

Abstract
Charcot-Marie-Tooth (CMT) disease is a peripheral neuropathy associated with gene duplication and point mutations in the peripheral myelin protein 22 (PMP22) gene. However, the role of PMP22 in Schwann cell physiology and the mechanisms by which PMP22 mutations cause CMT are not well-understood. On the basis of homology between PMP22 and proteins associated with modulation of ion channels, we hypothesized that PMP22 alters ion channel activity. Using whole-cell electrophysiology, we show here that heterologous PMP22 expression increases the amplitude of currents similar to those ascribed to store-operated calcium (SOC) channels, particularly those involving transient receptor canonical channel 1 (TrpC1). These channels help replenish Ca2+ in the endoplasmic reticulum (ER) following stimulus-induced depletion. Currents with similar properties were recorded in WT but not pmp22−/− mouse Schwann cells. Heterologous expression of the CMT-associated PMP22_L16P variant, which fails to reach the plasma membrane and localizes to the ER, led to larger currents than WT PMP22. Similarly, Schwann cells isolated from Trembler J (TrJ; PMP22_L16P) mice had larger currents than WT littermates. Calcium imaging in live nerves and cultured Schwann cells revealed elevated intracellular Ca2+ in TrJ mice compared with WT. Moreover, we found that PMP22 co-immunoprecipitated with stromal interaction molecule 1 (STIM1), the Ca2+ sensor SOC channel subunit in the ER. These results suggest that in the ER, PMP22 interacts with STIM1 and increases Ca2+ influx through SOC channels. Excess or mutant PMP22 in the ER may elevate intracellular Ca2+ levels, which could contribute to CMT pathology.
Keywords: Schwann cells, Charcot-Marie-Tooth disease (CMT), stromal interaction molecule 1 (STIM1), calcium release-activated calcium channel protein 1 (ORAI1), endoplasmic reticulum stress (ER stress), transient receptor potential channels (TRP channels), gas3, myelin protein, myelination, neuropathy, PMP22_L16P
Introduction
Peripheral myelin protein 22 (PMP22) is a tetra-span membrane protein that constitutes 2–5% of total peripheral myelin protein (1, 2). Duplication or point mutations in PMP22 are the most common cause of Charcot-Marie-Tooth (CMT)5 disease, an inherited peripheral neuropathy affecting 1 in 2500 people that causes severe disability associated with disrupted myelination (3, 4). In contrast, heterozygous deletion of PMP22 causes hereditary neuropathy with liability to pressure palsies (HNPP) (5). CMT1A, the most prevalent form of CMT disease, is caused by heterozygous duplication of chromosome 17p11.2, which contains PMP22, resulting in overexpression of WT PMP22 (6–8). Other forms of CMT disease are caused by single amino acid substitutions in PMP22 and are collectively referred to as CMT1E (4). The CMT1E-associated L16P mutation results in protein misfolding, accumulation in the ER, and formation of cytoplasmic aggresomes (9–15). Both duplication and point mutations in PMP22 lead to dys-/demyelination, increased Schwann cell number, and severe secondary axonal loss. However, the mechanisms by which PMP22 overexpression or mutations cause these diseases are poorly understood, as is the functional role of PMP22 in myelination.
Proposed roles for PMP22 include regulation of growth arrest (16), apoptosis (17, 18), myelin compaction (19), formation of epithelial intercellular junctions (20–22), and linking the actin-cytoskeleton to lipid rafts (23). However, these remain controversial (24), and the mechanisms by which PMP22 contributes to these functions remain to be elucidated. Recent analysis of pmp22+/− mice revealed an essential role for this protein in the formation of tight adherens and adhesion junctions in peripheral myelin (25). This may be related to the fact that PMP22 is homologous to the claudins and likely shares a similar structure (26, 27). Deficiency in PMP22 disrupted these junctions, resulting in leaky myelin and impaired action potential propagation. Because PMP22 also shows structural homology to proteins associated with modulation of ion channels (26), we hypothesized that PMP22 modifies ion channel activity in a manner that impacts cell function.
Our results suggest that PMP22 modulates store-operated calcium (SOC) channels. SOC channels are composed of a pore-forming protein (Orai1) in the plasma membrane and a calcium sensor (STIM1) in the ER (28, 29). STIM1 senses depletion of ER calcium stores and activates Orai1 to allow for Ca2+ replenishing (30, 31). Orai1 can also recruit the transient receptor canonical channel 1 (TrpC1) to the plasma membrane, where it associates with STIM1 in trans to facilitate Ca2+ entry (32).
Heterologous expression of PMP22 increased the magnitude of whole-cell currents with properties similar to those ascribed to TrpC1-containing SOC channels. In contrast, these currents were severely impaired in pmp22−/− Schwann cells. We also found that similar currents in Schwann cells from mice harboring the trafficking-deficient PMP22_L16P mutation (TrJ) were significantly enhanced relative to cells isolated from WT littermates. Additionally, biochemical analysis demonstrated an interaction between PMP22 and STIM1 in Schwann cells. Together, the functional and biochemical results suggest that PMP22 has a functional role in the ER, possibly contributing to the regulation of calcium homeostasis via SOC channels. This conclusion is further supported by our observation that intracellular Ca2+ levels in TrJ Schwann cells are significantly higher than in WT cells. Because high levels of intracellular Ca2+ in Schwann cells can induce demyelination (33), our results provide novel insights into the pathogenesis of CMT.
Results
Because peripheral myelin protein 22 (PMP22) shows homology to several proteins (e.g. connexins and the γ accessory subunits of Ca2+ channels) associated with ion transport across the plasma membrane (26), we hypothesized that expression of PMP22 could affect ion channel activity in mammalian cells. To test this possibility, human WT PMP22 (PMP22_WT) was transiently expressed in tsA201 cells (HEK293 cells stably transfected with SV40 large T antigen), and changes in membrane conductance were analyzed.
PMP22 expression increases SOC current magnitude in tsA201 cells
Because of the modest homology between PMP22 and connexins, we first tested whether PMP22 affected membrane conductance using protocols previously used to activate connexin hemi-channels: long depolarizations or removal of extracellular divalent cations (34–36). Long-depolarizing pulses (+60 mV, 8 s long) failed to generate distinct currents between PMP22_WT-expressing and control tsA201 cells (cells transiently expressing empty EGFP vector), but exposure to divalent-free solution elicited whole-cell currents in PMP22_WT-expressing cells that were much larger than in control cells (data not shown). Because the intracellular calcium concentration in those experiments was low (<10 nm), the latter results suggest that the currents observed after the removal of extracellular divalent cations may be associated with store-operated calcium (SOC) channels, which are expressed in HEK293 cells (37, 38). SOC channels can be activated by emptying ER calcium stores by dialyzing the cytoplasm with a calcium chelator, e.g. EGTA (30). SOC currents may be very small but removal of all extracellular divalent cations allows sodium ions to permeate the SOC channel, generating larger currents that can more easily be measured (30).
In our experiments, we measured SOC channel activity after inducing passive Ca2+ depletion from the ER by including EGTA in the pipette solution plus no added Ca2+ and perfusing the cells with either control (+Ca2+/Mg2+) or -free (−Ca2+/Mg2+, DVF) external solutions. Whole-cell currents were recorded from PMP22_WT-expressing and control tsA201 cells. The control cells exhibited small currents after exposure to DVF solution (Fig. 1A), whereas the PMP22_WT-expressing cells generated much larger currents (Fig. 1B). Fig. 1C shows the average current-voltage (IV) relationships, normalized for membrane capacitance, measured from control and PMP22_WT-expressing cells exposed to +Ca2+/Mg2+ and −Ca2+/Mg2+ external solutions. External Gd3+ (10 μm), a SOC channel blocker, completely abolished the DVF-induced currents in both control and PMP22_WT-expressing cells (Fig. 1, A and B). Fig. 1D shows the IV relationships for the Gd3+-sensitive current (−Ca2+/Mg2+ minus versus −Ca2+/Mg2+ + 10 μm Gd3+) measured for the DVF-induced currents recorded from control and PMP22_WT-expressing cells. Because HEK293 cells display SOC currents (37, 38) and PMP22 expression markedly augmented their amplitude, our results suggest that PMP22 modulates SOC channel activity in these cells.
Figure 1.

Transient expression of PMP22_WT augments Gd3+-sensitive, DVF-induced whole-cell currents in tsA201 cells. Average whole-cell currents were recorded from control (A) or PMP22–WT–expressing (B) cells exposed to +divalent solution (left, +Ca2+/Mg2+), divalent-free solution (center, −Ca2+/Mg2+), or divalent-free plus 10 μm Gd3+ solution (right). C, average current-voltage relationships (IVs) measured in +divalents (left) or −divalents (right) solutions. D, average Gd3+-sensitive current-voltage relationships for DVF-induced currents measured for control (○, n = 16) or PMP22_WT expressing (●, n = 14) cells. *, p < 0.05; **, p ≤0.02; ***, p < 0.01.
An alternative explanation for the observed currents is that PMP22 is activating P2X7 receptors. P2X7 receptors are ATP-gated cation channels that have significant permeability to Ca2+ and Na+ (39, 40). PMP22 was previously reported to associate with P2X7 receptors (41) and to increase their activity (42). However, the currents measured in our expression system were independent of P2X7 receptors. Perfusion of control and PMP22_WT-transfected tsA201 cells with 1 mm ATP failed to activate any currents (data not shown), which is in accordance with previous findings indicating that HEK293 cells do not express P2X7 receptors or respond to ATP (39, 43, 44).
SOC currents are elicited in pmp22+/+ but not in pmp22−/− mouse Schwann cells
To determine whether endogenous PMP22 could contribute to SOC currents, we isolated Schwann cells from WT and pmp22−/− mice. Whole-cell currents from Schwann cells were recorded using calcium-free intracellular solution plus EGTA and exposed to control and DVF external solutions. As shown in Fig. 2, A and B, removal of external divalent cations induced currents in WT Schwann cells that were fully blocked by 10 μm Gd3+, similar to the currents detected in tsA201 cells transfected with PMP22_WT (Fig. 1). Further evidence that PMP22 may function as a physiological modulator of SOC channels was obtained when recording from mouse pmp22−/− Schwann cells. Removal of external divalents failed to generate current following passive depletion of ER Ca2+ in ∼60% of pmp22−/− Schwann cells (Fig. 2C). In the remaining cells, divalent removal induced large currents, ∼10× larger than pmp22+/+ cells, which were relatively insensitive to Gd3+ (Fig. 2D), requiring at least 100 μm Gd3+ before they were inhibited. Fig. 2E shows the average current recorded at −60 mV obtained from 14 pmp22−/− Schwann cells. The large currents in pmp22−/− cells are likely not mediated by the same channels as in pmp22+/+ cells because of their distinct Gd3+ insensitivity. Lee et al. (23) reported previously that pmp22−/− Schwann cells had impaired adhesion as well as disrupted lipid rafts and cytoskeletal organization. Indeed, we did note that pmp22−/− cells exhibited reduced adhesion, although this was not quantified. Such alterations in adhesion and the cytoskeleton could lead to increased opening of large, nonselective cation channels, which is consistent with the currents we detected in some of the cells. Interestingly, currents with these properties were not observed in any PMP22-expressing Schwann cells. These results strongly suggest that PMP22 expression modulates SOC channel activity.
Figure 2.

Gadolinium-sensitive, DVF-induced whole-cell currents are observed in pmp22+/+ but not in pmp22−/− mouse Schwann cells. A, average whole-cell currents recorded from WT mouse Schwann cells exposed to +divalents (left, +Ca2+/Mg2+), divalent-free (center, −Ca2+/Mg2+), or divalent-free plus 10 μm Gd3+ (right) solutions. B, average IVs for Gd3+-sensitive DVF-induced currents (●) and for currents measured in the presence of divalents (○) (n = 6). C, representative whole-cell currents recorded from pmp22−/− mouse Schwann cells exposed to divalent-free (left) or divalent-free plus 10 μm Gd3+ (right) solutions. D, example of large currents recorded from some pmp22−/− mouse Schwann cells exposed to divalent-free (left) or divalent-free plus 10 μm Gd3+ (right) solutions. E, average whole-cell currents measured at −60 mV and normalized for membrane capacitance in the presence of divalents, in DVF solution, and in DVF solution + 10 μm Gd3+. Left, no DVF-induced current (n = 8); right, Gd3+-resistant DVF-induced currents (n = 6). *, p < 0.05, **, p = 0.003; ***, p < 0.0001.
SOC currents in Schwann cells exhibit characteristics of TrpC1 channels
SOC currents flow through the channel Orai1 in the plasma membrane and are activated by the ER Ca2+ sensor STIM1 (28, 29). In addition, STIM1 can also activate the TrpC1 channel to allow Ca2+ influx, although this requires Orai1-mediated Ca+ entry, which causes TrpC1 to traffic to the cell surface where it forms a trans-complex with STIM1 (32). The STIM1–Orai1 complex comprises the classic calcium release–activated Ca2+ (CRAC) channel, whereas TrpC1 and STIM1 form distinct channels with different properties. The CRAC channel is inwardly rectifying, whereas the TrpC1 channel is weakly rectifying. The relatively linear IV curves we measured in both mouse Schwann cells and PMP22-transfected tsA201 cells (Figs. 1C and 2B) suggest that TrpC1, not Orai1, is mediating the current recorded in these cells. To further interrogate a role for TrpC1 in these currents, we tested the permeability of DVF-induced currents in Schwann cells to cesium (Cs+). Orai1 channels have a very low permeability to Cs+ (45), although this cation readily flows through TrpC1 channels (46). Fig. 3, A and B, illustrates whole-cell currents, and the average IV relationships measured for currents recorded from rat Schwann cells in the presence and absence of external divalents demonstrate that these cells exhibit similar currents to those recorded from mouse Schwann cells and tsA201 cells transfected with PMP22 (Figs. 1, A and B, and 2, A and B). We next tested whether substituting Na+ for Cs+ as the main external cation generated whole-cell currents in the absence of external divalent cations. As shown in Fig. 3C, similar currents were recorded from Schwann cells with either Na+ (●) or Cs+ (□) as the main external cation. In both conditions, removal of external divalents elicited inward currents with equal amplitudes and reversal potentials of ∼0 mV, indicating similar permeability to these two cations, supporting a role for TrpC1 in these currents.
Figure 3.

Divalent-free induced current in Schwann cells is permeable to both Cs+ and Na+ and similar to TrpC channels. A, average whole-cell currents recorded from WT rat Schwann cells exposed to +divalents (left, +Ca2+/Mg2+), divalent-free (center, −Ca2+/Mg2+), or divalent-free plus 10 μm Gd3+ (right) solutions. B, average IVs for Gd3+-sensitive for divalent-free (●) and for currents recorded in +Ca2+/Mg2+ solution (○). n = 7. *, p < 0.05; **, p < 0.01. C, average IVs recorded from rat Schwann cells exposed to +divalents (left, +Ca2+/Mg2+) or divalent-free (right, −Ca2+/Mg2+) solution with Na+ (●, n = 6) or Cs+ (□, n = 7) as the main external cation.
ER-restricted PMP22_L16P mutant is sufficient for modulation of SOC channels
Under normal conditions only ∼20% of WT PMP22 reaches the cell surface (26, 47), whereas the rest is retained in the ER and is eventually targeted for degradation via the ER-associated degradation pathway (10, 48). Thus, WT PMP22 may regulate SOC channel activity when localized to either the plasma membrane through association with a channel protein or at the ER by interacting with STIM1. To distinguish between these two possibilities, we isolated Schwann cells from TrJ mice, a mouse model for CMT1E disease that expresses the PMP22 point mutation L16P (49). This mutant protein, in contrast to WT PMP22, fails to localize to the cell surface and accumulates in the ER and ER-Golgi intermediate compartment (9, 50). Moreover, PMP22_L16P also reduces the surface expression of WT PMP22 (50). However, the mechanism by which the mutation leads to disease pathology is poorly understood. It is not simply a loss of function, because heterozygous loss of function mutations result in a different disease state, HNPP (51).
Dialysis with calcium-free intracellular solution followed by exposure to DVF external solution yielded Gd3+-sensitive currents in Schwann cells isolated from littermate WT and TrJ heterozygous (TrJ-Het) mice (Fig. 4, A and B). Interestingly, the currents recorded from TrJ-Het Schwann cells were significantly larger than those recorded from WT cells. Fig. 4C shows the IV relationships for Gd3+-sensitive DVF-induced currents for WT and TrJ-Het cells. DVF-induced currents in both WT and TrJ-Het cells exhibited similar sensitivity to block by external Gd3+, WT IC50 ∼1.3 μm, and TrJ-Het IC50 ∼1.4 μm (Fig. 4D). These results suggest that SOC channel modulation by PMP22 does not require its localization to the plasma membrane.
Figure 4.

TrJ-Het mouse Schwann cells exhibit Gd3+-sensitive, DVF-induced whole-cell currents larger than littermate WT cells. Representative whole-cell currents were recorded from WT (A) or TrJ-Het (B) mouse Schwann cells exposed to +divalent (left, +Ca2+/Mg2+), divalent-free (center, −Ca2+/Mg2+), or divalent-free plus 10 μm Gd3+ (right) solutions. C, average IVs for Gd3+-sensitive DVF-induced currents measured from WT (♢, n = 21) or TrJ-Het (♦, n = 6) cells. D, concentration-response curves for Gd3+ block of DVF-induced currents recorded from WT (♢) or TrJ-Het (♦) Schwann cells. Solid lines represent data fit with Hill equation. n ≤4 for all concentrations. *, p < 0.05; **, p ≤0.02.
Because TrJ-Het Schwann cells can still express some PMP22_WT at the plasma membrane, albeit at much reduced levels, we further explored the possibility of current modulation by ER-bound PMP22 by transiently expressing the TrJ PMP22 mutant (L16P) in tsA201 cells. Ectopic expression of PMP22_L16P results in virtually all of the protein trapped inside the cell with no detectable PMP22 on the cell surface (15, 50). As illustrated in Fig. 5, expression of either PMP22_WT or PMP22_L16P produced Gd3+-sensitive, DVF-induced currents (Fig. 5, A and B). Similar to the results from TrJ-Het Schwann cells, the currents recorded from heterologous expression of PMP22_L16P were substantially larger than those from PMP22_WT-expressing tsA201 cells. Fig. 5C shows the IV relationships for Gd3+-sensitive DVF-induced currents for PMP22_WT type and PMP22_L16P-expressing cells. The DVF-induced currents recorded from tsA201 cells expressing either PMP22_WT or PMP22_L16P were equally sensitive to Gd3+ (IC50 ∼0.4 μm, Fig. 5D) and show similar Gd3+ sensitivity as the DVF-induced currents in mouse WT and TrJ-Het Schwann cells (Fig. 4D). These results support the notion that it is the ER-located population of PMP22 that modulates SOC function.
Figure 5.

Traffic-deficient PMP22_L16P mutant generates Gd3+-sensitive, DVF-induced whole-cell currents in tsA201 cells. Representative whole-cell currents recorded from tsA201 cells transiently expressing PMP22_WT (A) or PMP22_L16P (B) exposed to +divalents (left, +Ca2+/Mg2+), divalent-free (center, −Ca2+/Mg2+), or divalent-free plus 10 μm Gd3+ (right) solutions. C, average IVs for Gd3+-sensitive DVF-induced currents measured from cells expressing PMP22_WT (♢, n = 11) or PMP22_L16P (♦, n = 14). D, concentration-response curves for Gd3+ block of DVF-induced currents recorded from tsA201 cells transiently expressing PMP22_WT (♢) or PMP22_L16P (♦). Solid lines represent data fit with Hill equation. n ≤5 for all concentrations.
Further evidence that PMP22 at the plasma membrane is not required for SOC channel modulation was provided by heterologous expression of the PMP22 trembler mutation, G150D, which also fails to reach the cell surface (11). Transient expression of PMP22_G150D in tsA201 cells generated DVF-induced currents similar to those observed when PMP22_L16P was expressed. In the presence of external divalents, the currents measured at −80 mV were −1.6 ± 0.7 pA/pF (n = 5) for cells expressing PMP22_WT and −2.0 ± 1.5 pA/pF (n = 5) for cells expressing PMP22_G150D. Following the removal of external divalents, the Gd3+-sensitive current measured at −80 mV in cells expressing the WT protein was −16.0 ± 8.3 pA/pF (n = 5) and −24.0 ± 10.6 pA/pF (n = 5) in PMP22_G150D-expressing cells. Therefore, similar to PMP22_L16P, expression of the ER-retained PMP22_G150D mutant also resulted in DVF-induced currents that were larger than those measured when WT PMP22 was expressed. These findings further indicate that SOC channels are modulated by ER-localized PMP22.
Intracellular calcium levels are increased in TrJ nerves and Schwann cells
The larger currents observed in TrJ-Het Schwann cells suggested that there may be disrupted intracellular Ca2+ homeostasis in the nerves of Trembler J mice, which could contribute to demyelinating pathology. To test this possibility, isolated sciatic nerves from TrJ-Het or WT mice were incubated in cell culture media containing the Ca2+ indicator Calcium OrangeTM. After 20 min, the nerves were rinsed and rapidly imaged, and the fluorescence intensity was measured. Compared with WT nerves, those from TrJ-Het mice exhibited significantly higher levels of intracellular Ca2+ (Fig. 6, A and B). In addition, the distribution pattern for the Ca2+ was altered in the TrJ-Het nerves: in the WT, Ca2+ was primarily localized to the regions adjacent to the nodes of Ranvier, as described previously (52), whereas in TrJ-Het nerves, it was distributed throughout the internodal regions, suggesting disrupted Ca2+ homeostasis.
Figure 6.

Intracellular calcium levels are increased in TrJ nerves. A, sciatic nerves from 4- to 8-week-old WT and TrJ-Het mice. The perineurium was extracted, and fibers were teased apart before being incubated with Calcium Orange AM in DMEM. Nerves were visualized at ×400 on a fluorescent microscope. Scale bar, 5 μm. B, average signal intensities were obtained and normalized to the area of the regions of interest and the mean intensity of the neighboring background. The nodal regions in the WT nerves (arrows) were avoided because they exhibited higher levels of Ca2+ than the internodal regions. n = 3, **, p = 0.0018, based on a Student's t test.
To quantify intracellular Ca2+ levels specifically in Schwann cells, as opposed to whole-nerve fibers, we cultured Schwann cells from WT and TrJ-Het sciatic nerves. The cells were incubated with the Ca2+-sensitive dye FURA-2AM, and the resting intracellular Ca2+ concentration was determined using an in vitro calibration curve. Corroborating the results from the intact nerve analysis, there was a 32.5% increase in intracellular Ca2+ in TrJ-Het Schwann cells relative to WT (Fig. 7). These results are consistent with our finding that the L16P mutant PMP22 allows excess Ca2+ flux through SOC channels, which could contribute to dysmyelination.
Figure 7.
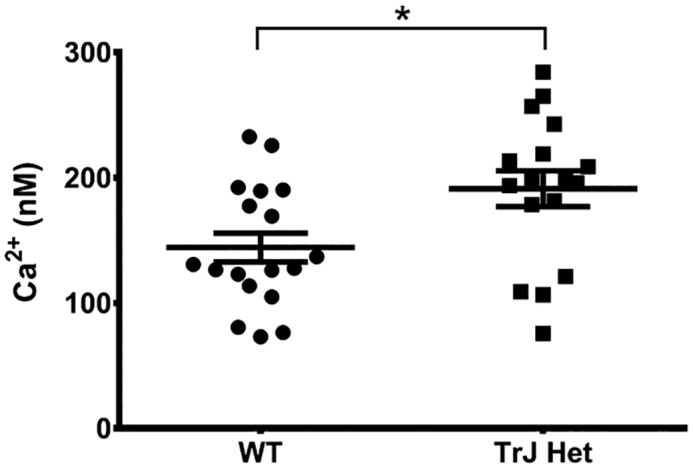
Intracellular calcium levels are increased in TrJ Schwann cells. Schwann cells cultured from WT or TrJ-Het mice were loaded with Fura-2AM, and the 340/380 fluorescence intensity ratio was measured and converted to [Ca2+]i using an in vitro calibration curve. Shown are the measurements for individual cells from two separate dissections with the average value ± S.E. (*, p = 0.015, based on a Student's t test).
PMP22 associates with STIM1
The most likely mechanism by which ER-located PMP22 could modulate SOC channel activity is by interacting with STIM1, the SOC channel subunit localized to the ER (28, 29). We tested this possibility by performing protein co-immunoprecipitation from rat sciatic nerve. As depicted in Fig. 8, STIM1 and PMP22 could be co-immunoprecipitated, suggesting that modulation of SOC currents by PMP22 occurs through its interaction with STIM1 in the ER.
Figure 8.
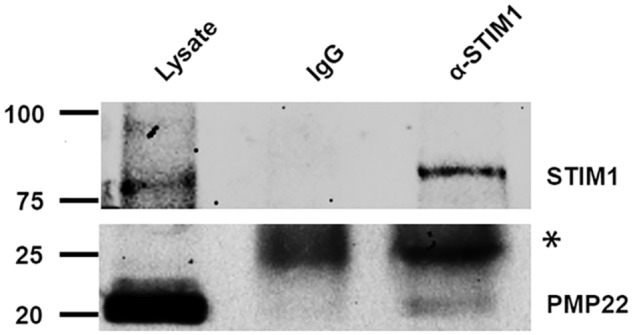
Co-immunoprecipitation of PMP22 and STIM1 from mouse sciatic nerve. The top and bottom panels illustrate whole-cell lysate proteins (left) and proteins immunoprecipitated with IgG (middle) or anti-STIM1 (right) antibody and then probed with anti-STIM1 antibody (top) or with anti-PMP22 antibody (bottom) (*, antibody light chain).
Discussion
CMT disease is the most common inherited neuropathy in humans (3, 24), yet there is currently no cure for this debilitating disease. CMT disease is associated with gene duplication (CMT1A) (6–8), dominant single-point mutations (CMT1E) (24), and heterozygous loss of function (HNPP) (5) of PMP22. Although the function of PMP22 is not well-understood, it clearly has an essential role in myelination: pmp22−/− mice exhibit tomacula and eventual loss of myelin, axon degeneration, and severe neuropathy (53). Both gene duplication and single-point mutations in PMP22 lead to dys-/demyelination, increased Schwann cell number, and secondary axon degeneration, but the mechanism leading to the pathology is not known (24, 54, 55). It is clear, however, that the disease involves more than a simple loss of PMP22 function because the phenotype of the pmp22 knockout is very distinct from that of CMT1A and CMT1E (53). Further evidence of the differences between the pathology caused by the absence, overexpression, or mutations in PMP22 is suggested by our ability to elicit SOC-like currents in WT, and even larger SOC-like currents in heterozygous TrJ Schwann cells, but not in Schwann cells isolated from pmp22−/− mice.
PMP22 expression increases SOC currents independently of P2X7 receptor expression
We hypothesized that PMP22 affects ion channel activity, thus modifying cell function. A previous study showed that PMP22 affects membrane conductance by modifying the activity of the ATP-gated P2X7 receptor, a calcium channel, leading to higher intracellular calcium and demyelination (42). However, our results show that PMP22 increases the magnitude of SOC-like currents independently of P2X7 receptors: HEK293 cells do not express P2X7 receptors or ATP-activated currents (39, 43, 44) and perfusion of ATP failed to activate currents in control or PMP22-expressing tsA201 cells. In addition, the interaction between PMP22 and P2X7 most likely occurs at the plasma membrane, because these proteins were shown to co-immunoprecipitate (40) and P2X7 mediates influx of extracellular Ca2+. In contrast, our results indicate that modulation of SOC channels by PMP22 occurs via interactions in the ER, not at the plasma membrane; SOC-like currents were detected upon expression of the PMP22_L16P mutant, which fails to reach the cell surface, and an interaction between the ER resident protein STIM1 and PMP22 was detected. Therefore, our findings together with previous studies suggest that PMP22 may affect cellular Ca2+ homeostasis by increasing the function of two pathways, P2X7 and SOC channels. Whether the PMP22 regulation occurs individually, or concurrently on both channels, is an interesting yet unaddressed question.
SOC-like currents regulated by PMP22 suggest the involvement of TrpC1
SOC channels are composed of a pore-forming protein (Orai1) in the plasma membrane and a calcium sensor in the ER (STIM1), which senses depletion of ER calcium stores following various stimuli (28, 29). Following Ca2+ efflux from the ER, the C-terminal region of STIM1 interacts with Orai1 in trans fashion, inducing channel opening and influx of extracellular Ca2+ to replenish the stores. STIM1 appears to be the primary activator of Orai1 (56). This CRAC channel is very Ca2+-selective and highly inwardly rectifying, which differs from the weakly rectifying, Na+ permeant currents that we observed. However, Orai1-mediated Ca2+ entry causes TrpC1 to traffic to the cell surface where it also interacts with STIM1 to function as a unique SOC channel (57). TrpC1 can form a channel on its own or in combination with other TrpC channels, although only TrpC1 has been detected at significant levels in cultured Schwann cells (GEO database, National Institutes of Health). Nevertheless, the channels formed in combination with TrpC1 mediate a Ca2+ flux that is weakly rectifying and is a relatively nonselective cation channel (58–60). Notably, TrpC1 is permeable to Cs+ (46), unlike the CRAC channel (45). The currents we found regulated by PMP22 in Schwann cells are most consistent with those mediated by TrpC1.
PMP22 interacts with the SOC channel subunit STIM1
Our results, both functional and biochemical, suggest that PMP22 modulates SOC channels by interacting with STIM1. However, it is possible that the PMP22 does not directly interact with STIM1. Previous results showed that STIM1 binds to the ER chaperone protein calnexin (61). Moreover, calnexin has been demonstrated to associate with PMP22 (62). Thus, the PMP22–STIM1 interaction may be mediated by calnexin. Interestingly, the mutant PMP22_L16P protein displays dramatically prolonged binding to calnexin in the ER (62). Hence, it is possible that the larger currents recorded from TrJ heterozygous Schwann cells and tsA201 cells transiently expressing PMP22_L16P, when compared with WT Schwann cells or tsA201 cells expressing PMP22_WT, may be due to a prolonged lifetime of the PMP22–calnexin–STIM1 complex, not just higher PMP22 protein levels in the ER.
PMP22 is a member of the claudin/EMP/voltage-gated Ca2+ channel γ-subunit superfamily of tetraspan membrane proteins, which include the transmembrane α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor regulatory proteins (TARP). Like many of these proteins, PMP22 has been shown to regulate the formation of various types of cell junctions, including various myelin junctions in Schwann cells (25) as well as epithelial and endothelial cell–cell junctions (22). Considering that the Orai1–STIM1–TrpC1 complex essentially forms a junction between the ER and the plasma membrane, it is not surprising that PMP22 would also regulate this type of junction. However, PMP22 has primarily been considered to function at the plasma membrane, yet only about 20% of the nascent WT protein successfully traffics to the cell surface, most of it being retained in the ER, where it is eventually targeted for degradation (47). The results presented here suggest that some portion of ER-retained PMP22 also has a role in regulating SOC channels.
SOC channels and demyelination
Currently, the mechanism by which PMP22 gene duplication or single-point mutations cause the pathology of CMT is not known (24, 54, 55). Studies with CMT1A rat Schwann cells, which overexpress PMP22 due to gene duplication, show that down-regulation (42) or block (63) of the P2X7 receptor promotes myelination, suggesting that altered intracellular calcium levels may contribute to defects in myelination in this model of disease. In contrast, single-point mutations in PMP22, such as the L16P mutation, cause the protein to accumulate in the Schwann cell ER (9, 50). Higher levels of PMP22 protein in the ER may lead to increased PMP22–STIM1 complexes and larger SOC channel activity. Indeed, the currents we detected in the TrJ Schwann cells under divalent free conditions were much larger than those in WT cells. In addition, there were significantly higher levels of intracellular Ca2+ in nerves from TrJ mice, relative to the WT. Calcium levels in the ER are tightly controlled, and perturbations in Ca2+ homeostasis via Ca2+ depletion or overload would alter cellular function (64). For example, changes in ER Ca2+ concentrations trigger ER stress, which activates the unfolded protein response (64). Indeed, markers of ER stress have been detected in TrJ nerves, which may also contribute to the nerve pathology (65).
PMP22 possibly affects intracellular Ca2+ levels by modulating both P2X7 and, based on the results presented here, SOC channels, depending on its cellular location. This divergent effect would indicate a difference between the pathology of CMT1A and CMT1E: increased P2X7 activity in CMT1A (due to PMP22 trisomy) but SOC channel activity in CMT1E. Moreover, the degree of Ca2+ disruption may differ between CMT1A and CMT1E: Schwann cells isolated from CMT1E-like TrJ mice (and tsA201 cells expressing PMP22_L16P) exhibit larger currents (with similar kinetics and Gd3+ sensitivity) than cells expressing the WT protein. Moreover, indicators of ER stress have only been detected in nerves from TrJ but not PMP22-overexpressing animals (65, 66).
In summary, our results suggest a new role for PMP22 in Schwann cell physiology and pathophysiology. PMP22 localized in the ER affects Ca2+-dependent cellular processes by modifying SOC channel activity and calcium influx. The observed association between PMP22 and STIM1 may explain how misfolding-based ER retention (26) (13) of mutant forms of PMP22 alters intracellular Ca2+ levels, contributing to CMT1E pathogenesis. Future studies will investigate the role of PMP22 on Ca2+ homeostasis and determine whether the variety of CMT disease phenotypes is the result of variations in the specific interactions between STIM1 with various disease-associated forms of PMP22.
Materials and methods
Cell culture
tsA201 cells (HEK293 cells stably transfected with SV40 large T antigen) were grown at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Norcross, GA), 2 mm l-glutamine, and penicillin (50 units/ml)/streptomycin (P/S) (50 μg/ml). Only cells from passage numbers of <13 were used. Unless otherwise stated, all tissue culture media were obtained from Life Technologies, Inc.
Mouse Schwann cells were isolated from sciatic nerves of CD1, C57BL/6J WT, pmp22−/− (53) backcrossed into C57BL/6 for nine generations, or TrJ heterozygous mice (49) on postnatal days 4–6, as described previously (67). The cells were plated on culture dishes coated with poly-d-lysine (Thermo Fisher Scientific) and maintained in DMEM with 10% FBS (Sigma) supplemented with 50 ng/ml NRG1 recombinant epidermal growth factor domain (R & D Sciences catalog no. 396-B).
Rat Schwann cells were isolated from postnatal days 4–6 pups and maintained in DMEM with 10% FBS and P/S, supplemented with forskolin (2 μm, Sigma catalog no. F86886).
All experiments conducted with tissues derived from animals were approved by the Animal Care and Use Committee at Vanderbilt University.
Plasmids and cell transfection
Full-length human PMP22 (GenBankTM accession no. NG_007949.1) cDNA was engineered into the mammalian expression vector pIRES2-EGFP (BD Biosciences–Clontech). The PMP22_L16P mutant was generated by introducing the mutation into PMP22_WT using the QuikChange site-directed mutagenesis system (Stratagene, La Jolla, CA). All recombinant cDNAs were sequenced in their entirety to confirm the presence of the desired modification and the absence of unwanted mutations. Expression of PMP22_WT and PMP22_L16P constructs were achieved by transient transfection using FuGENE 6 (Roche Applied Science) in which 0.7 μg of cDNA was transfected. To rule out nonspecific effects due to transfection or EGFP expression, control cells were transfected with the nonrecombinant pIRES2-EGFP vector. After transfection, tsA201 cells were incubated for 48 h as described above before use in electrophysiological experiments.
Immunoprecipitation and Western blotting
Sciatic nerves isolated from postnatal day 5 rats were lysed in RIPA buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, with cOmpleteTM protease inhibitor (Millipore Sigma)). The lysate was quantified using the Bradford reagent (Bio-Rad). 500 μg of total sciatic nerve lysate were incubated with protein G–Sepharose beads (Life Technologies, Inc.) to pre-clear nonspecific protein binding to the Sepharose beads. For immunoprecipitation, anti-STIM1 (Abnova, Walnut, CA) or mouse IgG was incubated with protein G–Sepharose beads. The precipitates were then washed three times with RIPA buffer followed by two washes with TBS. Immune complexes were then separated on a 9% polyacrylamide gel for STIM1 and a 12% polyacrylamide gel for PMP22 and Western-blotted using a rabbit anti-STIM1 antibody (Cell Signaling, Danvers, MA) at 1:1000 or rabbit anti-PMP22 antibody (Assay Biotech, Fremont, CA) at 1:2500. Following incubation with primary antibody, the blots were probed with horseradish peroxidase-conjugated anti-rabbit (Pierce) and detected by enhanced chemiluminescence on a Bio-Rad ChemiDoc MP imaging system and processed in ImageLab 6 (Bio-Rad).
Live calcium imaging
Sciatic nerves of adult (6–8 weeks) TrJ heterozygous and C57BL/6J mice were isolated and immediately transferred into DMEM without phenol red (Gibco/Invitrogen) on ice. The epineurium and perineurium were excised, and individual nerve fibers were teased apart. The nerves were incubated for 20 min (37 °C, 5% CO2) in DMEM containing 4 μm Calcium OrangeTM AM (Life Technologies, Inc., catalog no. C-3015). The nerves were then rinsed once with DMEM and incubated in low light at room temperature (20–23 °C) for 20 min. Immediately following incubation, the fibers were imaged on glycerol-mounted slides at ×400 magnification using a Zeiss Axioskop2 microscope (Zeiss Microimaging, Thornwood, NY) and an AxioCam MRc5 camera with Axiovision Imaging software. For analysis, FIJI was used to determine the total corrected fluorescence levels of internodal regions relative to background by collecting the integrated density of fluorescence levels of manually drawn regions of interest and subtracting the average background intensity (calculated from three adjacent selections, equal to the region of interest in area). Statistical significance among the two groups was determined using an unpaired Student's t test.
Intracellular calcium level measurement
The fluorescent calcium (Ca2+) dye Fura-2AM (Setareh Biotech catalog no. 6101, Eugene, OR) was used to measure cytosolic calcium concentration. Primary mouse Schwann cells were harvested and plated on poly-d-lysine (Thermo Fisher Scientific)-coated glass coverslips 2–3 days prior to recording. Cells were washed twice with HEPES-buffered Hanks' balanced salt solution (HBSS) and incubated for 30–45 min with 3 μm Fura-2 acetoxymethyl ester at 37 °C and then washed in Fura-free HBSS solution for 30–60 min before recording. The coverslips with the cells were transferred to a recording chamber mounted on the stage of a Nikon TE2000 fluorescence microscope (Nikon, Tokyo, Japan). The total volume of the recording chamber was 300–400 μl with constant perfusion from gravity-fed chambers with an extracellular solution containing 145 mm NaCl, 2 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm glucose, 10 mm HEPES, pH 7.3, osmolarity 305 mosm at a rate of 4 ml/min. An InCyt IM2 fluorescence imaging system (Intracellular Imaging Inc., Cincinnati, OH) was used to monitor [Ca2+]i. Cells were alternately excited at wavelengths of 340 and 380 nm, and emission at 510 nm was detected using a PixelFly digital camera. Ratios were collected every 1 s throughout the experiment and converted to [Ca2+]i using an in vitro calibration curve, generated by adding 15.8 μm Fura-2 pentapotassium salt to solutions from a calibration kit containing 1 mm MgCl2 and known concentrations of Ca2+ (0–1350 nm) (Invitrogen). Cells with an unstable baseline were excluded from the analysis.
Electrophysiology
On the day of the experiment, all cells were dissociated by brief exposure to trypsin/EDTA, resuspended in DMEM supplemented with 10% FBS, l-glutamine, and penicillin/streptomycin, plated on glass coverslips, and allowed to recover for ∼2 h at 37 °C in 5% CO2. Cells were kept under this condition for ≤6 h. Only green fluorescent tsA201 cells (i.e. positive for PMP22 or empty vector) were analyzed. For electrophysiological analysis of cells isolated from TrJ or WT mice, the experimenter was blinded to the genotype of the cells. Whole-cell currents were recorded at room temperature using Axopatch 200B amplifiers (Molecular Devices Corp., Sunnyvale, CA) in the whole-cell configuration of the patch-clamp technique (68). Pulse generation was carried out via Clampex 8.0 (Molecular Devices Corp.), and whole-cell currents were filtered at 1 kHz and acquired at 5 kHz. The access resistance and apparent membrane capacitance were estimated using an established method (69). Whole-cell currents were not leak-subtracted. Whole-cell currents were measured from −80 to +60 mV (in 20-mV steps) from a holding potential of 0 mV. We used solutions similar to those previously employed in studies of SOC channels (37, 38). The control external solution contained (in mm): NaCl 140, MgCl2 1.2, CaCl2 20, glucose 10, HEPES 10, pH 7.4. The divalent-free external solution contained (in mm): NaCl 140, glucose 10, HEPES 10, sucrose 30, pH 7.4. The internal solution contained (in mm): CsOH 140, aspartic acid 140, MgCl2 1.22, EGTA 5, HEPES 10, pH 7.2. The pipette solution was diluted 5–10% to prevent activation of swelling-activated currents. Patch pipettes were pulled from thick-wall borosilicate glass (World Precision Instruments, Inc., Sarasota, FL) with a multistage P-97 Flaming-Brown micropipette puller (Sutter Instruments Co., San Rafael, CA) and heat-polished with a Microforge MF 830 (Narashige, Japan). After heat polishing, the resistance of the patch pipettes was 3–5 MΩ in the standard extracellular solution. The access resistance varied from to 5 to 9 MΩ. As a reference electrode, a 2% agar bridge with composition similar to the control bath solution was utilized. Junction potentials were zeroed with the filled pipette in the bath solution. Unless otherwise stated, all chemicals were obtained from Sigma.
Data analysis
Data were collected for each experimental condition from at least three transient transfections or two Schwann cell isolations, analyzed, and plotted using a combination of Clampfit (Molecular Devices Corp.), SigmaPlot 2000 (Systat Software, Inc., San Jose, CA), and Origin 7.0 (OriginLab, Northampton, MA). Statistical analyses were carried out using SigmaStat 2.03 (Systat Software, Inc.) or Prism 6/7 (Graphpad Software, San Diego, CA), and p values are listed in the figure legends. Statistical significance between the two groups was determined using unpaired Student's t tests. Whole-cell currents are normalized for membrane capacitance, and results are expressed as mean ± S.E. The number of cells used for each experimental condition is given in the figure legends.
Author contributions
C. G. V., M. S., C. R. S., and B. D. C. conceptualization; C. G. V., M. S., R. M. F., and M. N. data curation; C. G. V., R. M. F., and A. J. T. formal analysis; C. G. V., M. S., R. M. F., and M. N. investigation; C. G. V., M. S., R. M. F., A. J. T., M. N., and J. L. methodology; C. G. V. writing-original draft; C. G. V., R. M. F., J. L., C. R. S., and B. D. C. writing-review and editing; J. L., C. R. S., and B. D. C. resources; J. L., C. R. S., and B. D. C. funding acquisition; C. R. S. and B. D. C. supervision; C. R. S. and B. D. C. project administration.
This work was supported by National Institutes of Health Grants R01 NS058815 and R01 NS095989 (to C. R. S. and B. C.), R01NS038220 and R01NS102365 (to B. C.), and R01NS066927 (to J. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CMT
- Charcot-Marie-Tooth
- ER
- endoplasmic reticulum
- SOC
- store-operated calcium
- HNPP
- hereditary neuropathy with liability to pressure palsy
- FBS
- fetal bovine serum
- HBSS
- Hanks' balanced salt solution
- DMEM
- Dulbecco's modified Eagle's medium
- EGFP
- enhanced GFP
- TrJ
- Trembler J
- TrJ-Het
- Trembler-J heterozygous
- CRAC
- calcium release-activated Ca2+
- P/S
- penicillin/streptomycin
- MΩ
- megohm
- DVF
- divalent-free
- pF
- picofarad.
References
- 1. Hayasaka K., Himoro M., Nanao K., Sato W., Miura M., Uyemura K., Takahashi E., and Takada G. (1992) Isolation and sequence determination of cDNA encoding PMP-22 (PAS-II/SR13/Gas-3) of human peripheral myelin. Biochem. Biophys. Res. Commun. 186, 827–831 10.1016/0006-291X(92)90820-B [DOI] [PubMed] [Google Scholar]
- 2. Snipes G. J., Suter U., Welcher A. A., and Shooter E. M. (1992) Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J. Cell Biol. 117, 225–238 10.1083/jcb.117.1.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grandis M., and Shy M. E. (2005) Current therapy for Charcot-Marie-Tooth disease. Curr. Treat. Options Neurol. 7, 23–31 10.1007/s11940-005-0003-5 [DOI] [PubMed] [Google Scholar]
- 4. Li J. (2012) Inherited neuropathies. Semin. Neurol. 32, 204–214 10.1055/s-0032-1329198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J., Krajewski K., Lewis R. A., and Shy M. E. (2004) Loss-of-function phenotype of hereditary neuropathy with liability to pressure palsies. Muscle Nerve 29, 205–210 10.1002/mus.10521 [DOI] [PubMed] [Google Scholar]
- 6. Timmerman V., Raeymaekers P., De Jonghe P., De Winter G., Swerts L., Jacobs K., Gheuens J., Martin J. J., Vandenberghe A., and Van Broeckhoven C. (1990) Assignment of the Charcot-Marie-Tooth neuropathy type 1 (CMT 1a) gene to 17p11.2-p12. Am. J. Hum. Genet. 47, 680–685 [PMC free article] [PubMed] [Google Scholar]
- 7. Lupski J. R., de Oca-Luna R. M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B. J., Saucedo-Cardenas O., Barker D. F., Killian J. M., Garcia C. A., Chakravarti A., and Patel P. I. (1991) DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219–232 10.1016/0092-8674(91)90613-4 [DOI] [PubMed] [Google Scholar]
- 8. Raeymaekers P., Timmerman V., Nelis E., Van Hul W., De Jonghe P., Martin J. J., and Van Broeckhoven C. (1992) Estimation of the size of the chromosome 17p11.2 duplication in Charcot-Marie-Tooth neuropathy type 1a (CMT1a). HMSN Collaborative Research Group. J. Med. Genet. 29, 5–11 10.1136/jmg.29.1.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Urso D., Prior R., Greiner-Petter R., Gabreëls-Festen A. A., and Müller H. W. (1998) Overloaded endoplasmic reticulum-Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J. Neurosci. 18, 731–740 10.1523/JNEUROSCI.18-02-00731.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Naef R., and Suter U. (1999) Impaired intracellular trafficking is a common disease mechanism of PMP22 point mutations in peripheral neuropathies. Neurobiol. Dis. 6, 1–14 10.1006/nbdi.1998.0227 [DOI] [PubMed] [Google Scholar]
- 11. Colby J., Nicholson R., Dickson K. M., Orfali W., Naef R., Suter U., and Snipes G. J. (2000) PMP22 carrying the trembler or trembler-J mutation is intracellularly retained in myelinating Schwann cells. Neurobiol. Dis. 7, 561–573 10.1006/nbdi.2000.0323 [DOI] [PubMed] [Google Scholar]
- 12. Tobler A. R., Liu N., Mueller L., and Shooter E. M. (2002) Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc. Natl. Acad. Sci. U.S.A. 99, 483–488 10.1073/pnas.012593399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Myers J. K., Mobley C. K., and Sanders C. R. (2008) The peripheral neuropathy-linked Trembler and Trembler-J mutant forms of peripheral myelin protein 22 are folding-destabilized. Biochemistry 47, 10620–10629 10.1021/bi801157p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakakura M., Hadziselimovic A., Wang Z., Schey K. L., and Sanders C. R. (2011) Structural basis for the Trembler-J phenotype of Charcot-Marie-Tooth disease. Structure 19, 1160–1169 10.1016/j.str.2011.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schlebach J. P., Narayan M., Alford C., Mittendorf K. F., Carter B. D., Li J., and Sanders C. R. (2015) Conformational stability and pathogenic misfolding of the integral membrane protein PMP22. J. Am. Chem. Soc. 137, 8758–8768 10.1021/jacs.5b03743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zoidl G., Blass-Kampmann S., D'Urso D., Schmalenbach C., and Müller H. W. (1995) Retroviral-mediated gene transfer of the peripheral myelin protein PMP22 in Schwann cells: modulation of cell growth. EMBO J. 14, 1122–1128 10.1002/j.1460-2075.1995.tb07095.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fabbretti E., Edomi P., Brancolini C., and Schneider C. (1995) Apoptotic phenotype induced by overexpression of wild-type gas3/PMP22: its relation to the demyelinating peripheral neuropathy CMT1A. Genes Dev. 9, 1846–1856 10.1101/gad.9.15.1846 [DOI] [PubMed] [Google Scholar]
- 18. Mittendorf K. F., Marinko J. T., Hampton C. M., Ke Z., Hadziselimovic A., Schlebach J. P., Law C. L., Li J., Wright E. R., Sanders C. R., and Ohi M. D. (2017) Peripheral myelin protein 22 alters membrane architecture. Sci. Adv. 3, e1700220 10.1126/sciadv.1700220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'Urso D., Ehrhardt P., and Müller H. W. (1999) Peripheral myelin protein 22 and protein zero: a novel association in peripheral nervous system myelin. J. Neurosci. 19, 3396–3403 10.1523/JNEUROSCI.19-09-03396.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jetten A. M., and Suter U. (2000) The peripheral myelin protein 22 and epithelial membrane protein family. Prog. Nucleic Acid Res. Mol. Biol. 64, 97–129 10.1016/S0079-6603(00)64003-5 [DOI] [PubMed] [Google Scholar]
- 21. Notterpek L., Roux K. J., Amici S. A., Yazdanpour A., Rahner C., and Fletcher B. S. (2001) Peripheral myelin protein 22 is a constituent of intercellular junctions in epithelia. Proc. Natl. Acad. Sci. U.S.A. 98, 14404–14409 10.1073/pnas.251548398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roux K. J., Amici S. A., Fletcher B. S., and Notterpek L. (2005) Modulation of epithelial morphology, monolayer permeability, and cell migration by growth arrest specific 3/peripheral myelin protein 22. Mol. Biol. Cell 16, 1142–1151 10.1091/mbc.e04-07-0551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee S., Amici S., Tavori H., Zeng W. M., Freeland S., Fazio S., and Notterpek L. (2014) PMP22 is critical for actin-mediated cellular functions and for establishing lipid rafts. J. Neurosci. 34, 16140–16152 10.1523/JNEUROSCI.1908-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J., Parker B., Martyn C., Natarajan C., and Guo J. (2013) The PMP22 gene and its related diseases. Mol. Neurobiol. 47, 673–698 10.1007/s12035-012-8370-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo J., Wang L., Zhang Y., Wu J., Arpag S., Hu B., Imhof B. A., Tian X., Carter B. D., Suter U., and Li J. (2014) Abnormal junctions and permeability of myelin in PMP22-deficient nerves. Ann. Neurol. 75, 255–265 10.1002/ana.24086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanders C. R., Ismail-Beigi F., and McEnery M. W. (2001) Mutations of peripheral myelin protein 22 result in defective trafficking through mechanisms which may be common to diseases involving tetraspan membrane proteins. Biochemistry 40, 9453–9459 10.1021/bi010894f [DOI] [PubMed] [Google Scholar]
- 27. Mittendorf K. F., Kroncke B. M., Meiler J., and Sanders C. R. (2014) The homology model of PMP22 suggests mutations resulting in peripheral neuropathy disrupt transmembrane helix packing. Biochemistry 53, 6139–6141 10.1021/bi500809t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soboloff J., Spassova M. A., Tang X. D., Hewavitharana T., Xu W., and Gill D. L. (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 281, 20661–20665 10.1074/jbc.C600126200 [DOI] [PubMed] [Google Scholar]
- 29. Mercer J. C., Dehaven W. I., Smyth J. T., Wedel B., Boyles R. R., Bird G. S., and Putney J. W. Jr. (2006) Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J. Biol. Chem. 281, 24979–24990 10.1074/jbc.M604589200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bird G. S., DeHaven W. I., Smyth J. T., and Putney J. W. Jr. (2008) Methods for studying store-operated calcium entry. Methods 46, 204–212 10.1016/j.ymeth.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parekh A. B., and Putney J. W. Jr. (2005) Store-operated calcium channels. Physiol. Rev. 85, 757–810 10.1152/physrev.00057.2003 [DOI] [PubMed] [Google Scholar]
- 32. Ong H. L., and Ambudkar I. S. (2017) STIM-TRP pathways and microdomain organization: contribution of TRPC1 in store-operated Ca2+ entry: impact on Ca2+ signaling and cell function. Adv. Exp. Med. Biol. 993, 159–188 10.1007/978-3-319-57732-6_9 [DOI] [PubMed] [Google Scholar]
- 33. Smith K. J., Hall S. M., and Schauf C. L. (1985) Vesicular demyelination induced by raised intracellular calcium. J. Neurol. Sci. 71, 19–37 10.1016/0022-510X(85)90034-6 [DOI] [PubMed] [Google Scholar]
- 34. Paul D. L., Ebihara L., Takemoto L. J., Swenson K. I., and Goodenough D. A. (1991) Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 115, 1077–1089 10.1083/jcb.115.4.1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y., McBride D. W. Jr, and Hamill O. P. (1998) The ion selectivity of a membrane conductance inactivated by extracellular calcium in Xenopus oocytes. J. Physiol. 508, 763–776 10.1111/j.1469-7793.1998.763bp.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vanoye C. G., Vergara L. A., and Reuss L. (1999) Isolated epithelial cells from amphibian urinary bladder express functional gap junctional hemichannels. Am. J. Physiol. 276, C279–C284 10.1152/ajpcell.1999.276.1.C279 [DOI] [PubMed] [Google Scholar]
- 37. Mignen O., and Shuttleworth T. J. (2001) Permeation of monovalent cations through the non-capacitative arachidonate-regulated Ca2+ channels in HEK293 cells. Comparison with endogenous store-operated channels. J. Biol. Chem. 276, 21365–21374 10.1074/jbc.M102311200 [DOI] [PubMed] [Google Scholar]
- 38. Bugaj V., Alexeenko V., Zubov A., Glushankova L., Nikolaev A., Wang Z., Kaznacheyeva E., Bezprozvanny I., and Mozhayeva G. N. (2005) Functional properties of endogenous receptor- and store-operated calcium influx channels in HEK293 cells. J. Biol. Chem. 280, 16790–16797 10.1074/jbc.M500192200 [DOI] [PubMed] [Google Scholar]
- 39. Valera S., Hussy N., Evans R. J., Adami N., North R. A., Surprenant A., and Buell G. (1994) A new class of ligand-gated ion channel defined by P2x receptor for extracellular ATP. Nature 371, 516–519 10.1038/371516a0 [DOI] [PubMed] [Google Scholar]
- 40. Jarvis M. F., and Khakh B. S. (2009) ATP-gated P2X cation-channels. Neuropharmacology 56, 208–215 10.1016/j.neuropharm.2008.06.067 [DOI] [PubMed] [Google Scholar]
- 41. Wilson H. L., Wilson S. A., Surprenant A., and North R. A. (2002) Epithelial membrane proteins induce membrane blebbing and interact with the P2X7 receptor C terminus. J. Biol. Chem. 277, 34017–34023 10.1074/jbc.M205120200 [DOI] [PubMed] [Google Scholar]
- 42. Nobbio L., Sturla L., Fiorese F., Usai C., Basile G., Moreschi I., Benvenuto F., Zocchi E., De Flora A., Schenone A., and Bruzzone S. (2009) P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J. Biol. Chem. 284, 23146–23158 10.1074/jbc.M109.027128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Worthington R. A., Dutton J. L., Poronnik P., Bennett M. R., and Barden J. A. (1999) Localisation of P2X receptors in human salivary gland epithelial cells and human embryonic kidney cells by sodium dodecyl sulfate-polyacrylamide gel electrophoresis/Western blotting and immunofluorescence. Electrophoresis 20, 2065–2070 [DOI] [PubMed] [Google Scholar]
- 44. Milius D., Gröger-Arndt H., Stanchev D., Lange-Dohna C., Rossner S., Sperlagh B., Wirkner K., and Illes P. (2007) Oxygen/glucose deprivation increases the integration of recombinant P2X7 receptors into the plasma membrane of HEK293 cells. Toxicology 238, 60–69 10.1016/j.tox.2007.05.028 [DOI] [PubMed] [Google Scholar]
- 45. Lepple-Wienhues A., and Cahalan M. D. (1996) Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys. J. 71, 787–794 10.1016/S0006-3495(96)79278-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Voets T., Prenen J., Fleig A., Vennekens R., Watanabe H., Hoenderop J. G., Bindels R. J., Droogmans G., Penner R., and Nilius B. (2001) CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. J. Biol. Chem. 276, 47767–47770 10.1074/jbc.C100607200 [DOI] [PubMed] [Google Scholar]
- 47. Pareek S., Notterpek L., Snipes G. J., Naef R., Sossin W., Laliberté J., Iacampo S., Suter U., Shooter E. M., and Murphy R. A. (1997) Neurons promote the translocation of peripheral myelin protein 22 into myelin. J. Neurosci. 17, 7754–7762 10.1523/JNEUROSCI.17-20-07754.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Notterpek L., Ryan M. C., Tobler A. R., and Shooter E. M. (1999) PMP22 accumulation in aggresomes: implications for CMT1A pathology. Neurobiol. Dis. 6, 450–460 10.1006/nbdi.1999.0274 [DOI] [PubMed] [Google Scholar]
- 49. Suter U., Moskow J. J., Welcher A. A., Snipes G. J., Kosaras B., Sidman R. L., Buchberg A. M., and Shooter E. M. (1992) A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc. Natl. Acad. Sci. U.S.A. 89, 4382–4386 10.1073/pnas.89.10.4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tobler A. R., Notterpek L., Naef R., Taylor V., Suter U., and Shooter E. M. (1999) Transport of Trembler-J mutant peripheral myelin protein 22 is blocked in the intermediate compartment and affects the transport of the wild-type protein by direct interaction. J. Neurosci. 19, 2027–2036 10.1523/JNEUROSCI.19-06-02027.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chance P. F., Alderson M. K., Leppig K. A., Lensch M. W., Matsunami N., Smith B., Swanson P. D., Odelberg S. J., Disteche C. M., and Bird T. D. (1993) DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 72, 143–151 10.1016/0092-8674(93)90058-X [DOI] [PubMed] [Google Scholar]
- 52. Lev-Ram V., and Ellisman M. H. (1995) Axonal activation-induced calcium transients in myelinating Schwann cells, sources, and mechanisms. J. Neurosci. 15, 2628–2637 10.1523/JNEUROSCI.15-04-02628.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adlkofer K., Martini R., Aguzzi A., Zielasek J., Toyka K. V., and Suter U. (1995) Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat. Genet. 11, 274–280 10.1038/ng1195-274 [DOI] [PubMed] [Google Scholar]
- 54. Roa B. B., Garcia C. A., Suter U., Kulpa D. A., Wise C. A., Mueller J., Welcher A. A., Snipes G. J., Shooter E. M., Patel P. I., and Lupski J. R. (1993) Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 gene. N. Engl. J. Med. 329, 96–101 10.1056/NEJM199307083290205 [DOI] [PubMed] [Google Scholar]
- 55. Gabreels-Festen A., and Wetering R. V. (1999) Human nerve pathology caused by different mutational mechanisms of the PMP22 gene. Ann. N.Y. Acad. Sci. 883, 336–343 10.1111/j.1749-6632.1999.tb08595.x [DOI] [PubMed] [Google Scholar]
- 56. Soboloff J., Rothberg B. S., Madesh M., and Gill D. L. (2012) STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565 10.1038/nrm3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ambudkar I. S., de Souza L. B., and Ong H. L. (2017) TRPC1, Orai1, and STIM1 in SOCE: friends in tight spaces. Cell Calcium 63, 33–39 10.1016/j.ceca.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu X., Bandyopadhyay B. C., Singh B. B., Groschner K., and Ambudkar I. S. (2005) Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J. Biol. Chem. 280, 21600–21606 10.1074/jbc.C400492200 [DOI] [PubMed] [Google Scholar]
- 59. Liu X., Groschner K., and Ambudkar I. S. (2004) Distinct Ca2+-permeable cation currents are activated by internal Ca2+-store depletion in RBL-2H3 cells and human salivary gland cells, HSG and HSY. J. Membr. Biol. 200, 93–104 10.1007/s00232-004-0698-3 [DOI] [PubMed] [Google Scholar]
- 60. Liu X., Singh B. B., and Ambudkar I. S. (2003) TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5–S6 region. J. Biol. Chem. 278, 11337–11343 10.1074/jbc.M213271200 [DOI] [PubMed] [Google Scholar]
- 61. Saitoh N., Oritani K., Saito K., Yokota T., Ichii M., Sudo T., Fujita N., Nakajima K., Okada M., and Kanakura Y. (2011) Identification of functional domains and novel binding partners of STIM proteins. J. Cell. Biochem. 112, 147–156 10.1002/jcb.22910 [DOI] [PubMed] [Google Scholar]
- 62. Dickson K. M., Bergeron J. J., Shames I., Colby J., Nguyen D. T., Chevet E., Thomas D. Y., and Snipes G. J. (2002) Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc. Natl. Acad. Sci. U.S.A. 99, 9852–9857 10.1073/pnas.152621799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sociali G., Visigalli D., Prukop T., Cervellini I., Mannino E., Venturi C., Bruzzone S., Sereda M. W., and Schenone A. (2016) Tolerability and efficacy study of P2X7 inhibition in experimental Charcot-Marie-Tooth type 1A (CMT1A) neuropathy. Neurobiol. Dis. 95, 145–157 10.1016/j.nbd.2016.07.017 [DOI] [PubMed] [Google Scholar]
- 64. Stutzmann G. E., and Mattson M. P. (2011) Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol. Rev. 63, 700–727 10.1124/pr.110.003814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Okamoto Y., Pehlivan D., Wiszniewski W., Beck C. R., Snipes G. J., Lupski J. R., and Khajavi M. (2013) Curcumin facilitates a transitory cellular stress response in Trembler-J mice. Hum. Mol. Genet. 22, 4698–4705 10.1093/hmg/ddt318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chittoor V. G., Sooyeon L., Rangaraju S., Nicks J. R., Schmidt J. T., Madorsky I., Narvaez D. C., and Notterpek L. (2013) Biochemical characterization of protein quality control mechanisms during disease progression in the C22 mouse model of CMT1A. ASN Neuro 5, e00128 10.1042/AN20130024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yeiser E. C., Rutkoski N. J., Naito A., Inoue J., and Carter B. D. (2004) Neurotrophin signaling through the p75 receptor is deficient in traf6−/− mice. J. Neurosci. 24, 10521–10529 10.1523/JNEUROSCI.1390-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hamill O. P., Marty A., Neher E., Sakmann B., and Sigworth F. J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100 10.1007/BF00656997 [DOI] [PubMed] [Google Scholar]
- 69. Lindau M., and Neher E. (1988) Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 411, 137–146 10.1007/BF00582306 [DOI] [PubMed] [Google Scholar]