. Baker S. J., and Reddy E. P. (2012) CDK4: a key player in the cell cycle, development, and cancer. Genes Cancer 3, 658–669 10.1177/1947601913478972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barretina J., Taylor B. S., Banerji S., Ramos A. H., Lagos-Quintana M., Decarolis P. L., Shah K., Socci N. D., Weir B. A., Ho A., Chiang D. Y., Reva B., Mermel C. H., Getz G., Antipin Y., et al. (2010) Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 42, 715–721 10.1038/ng.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Italiano A., Bianchini L., Gjernes E., Keslair F., Ranchere-Vince D., Dumollard J. M., Haudebourg J., Leroux A., Mainguene C., Terrier P., Chibon F., Coindre J. M., and Pedeutour F. (2009) Clinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomas. Clin. Cancer Res. 15, 5696–5703 10.1158/1078-0432.CCR-08-3185 [DOI] [PubMed] [Google Scholar]
- 52. Sheppard K. E., and McArthur G. A. (2013) The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin. Cancer Res. 19, 5320–5328 10.1158/1078-0432.CCR-13-0259 [DOI] [PubMed] [Google Scholar]
- 53. Feng Y. Q., Baig T. T., Love R. P., and Chelico L. (2014) Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front. Microbiol. 5, 450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bransteitter R., Prochnow C., and Chen X. S. (2009) The current structural and functional understanding of APOBEC deaminases. Cell. Mol. Life Sci. 66, 3137–3147 10.1007/s00018-009-0070-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee C. R., Park Y. H., Min H., Kim Y. R., and Seok Y. J. (2019) Determination of protein phosphorylation by polyacrylamide gel electrophoresis. J. Microbiol. 57, 93–100 10.1007/s12275-019-9021-y [DOI] [PubMed] [Google Scholar]
- 56. Wegener A. D., and Jones L. R. (1984) Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels: evidence for a protein structure consisting of multiple identical phosphorylatable subunits. J. Biol. Chem. 259, 1834–1841 [PubMed] [Google Scholar]
- 57. Xue Y., Ren J., Gao X., Jin C., Wen L., and Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 10.1074/mcp.M700574-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Obenauer J. C., Cantley L. C., and Yaffe M. B. (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 10.1093/nar/gkg584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stevenson-Lindert L. M., Fowler P., and Lew J. (2003) Substrate specificity of CDK2-cyclin A: what is optimal? J. Biol. Chem. 278, 50956–50960 10.1074/jbc.M306546200 [DOI] [PubMed] [Google Scholar]
- 60. Nigg E. A. (1991) The substrates of the cdc2 kinase. Semin. Cell Biol. 2, 261–270 [PubMed] [Google Scholar]
- 61. Moreno S., and Nurse P. (1990) Substrates for p34cdc2: in vivo veritas? Cell 61, 549–551 10.1016/0092-8674(90)90463-O [DOI] [PubMed] [Google Scholar]
- 62. Grafstrom R. H., Pan W., and Hoess R. H. (1999) Defining the substrate specificity of cdk4 kinase-cyclin D1 complex. Carcinogenesis 20, 193–198 10.1093/carcin/20.2.193 [DOI] [PubMed] [Google Scholar]
- 63. Pan W., Sun T., Hoess R., and Grafstrom R. (1998) Defining the minimal portion of the retinoblastoma protein that serves as an efficient substrate for cdk4 kinase/cyclin D1 complex. Carcinogenesis 19, 765–769 10.1093/carcin/19.5.765 [DOI] [PubMed] [Google Scholar]
- 64. Kouno T., Silvas T. V., Hilbert B. J., Shandilya S. M. D., Bohn M. F., Kelch B. A., Royer W. E., Somasundaran M., Kurt Yilmaz N., Matsuo H., and Schiffer C. A. (2017) Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 8, 15024 10.1038/ncomms15024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi K., Carpenter M. A., Banerjee S., Shaban N. M., Kurahashi K., Salamango D. J., McCann J. L., Starrett G. J., Duffy J. V., Demir Ö., Amaro R. E., Harki D. A., Harris R. S., and Aihara H. (2017) Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139 10.1038/nsmb.3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xiao X., Yang H., Arutiunian V., Fang Y., Besse G., Morimoto C., Zirkle B., and Chen X. S. (2017) Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res. 45, 7494–7506 10.1093/nar/gkx362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Land A. M., Law E. K., Carpenter M. A., Lackey L., Brown W. L., and Harris R. S. (2013) Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J. Biol. Chem. 288, 17253–17260 10.1074/jbc.M113.458661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ayaz P., Andres D., Kwiatkowski D. A., Kolbe C. C., Lienau P., Siemeister G., Lücking U., and Stegmann C. M. (2016) Conformational adaption may explain the slow dissociation kinetics of Roniciclib (BAY 1000394), a type I CDK inhibitor with kinetic selectivity for CDK2 and CDK9. ACS Chem. Biol. 11, 1710–1719 10.1021/acschembio.6b00074 [DOI] [PubMed] [Google Scholar]
- 69. Peng G., Lei K. J., Jin W., Greenwell-Wild T., and Wahl S. M. (2006) Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 203, 41–46 10.1084/jem.20051512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bonvin M., Achermann F., Greeve I., Stroka D., Keogh A., Inderbitzin D., Candinas D., Sommer P., Wain-Hobson S., Vartanian J. P., and Greeve J. (2006) Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43, 1364–1374 10.1002/hep.21187 [DOI] [PubMed] [Google Scholar]
- 71. Hultquist J. F., Lengyel J. A., Refsland E. W., LaRue R. S., Lackey L., Brown W. L., and Harris R. S. (2011) Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234 10.1128/JVI.05238-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Refsland E. W., Hultquist J. F., Luengas E. M., Ikeda T., Shaban N. M., Law E. K., Brown W. L., Reilly C., Emerman M., and Harris R. S. (2014) Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet. 10, e1004761 10.1371/journal.pgen.1004761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vieira V. C., Leonard B., White E. A., Starrett G. J., Temiz N. A., Lorenz L. D., Lee D., Soares M. A., Lambert P. F., Howley P. M., and Harris R. S. (2014) Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 5, e02234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Starrett G. J., Serebrenik A. A., Roelofs P. A., McCann J. L., Verhalen B., Jarvis M. C., Stewart T. A., Law E. K., Krupp A., Jiang M., Martens J. W. M., Cahir-McFarland E., Span P. N., and Harris R. S. (2019) Polyomavirus T antigen induces APOBEC3B expression using an LXCXE-dependent and TP53-independent mechanism. MBio 10, e02690–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Salamango D. J., Becker J. T., McCann J. L., Cheng A. Z., Demir Ö., Amaro R. E., Brown W. L., Shaban N. M., and Harris R. S. (2018) APOBEC3H subcellular localization determinants define zipcode for targeting HIV-1 for restriction. Mol. Cell. Biol. 38, e00356–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Paladino P., Marcon E., Greenblatt J., and Frappier L. (2014) Identification of herpesvirus proteins that contribute to G1/S arrest. J. Virol. 88, 4480–4492 10.1128/JVI.00059-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Desimmie B. A., Delviks-Frankenberrry K. A., Burdick R. C., Qi D., Izumi T., and Pathak V. K. (2014) Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 426, 1220–1245 10.1016/j.jmb.2013.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Izumi T., Io K., Matsui M., Shirakawa K., Shinohara M., Nagai Y., Kawahara M., Kobayashi M., Kondoh H., Misawa N., Koyanagi Y., Uchiyama T., and Takaori-Kondo A. (2010) HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. U.S.A. 107, 20798–20803 10.1073/pnas.1008076107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ma W., Ho D. W., Sze K. M., Tsui Y. M., Chan L. K., Lee J. M., and Ng I. O. (2019) APOBEC3B promotes hepatocarcinogenesis and metastasis through novel deaminase-independent activity. Mol. Carcinogen. 58, 643–653 10.1002/mc.22956 [DOI] [PubMed] [Google Scholar]
- 80. Nikkilä J., Kumar R., Campbell J., Brandsma I., Pemberton H. N., Wallberg F., Nagy K., Scheer I., Vertessy B. G., Serebrenik A. A., Monni V., Harris R. S., Pettitt S. J., Ashworth A., and Lord C. J. (2017) Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br. J. Cancer 117, 113–123 10.1038/bjc.2017.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ohba K., Ichiyama K., Yajima M., Gemma N., Nikaido M., Wu Q., Chong P., Mori S., Yamamoto R., Wong J. E., and Yamamoto N. (2014) In vivo and in vitro studies suggest a possible involvement of HPV infection in the early stage of breast carcinogenesis via APOBEC3B induction. PLoS ONE 9, e97787 10.1371/journal.pone.0097787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Parker G. A., Crook T., Bain M., Sara E. A., Farrell P. J., and Allday M. J. (1996) Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene 13, 2541–2549 [PubMed] [Google Scholar]
- 83. Yim E. K., and Park J. S. (2005) The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res. Treat. 37, 319–324 10.4143/crt.2005.37.6.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Martin L. G., Demers G. W., and Galloway D. A. (1998) Disruption of the G1/S transition in human papillomavirus type 16 E7-expressing human cells is associated with altered regulation of cyclin E. J. Virol. 72, 975–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nascimento R., Dias J. D., and Parkhouse R. M. (2009) The conserved UL24 family of human α, β and γ herpesviruses induces cell cycle arrest and inactivation of the cyclinB/cdc2 complex. Arch. Virology 154, 1143–1149 10.1007/s00705-009-0420-y [DOI] [PubMed] [Google Scholar]
- 86. Davy C. E., Jackson D. J., Raj K., Peh W. L., Southern S. A., Das P., Sorathia R., Laskey P., Middleton K., Nakahara T., Wang Q., Masterson P. J., Lambert P. F., Cuthill S., Millar J. B., and Doorbar J. (2005) Human papillomavirus type 16 E1 E4-induced G2 arrest is associated with cytoplasmic retention of active Cdk1/cyclin B1 complexes. J. Virol. 79, 3998–4011 10.1128/JVI.79.7.3998-4011.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Andersen J. L., Le Rouzic E., and Planelles V. (2008) HIV-1 Vpr: mechanisms of G2 arrest and apoptosis. Exp. Mol. Pathol. 85, 2–10 10.1016/j.yexmp.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. McNamara R. P., McCann J. L., Gudipaty S. A., and D'Orso I. (2013) Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 5, 1256–1268 10.1016/j.celrep.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Alt J. R., Cleveland J. L., Hannink M., and Diehl J. A. (2000) Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 14, 3102–3114 10.1101/gad.854900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Alt J. R., Gladden A. B., and Diehl J. A. (2002) p21(Cip1) promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. J. Biol. Chem. 277, 8517–8523 10.1074/jbc.M108867200 [DOI] [PubMed] [Google Scholar]
- 91. Dhar K. K., Branigan K., Parkes J., Howells R. E., Hand P., Musgrove C., Strange R. C., Fryer A. A., Redman C. W., and Hoban P. R. (1999) Expression and subcellular localization of Cyclin D1 protein in epithelial ovarian tumour cells. Br. J. Cancer 81, 1174–1181 10.1038/sj.bjc.6690826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Musgrove E. A., Caldon C. E., Barraclough J., Stone A., and Sutherland R. L. (2011) Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 11, 558–572 10.1038/nrc3090 [DOI] [PubMed] [Google Scholar]
- 93. Diehl J. A., and Sherr C. J. (1997) A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol. Cell. Biol. 17, 7362–7374 10.1128/MCB.17.12.7362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Holland T. A., Elder J., McCloud J. M., Hall C., Deakin M., Fryer A. A., Elder J. B., and Hoban P. R. (2001) Subcellular localisation of cyclin D1 protein in colorectal tumours is associated with p21(WAF1/CIP1) expression and correlates with patient survival. Int. J. Cancer 95, 302–306 [DOI] [PubMed] [Google Scholar]
- 95. Hoopes J. I., Cortez L. M., Mertz T. M., Malc E. P., Mieczkowski P. A., and Roberts S. A. (2016) APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 14, 1273–1282 10.1016/j.celrep.2016.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kinomoto M., Kanno T., Shimura M., Ishizaka Y., Kojima A., Kurata T., Sata T., and Tokunaga K. (2007) All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 35, 2955–2964 10.1093/nar/gkm181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Leonard B., Hart S. N., Burns M. B., Carpenter M. A., Temiz N. A., Rathore A., Vogel R. I., Nikas J. B., Law E. K., Brown W. L., Li Y., Zhang Y., Maurer M. J., Oberg A. L., Cunningham J. M., et al. (2013) APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 73, 7222–7231 10.1158/0008-5472.CAN-13-1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Carpenter M. A., Law E. K., Serebrenik A., Brown W. L., and Harris R. S. (2019) A lentivirus-based system for Cas9/gRNA expression and subsequent removal by Cre-mediated recombination. Methods 156, 79–84 10.1016/j.ymeth.2018.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Brown W., Law E., Carpenter M., Argyris P., Levin-Klein R., Ranum A., Molan A., Forster C., Anderson B., Lackey L., and Harris R. (2019) A rabbit monoclonal antibody against the antiviral and cancer genomic DNA mutating enzyme APOBEC3B. BioRxiv 10.1101/513341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Refsland E. W., Stenglein M. D., Shindo K., Albin J. S., Brown W. L., and Harris R. S. (2010) Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38, 4274–4284 10.1093/nar/gkq174 [DOI] [PMC free article] [PubMed] [Google Scholar]