

Abstract
Lysine methyltransferase 2D (KMT2D; OMIM 602113) encodes a histone methyltransferase involved in transcriptional regulation of the beta-globin and estrogen receptor as part of a large protein complex known as activating signal cointegrator-2 -containing complex (ASCOM). Heterozygous germline mutations in the KMT2D gene are known to cause Kabuki syndrome (OMIM 147920), a developmental multisystem disorder. Neither holoprosencephaly nor other defects in human forebrain development have been previously associated with Kabuki syndrome. Here we report two patients diagnosed with alobar holoprosencephaly in their antenatal period with de novo monoallelic KMT2D variants identified by trio-based exome sequencing. The first patient was found to have a stop-gain variant c.12565G>T (p.Gly4189*), while the second patient had a missense variant c.5A>G (p.Asp2Gly). Phenotyping of each patient did not reveal any age-related feature of Kabuki syndrome. These two cases represent the first report on association between KMT2D and holoprosencephaly.
Keywords: KMT2D, Holoprosencephaly, HPE, Human forebrain development, Whole exome sequencing.
Graphical Abstract
1. INTRODUCTION
Holoprosencephaly (HPE) is a structural brain defect characterized by incomplete cleavage of the developing forebrain during embryogenesis.1 It is considered as one of the most common developmental abnormalities in humans, with an estimated prevalence of approximately 1 in 250 conceptuses and 1 in 10,000 live births.2 Classically, HPE can occur in a non-syndromic form or as part of recognizable monogenic conditions and chromosomal anomalies. To date, non-syndromic HPE has been associated with pathogenic variants in nearly 17 genes, while syndromic HPE features in at least 25 monogenic conditions.3,4 The gene KMT2D has not been shown to play a major role in the regulation of human forebrain development. KMT2D encodes a histone methyltransferase responsible for the trimethylation of the fourth lysine residue of histone H3 protein, which is an epigenetic mark associated with transcriptionally active genes.5 The encoded protein is part of a large protein complex known as activating signal cointegrator-2-containing complex (ASCOM) that is involved in transcriptional regulation of the beta-globin and estrogen receptor genes.6 Germline pathogenic variants in KMT2D are known to cause Kabuki syndrome (KS), a condition characterized by specific craniofacial features, infantile hypotonia, mild-to-moderate intellectual disability, skeletal defects and dermatoglyfic abnormalities.7,8 Various structural brain anomalies have been occasionally described in individuals affected with KS;9 however, no association with HPE has ever been reported, regardless of the gene/variant involved. Likewise, HPE has not been previously associated with KMT2D. In this study, we report on two patients with HPE and alterations in KMT2D identified using exome sequencing.
2. MATERIALS AND METHODS
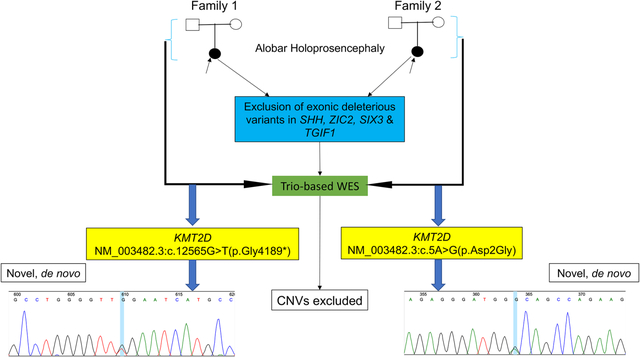
All research procedures were conducted in accordance with the Declaration of Helsinki. This study was approved by the National Human Genome Research Institute (NHGRI) Institutional Review Board. Written informed consent was obtained from each family, including consent to publish photographs. The two patients with HPE included in this study were part of our NHGRI cohort of 277 individuals with HPE (135 trios and 142 singletons). Patients were evaluated by brain imaging (prenatal ultrasound) and postnatal clinical examination. The two patients were initially screened for exonic variants in the four most common genes known to cause HPE, namely SHH (OMIM 600725), ZIC2 (OMIM 603073), SIX3 (OMIM 603714) and TGIF1 (OMIM 602630) using Sanger sequencing as previously recommended.10 Trio-based exome paired-end sequencing was performed at the NIH Intramural Sequencing Center (NISC) following the HiSeq2000 Sequencer protocol (Illumina, San Diego, CA) as previously described.11 The mean read depth for each sample was 79.8x. Novel variants were filtered using stringent criteria, including ExAC database allele frequency of zero, gene constraint based on a probability of being loss-of-function intolerant (pLI) score higher than 0.95 or missense z-score higher than 3.09, and de novo inheritance. Sequence variants identified were validated by Sanger sequencing on an ABI 3730xl Sequencer (Life Technologies, Grand Island, NY) according to the manufacturer’s protocol. Copy number variation (CNV) prediction from exome data was done using the XHMM (eXome-Hidden Markov Model) caller. We used GATK to generate the depth of coverage statistics required for XHMM from the BAM files of our HPE cohort and a control set. GATK output was then run through the XHMM pipeline, generating a VCF file containing each predicted CNV. We then annotated each CNV for genes contained and cytogenetic region using Annovar. A custom Perl script was used to evaluate for any de-novo CNVs in the HPE probands. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
3. RESULTS
3.1. Clinical report
3.1.1. Case 1
The patient was a female fetus (Figure 1: A), first of three pregnancies of a non-consanguineous union. Her two living siblings were healthy and there was no relevant family history. Her healthy mother and father were respectively aged 27 and 23 at the time of the pregnancy, and there was no reported history of teratogen exposure. The pregnancy with her was uneventful until 19 weeks’ gestation when a diagnosis of alobar HPE was made on ultrasound examination. Screening for common fetal aneuploidies by means of amniocentesis and G-banding karyotype was negative for trisomies 13, 18, 21 and sex chromosomes aneuploidies (46,XX). No other prenatal genetic investigation was performed. The pregnancy ended at 22 weeks of gestation following preterm labor and delivery of a nonviable fetus. No post-mortem examination was performed. The clinical features of the patient are summarized in Table 1.
Figure1:

Clinical photographs of the two patients with alobar HPE A: postmortem image of case 1 showing an apparently well-developed fetus with craniofacial dysmorphism, including closely spaced eyes, very depressed nasal bridge with flat nose, and cleft of the upper lip and palate. B1 and B2: case 2 facial images showing a metopic ridge and features characteristic of cebocephaly. Publication of these photographs is supported by consents from the families.
Table 1:
Summary of clinical and molecular data of the two patients with HPE
| Case 1 | Case 2 | |
|---|---|---|
| Clinical features\KMT2D variant | NM_003482.3:c.12565G>T (p.Gly4189*) | NM_003482.3:c.5A>G (p.Asp2Gly) |
| Gender | Female | Female |
| Ethnicity | Caucasian | Caucasian |
| Antenatal | ||
| Gestational age at diagnosis of HPE (weeks) | 18 | 19 |
| Termination of pregnancy | +(†) | −(‡) |
| Intrauterine Growth Restriction | − | + |
| Polyhydramnios | − | + |
| Brain abnormalities | ||
| Completely absent interhemispheric fissure | + | + |
| Single ventricle | + | + |
| Fused thalami | + | + |
| Agenesis of the corpus callosum | + | + |
| Craniofacial features | ||
| Microcephaly | + | + |
| Hypotelorism | + | + |
| Midface hypoplasia | + | + |
| Depressed nasal bridge | + | − |
| Flat nose | + | − |
| Proboscis-like nose | − | + |
| Median cleft lip | + | − |
| Cleft palate | + | − |
| Cardiovascular features | ||
| Right ventricular dilation and hypertrophy with decreased systolic function | − | + |
| Other extra-cephalic features | − | − |
+ indicates presence of the feature;
- indicates absence of the feature
3.1.2. Case 2
The patient is a 5-day-old female (Figure 1: B1& B2), fifth of six siblings from a non-consanguineous union between her 39-year-old mother and her 40-year-old father. The mother had two first trimester miscarriages and there is a family history of autism spectrum disorder, with a brother, a sister and a maternal half-sister affected. The pregnancy with her was uneventful until 18 weeks when she was diagnosed with alobar HPE by means of ultrasound examination. No history of teratogen exposure was recorded. Screening for common fetal aneuploidies using cell-free DNA testing, revealed diploid chromosomes 13, 18 and 21 with two X chromosomes. No other prenatal genetic investigation was performed. She was born at 38 weeks 5 days by normal vaginal delivery with Apgar scores of two and seven at 1 and 5 minutes respectively. Her birth weight was 2490 g (5th percentile), Length of 44 cm (<5th percentile) and head circumference 29.5 cm (<5th percentile). The patient was resuscitated soon after birth for absent spontaneous breathing or chest movement and was admitted afterward in the neonatal intensive care unit for respiratory failure. Postnatal imaging performed were limited to head ultrasound, repeated echocardiographies and chest X-rays. She died on postnatal day 5 in the context of body temperature instability, recurrent generalized tonic-clonic seizures, myocardial dysfunction and respiratory failure. The family opted not to have post-mortem examination. The patient’s prenatal and postnatal clinical features identified are summarized in Table 1.
3.2. Molecular analysis
Using trio-based exome sequencing, a novel KMT2D heterozygous variant was identified in each patient (Table 1). Both variants were confirmed by Sanger sequencing. The variant occurred de novo in each family, as none of the parents was found to be a carrier. Case 1 was found to have a deleterious stop-gain variant, NM_003482.3:c.12565G>T(p.Gly4189*) on exon 39 (CADD scaled score: 47; pLI score: 1). This variant is predicted to result in a truncated protein missing amino acids 4190–5537. The missing region contains all functional domains (N- and C-terminal FYR, SET and Post-SET), LXXLL motifs 3 to 6, the last two zinc fingers, and several predicted post-translational modification residues. The predicted S-adenosyl-L-methionine (SAM) binding site at position 5474–5475 which is essential for the methyltransferase activity is also lost. Case 1 had one other de novo variant, a missense variant of unknown significance: FMC1 (NM_001244584.2):c.8>T (p.Ala3Val). There is no known human disease associated with FMC1. Case 2 had a deleterious missense variant, NM_003482.3:c.5A>G(p.Asp2Gly) on exon 1 (CADD scaled score: 12.9; missense z-score of 3.73). In addition to ExAC database, this missense variant was not found in other population databases including 1000 Genomes Project and dbSNP. This position does not involve any known or predicted functional domain, but it represents a non-conservative amino acid change expected to alter the conformation of the protein. The variants were respectively classified as pathogenic (c.12565G>T; p.Gly4189*) and likely-pathogenic (c.5A>G; p.Asp2Gly) based on the 2015 American College of Medical Genetics and Genomics-Association for Molecular Pathology criteria.12 One additional de novo variant was found in Case 2, ADAM11 (NM_002390.6), c.570G>A:p.Arg190Arg, a synonymous change not considered significant. No pathogenic CNV was identified in both patients. In the Supplementary Materials, variants filtered in Cases 1 and 2 are listed (Table S1 & Table S2). In addition to the two de novo variants that are discussed above, no other variants that may explain the HPE phenotype in both patients were found.
4. DISCUSSION
The etiology of HPE is complex and heterogeneous, involving genetic and environmental determinants. HPE is known as a feature of several developmental disorders, including numeric and structural chromosomal abnormalities, CNVs, as well as at least 25 monogenic conditions.13,14 HPE also occurs in a nonsyndromic form with various modes of inheritance reported, including autosomal dominant with reduced penetrance and variable expressivity, autosomal recessive and a growing evidence of oligogenic inheritance.4,15 The two patients reported in this study were diagnosed with alobar HPE in utero by ultrasound examination. Alobar HPE, which is the most severe subtype in the HPE spectrum, is classically characterized by agenesis of the corpus callosum and olfactory bulbs, fused thalami, a midline monoventricle and complete absence of the interhemispheric fissure with or without associated craniofacial defects.16 Development of alobar HPE is thought to be promoted by defective signals from the prechordal plate that are essential for the patterning and morphogenesis of the forebrain.17 Both patients presented with craniofacial features known to be part of the clinical spectrum of alobar HPE, which could be classified as type IV (case 1) and type III (case 2) of DeMyer’s categories of facial phenotypes in severe forms of HPE.18 The clinical diagnosis of KS is hardly achievable in fetuses or newborns since recognizable physical features of the condition evolve over time.8 One can argue that in both patients, the severity of craniofacial defects associated with HPE could have concealed any craniofacial characteristic suggestive of KS. However, age-related extra-cephalic features of KS that could be assessed in each patient, such as prominent fingertip pads, were absent. Cleft palate and/or occasionally cleft lip and congenital heart defects, most commonly left-sided obstructive defects, are among the structural defects associated with KS.8,19 While the cardiovascular phenotype of Case 2 includes hypertrophic cardiomyopathy which has been sporadically reported in KS, the characteristic HPE-associated median cleft lip and cleft palate present in Case 1 are rather not typical to KS.20 Considering the age of the two patients, it would be cumbersome to attempt a syndromic classification of both HPE cases. Genes involved in the pathogenesis of HPE are expected to be expressed in the prosencephalic neural folds that give rise to the forebrain during primary neurulation.17 The two patients reported here were indentified with novel deleterious variants in KMT2D, a gene not previously associated with defects in human forebrain development. Investigations of the mutational spectrum of KMT2D have shown that truncating mutations including nonsense and frameshift are the most common mutations types, followed by missense mutations. Exons 10, 31, 34, 39 and 48 are known as hotspots, with the majority of trucating mutations involving exon 39 and 48.21,22 These data support our finding in Case 2 who was identified with a nonsense variant (c.12565G>T (p.Gly4189*)) in exon 39. Most KMT2D missense variants are confined in regions encoding C-terminal domains.21,7 Though, some of these variants do not involve any known functional domain of the protein. The missense variant (c.5A>G (p.Asp2Gly)) identified in Case 1, although located in the first exon of the gene and not involving any known domain of the protein, is predicted in sillico to be damaging. The role of KMT2D in several developmental processes remains to be clarified. KMT2D morpholino knockdown in zebrafish results in reduced overall brain size and defect in neural progenitor cells’ ability to differentiate in the forebrain and midbrain.23 Similarly, Lee and collaborators did not report features of HPE in Kmt2d knockout mice embryos, which died at embryonic day 9.5.24 The contrast between the brain phenotype of our two patients and the results of the above animal studies may be explained by their respective study method which was not designed to comprehensively explore the development of the forebrain. Therefore, there is a need for further investigation of the role of KMT2D in HPE pathogenesis. Based on previous evidences, many individuals affected with HPE carry a heterozygous mutation in an HPE-associated gene, which either occurs de novo or is transmitted following an autosomal dominant mode of inheritance.14 Consistently, the two patients reported in this study were found to carry a de novo heterozygous variant in KMT2D, predicted to be highly deleterious.
This report presents for the first time an association between HPE and heterozygous mutations in KMT2D, a gene known to cause KS. Further studies will attempt to clarify the role of KMT2D in the regulation of forebrain development and pathogenesis of HPE.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the patients and families for their participation in this study. This work was supported by fund from the Intramural Research Program at the National Human Genome Research Institute, NIH.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
REFERENCES
- 1.Muenke M Holoprosencephaly as a genetic model for normal craniofacial development. Semin Dev Biol. 1994;5:293–301. [Google Scholar]
- 2.Orioli IM, Castilla EE. Epidemiology of holoprosencephaly: Prevalence and risk factors. Am J Med Genet Part C Semin Med Genet. 2010;154C(1):13–21. [DOI] [PubMed] [Google Scholar]
- 3.Kruszka P, Martinez AF, Muenke M. Molecular testing in holoprosencephaly. Am J Med Genet Part C Semin Med Genet. 2018;178(2):187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dubourg C, Kim A, Watrin E, et al. Recent advances in understanding inheritance of holoprosencephaly. Am J Med Genet Part C Semin Med Genet. 2018;178(2):258–269. [DOI] [PubMed] [Google Scholar]
- 5.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. [DOI] [PubMed] [Google Scholar]
- 6.Demers C, Chaturvedi CP, Ranish JA, et al. Activator-Mediated Recruitment of the MLL2 Methyltransferase Complex to the β-Globin Locus. Mol Cell. 2007;27(4):573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adam MP, Banka S, Bjornsson HT, et al. Kabuki syndrome: International consensus diagnostic criteria. J Med Genet. 2019;56(2):89–95. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Omran T, Teebi AS. Structural Central Nervous System (CNS) anomalies in Kabuki syndrome. Am J Med Genet. 2005;137 A(1):100–103. [DOI] [PubMed] [Google Scholar]
- 10.Kauvar EF, Muenke M. Holoprosencephaly: Recommendations for diagnosis and management. Curr Opin Pediatr. 2010;22(6):687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murdock DR, Donovan FX, Chandrasekharappa SC, et al. Whole-Exome sequencing for diagnosis of turner syndrome: Toward next-generation sequencing and newborn screening. J Clin Endocrinol Metab. 2017;102(5):1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007;2(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solomon BD, Gropman A, Muenke M. Holoprosencephaly Overview. 2000 Dec 27 [Updated 2013 Aug 29]. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1530/ [Google Scholar]
- 15.Kim A, Savary C, Dubourg C, et al. Integrated clinical and omics approach to rare diseases: Novel genes and oligogenic inheritance in holoprosencephaly. Brain. 2019;142(1):35–49. [DOI] [PubMed] [Google Scholar]
- 16.DeMyer W, Zeman W. Alobar Holoprosencephaly (Arhinencephaly) with Median Cleft Lip and Palate : Clinical, Electroencephalographic and Nosologic Considerations. Confin Neurol. 1963;23(1):1–36. [DOI] [PubMed] [Google Scholar]
- 17.Geng X, Oliver G. Pathogenesis of holoprosencephaly. Sci Med. 2009;119(6):1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeMyer W, Zeman W, Palmer CG. The Face Predicts the Brain: Diagnostic Significance of Median Facial Anomalies for Holoprosencephaly (Arhinencephaly). Pediatrics. 1964;34(1):256–263. [PubMed] [Google Scholar]
- 19.Digilio MC, Gnazzo M, Lepri F, et al. Congenital heart defects in molecularly proven Kabuki syndrome patients. Am J Med Genet Part A. 2017;173(11):2912–2922. [DOI] [PubMed] [Google Scholar]
- 20.Paik JM, Lim SY. Kabuki Syndrome with Cleft Palate. Arch Plast Surg. 2016;43(5):474–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banka S, Veeramachaneni R, Reardon W, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2011;20(4):381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S, Shen C, Wang J, et al. Kabuki syndrome: a Chinese case series and systematic review of the spectrum of mutations. BMC Med Genet. 2015;16(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Laarhoven PM, Neitzel LR, Quintana AM, et al. Kabuki syndrome genes KMT2D and KDM6A : functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum Mol Genet. 2015;24(15):4443–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J-E, Wang C, Xu S, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2:e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.