

Abstract
Cytochrome bc1 complexes (cyt bc1), also known as complex III in mitochondria, are components of the cellular respiratory chain and of the photosynthetic apparatus of non-oxygenic photosynthetic bacteria. They catalyze electron transfer (ET) from ubiquinol to cytochrome c and concomitantly translocate protons across the membrane, contributing to the cross-membrane potential essential for a myriad of cellular activities. This ET-coupled proton translocation reaction requires a gating mechanism that ensures bifurcated electron flow. Here, we report the observation of the Rieske iron–sulfur protein (ISP) in a mobile state, as revealed by the crystal structure of cyt bc1 from the photosynthetic bacterium Rhodobacter sphaeroides in complex with the fungicide azoxystrobin. Unlike cyt bc1 inhibitors stigmatellin and famoxadone that immobilize the ISP, azoxystrobin causes the ISP-ED to separate from the cyt b subunit and to remain in a mobile state. Analysis of anomalous scattering signals from the iron–sulfur cluster of the ISP suggests the existence of a trajectory for electron delivery. This work supports and solidifies the hypothesis that the bimodal conformation switch of the ISP provides a gating mechanism for bifurcated ET, which is essential to the Q-cycle mechanism of cyt bc1 function.
Keywords: cytochrome, conformational change, respiratory chain, electron transfer, X-ray crystallography, conformation switch, cytochrome bc1, electron transfer, electron transfer mechanism, respiratory inhibitors
Introduction
The cytochrome bc1 complex (cyt bc1 or bc1),2 also known as mitochondrial complex III, forms the mid-section of the cellular respiratory chain (1). It is also an essential component of the photosynthetic apparatus of purple bacteria (2). A major function of cyt bc1 is the pumping of protons across the membrane, which is coupled to electron transfer (ET) from its substrate, ubiquinol, to cyt c (cyt c2 in bacteria). Depending on the organism, protein compositions of cyt bc1 vary from 11 subunits in humans (Homo sapiens, Hsbc1) (3) and cows (Bos taurus, Btbc1) (4) to three or four subunits in photosynthetic bacteria such as Rhodobacter capsulatus (R. capsulatus, Rcbc1) (5) or Rhodobacter sphaeroides (R. sphaeroides, Rsbc1) (6). Common to all cyt bc1 complexes are three catalytic subunits that contain prosthetic groups: cyt b, containing hemes bL and bH; cyt c1, containing a c-type heme; and the Rieske iron–sulfur protein (ISP), containing an Fe2S2 cluster.
The mechanism by which the cyt bc1 carries out its ET-coupled proton translocation is known as the Q-cycle mechanism (Fig. 1) (7). This mechanism requires a quinol oxidation site (Qo or QP site) near the electrochemically-positive side of the membrane and a quinone reduction site (Qi or QN site) near the negative side of the membrane. The presence of the two sites has been verified by crystal structures in the presence of various QP and QN site inhibitors and by mutational studies (8–11). Essential to the Q-cycle mechanism is the bifurcated ET at the QP site, in which the two electrons liberated from a ubiquinol substrate follow two different paths: one electron enters the high-potential chain (ISP, cyt c1, and cyt c) and the other follows the low-potential chain (bL, bH, and ubiquinone/ubisemiquinone). Despite the peculiarity of this proposed bifurcated ET, the Q-cycle mechanism is able to explain various experimental observations, including the H+/e ratio, and the oxidant-induced cyt b reduction (12, 13). Numerous hypotheses have been proposed to provide chemical explanations for the bifurcated ET (14).
Figure 1.
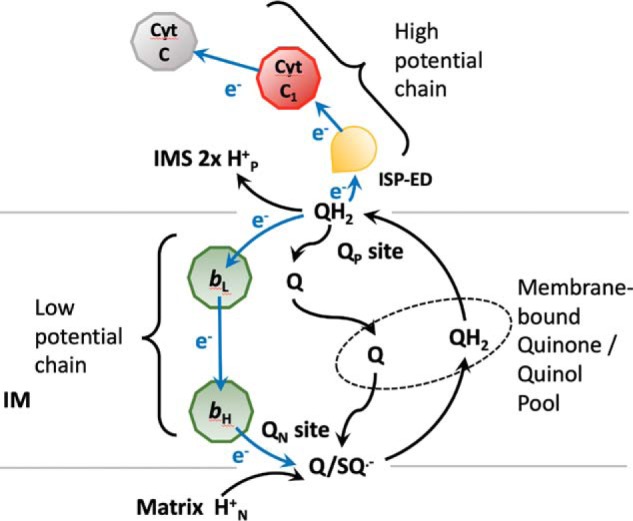
Q-cycle mechanism The cyt bc1 complex can be seen as made of a high-potential chain (ISP, cyt c1, and cyt c) located in the intermembrane space (IMS) of mitochondria and a low-potential chain (hemes bL and bH) embedded in the inner membrane (IM). There are two reaction sites: quinone reduction (QN) and quinol oxidation (QP) sites. At the QP site, the two electrons from a ubiquinol (QH2) diverge: the first one taking the high-potential chain and the second one going into the low-potential chain, reducing ubiquinone/ubisemiquinone (Q/SQ˙̄) at the QN site. The Q and QH2 at the QN and QP sites are in free exchange with those present in the membrane-bound quinone/quinol pool shown inside the ellipse in the dashed line.
The crystal structures of mitochondrial cyt bc1 reveal several important features. 1) In the native enzyme, the extrinsic domain of the ISP (ISP–ED), a single electron carrier mediating ET from the membrane-embedded cyt b to cyt c1 in the aqueous phase, exhibits a high degree of mobility and appears in distinctly different positions in different crystal forms (4, 10, 15–17). 2) In a large number of structures, ISP–ED is clustered at the b-site: an ISP-binding site on the surface of the cyt b subunit near the QP site. The distance between the Fe2S2 of the ISP and the cyt c1 heme is ∼31 Å, which is too great for efficient ET to the high-potential chain (18). 3) In a small number of structures, the ISP–ED is found close to cyt c1 (c1-site) and somewhere between the c1-site and the b-site (intermediate positions) (16, 17). 4) The conformation of the ISP–ED can be modulated by two classes of cyt bc1 inhibitors targeting the QP site: one class is the Pf-type, which fixes the ISP–ED at the b-site, and the other class is the Pm-type, which mobilizes the ISP–ED (10, 15, 19). It was thus hypothesized that the ISP–ED functions as a controlled, swiveling single electron carrier that bridges the distance from ubiquinol to cyt c1 in the aqueous phase, accomplishing the bifurcated ET at the QP site (15, 20).
The ISP–ED is roughly cone-shaped and at its tapered end (the tip) is an Fe2S2 cluster coordinated by two internal cysteines and two surface-accessible histidine residues. This structural feature enables the ISP–ED, once oxidized, to dip into the volcanic crater-shaped binding site on the cyt b subunit (b-site), gaining access to the QP site. Although the ISP–ED is bound at the b-site, it extracts one electron as well as one proton from ubiquinol, producing transiently a ubisemiquinol free radical at the QP site and a reduced ISP–ED. To force a bifurcated ET to occur, the reduced ISP–ED is postulated to stay bound at the QP site until the remaining electron associated with the ubisemiquinol enters the low-potential chain. Subsequently, with the removal of the reaction product ubiquinone into the membrane phase, the reduced ISP–ED is released and delivers the electron to cyt c1, while the proton enters the aqueous phase (15).
The requirement of a mobile ISP–ED for the function of cyt bc1 was elegantly demonstrated in bacterial cyt bc1 systems by introducing mutations that altered the mechanical flexibility of residues in the “neck” region of ISP (21, 22) and by engineering a disulfide bridge between cyt b and ISP–ED (23). Foreshortening the neck region or substituting flexible residues with rigid ones (Ala → Pro and Gly → Val) in the ISP inactivated the entire complex without affecting the assembly or complex integration into the membrane (24). Likewise, using a mutant cyt bc1 that carries a K70C mutation in the ISP subunit and an A185C mutation in the cyt b subunit (ISPK70CCytbA185C Rsbc1), Xiao et al. (23) showed that in an oxidizing environment that forms and maintains the disulfide bridge the enzyme's function was abolished but could be restored by reduction.
It was demonstrated that the ISP–ED of bacterial cyt bc1 can be immobilized at the b-site by Pf-type inhibitors such as stigmatellin or famoxadone (6, 25). Despite the fact that a large number of mechanistic studies of the cyt bc1 complex were conducted using photosynthetic bacteria such as R. sphaeroides and R. capsulatus for their ease of molecular manipulability and the demonstration that their enzyme's function requires the ISP–ED to be conformationally flexible, this very transition from a fixed to a mobile conformation has never been structurally demonstrated for any bacterial cyt bc1 complex. Furthermore, the sequence of events for the ISP–ED to transition from a fixed state in the b-site to a mobile state and subsequently to approach the c1-site has not been clearly defined. Although the functional significance for the existence of the b-site is well-established, that for the c1-site has been controversial. The question is whether a fixed c1-site is truly a requirement for the function of cyt bc1. In this work, we demonstrate the existence of an ISP–ED conformation switch in bacterial cyt bc1 by solving the crystal structure of cyt bc1 in complex with azoxystrobin (Rsbc1/azo). We further show that the ISP–ED adopts multiple positions when it is in the mobile state. This was accomplished by enhancing the anomalous signal of iron atoms by collecting data from a crystal of bovine mitochondrial cyt bc1 in complex with azoxystrobin (Btbc1/azo) at an incident wavelength of 1.739 Å near the Fe absorption edge. Our data corroborate with the highly transient nature of the c1-state when ISP–ED is mobile (26).
Results
Structure of Rsbc1 arrested in the b-state reveals the correct position of the engineered disulfide bond
We previously introduced a double-cysteine mutation in Rsbc1 to show that the flexibility of the ISP–ED is important for the cyt bc1 function (23). The mutation sites were K70C in the ISP subunit and A185C in the cyt b subunit, resulting in a mutant designated ISPK70CcytbA185CRsbc1. To verify the correct formation of the disulfide bond, we crystallized ISPK70CcytbA185CRsbc1 in the presence of stigmatellin, a known Pf-type inhibitor that arrests the ISP–ED at the b-site. The structure (Fig. 2A) of ISPK70CcytbA185CRsbc1/stg was determined with three independent dimers per crystallographic asymmetric unit (Table 1). In the structure, the ISP–ED is trapped in the b-site, and the disulfide bridge between residues Cys-70 of the ISP and Cys-185 of cyt b is correctly formed (Fig. 2B). All three dimeric cyt bc1 molecules, with a total of six disulfide bridges, are superimposable. Importantly, the structure proves that the residues chosen for mutation to cysteine are properly aligned when the ISP–ED is at the b-site and the disulfide bridges are formed under normal oxidizing conditions. The engineered inter-subunit disulfide bond changes the position of the ISP–ED domain only slightly from its position in the WT Rsbc1/stg complex published earlier (Fig. S1) (6).
Figure 2.
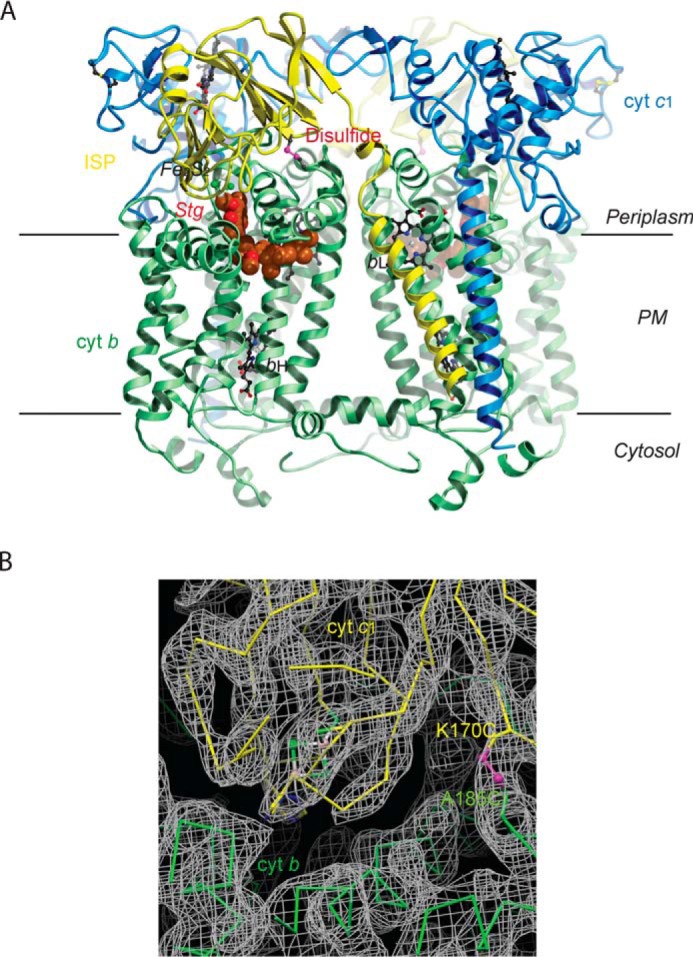
Structure of Rsbc1/stg with the ISP–ED in the b-site. A, overall structure of the dimeric Rsbc1 with the ISP–ED trapped in the b-site. The cyt b subunits are shown in green, cyt c1 in blue, and the ISP in yellow. Stigmatellin is rendered as a CPK model with carbon atoms colored brown. The engineered disulfide bridge (ISP) Cys-70–Cys-185 (cyt b) is shown in magenta. Heme groups are depicted as ball-and-stick models with carbon atoms in black, nitrogen in blue, and oxygen in red. The Fe2S2 cluster of ISP is shown as balls with iron atoms in orange and sulfur in green. B, electron density in the vicinity of the disulfide bridge (magenta) between ISP (shown in yellow) and cyt b (shown in green). The continuous density around this bridge (K170C of ISP and A185C of cyt b) shows that it formed correctly to arrest ISP–ED in the b-state.
Table 1.
Statistics on the quality of diffraction data sets of bc1 crystals and structural models
| Rsbc1/azoa | ISPK70CcytbA185C Rsbc1/stga | Btbc1/azoa | |
|---|---|---|---|
| Diffraction data | |||
| Space group | P21 | C2 | I4122 |
| Cell parameter (Å,°) (angles not listed equal 90°) | a = 89.31, b = 154.7, c = 100.9, β = 95.36 | a = 356.7, b = 145.8, c = 162.2, β = 104.97 | a = b = 154.2, c = 598.1 |
| Resolution (Å) (outer shell) | 39.29–3.00 (3.02–3.00)b | 28.79–3.60 (3.728–3.60) | 27.84–2.80 (2.90–2.80) |
| Wavelength (Å) | 1.00 | 0.827 | 1.739 |
| Rmerge | 0.195 (0.91) | 0.114 (0.73) | 0.144 (1.65) |
| Completeness (%) | 99.54 (96.14) | 97.05 (95.48) | 94.60 (95.74) |
| 〈I/σI〉 | 8.1 (1.9) | 6.72 (1.2) | 32.36 (2.16) |
| No. of unique observations | 54,666 (5,228) | 90,139 (8,819) | 88,694 (8,314) |
| Refinement | |||
| Resolution (Å) | 39.29–3.00 | 28.79–3.60 | 27.84–2.80 |
| Rfree | 0.288 (0.382) | 0.293 (0.378) | 0.284 (0.397) |
| Rwork | 0.266 (0.377) | 0.250 (0.348) | 0.252 (0.362) |
| No. of atoms | 13,927 | 40,362 | 16,342 |
| No. of residues | 1,738 | 5,213 | 2,085 |
| No. of ligand atoms | 423 | 1,146 | 537 |
| No. of solvent molecules | 0 | 0 | 119 |
| rms deviations | |||
| Bond length (Å) | 0.005 | 0.003 | 0.006 |
| Bond angles (°) | 0.71 | 0.61 | 0.67 |
| Ramachandran plot | |||
| Favored, allowed (%) | 94.9, 4.9 | 97.6, 2.4 | 96.0, 3.4 |
| Outliers (%) | 0.2 | 0.0 | 0.6 |
| PDB entry | 6NHH | 6NIN | 6NHG |
a azo is azoxystrobin; stg is stigmatellin.
b Statistics for the highest-resolution shell are shown in parentheses.
ISP–ED is in the mobile state as revealed by the structure of Rsbc1 inhibited by azoxystrobin
To demonstrate the ISP–ED conformation switch in bacterial cyt bc1, we used a Pm-type inhibitor, namely azoxystrobin, in a crystallization experiment with Rsbc1 (Rsbc1/azo), as binding of azoxystrobin in Btbc1 promotes the mobile state of the ISP–ED. This experiment was expected to be challenging, due to the enhanced mobility of the ISP–ED and with it the loss of a rigid hydrophilic surface that could contribute to crystal contacts. With just cyt c1 remaining in a rigid conformation, the chances of crystallizing Rsbc1/azo were considered small. Nevertheless, crystallization conditions were established after extensive screening, and the resulting crystals of Rsbc1/azo diffracted X-rays to 3 Å resolution (Table 1). Consistent with the structures reported previously (6), crystal contacts are formed between neighboring ef1 helices through pairs of π-stacked tryptophan residues (Trp-313) flanked by molecules that appear to be lipids.
The structure of Rsbc1/azo has at its core a cyt b homodimer with a total of 16 membrane-spanning helices (Fig. 3A) that are flanked by pairs of C-terminal helices of cyt c1 and N-terminal helices of the ISP. The large functional head domains of both cyt c1 and the ISP extend into the periplasm. Unlike the static head domain of cyt c1, the ISP–ED is mobile and disordered in both halves of the cyt bc1 dimer, with no clearly defined electron density to model the polypeptide chain (Fig. 3B). In fact, both ISP–EDs are so disordered that the Fe2S2 cluster can only be located with the help of its anomalous signal, which is obtained from the anomalous difference Fourier map using the coefficients F(h,k,l) – F(-h,-k,-l) with a phase of φmodel −90°. This calculation produced several peaks >3σ near the subunits cyt b and cyt c1 in addition to the expected peaks for the bL, bH, and c1 heme iron atoms (Table 2). However, in each case their occupancies are low compared with those of the heme groups bL, bH, and c1. The highest nonheme iron peak is 6σ above average and is 19.2 Å from the c1 heme iron. We assigned this peak to the Fe2S2 cluster of the ISP–ED of the second monomer (model B). A model for this ISP–ED was constructed but had a low occupancy of 0.25. It should be noted that this ISP–ED conformation is only one of many conformations that exist (see below). Indeed, structure refinement of Rsbc1/azo with the ISP–ED included produced no better refinement statistics than without it. Furthermore, the highest Fe2S2 anomalous peak of model B is slightly offset (∼2 Å) from that of model A (Table 2). Taken together, the binding of azoxystrobin allows the ISP–ED to be in a mobile state with no specific positional preference. This degree of mobility is also observed in mitochondrial cyt bc1 in the structures Btbc1/azo (PDB code 1SQB) and Ggbc1/azo (PDB code 3L71). The latter is a homodimer with one ISP–ED (chain E) disordered and the other (chain R) ordered. It occupies the c1′ position, far from cyt b. It should be mentioned that the disorder of the ISP–ED is not an indication of the absence of the ISP subunit as a whole, because the N-terminal helix of the ISP is clearly visible in the electron density from residues 13 to 52 of the neck region (Fig. 3B).
Figure 3.
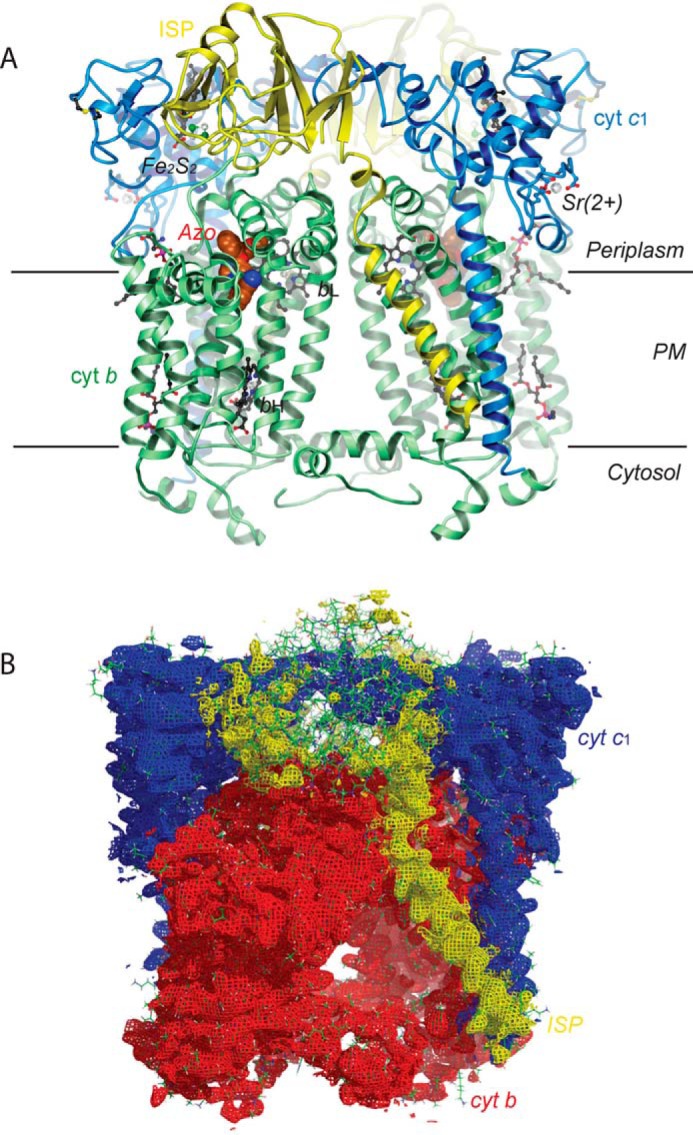
Structure of Rsbc1/azo with ISP–ED disordered. A, two cyt b subunits are shown as green ribbons containing the bH and bL heme groups. Azoxystrobin is shown as a CPK model and lipid molecules as ball-and-stick models. The two cyt c1 subunits with covalently linked c-type hemes are shown in blue. The bound Sr2+ ions (silver spheres) stem from a crystallization additive. Both yellow ribbon models of the iron–sulfur protein are highly disordered and are shown in a position that places their Fe2S2 clusters in or near a position with detectable anomalous signal from their iron atoms. The parallel horizontal lines delineate the boundary of the membrane bilayer. B, electron density for the Rsbc1/azo dimer with cyt b in red, cyt c1 in blue, and ISP in yellow. Although the N-terminal helix of ISP has a good electron density, the head domain is largely disordered.
Table 2.
Peak positions from anomalous difference Fourier correspond to iron positions in crystal structures of Btbc1 and Rsbc1
| Iron center | Peaka height (σ)a | Coordinate |
Modeled | Distance to c1 heme iron | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Å | Å | |||||
| Btbc1/azob data collected at 1.739 Å wavelength | ||||||
| bL | 39.0 | 68.81 | 70.76 | 166.09 | Cyt b | 34.6 |
| bH | 43.5 | 66.93 | 63.74 | 146.91 | Cyt b | 51.4 |
| c1 | 28.0 | 55.93 | 61.79 | 196.89 | Cyt c1 | 0.0 |
| Fe2S2-1 | 12.5 | 72.94 | 44.11 | 178.61 | Yes as ISP | 30.6 |
| Fe2S2-2 | 7.8 | 68.96 | 45.51 | 189.72 | No | 22.1 |
| Fe2S2-3 | 4.7 | 76.64 | 37.05 | 178.97 | No | 36.9 |
| Fe2S2-4 | 3.5 | 64.79 | 50.95 | 193.99 | No | 14.3 |
| Rsbc1/azoa data collected at 1.0 Å wavelength | ||||||
| Monomer A | ||||||
| bLA | 8.1 | 1.38 | 3.77 | 32.76 | Cyt bA | 34.1 |
| bHA | 4.6 | 19.78 | 0.88 | 41.62 | Cyt bA | 50.6 |
| c1A | 6.2 | −30.18 | −6.78 | 40.06 | Cyt c1A | 0.0 |
| Fe2S2A-1 | 3.2 | −17.37 | 16.70 | −9.37 | Yes as ISP | 23.5 |
| Fe2S2A-2 | 3.2 | −25.66 | 22.11 | −2.47 | No | 13.9 |
| Fe2S2A-3 | 3.2 | −12.23 | 9.70 | −1.23 | No | 27.4 |
| Fe2S2A-4 | 3.2 | −26.17 | 4.28 | 1.27 | No | 29.3 |
| Monomer B | ||||||
| bLB | 5.5 | 3.64 | 19.04 | 18.87 | Cyt bB | 34.1 |
| bHB | 5.7 | 23.58 | 19.40 | 13.60 | Cyt bB | 50.6 |
| c1B | 6.4 | −24.51 | 33.15 | 5.84 | Cyt c1B | 0.0 |
| Fe2S2B-1 | 6.0 | −26.04 | 6.95 | 52.77 | Yes as ISP | 19.2 |
| Fe2S2B-2 | 3.5 | −28.59 | 12.44 | 51.40 | No | 22.4 |
| Fe2S2B-3 | 3.6 | −22.63 | 4.01 | 55.99 | No | 25.1 |
a The crystal of Btbc1/azo has a space group symmetry of I4122 with one monomer per crystallographic asymmetric unit. The crystal of Rsbc1/azo has a space group symmetry of P21 with a dimer in the asymmetric unit.
b All anomalous difference Fourier maps were calculated using the data in the resolution range between 25 and 5 Å.
Binding of azoxystrobin to the QP site of Rsbc1
In each of the cyt b subunits, a single azoxystrobin molecule (Fig. 4A) is firmly bound at the QP site, displacing ubiquinol and thus preventing any direct reduction of the Fe2S2 cluster of the ISP. The toxophore of azoxystrobin is a planar E-β-methoxy methyl acrylate group forming a hydrogen bond (2.79 Å) through the ester's carbonyl group with the backbone amide of Glu-295 (Fig. 4B), which is part of the highly conserved PEWY motif. At the same time, the side chain of Glu-295 is rotated toward the aqueous phase, compared with its inward conformation when Pf-type inhibitors such as UHDBT or stigmatellin are bound (19). The flat toxophore (3-methoxy methylacrylate) is positioned between Phe-144 and Tyr-147. The attached o-phenyloxy ring is nearly orthogonal to the acrylate moiety due to steric constraints (Fig. 4B), but it is also ideally positioned to wedge between Pro-294 of the PEWY motif and Gly-158 of the cd1 helix. The pyrimidine ring forms an aromatic–aromatic (Ar–Ar) pair with Phe-298 and prevents Ile-162 of the cd1 helix from returning to a fully relaxed position. The o-cyano-phenoxy group is surrounded by the hydrophobic residue Met-140 and the aromatic groups of Phe-337 and Phe-301 at ideal distances for Ar–Ar interactions.
Figure 4.

Chemical structure of azoxystrobin and its binding environment in Rsbc1. A, chemical structure of azoxystrobin. B, stereographic diagram of the bound azoxystrobin in stick model in the QP pocket overlaid with the blue difference electron density cage for azoxystrobin. The electron density was generated with phenix.polder and contoured at 5σ. Residues lining the QP pocket are shown as a stick model. The hydrogen bond between azoxystrobin and the main-chain amide N atom of Glu-295 is shown as the dotted line in red. C, superposition of the QP site between the Btbc1/azo and Rsbc1/azo structures. The poses of azoxystrobin molecules bound to the QP sites of Rsbc1 (yellow sticks) or Btbc1 (cyan sticks) are nearly identical.
Multiple positions of the ISP–ED are also observed in the structure of mitochondrial cyt bc1 in complex with azoxystrobin
The fact that iron atoms can appear as significant peaks in anomalous difference Fourier maps and thus mark the positions of the Fe2S2 clusters in Rsbc1/azo inspired us to re-visit the structure of mitochondrial cyt bc1 with the Pm-type inhibitor azoxystrobin bound. To study this, we grew crystals of Btbc1 in the presence of azoxystrobin (Btbc1/azo) and collected a diffraction data set using an incident wavelength of λ = 1.739 Å near the iron K absorption edge (FeK: 1.743 Å) (Table 1). Using a Btbc1 crystal is particularly advantageous over other alternatives because of its stability, very high space group symmetry of I4122 (highly redundant data), and non involvement of the ISP-ED in crystal contacts. The crystal structure not only shows enhanced anomalous signals for the element iron (Table 2), but indeed it dramatically improves the signal for all sulfur atoms in cofactors, standard cysteine and methionine residues, as well as those of phosphorus atoms in lipids (Fig. 5A). Although our re-investigation of the structure of Btbc1/azo does not substantially differ from our previously published structure (PDB code 1SQB) or its avian counterpart (PDB code 3L71) (both were collected at 1.0 Å incident radiation), it does, however, allow better visualization of the ISP–ED in its mobile state (Table 2). Although the ISP–EDs are disordered, the presence of four >3.5σ peaks for the Fe2S2 clusters indicate that, despite the absence of obvious restraints (domain–domain interactions, crystal contacts), the ISP–ED still finds local minima between cyt b and cyt c1 (Fig. 5A).
Figure 5.

ISP–ED mobility of Btbc1/azo revealed by anomalous difference Fourier. A, peaks from anomalous difference Fourier using X-ray diffraction data collected near the iron-absorption edge. The enhanced anomalous peaks in Btbc1/azo (magenta) contoured at 3.5σ reveal four prominent sites for Fe2S2-labeled b′-, b″-, c1′-, and c1-sites, respectively. The stronger signal of the b′-site led to a model of ISP–ED with high-atomic displacement factors. The outline of the ISP–ED at the c1′-position, which was not modeled in the crystal structure, is indicated by dotted lines. Similarly, the weaker sites b″ and c1′ were not modeled. B, alignment of cyt bc1 structures based on superpositions of the cyt b subunits to a single monomer (PDB code 6NHG and Btbc1/azo, this work). The cyt b and cyt c1 subunits superimpose well and are rendered here as the green and blue molecular surfaces, respectively. The positions of the ISP subunit are represented by positions of the Fe2S2 cluster (from available experimental coordinates in PDB in stick models) and by anomalous difference Fourier peaks, contoured at 3.5–3.8σ values in red (PDB code 1SQX, Btbc1/stg), white (PDB code 2FYU, Btbc1/jg144), dark cyan (PDB code 1NTZ, Btbc1/native), yellow (PDB code 1SQQ, Btbc1/MOAS), magenta (6NHG, Btbc1/azo), and blue (PDB code 6NHH, Rsbc1/azo). PDB identifiers correspond to the published position of the ISP–ED. The arc in black represents a possible trajectory for ISP movement. The inset features a magnified view of the ISP-docking site showing the b-site represented by the Fe2S2 position from the structure of cyt bc1 bound with stigmatellin, the b′-site from structures of cyt bc1 in the apo or Pm-type inhibitor-bound state.
Discussion
ISP–ED conformation switch is characterized by the change from a fixed to a mobile state and induced by different types of QP site inhibitors
The ISP–ED conformation switch was first suggested in a structural study of mitochondrial cyt bc1, in which different inhibitors gave rise to dramatically different peak heights in anomalous difference Fourier maps for the Fe2S2 cluster at its cyt b–binding site (10). This phenomenon was subsequently used as a basis for classification of cyt bc1 inhibitors that target the QP site (19); those that give rise to strong peaks for the Fe2S2 cluster are called Pf-type inhibitors, and those that lead to no or weak anomalous peaks are termed Pm-type inhibitors. However, this switch between conformations is often misinterpreted as evidence that the ISP–ED moves from the fixed b-site to a similarly rigid c1-site (27–29), and thus it deserves further clarification.
The accumulation of a large number of homologous structures of cyt bc1 from different species, including bacteria, yeast, birds, and mammals, that were crystallized in different space groups and under various conditions allowed comparisons aimed at distinguishing lattice-induced stabilization of the ISP–ED from natural, low-affinity sites. However, this comparison is often biased toward ISP–ED models built by crystallographers, who naturally select the most prominent conformation and consequently ignore all the others. To overcome this deficiency, in this work we performed structure superposition using not only the positions of Fe2S2 from different models but also anomalous difference density maps, if available, resulting in a distribution of Fe2S2 locations in 3D space (Fig. 5B and Table 3). From the distribution, we observed the following. 1) When the QP site is occupied by the Pf-type inhibitors (stigmatelin, UHDBT, etc.), there is one strong Fe2S2 anomalous peak, located at the bottom of the ISP-docking site. This position is frequently referred to as the b-site. 2) When the QP site is not occupied by substrates or inhibitors, Fe2S2 can appear in three different positions, and its corresponding anomalous peaks are weak. The most frequent position is near the b-site and is displaced on average from it by 1.5 Å (Nat site, Fig. 5B). A second position, represented by one model (PDB code 1BE3), has the Fe2S2 near the heme c1, a position referred to as the c1-site. The same position was found to overlap with weak anomalous peaks from Pm-type inhibitor-bound cyt bc1 crystals. A third position, also represented by only one model (PDB code 1BGY), is 7 Å from the second site and is referred to as the intermediate site (Int site, Fig. 5B). Again, this site also overlaps with anomalous peaks from apo- and Pm-type inhibitor-bound cyt bc1 crystals. 3) When the QP site is bound with Pm-type inhibitors (azoxystrobin, MOAS, etc.), the Fe2S2 positions are further displaced from the apo-site, spanning the distance between the “Int” site and the “Nat” site, as exemplified by the 3.8 Å shift from the apo-site for the azoxystrobin-bound structure (Fig. 5B). We term this site the b′-site. It has a rms deviation from its mean position of 7.3 Å. There are three additional sites marked also by weak anomalous peaks. One overlaps with the c1-site. Another was previously observed in a number of avian cyt bc1 structures inhibited by Pm-type inhibitors (PDB code 6NHH, 3L70, etc.), referred to as the c1′-site. The third Fe2S2 position has not previously been reported and is designated the b″-site.
Table 3.
Survey of additional peak positions in anomalous difference Fourier maps
| PDB | Species | Inhibitor P/Na | Wavelength | Anomalous map residue range | Anomalous peak height (S/N)b |
ISPc state | Additional ISP anomalous peak height (position) | Comments | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| bL | bH | cyt c1 | ISP | ||||||||
| Å | Å | ||||||||||
| 1L0N | B. taurus | −/− | 1.0 | 30–5.0 | 15.5 | 18.2 | 10.8 | 5.8 | b′ | Native, no new Fe2S2 anomalous peak | |
| 1NTM | B. taurus | −/− | 1.0 | 30–5.0 | 15.3 | 17.5 | 10.9 | 9.1 | b′ | Native, no new Fe2S2 anomalous peak | |
| 1SQB | B. taurus | Azo Pm/− | 0.9189 | 30–5.0 | 10.7 | 13.6 | 8.5 | 5.3 | b′ | No new Fe2S2 anomalous peak | |
| 6NHGd | B. taurus | Azo Pm/− | 1.736 | 30- 3.5 | 47.7 | 58.4 | 30.9 | 10.0 | b′ | 4.5 (76.04, 37.51, 179.84) 6.5 (68.58, 46.21, 190.19) |
Three additional anomalous peaks for Fe2S2, very strong anomalous signal for S and P atoms |
| 3.7 (65.40, 50.47, 193.93) | |||||||||||
| 6NHHd | R. sphaeroides | Azo Pm/− | 1.0 | 30–5.0 | 8.1 | 4.6 | 6.1 | 5.9 | c1′ | No new peaks above 3.5σ cutoff. There are several peaks above 3σ (Table 1) | |
| 5.5 | 5.6 | 6.6 | 3.3 | c1′ | |||||||
| 3H1K | Gallus gallus | IKR Pm/UQ | 1.283 | 30–4.5 | 9.1 | 8.7 | 7.9 | 4.5 | c1′ | Fairly weak anomalous signal, except for Zn2+ and iodine of the inhibitor | |
| 6.5 | 6.2 | 6.1 | 1.0 | c1′ | |||||||
| 1SQP | B. taurus | Myx Pm/− | 1.0 | 30–5.0 | 11.5 | 10.9 | 7.9 | 5.3 | b′ | No new Fe2S2 anomalous peak | |
| 1SQQ | B. taurus | MOAS Pm /UQ | 1.736 | 30–4.5 | 24.4 | 28.2 | 18.8 | 6.1 | b′ | 4.20 (77.47, 36.66, 178.10) | Two new Fe2S2 anomalous peaks |
| 3.70 (70.21, 45.29, 189.92) | |||||||||||
| 1NU1 | B. taurus | −/NQNO | 1.2 | 30–4.3 | 11.9 | 15.9 | 7.6 | 13.0 | b′ | No significant new Fe2S2 anomalous peak | |
| 4D6T | B. taurus | −/GW 844520 | 0.979 | 30–5.0 | 5.8 | 4.0 | 5.4 | 2.7 | b′ | No extra anomalous Fe2S2 peak, very noisy, weak | |
| 5.0 | 3.1 | 4.6 | 0 | ||||||||
| 4D6U | B. taurus | −/GSK 932121 | 0.979 | 30–5.0 | 2.9 | 3.1 | 3.3 | 3.5 | b′ | No extra anomalous Fe2S2 peak, very noisy, weak | |
| 3.2 | 3.8 | 2.9 | 1.0 | b′ | |||||||
| 1SQV | B. taurus | UHDBT Pf/- | 1.2 | 30–4.5 | 13.8 | 16.6 | 9.8 | 17.8 | b | Very clean, no extra anomalous peak | |
| 1L0L | B. taurus | Fam Pf/− | 1.0 | 30–5.0 | 20.7 | 24.2 | 16.9 | 29.4 | b | Very clean, no extra anomalous peak | |
| 2FYU | B. taurus | JG144 Pf/− | 1.009 | 30–5.0 | 15.7 | 17.6 | 13.0 | 24.5 | b | Very clean, no extra anomalous peak | |
| 5KLV | B. taurus | Fen Pf/− | 1.0 | 30- 5.0 | 13.9 | 17.8 | 11.5 | 16.9 | b | No additional anomalous peak, very clean | |
| 5KKZ | R. sphaeroides | Fam Pf/− | 1.0 | 30–5.0 | 5.5 | 5.7 | 8.0 | 9.8 | b | No additional anomalous peak, very clean | |
| 6.9 | 6.9 | 6.9 | 8.0 | b | |||||||
| 6.3 | 7.4 | 7.1 | 7.7 | b | |||||||
| 7.5 | 5.8 | 5.5 | 7.6 | b | |||||||
| 1SQX | B. taurus | Stig Pf/UQ | 1.0 | 30–5.0 | 12.0 | 11.2 | 11.1 | 15.7 | b | Very clean, no extra anomalous peak | |
| 2YIU | B. taurus | Stig Pf/− | 0.9763 | 30- 5.0 | 3.6 | 4.7 | 5.4 | 5.9 | b | No additional anomalous peak | |
| 5.3 | 2.7 | 5.0 | 6.4 | b | |||||||
a Inhibitor or substrate bound at the P- and/or N-site.
b Anomalous peak height is given as number of standard deviations above mean value of the map, and a 3.5 σ cutoff value is imposed.
c ISP states include the b-state and the b′-state.
d PDB code is from this work.
Based on the distribution of the anomalous peaks and following published conventions, we propose the following. 1) ISP–ED is at the b-site (therefore, the cyt bc1 is in the b-state) when it associates tightly with the cyt b subunit, permitting the first electron and proton transfer from the substrate to the ISP. The b-site is approximated by the binding of Pf-type inhibitors. At this position, the ISP–ED features a very large anomalous peak, and the rms deviation for the Fe2S2 position is 0.72 Å. 2) In the absence of substrate at the QP site, the ISP–ED switches to a mobile conformation, which is facilitated by the binding of Pm-type inhibitors at the QP site. The Fe2S2 cluster can appear in either the b′-site, b″-site, c1-site, or c1′-site, indicating that the positions of the ISP–ED are not well-determined. Thus, we further define the conformation switch of the ISP–ED as the change from a fixed state at the b-site to a mobile state, detached from the b-site (Fig. 5B) (15). In the fixed state, the ISP–ED docks firmly at the b-site with its Fe2S2 cluster penetrating into and being part of the QP site, where the substrate ubiquinol and inhibitors bind. By contrast, the ISP–ED is in the mobile state that requires the ISP–ED to be detached by at least 1.5 Å from the b-site, which is triggered by the displacement of the cd1/cd2 helices. It becomes apparent that the liberated ISP–ED is very mobile but may still favor distinct positions on its path to the cyt c1 for the electron transfer. Interestingly, the c1′-, b′-, and the b″-sites are located along the path of an arc that may define the preferred trajectory for the movement of the ISP–ED (Fig. 5B).
ISP–ED conformation switch is critical to the Q-cycle mechanism
The key question surrounding the bifurcated ET at the QP site is how the second electron of ubiquinol is prevented from following the first electron, which enters the high-potential chain consisting of ISP (Em, 7 = +285 mV for Rsbc1), cyt c1 (Em, 7 = +295 mV), and soluble cyt c2 (Em, 7 = +295 mV). The second electron actually enters the low-potential chain that is formed by two b-type heme groups (bL, Em, 7 = −90 mV; bH, Em, 7 = +50 mV) (30–32). The distance of ∼31 Å between Fe2S2 and cyt c1 heme iron, as seen in crystal structures of cyt bc1 bound with established Pf-type inhibitors, precludes an efficient ET between the two redox groups (Fig. 6A and Table S1). Thus, it was proposed that the ET between Fe2S2 and cyt c1 is achieved by the oscillatory motion of the iron–sulfur protein's extrinsic domain (ISP–ED). At the end point of this motion, the Fe2S2 and cyt c1 are brought close enough for efficient ET (Table S1). Indeed, the ISP–ED has been modeled in crystal structures as close as 15–21 Å from the Fe-center of the c-type heme group (Fig. 6A) (17). Furthermore, the flexibility of the ISP–ED was shown to be essential for the function of cyt bc1 in an elegant biochemical study (22). However, such a model is at odds with the kinetic measurements because the ET rate between ISP and cyt c1 was experimentally measured at 60,000 s−1 for Rsbc1 (Sadoski et al. (26)), whereas the enzymatic turnover rate of Rsbc1 is only 83 s−1 (2.5 μmol of cyt c/min/nmol of cyt b) (33). In other words, it is not a lack of flexibility of the ISP–ED that prevents the second electron from entering the high-potential chain. Furthermore, it is unable to explain the observation that cyt b reduction precedes that of cyt c1 in pre-steady–state quinol oxidation experiments (34, 35).
Figure 6.

Controlled ISP–ED motion switch and the bifurcated ET mechanism. A, structure of the dimeric Rsbc1/azo represented by the prosthetic groups bL, bH, c1, and Fe2S2 in stick models. The bound azoxystrobin molecules are shown as ball models. The ISP–ED subunit was mobile and is shown as a molecular surface in the two extreme conformations: one is at the b-site (gray) and another is near the c1′-site (orange). Distances between pairs of prosthetic groups are also indicated. B, Q-cycle mechanism is modified to incorporate the conformation switch of ISP–ED, which allows the enforcement of the bifurcated ET at the QP site.
The ISP–ED conformation switch offers a satisfactory mechanism for bifurcated ET at the QP site (Esser et al. (15)) (Fig. 6B), utilizing the architecture of the enzyme that has the ability to control the docking and undocking of the ISP–ED from the cyt b subunit. Thus, although the single-electron carrier ISP–ED remains immobilized at the b-site, the electron it carries cannot be delivered to the cyt c1 and neither can it accept a second electron; therefore, the high-potential chain is disconnected. The initial one-electron oxidation creates an unstable ubisemiquinone radical with a very-low redox potential (Em, 7 = −60 mV) (36), leaving the low-potential chain (bL, bH, and Q/Q•) as the only choice for the second electron.
We previously showed that the conformation switch of the ISP–ED is achieved by modulating its affinity to cyt b through substrate-induced movements of the cd1/cd2 helices. Fixation of the ISP–ED at the b-site is due to the increased affinity of the ISP–ED to the b-site in the presence of Pf-type inhibitors as a result of cd1 helix movement, which alters the ISP–ED-binding site. By contrast, the Pm-type inhibitors move the cd1 helix in the opposite direction, reducing the affinity for the ISP–ED (15).
Implications concerning development of novel inhibitors that target cyt bc1
Azoxystrobin is an effective cyt bc1 inhibitor of both Btbc1 and Rsbc1. As a classical QP-site inhibitor of the Pm-type, the measured IC50 values for azoxystrobin in Rsbc1 and Btbc1 are 1.34 and 1.03 nm, respectively (Fig. S2), which is corroborated by the nearly identical binding mode of the inhibitor revealed by comparison of the structures of Btbc1 and Rsbc1 in complex with azoxystrobin (Fig. 3B). High-resolution structures of the complexes afford us a detailed analysis for the observed resistance against azoxystrobin in certain mutant strains of the yeast. One frequently observed mutation (G143A) in cyt b impedes binding and thus renders pathogens resistant to azoxystrobin (Fig. 3C), which is caused by the smaller gap between the highly conserved residues Pro-294 and Gly-158 (the residues Pro-271 and Gly-143 in yeast), into which the aromatic ring of azoxystrobin intercalates. Although the G143A mutation is often the sole reason for azoxystrobin resistance in fungal pathogens (with cross-resistance to numerous other strobilurin-derived pesticides), a performance penalty for the cyt bc1 activity is incurred, as the binding of ubiquinol is also negatively affected.
The binding pose of azoxystrobin in Rsbc1 compares well with those found in the structures of mitochondrial cyt bc1 (Btbc1, PDB code 1SQB) (Fig. 4C) and (Ggbc1, PDB code 3L71). In the first crystal structure (PDB code 1SQB) with bound azoxystrobin, the methyl acrylate ester was assigned the s-cis conformation because of ambiguity in the electron density. Although the hydrogen bond to the Glu-295 amide exists even in the s-cis conformation, the energetically more favorable s-trans conformation was found in the higher-resolution structure of Ggbc1 (PDB code 3L71) and was confirmed in our current Btbc1/azo structure. In Rsbc1, the bound azoxystrobin molecule adopts an overall conformation that displays a very small rms deviation from that in mitochondrial Btbc1 after careful superposition of the cyt b subunits. However, despite impressive overall sequence conservation of cyt b between species, Rsbc1 features additional hydrophobic residues in the QP area of which azoxystrobin takes advantage. Residues Phe-301 and Phe-337 are large aromatic residues not found in mitochondrial cyt b, which feature instead Ala-277 and Ala-295, respectively, in Btbc1 and Ala-278 and Ala-296, respectively, in Ggbc1. These differences could be exploited in the future in the development of species-specific strobilurin-derived compounds.
Experimental procedures
Protein purification of Rsbc1 and Mtbc1
The purification procedure for Rsbc1, published earlier (37), was used with minor changes to obtain protein suitable for crystallization. Briefly, chromatophore membranes were prepared from cells harboring the plasmid pRKDfbcFBCH, which codes for a WT Rsbc1 lacking subunit IV (Δ-IVRsbc1), by disrupting cells with a French press followed by differential centrifugations. To purify the His6-tagged Rsbc1, the chromatophore suspensions were adjusted to a cyt b concentration of 25 μm with 50 mm Tris-HCl (pH 8.0 at 4 °C), containing 20% glycerol, 1 mm MgSO4 and 1 mm phenylmethylsulfonyl fluoride. Dodecyl-d-maltopyranoside (DDM) solution (10% w/v) was added dropwise to a final detergent–to–protein ratio of 0.57 (mg/mg). After centrifugation, the supernatant was loaded onto a nickel-nitrilotriacetic acid column, which was washed with 6 column volumes of buffer A (50 mm Tris-HCl, pH 8.0, at 4 °C, 200 mm NaCl, 0.01% DDM), 6 column volumes of buffer A in the presence of 5 mm histidine and 4 column volumes of buffer B (50 mm Tris-HCl, pH 8.0, at 4 °C, 200 mm NaCl, 0.5% β-octyl glucoside (β-OG)) in the presence of 5 mm histidine. The desired protein fractions were eluted with buffer B containing 200 mm histidine and concentrated with Centriprep-50 to a final concentration of 300 μm.
The Btbc1 was prepared starting from highly-purified succinate–cyt c reductase, as reported previously (38). The cyt bc1 particles were solubilized by deoxycholate, and contaminants were removed by a 15-step ammonium acetate fractionation. The purified cyt bc1 complex was recovered in the oxidized state from the precipitates formed between 18.5 and 33.5% ammonium acetate saturation. The final product was dissolved in a buffer containing 50 mm Tris-HCl, pH 7.8, and 0.66 m sucrose. The protein stock solution with a protein concentration of 30 mg/ml was stored at −80 °C. The concentrations of cyt b and c1 were determined spectroscopically using millimolar extinction coefficients of 28.5 and 17.5 mm−1 cm−1 for cyt b and c1, respectively.
Crystallization of Rsbc1 in complex with azoxystrobin and the double-cysteine mutant (ISPK70CcytbA185CRsbc1) inhibited by stigmatellin
The Δ-IVRsbc1 variant at a concentration of 57 mg/ml was incubated with 3.5 mm azoxystrobin in the presence of 3.5 mm fresh sodium ascorbate for 24 h at 4 °C. The solution was diluted with a buffer containing 50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 200 mm histidine, 0.5% β-octyl glucoside, 10% glycerol, 5 mm NaN3 and 1.25 mm sodium ascorbate. This produced a 14.25 mm solution of the Δ-IVRsbc1/azo complex, which was screened for optimal crystal growth in four different concentrations of sucrose monocaprate (SMC) (0.12, 0.18, 0.24, or 0.30%) and at different concentrations of PEG 400 (5, 6, 7 or 8%). Prior to the addition of the PEG 400, each drop was augmented with 10 mm Sr(NO3)2. The best crystals grew in standard 24-well, sitting drop plates at 16 °C in the presence of 7% PEG 400 and 0.12% SMC over a reservoir of 100 mm Tris-HCl, pH 7.0, 384 mm NaCl, 16% PEG 400, 12.8% glycerol, 5 mm NaN3, and 9 mm tris(carboxymethyl)phosphine.
The double-cysteine mutant ISPK70CcytbA185CRsbc1, at a concentration of 47 mg/ml, was incubated with stigmatellin (1.4 mm final concentration) overnight at 4 °C. The resulting Rsbc1-mutant/stigmatellin complex was diluted 3-fold (to ∼16 mg/ml) with a buffer containing 50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 200 mm histidine, 0.5% β-OG, and 10% glycerol. As before, 10 mm Sr(NO3)2 was added. Three different amounts of SMC (0.06, 0.12, or 0.18%) were screened for optimal crystallization with PEG 400 as the main precipitant in the range of 4–10%. Best crystal growth occurred at a temperature of 16 °C in 8% PEG 400 and 0.12% SMC.
Crystallization of Btbc1 in complex with azoxystrobin
A solution of the purified Btbc1 was adjusted to a final concentration of 20 mg/ml in a buffer containing 50 mm MOPS at pH 7.2, 20 mm ammonium acetate, 20% (w/v) glycerol, and 0.16% sucrose monocaprate. The inhibitor azoxystrobin (ChemService) was dissolved in DMSO to a concentration of 100 mm and mixed with native Btbc1 in a molar ratio of 5:1. The inhibitor/Btbc1 solution was incubated at 4 °C overnight before it was used for crystallization. This solution was set up for crystallization as described previously (Xia et al. (4)). Crystals of cyt bc1 with bound azoxystrobin appeared within 3–4 weeks and were cryo-protected at a glycerol concentration of 30–40% (w/v).
X-ray diffraction data collection, structure determination, and refinement
Rsbc1 crystals were flash-frozen in liquid propane without additional cryoprotectant. A crystal of ISPK70CcytbA185C Rsbc1/stg diffracted X-rays to about 3.6 Å Bragg spacing at beamline 22 ID (Ser-CAT) of the Advanced Photon Source at the Argonne National Laboratory (APS, ANL) and displayed the symmetry of space group C2. Structure solution proceeded by rigid-body refinement of the isomorphous high-resolution trimer of Rsbc1/stg (PDB code 2QJY). The refinement protocol was carried out initially in the absence of a disulfide bridge, resulting in maps with clear evidence for electron density between the residues forming an inter-subunit Cys–Cys bridge. The structure of the mutant was refined and rebuilt in Phenix (phenix-online.org) and COOT (Crystallographic Object Oriented Toolkit Software), respectively.
For the Rsbc1/azo crystal, a complete data set to 3.0 Å Bragg spacing was collected. The crystal belonged to the space group P21 and contained one Rsbc1 dimer per asymmetric unit. The structure was solved by molecular replacement (Molrep, CCP4) using a trimmed dimer from a high-resolution structure (PDB code 2QJY). While having very poor densities, the mobile head domains of ISP (residues 52 to 187) were positioned in the near-“c1” positions based on the anomalous signal for the Fe2S2 cluster. The low occupancies of the mobile ISP extrinsic domains were fixed at 0.25 and grouped b-factor refinement was applied.
Diffraction images of a Btbc1/azo crystal were collected at beamline 5 ID (DND-CAT, APS, ANL) specifically using an X-ray wavelength of λ = 1.739 Å at the K absorption edge of iron. The crystal diffracted initially well beyond the edge of the detector, limiting the extent of the recordable data to 2.8 Å Bragg spacing. The highly-redundant data set has strong anomalous data stemming from iron with contributions from sulfur and phosphorus atoms.
Author contributions
L. E. and D. X. conceptualization; L. E., F. Z., and D. X. data curation; L. E., F. Z., and D. X. formal analysis; L. E. and D. X. validation; L. E. and D. X. investigation; L. E. and D. X. visualization; L. E., F. Z., C.-A. Y., and D. X. methodology; L. E. writing-original draft; L. E. and D. X. writing-review and editing; C.-A. Y. and D. X. resources; D. X. supervision; D. X. funding acquisition; D. X. project administration.
Supplementary Material
Acknowledgments
We thank the staff of the beamlines SER-CAT, GM/CA, and DND at Advanced Photon Source, ANL, for assistance in X-ray diffraction data collection. We also thank George Leiman for editorial assistance. This work utilized the computational resources of the NIH HPC Biowulf Linux cluster (https://hpc.nih.gov).
This work was supported by the Intramural Research Program of the National Institutes of Health, NCI, Center for Cancer Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Table 1.
The atomic coordinates and structure factors (codes 6NHH, 6NIN, and 6NHG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- bc1
- cytochrome bc1
- cyt
- cytochrome
- azo (azoxystrobin)
- methyl (E)-2-[2-[6-(2-cyano-phenoxy) pyrimidin-4-yl] oxyphenyl]-3-methoxyprop-2-enolate
- bH
- high-potential heme b
- bL
- low-potential heme b
- Btbc1
- Bos taurus mitochondrial cyt bc1
- ET
- electron transfer
- famoxadone
- 5-methyl-5-(4-phenoxy-phenyl)-3-phenylamino-2,4-oxazolidinedione
- UHDBT
- 6-hydroxy-5-undecyl-4,7-benzothiazoledione
- MOAS
- methyl (2Z)-3-methoxy-2-(2-[(E)-2-phenylvinyl] phenyl)acrylate
- Pf
- QP site inhibitors that fix ISP–ED conformation
- Pm
- QP site inhibitors that mobilize ISP–ED conformation
- stg (stigmatellin)
- 2-[(3S,4S,5S,6S,7E,9E,11E)-4,6-dimethoxy-3,5,11-trimethyltrideca-7,9,11-trienyl]-8-hydroxy-5,7-dimethoxy-3-methylchromen-4-one
- SMC
- sucrose monocaprate
- ISP
- iron–sulfur protein
- ISP–ED
- extrinsic domain of ISP
- Ggbc1
- Gallus gallus mitochondrial cyt bc1
- QP or Qo
- ubiquinol oxidation
- QN or Qi
- ubiquinone reduction
- rms
- root-mean square
- Rsbc1
- cyt bc1 from photosynthetic bacterium Rhodobacter sphaeroides
- PDB
- Protein Data Bank
- ANL
- Argonne National Laboratory
- APS
- Advanced Photon Source
- DDM
- dodecyl-d-maltopyranoside
- β-OG
- β-octyl glucoside
- Ar–Ar
- aromatic–aromatic.
References
- 1. Keilin D. (1925) On cytochrome, a respiratory pigment, common to animals, yeast, and higher plants. Proc. R. Soc. Lond. B 98, 312–399 10.1098/rspb.1925.0039 [DOI] [Google Scholar]
- 2. Trumpower B. L. (1990) Cytochrome bc1 complexes of microorganisms. Microbiol. Rev. 54, 101–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guo R., Zong S., Wu M., Gu J., and Yang M. (2017) Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell 170, 1247–1257.e12 10.1016/j.cell.2017.07.050 [DOI] [PubMed] [Google Scholar]
- 4. Xia D., Yu C. A., Kim H., Xia J. Z., Kachurin A. M., Zhang L., Yu L., and Deisenhofer J. (1997) Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 277, 60–66 10.1126/science.277.5322.60 [DOI] [PubMed] [Google Scholar]
- 5. Berry E. A., Huang L. S., Saechao L. K., Pon N. G., Valkova-Valchanova M., and Daldal F. (2004) X-ray structure of Rhodobacter capsulatus cytochrome bc1: comparison with its mitochondrial and chloroplast counterparts. Photosynth. Res. 81, 251–275 10.1023/B:PRES.0000036888.18223.0e [DOI] [PubMed] [Google Scholar]
- 6. Esser L., Elberry M., Zhou F., Yu C. A., Yu L., and Xia D. (2008) Inhibitor complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides at 2.40 Å resolution. J. Biol. Chem. 283, 2846–2857 10.1074/jbc.M708608200 [DOI] [PubMed] [Google Scholar]
- 7. Trumpower B. L. (1990) The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 265, 11409–11412 [PubMed] [Google Scholar]
- 8. Brandt U., Schägger H., and von Jagow G. (1988) Characterisation of binding of the methoxyacrylate inhibitors to mitochondrial cytochrome c reductase. Eur. J. Biochem. 173, 499–506 10.1111/j.1432-1033.1988.tb14026.x [DOI] [PubMed] [Google Scholar]
- 9. Gao X., Wen X., Esser L., Quinn B., Yu L., Yu C. A., and Xia D. (2003) Structural basis for the quinone reduction in bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors. Biochemistry 42, 9067–9080 10.1021/bi0341814 [DOI] [PubMed] [Google Scholar]
- 10. Kim H., Xia D., Yu C. A., Xia J. Z., Kachurin A. M., Zhang L., Yu L., and Deisenhofer J. (1998) Inhibitor binding changes domain mobility in the iron–sulfur protein of the mitochondrial bc1 complex from bovine heart. Proc. Natl. Acad. Sci. U.S.A. 95, 8026–8033 10.1073/pnas.95.14.8026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schnaufer A., Sbicego S., and Blum B. (2000) Antimycin A resistance in a mutant Leishmania tarentolae strain is correlated to a point mutation in the mitochondrial apocytochrome b gene. Curr. Genet. 37, 234–241 10.1007/s002940050525 [DOI] [PubMed] [Google Scholar]
- 12. Lawford H. G., and Garland P. B. (1973) Proton translocation coupled to quinol oxidation in ox heart mitochondria. Biochem. J. 136, 711–720 10.1042/bj1360711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wikström M. K., and Berden J. A. (1972) Oxidoreduction of cytochrome b in the presence of antimycin. Biochim. Biophys. Acta 283, 403–420 10.1016/0005-2728(72)90258-7 [DOI] [PubMed] [Google Scholar]
- 14. Xia D., Esser L., Tang W. K., Zhou F., Zhou Y., Yu L., and Yu C. A. (2013) Structural analysis of cytochrome bc1 complexes: implications to the mechanism of function. Biochim. Biophys. Acta 1827, 1278–1294 10.1016/j.bbabio.2012.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Esser L., Gong X., Yang S., Yu L., Yu C. A., and Xia D. (2006) Surface-modulated motion switch: capture and release of iron–sulfur protein in the cytochrome bc1 complex. Proc. Natl. Acad. Sci. U.S.A. 103, 13045–13050 10.1073/pnas.0601149103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iwata S., Lee J. W., Okada K., Lee J. K., Iwata M., Rasmussen B., Link T. A., Ramaswamy S., and Jap B. K. (1998) Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex [see comments]. Science 281, 64–71 10.1126/science.281.5373.64 [DOI] [PubMed] [Google Scholar]
- 17. Zhang Z., Huang L., Shulmeister V. M., Chi Y. I., Kim K. K., Hung L. W., Crofts A. R., Berry E. A., and Kim S. H. (1998) Electron transfer by domain movement in cytochrome bc1. Nature 392, 677–684 10.1038/33612 [DOI] [PubMed] [Google Scholar]
- 18. Moser C. C., Page C. C., Farid R., and Dutton P. L. (1995) Biological electron transfer. J. Bioenerg. Biomembr. 27, 263–274 10.1007/BF02110096 [DOI] [PubMed] [Google Scholar]
- 19. Esser L., Quinn B., Li Y. F., Zhang M., Elberry M., Yu L., Yu C. A., and Xia D. (2004) Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc1 complex. J. Mol. Biol. 341, 281–302 10.1016/j.jmb.2004.05.065 [DOI] [PubMed] [Google Scholar]
- 20. Esser L., Yu L., Yu C., and Xia D. (2008) Insights into the mechanisms of quinol oxidation in cytochrome bc1 in light of the structure of bacterial complex. Biophys. Rev. Lett. 2, 229–257 10.1142/S1793048007000532 [DOI] [Google Scholar]
- 21. Tian H., White S., Yu L., and Yu C. A. (1999) Evidence for the head domain movement of the Rieske iron–sulfur protein in electron transfer reaction of the cytochrome bc1 complex. J. Biol. Chem. 274, 7146–7152 10.1074/jbc.274.11.7146 [DOI] [PubMed] [Google Scholar]
- 22. Tian H., Yu L., Mather M. W., and Yu C. A. (1998) Flexibility of the neck region of the Rieske iron–sulfur protein is functionally important in the cytochrome bc1 complex. J. Biol. Chem. 273, 27953–27959 10.1074/jbc.273.43.27953 [DOI] [PubMed] [Google Scholar]
- 23. Xiao K., Yu L., and Yu C. A. (2000) Confirmation of the involvement of protein domain movement during the catalytic cycle of the cytochrome bc1 complex by the formation of the intersubunit disulfide bond between cytochrome b and the iron–sulfur protein. J. Biol. Chem. 275, 38597–38604 10.1074/jbc.M007444200 [DOI] [PubMed] [Google Scholar]
- 24. Darrouzet E., Valkova-Valchanova M., and Daldal F. (2000) Probing the role of the Fe-S subunit hinge region during Q(o) site catalysis in Rhodobacter capsulatus bc(1) complex. Biochemistry 39, 15475–15483 10.1021/bi000750l [DOI] [PubMed] [Google Scholar]
- 25. Esser L., Zhou F., Zhou Y., Xiao Y., Tang W. K., Yu C. A., Qin Z., and Xia D. (2016) Hydrogen bonding to the substrate is not required for Rieske iron–sulfur protein docking to the quinol oxidation site of complex III. J. Biol. Chem. 291, 25019–25031 10.1074/jbc.M116.744391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sadoski R. C., Engstrom G., Tian H., Zhang L., Yu C. A., Yu L., Durham B., and Millett F. (2000) Use of a photoactivated ruthenium dimer complex to measure electron transfer between the Rieske iron–sulfur protein and cytochrome c(1) in the cytochrome bc(1) complex. Biochemistry 39, 4231–4236 10.1021/bi000003o [DOI] [PubMed] [Google Scholar]
- 27. Crofts A. R. (2004) The cytochrome bc1 complex: function in the context of structure. Annu. Rev. Physiol. 66, 689–733 10.1146/annurev.physiol.66.032102.150251 [DOI] [PubMed] [Google Scholar]
- 28. Iwata M., Björkman J., and Iwata S. (1999) Conformational change of the Rieske [2Fe-2S] protein in cytochrome bc1 complex. J. Bioenerg. Biomembr. 31, 169–175 10.1023/A:1005407410005 [DOI] [PubMed] [Google Scholar]
- 29. Izrailev S., Crofts A. R., Berry E. A., and Schulten K. (1999) Steered molecular dynamics simulation of the Rieske subunit motion in the cytochrome bc1 complex. Biophys. J. 77, 1753–1768 10.1016/S0006-3495(99)77022-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dutton P. L., and Jackson J. B. (1972) Thermodynamic and kinetic characterization of electron-transfer components in situ in Rhodopseudomonas sphaeroides and Rhodospirillum rubrum. Eur. J. Biochem. 30, 495–510 10.1111/j.1432-1033.1972.tb02121.x [DOI] [PubMed] [Google Scholar]
- 31. Prince R. C., and Dutton P. L. (1977) Single and multiple turnover reactions in the ubiquinone-cytochrome b-c2 oxidoreductase of Rhodopseudomonas sphaeroides: the physical chemistry of the major electron donor to cytochrome c2, and its coupled reactions. Biochim. Biophys. Acta 462, 731–747 10.1016/0005-2728(77)90114-1 [DOI] [PubMed] [Google Scholar]
- 32. Prince R. C., Lindsay J. G., and Dutton P. L. (1975) The Rieske iron–sulfur center in mitochondrial and photosynthetic systems: Em/pH relationships. FEBS Lett. 51, 108–111 10.1016/0014-5793(75)80864-7 [DOI] [PubMed] [Google Scholar]
- 33. Xiao K., Liu X., Yu C. A., and Yu L. (2004) The extra fragment of the iron–sulfur protein (residues 96–107) of Rhodobacter sphaeroides cytochrome bc1 complex is required for protein stability. Biochemistry 43, 1488–1495 10.1021/bi035378z [DOI] [PubMed] [Google Scholar]
- 34. Hansen K. C., Schultz B. E., Wang G., and Chan S. I. (2000) Reaction of Escherichia coli cytochrome bo3 and mitochondrial cytochrome bc1 with a photoreleasable decylubiquinol. Biochim. Biophys. Acta 1456, 121–137 10.1016/S0005-2728(99)00107-3 [DOI] [PubMed] [Google Scholar]
- 35. Zhu J., Egawa T., Yeh S. R., Yu L., and Yu C. A. (2007) Simultaneous reduction of iron–sulfur protein and cytochrome bL during ubiquinol oxidation in cytochrome bc1 complex. Proc. Natl. Acad. Sci. U.S.A. 104, 4864–4869 10.1073/pnas.0607812104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robertson D. E., Prince R. C., Bowyer J. R., Matsuura K., Dutton P. L., and Ohnishi T. (1984) Thermodynamic properties of the semiquinone and its binding site in the ubiquinol-cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems. J. Biol. Chem. 259, 1758–1763 [PubMed] [Google Scholar]
- 37. Xiao Y. M., Esser L., Zhou F., Li C., Zhou Y. H., Yu C. A., Qin Z. H., and Xia D. (2014) Studies on inhibition of respiratory cytochrome bc1 complex by the fungicide pyrimorph suggest a novel inhibitory mechanism. PLoS ONE 9, e93765 10.1371/journal.pone.0093765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu L., Yang S., Yin Y., Cen X., Zhou F., Xia D., and Yu C. A. (2009) Analysis of electron transfer and superoxide generation in the cytochrome bc1 complex. Methods Enzymol. 456, 459–473 10.1016/S0076-6879(08)04425-X [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.