

SUMMARY
Deregulated signal transduction is a cancer hallmark, and its complexity and interconnectivity imply that combination therapy should be considered, but large data volumes that cover the complexity are required in user-friendly ways. Here, we present a searchable database resource of synthetic lethality with a PI3 kinase signal transduction inhibitor by performing a saturation screen with an ultra-complex shRNA library containing 30 independent shRNAs per gene target. We focus on Ras-PI3 kinase signaling with T cell leukemia as a screening platform for multiple clinical and experimental reasons. Our resource predicts multiple combination-based therapies with high fidelity, ten of which we confirmed with small molecule inhibitors. Included are biochemical assays, as well as the IPI145 (duvelisib) inhibitor. We uncover the mechanism of synergy between the PI3 kinase inhibitor GDC0941 (pictilisib) and the tubulin inhibitor vincristine and demonstrate broad synergy in 28 cell lines of 5 cancer types and efficacy in preclinical leukemia mouse trials.
In Brief
Mues et al. present a web browser-based, searchable database of their synthetic lethal screen to identify a potentially potent combination therapy in cancer. They validated their screen with ten small molecule inhibitors in leukemia and four solid tumor types and in a T cell leukemia mouse model preclinical trial.
Graphical Abstract
INTRODUCTION
Cancer is a genetically heterogeneous disease characterized by diverse patient-specific mutations that combine to confer “hallmark” biologic properties (Hanahan and Weinberg, 2011). One of these hallmarks—deregulated signal transduction — is implicated in driving cancer initiation, progression, and metastasis in different tissue contexts (Giancotti, 2014). In addition, these signals are interconnected in circuits or networks and not in neatly defined, linear pathways. The unknown identity of cancer signaling networks poses substantial challenges for the Precision Medicine Initiative (Collins and Varmus, 2015). The interconnectivity of cancer signaling networks is daunting and implies that combination therapy rather than monotherapy should be considered (Bozic et al., 2013), but what combination?
Synthetic lethality is formally defined in terms of molecular perturbation, where the co-occurrence of at least two genetic alterations results in cell death (Hartman et al., 2001). Cancer cells acquire multiple molecular changes, distinct from their wild-type counterparts, which can lead to unique genetic vulnerabilities of the cancer. Such cancer-specific synthetic lethal interactions offer therapeutic opportunities (Hartwell et al., 1997) and have been pursued through genome-scale synthetic lethal screens, an approach that became technically feasible in mammalian cells after the discovery of small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) (Wilson and Doudna, 2013). Pooled methods in which shRNAs can be accurately identified in mixed populations of cells by next-generation sequencing were developed (Corcoran et al., 2013; Sims et al., 2011; Xie et al., 2012), but challenges to reach full coverage and complete target inhibition, off-target effects by individual siRNAs or shRNAs, and difficulties in reproducing synthetic lethal interactions across different laboratories and cell lines dampened the initial excitement (Babij et al., 2011; Luo et al., 2012; Scholl et al., 2009; Weїwer et al., 2012). Whereas more recent CRISPR/Cas9 approaches have made it feasible to systematically abrogate gene function, it has been argued that the incomplete shRNA-mediated gene knockdown better mimics the incomplete target inhibition achieved by many anti-cancer drugs (Boettcher and McManus, 2015).
Hyperactive Ras signaling is one of the most common molecular alterations in human cancer (Bos, 1989). The small guanosine triphosphatase (GTPase) Ras is activated by many growth factors and cytokines and acts as a central regulator of cell signaling by coupling these extracellular stimuli to kinase effector pathways (Lu et al., 2016). KRASG12D and other somatic oncogenic mutations constitutively increase Ras signal output, which deregulates apoptotic, proliferative, and differentiation decisions (Miller and Miller, 2012). Oncogenic Ras mutations are present in ~30% of all human cancers, but aberrant Ras signaling can also be driven by the overexpression of Ras activators or the attenuation of Ras inhibitors (Ksionda et al., 2013; Pylayeva-Gupta et al., 2011). Despite recent progress in developing KRASG12C-specific inhibitors, oncogenic Ras is an exceedingly difficult therapeutic target (Cox et al., 2014; Ostrem and Shokat, 2016; Schubbert et al., 2007; Simanshu et al., 2017).
Normal and mutant Ras proteins bind to and directly activate phosphoinositide 3-kinase (PI3K) (Downward, 2006; Schubbert et al., 2007; Vanhaesebroeck et al., 2012; Vivanco and Sawyers, 2002). Frequent mutations in PIK3CA, PTEN, and other genes encoding components of the PI3K pathway in many cancers, the role of PI3K in oncogenic cell growth and protein translation, and the “druggable” nature of kinases identified PI3K an attractive target for targeted cancer therapy (Bader et al., 2005). Many different PI3K inhibitors have been developed in recent years, and efforts are continuing to optimize the efficiency and specificity of these inhibitors for the treatment of cancer (Dienstmann et al., 2014; Holmes, 2011; Thorpe et al., 2015). However, monotherapy with PI3K inhibitors may be insufficient, given the aforementioned interconnectivity of cancer signaling networks (Bozic et al., 2013; Brown and Toker, 2015). Furthermore, PI3K inhibition is generally cytostatic (García-Martinez et al., 2011; Gautam et al., 2016; Martini et al., 2013), and the PI3K inhibitor GDC0941 (pictilisib) (Folkes et al., 2008) causes cytotoxicity in only a few cancer cell lines (Ehrhardt et al., 2015; O’Brien et al., 2010; Ross et al., 2016). However, GDC0941 did show promising efficacy when combined with other drugs inhibiting complementary pathways (Floris et al., 2013; Munugalavadla et al., 2014; Wallin et al., 2012), yet systematic and unbiased screens are essential in finding novel and effective combination therapies (Al-Lazikani et al., 2012; Kummar et al., 2010). An alternative to GDC0941 is IPI145 (duvelisib) (Winkler et al., 2013), a potent PI3K-δ and -γ inhibitor that shows activity in T cell acute lymphoblastic leukemia (T-ALL) and is in clinical development for hematologic malignancies (Flinn et al., 2017; Proctor et al., 2013).
Here, we chose T-ALL as a cancer model to perform a synthetic lethal screen with the PI3K inhibitor GDC0941 for multiple reasons. T-ALL is an aggressive blood cancer of children and adults (Wiemels et al., 2005). While modern chemotherapy has improved clinical outcome (Pui and Evans, 2006), these regimens are toxic to normal cells, and no mechanism-based treatments exist (Collins and Varmus, 2015). Oncogenic Ras/PI3K signaling occurs in ~65% of T-ALL patients (Hartzell et al., 2013; von Lintig et al., 2000) due to oncogenic RAS mutations; overexpression of the Ras activator RasGRP1 (Ras guanine nucleotide releasing protein 1); or somatic mutations in PTEN, NF1, and other genes. Murine and human T-ALLs make frequent but heterogeneous use of PI3K signals (Gutierrez et al., 2009; Hartzell et al., 2013; Subramaniam et al., 2012; Zhang et al., 2012; Ksionda et al., 2018). Signals from the PI3K-δ, -γisoforms appear to dominate in T-ALL, but the PI3K-α and -β isoforms contribute as well (Lonetti et al., 2015; Subramaniam et al., 2012). Human T-ALL cell lines can be cultured in large quantities in suspension and efficiently infected with lentivirus, allowing for incredibly complex synthetic lethal screens containing thousands to millions of genetic perturbations. Finally, candidate synthetic lethal interactions identified by screening T-ALL cell lines in vitro can be directly tested by transplanting and treating primary leukemias in vivo (Dail et al., 2010, 2014).
Here, we present a searchable database resource of synthetic lethality with a PI3K inhibitor that we generated by performing a saturation screen in two independent T-ALL lines with an ultra-complex shRNA library containing ~30 independent shRNAs per gene target.
RESULTS
PI3K Signaling in T-ALL, Limited Effects of GDC0941, and Design of Synthetic Lethal Screen
Frequent somatic mutations in PTEN, NRAS, and NF1 provide clear genetic evidence that deregulated receptor-Ras-PI3K signals contribute to aberrant growth in T-ALL (Figure S1A) (Fransecky et al., 2015; Gutierrez et al., 2009). As GDC0941 (Figure S1B) and other PI3K inhibitors are being developed as anti-cancer therapies, we took an unbiased approach to systematically investigate proteins and pathways that may be co-targeted to enhance the efficacy of this approach (Figure 1A).
Figure 1. PI3K Signaling and Synthetic Lethal Screen Setup in T Cell Leukemia.
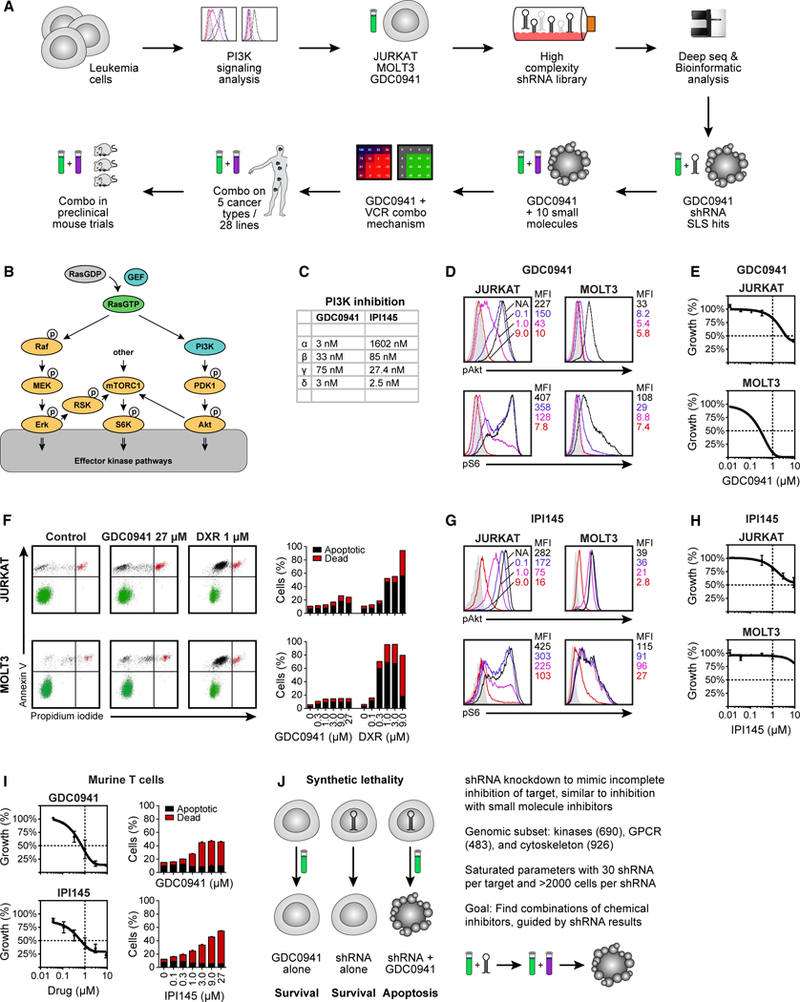
(A) Pipeline of experimental flow in this study.
(B) Schematic representation of RAS and PI3K signaling and activation of the mTORC1-S6K-S6 pathway.
(C) The half-maximal inhibitory concentration (IC50) values of GDC0941 and IPI145.
(D) Phospho-flow analysis of baseline phospho-Akt (top) and phospho-S6 (bottom) levels in JURKAT and MOLT3 cells, exposed to increasing concentrations of GDC0941 (0.1, 1.0, and 9.0 μM).
(E) GDC0941 titration on JURKAT and MOLT3 cells in 3-day growth assays.
(F) Flow cytometric analysis of JURKAT and MOLT3 cells treated with GDC0941 or doxorubicin (DXR), stained for apoptotic cells with annexin V and dead cells with propidium iodide, and depicted in bar graphs.
(G) Phospho-flow analysis of baseline phospho-Akt (top) and phospho-S6 (bottom) levels in JURKAT and MOLT3 cells, exposed to increasing concentrations of IPI145 (0.1, 1.0, and 9.0 μM) (n = 2).
(H) IPI145 titration on JURKAT and MOLT3 cells in 3-day growth assays (n = 4).
(I) Flow cytometric analysis of primary, non-transformed T cells treated with GDC0941 or IPI145, stained for apoptotic cells with annexin V and dead cells with propidium iodide, and depicted in bar graphs.
(J) Schematic representation of synthetic lethality and parameters of the shRNA screen.
(D)–(I) are representative examples of ≥3 independent experiments (n = 4).
Previous studies showed that T-ALL cell lines are heterogeneous in their strength of PI3K signaling (Dail et al., 2010; Gutierrez et al., 2009; Hartzell et al., 2013; Zhang et al., 2012). PI3K signaling results in the phosphorylation of Akt, which can turn on a mammalian target of rapamycin complex 1-S6 kinase-S6 (mTORC1-S6K-S6) pathway (Weigelt and Downward, 2012) (Figure 1B). The GDC0941 compound strongly inhibits PI3K-α and -δ, moderately inhibits the PI3K-β isoform, and is least effective against PI3K-γ (Figure 1C). For simplicity, we call GDC0941 a pan-PI3K inhibitor. Using quantitative fluorescence-activated cell sorting (FACS) staining for phosphorylated Akt (pAkt) as a proxy for PI3K pathway activation, we observed varying baseline levels of pAkt in a panel of 10 human T-ALL cell lines (Figure S1C). Exposing T-ALL to increasing concentrations of GDC0941 revealed variable but dose-dependent pAkt signal attenuation, with 9.0 μM GDC0941 extinguishing all of the pAkt signal (Figures 1D and S1C). Using phosphorylation of the ribosomal protein S6 (pS6) as a proxy for the activation of the mTORC1-S6K-S6 pathway, we reported on this pathway that has Akt-dependent and -independent inputs (Figures 1D and S1C). Highlighting the potential challenge of using PI3K inhibition as monotherapy and in agreement with the measured effects of GDC0941 on pAkt and pS6 levels, the 10 T-ALL lines demonstrated variable levels of growth inhibition (Figures 1E and S1D). We selected JURKAT and MOLT3 cells as they represent two extremes in our T-ALL panel: each line is characterized by a very distinct pattern of pAkt and pS6 expression and cell proliferation in the context of GDC0941 treatment. Exposing JURKAT and MOLT3 T-ALL cells to concentrations of GDC0941 known to inhibit pAkt and pS6 signals did not induce substantial apoptosis in either cell line. In contrast, the anthracycle doxorubicin (DXR), which is a component of T-ALL treatment protocols, robustly induced cell death (Figure 1F). These results are largely consistent with the limited and predominantly cytostatic effects of PI3K inhibition reported in other cancer types (García-Martínez et al., 2011; Gautam et al., 2016; Martini et al., 2013); the observation that GDC0941 efficiently inhibited PI3K signaling without inducing apoptosis motivated us to explore GDC0941-based synthetic lethality in combination with the shRNA-mediated knockdown of gene expression. IPI145 is a PI3K inhibitor that is in clinical trials for leukemias (Flinn et al., 2017; Proctor et al., 2013). IPI145 strongly inhibits PI3K-δ, has moderate effects on PI3K-γand PI3K-β, and does not notably inhibit the α isoform of PI3K (Figure 1C). IPI145 was able to inhibit Akt and S6 phosphorylation (Figure 1G) but did so somewhat less efficiently than GDC0941 (Figure 1D). IPI145 modestly affected growth in JURKAT but barely affected the growth of MOLT3 T-ALL (Figure 1H).
We also determined the effects of PI3K inhibitors on the growth characteristics of primary, non-transformed T cells that are isolated and purified from the lymph nodes of mice and cultured in vitro in the presence of serum and cytokines. Increasing concentrations of GDC0941 effectively inhibited the growth of primary murine T cells compared to Jurkat T-ALL cells (Figure 1I). Similarly, the IPI145 inhibitor dampened growth more effectively in primary murine T cells as compared to Jurkat T-ALL (Figure 1I). Growth inhibition was accompanied by dose-dependent increases in apoptotic cells and the cell death of primary T cells (Figure 1I).
Screening Procedure and Downstream Data Processing Pipeline
The combinatorial effects of GDC0941 with other compounds tested to date have been identified based largely on known pathway interactions and have not been identified by systematic screening (Badinloo and Esmaeili-Mahani, 2014; Dail et al., 2014; Floris et al., 2013; Haritunians et al., 2007; Munugalavadla et al., 2014; Opel et al., 2008; Shingu et al., 2003; Wallin et al., 2012). Here, we sought to identify a more complete compendium of synthetic lethal interactions, including unanticipated combinations (Figure 1J). From a technological approach, we specifically chose to perform an shRNA screen over a CRISPR/Cas9 screen to possibly better mimic the commonly observed incomplete blockade of molecules by chemical inhibitors (Boettcher and McManus, 2015). We used the University of California, San Francisco (UCSF) EXPANDed RNAi library resource, which dramatically improves RNAi screening, compared to many commercial resources. This ultra-complex shRNA library targets each gene with approximately 30 independent shRNAs per gene, minimizing experimental noise and allowing us to overcome common RNAi-centric problems related to high false-negative rates as well as high false-positive rates (Bassik et al., 2009). To avoid population-skewing effects and target over- or underestimation, each individual shRNA was represented by at least 2,000 infected cells throughout the screen. In this way, we targeted ~1,800 genes with 55,000 shRNAs, with an emphasis on kinases, G protein-coupled receptors (GPCRs), and cytoskeletal proteins in 120 million infected JURKAT or MOLT3 T-ALL cells. The targets of shRNAs that are depleted from T-ALL cell pools represent genes that show synthetic lethality in combination with GDC0941 treatment, and these hits were validated by determining the effectiveness of combinations of chemical inhibitors (Figure 1J).
Two independent synthetic lethal screens were executed in replicate using JURKAT and MOLT3 T-ALL cells. Following transduction with lentiviral particles containing the shRNA library and antibiotic selection to enrich for transduced cells, cell pools were split for subsequent treatment with the PI3K inhibitor GDC0941 or a DMSO control in roller bottles (Figure 2A). Cells were grown for 22 days under these conditions, with continuous passaging every 2 or 3 days. At the end of this period, ~120 million cells were harvested, and the shRNA signatures were amplified from genomic DNA and submitted for deep sequencing (Figure 2A). We administered a fixed concentration of GDC0941 at the previously determined EC20 to obtain slight growth retardation compared to the control. During the 22-day time course, these concentrations resulted in a reduced growth rate for the pools of JURKAT and MOLT3 cells, as well as a modest and transient decrease in the viability of the entire pool during the first 7 days, followed by a recovery in viability, indicating that the cells as a pool went through a selection process (Figure 2B).
Figure 2. Synthetic Lethal shRNA Screens with a Leukemia Platform.
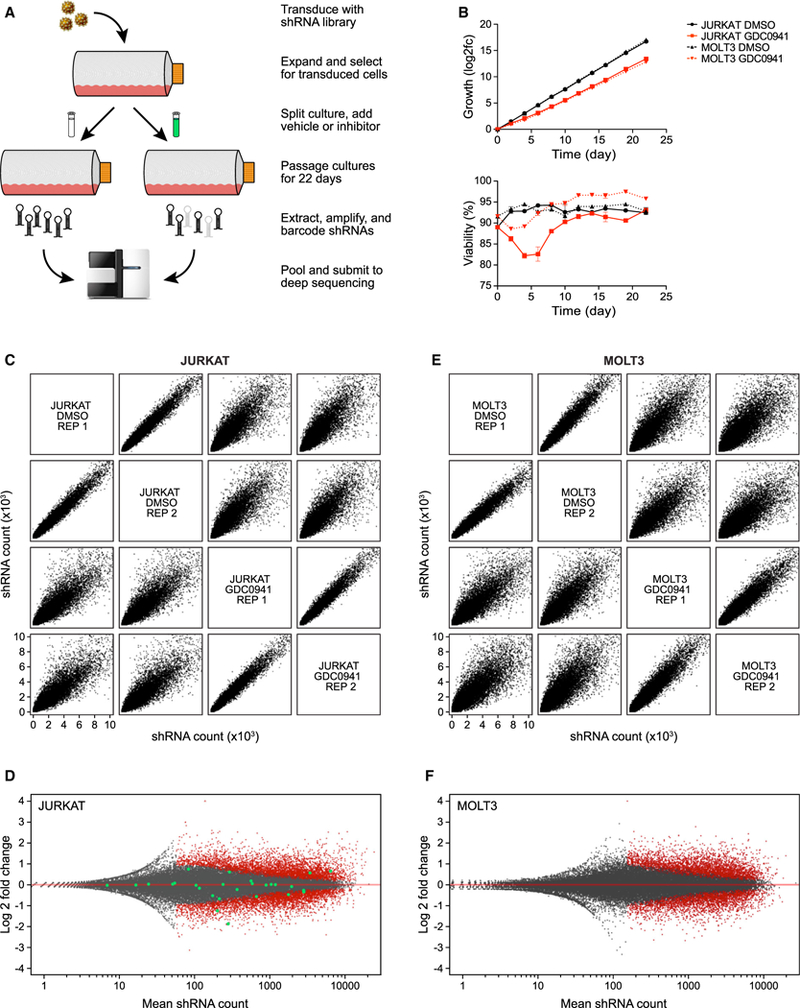
(A) Schematic representation of shRNA synthetic lethal screen screening procedure.
(B) Growth (top) and viability (bottom) of JURKAT and MOLT3 cells during screen.
(C) Comparisons of deep sequencing results from JURKAT screen, plotting all shRNA read counts of replicates from control and GDC0941-treated samples against one another.
(D) Distribution of individual shRNAs from JURKAT screen, with significant shRNAs labeled in red; shRNAs targeting MAPK9 as an example are highlighted in green.
(E) Same as (C), but for MOLT3 screen.
(F) Same as (D), but for MOLT3 screen.
The raw data sequence reads obtained from deep sequencing were bioinformatically processed to yield lists of counts for each shRNA from each condition. Those values were then statistically analyzed to define significantly depleted or enriched shRNAs and to determine the overall shRNA distribution for each gene represented in the screen (Figure S2A). When plotting shRNA distributions between conditions, the replicates display a narrow distribution of counts, indicating high reproducibility (Figures 2C and 2E). In contrast, when comparing controls with GDC0941-treated samples, the distribution of shRNA counts was quite broad, indicating significant enrichment and depletion of specific shRNAs. This observation was true for both T-ALL lines tested, JURKAT and MOLT3, which is indicative of successful screening (Figures 2C and 2E). To further analyze the significantly depleted and enriched shRNAs, counts from control replicates were compared to counts from drug-treated replicates. Plotting total shRNAs from each screen for log2 fold change revealed a substantial portion of significant alterations (Figures 2D and 2F). To illustrate the abundance of data points obtained for a single gene target, we highlighted for the JURKAT screen all of the shRNAs targeting the arbitrarily chosen MAPK9 gene (Figure 2D). Having performed quality control of our synthetic lethal screens, we next focused on generating a resource for the community: a user-friendly and searchable platform enabling customized interrogation of the data.
Consolidation of Ultra-complex shRNAs in a Searchable Synthetic Lethal Screen Database
We collapsed the information obtained for each set of shRNAs targeting the same gene and plotted all of the shRNAs recovered from the screen per single target. We displayed fold change in log2 value and color-coded the significance of individual shRNAs: red for significant, black for non-significant, and gray for excluded data point due to low sequence reads (Figure 3A). Next, we computed the collective values of the significant shRNAs per gene. We calculated the count difference between significantly depleted and enriched shRNAs per single target gene (Δ enr versus dep) and also projected the mean fold change of all significant shRNAs (μ of log2 fold change).
Figure 3. A Searchable Resource of Highly Saturated Synthetic Lethal Screen shRNA Screens.
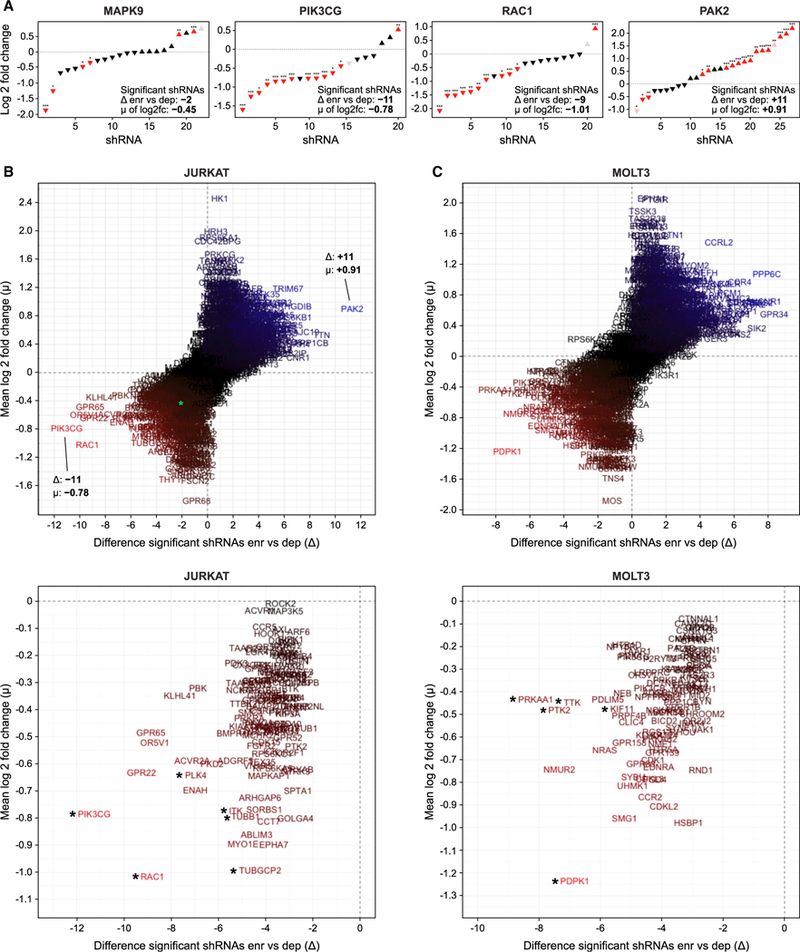
(A) Distribution of all shRNAs targeting selected genes from the JURKAT synthetic lethal screen, with difference △ of significantly depleted versus enriched shRNAs, and mean m of the significance of all of the shRNAs per single target.
(B) Dispersion of all of the genes represented in the JURKAT screen, with MAPK9 as an example highlighted in green (top), and magnified illustration of the depleted genes (bottom), with asterisks indicating targets selected for further verification with inhibitors.
(C) Same as (B), but for MOLT3 screen. All individual targets can be explored in our searchable database of our synthetic lethal screen with GDC0941 (https://mmues.shinyapps.io/K7screen/).
This analysis immediately revealed the necessity of performing shRNA synthetic lethal screens in full saturation and with ultra-complex shRNA libraries, and it helps explain why previous screens with a lower coverage of shRNAs per gene may have been difficult to interpret (Babij et al., 2011; Luo et al., 2012; Scholl et al., 2009; Weїwer et al., 2012). If we take MAPK9 as our specific example, 4 shRNAs displayed significant depletion, 2 shRNAs showed significant enrichment, 15 shRNAs were non-significant, and 1 shRNA did not provide sufficient reads, leading to a compiled D of –2 and a μ of –0.46 (Figure 3A). Thus, the large number of shRNAs targeting MAPK9 allowed us to confidently conclude that it is not synthetically lethal with GDC0941, which could have been problematic with lower coverage. In contrast, PIK3CG (Δ of –11 and a μ of –0.78) and RAC1 (Δ of –9 and a μ of –1.01) are examples of strong hits for highly depleted genes and PAK2 (Δ of +11 and a μ of +0.91) is an example of a highly enriched target (Figure 3A).
We next plotted D as a function of μ to display all of the targets for the JURKAT synthetic lethal screen, with depleted genes in the lower-left quadrants and enriched targets in the upper-right quadrants (Figure 3B). Similar results were obtained for the MOLT3 screen, yet with a different composition of depleted or enriched targets (Figure 3C). Moreover, we generated a searchable database of our synthetic lethal screen with GDC0941 (https://mmues.shinyapps.io/K7screen/) as a resource. The entry of a gene name will automatically generate the data presented in Figures 2D and 2F as well as the entire Figure 3, with the gene-specific information highlighted for the user. The most depleted genes revealed interesting candidates, and we validated 10 of these in the remainder of the study (Figures 3B and 3C).
Circuitry of the T-ALL Network and Pharmacological Validation of Synthetic Lethal Screen Hits
To better understand the circuitry between the signaling molecules selected by the GDC0941 and shRNA combinations, we performed a comprehensive pathway analysis and overlaid the results onto our initial RAS-PI3K signaling schematic (Figure S2B). These results revealed many connections to the Ras-PI3K network, suggesting that this Ras-PI3K network becomes sensitive when T-ALL cells are exposed to GDC0941 at effective concentration for 20% of the maximal effect (EC20). In Figure 1 we showed that JURKAT and MOLT3 T-ALL cells are two extremes in terms of PI3K-Akt and S6 signaling as well as growth retardation by GDC0941. Whereas JURKAT and MOLT3 cells showed some differences in the circuitry uncovered by our synthetic lethal screen, the majority of the genes enriched or depleted were still shared among both (Figure S2C). Thus, the sensitivity for synthetic lethality appears irrespective of absolute activity through the PI3K node. More surprising was that similar results were also obtained when we used INGENUITY to focus on other signaling pathways (Supplemental Data). We observed that genes linked to the mitotic machinery or cell-cycle progression were often affected, predicting that GDC0941 will render cells more sensitive to the inhibition of mitotic and cell-cycle processes. JURKAT and MOLT3 T-ALL cells demonstrated similar patterns, and we explored synthetic lethality with inhibitors of these processes next.
We took a pharmacological approach with the available chemical inhibitors to validate and translate the results obtained by our shRNA synthetic lethal screen (Figure 4A) (see Figure S3 for shRNA distributions of those targets and Figure S4A for full and alternate names and corresponding inhibitor characteristics). Before testing combinations of inhibitors, we subjected JURKAT and MOLT3 cells to single inhibitor titrations to establish the sensitive range for growth retardation by the 10 compounds in the 2 T-ALL cell lines (Figure S4B).
Figure 4. Pharmacological Inhibition in Combination with GDC0941 Validates Synthetic Lethal Screen Hits.
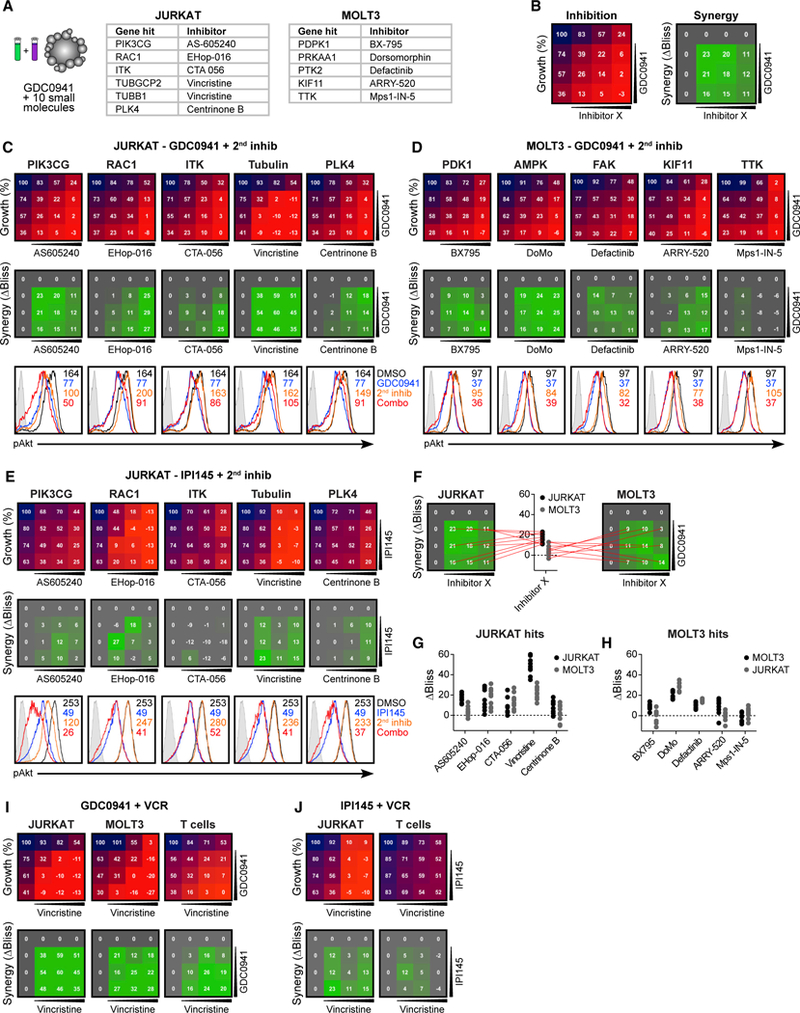
(A) Candidate genes selected from synthetic lethal screen for verification by Inhibitor combinations.
(B) Schematic representation of testing for inhibitor combinations, depicting cellular growth inhibition by inhibitors alone or combined (left), and resulting ΔBliss values indicating synergistic efficacy as calculated from expected and observed growth inhibition (right).
(C) 4 × 4 grids with combinatorial inhibitor titrations to test for the synthetic lethality of candidates from JURKAT screen on JURKAT cells, depicting growth (top) and inhibitor synergy (center). Phospho-flow analysis of baseline (black) phospho-Akt levels and treatment with GDC0941 (blue), second inhibitor alone (orange), or a combination of both (red) (bottom).
(D) Same as (C), but for the candidates from the MOLT3 screen tested on MOLT3 cells. For reciprocal tests, see Figure S5.
(E) Same as (C), but using IPI145 to test candidates from the JURKAT screen on JURKAT cells (n = 2).
(F) Example plots depicting the selection of ΔBliss values for each inhibitor tested.
(G and H) Summary of all 9 ΔBliss values from inhibitor combinations for candidates from the JURKAT screen (G) and from the MOLT3 screen (H).
(I) 4 × 4 grids with combinatorial GDC0941 and vincristine titrations, depicting growth (top) and inhibitor synergy (bottom) in JURKAT, MOLT3, and normal primary T cells (n = 4).
(J) Same as (I), but using IPI145 and vincristine.
(C)–(E), (I), and (J) are representative examples of ≥3 independent experiments (n = 4).
Using ten 4 × 4 grids, we tested the growth inhibition of JURKAT and MOLT3 T-ALL cells in 3-day growth assays with untreated samples set at 100% growth. For all of the combinations, we determined the ΔBliss value, which is a readout for synergistic inhibition and depicts the difference between expected inhibition (assuming the additive effects of inhibitor combinations) and observed inhibition. ΔBliss values >0 indicate synergistic effects (Figure 4B; see Figure S4C for inhibitor concentration ranges). As a control, we combined GDC0941 with itself and observed only additive or opposing effects, never synergy (Figure S4E). Nine of 10 combinations predicted by our shRNA synthetic lethal screen yielded strong synergistic inhibitory effects of treatment with GDC0941 and the additional compound, with the Mps1-IN-5 inhibitor for TTK as the exception (Figures 4C and 4D). Biochemically, only GDC0941 and AS605240 but not the other 9 inhibitors affected the level of Akt and S6 phosphorylation (Figures 4C, 4D, S5E, and S5F; see Figure S4D for the inhibitor concentrations). Thus, the synergistic effects on growth inhibition for these 9 other inhibitors came from hitting pathways others than those revolving around Akt and S6.
We also analyzed the synergy of the inhibitors in a crisscross manner by testing the hits from the JURKAT screen on MOLT3 cells and vice versa. For 6 of 9 inhibitor combinations, we observed synergy with GDC0941 in the crisscrossed tests (Figures S5A and S5B). For hits derived from the JURKAT screen, only centrinone B did not show synergy with GDC0941 when tested on MOLT3 cells, while BX795 and ARRY-520 from the MOLT3 screen did not show synergy when tested with GDC0941 on JURKAT cells. Overall, the hits from the JURKAT cell screen provided stronger candidates for combination therapy with GDC0941, both in terms of degree of synergy measured by ΔBliss value and generalizability to another T-ALL cell line (Figures 4F–4H).
To validate the synergistic effects of GDC0941 with the 10 compounds more thoroughly, we repeated the cell growth and biochemical assays with IPI145. As already demonstrated in Figure 1H, IPI145 had a milder growth-inhibitory effect on Jurkat by itself, compared to GDC0941. Nevertheless, the combination of IPI145 with the other 5 compounds revealed synergistic effects (Figure 4E). MOLT3 is resistant to growth inhibition by IPI145 (Figures 1H), even when 4-fold higher concentrations of IPI145 are used compared to the GDC0941 concentrations in Figure 4D. Still, synergy could be detected in IPI145 with BX795, DoMo, defaticinib, and ARRY-520 (Figure S5C). Biochemical inhibition of Akt and S6 phosphorylation by IPI145 in both JURKAT and MOLT3 was very robust (Figures 4E, S5G, and S5H). Therefore, it is possible that IPI145 is slightly underperforming in the 3-day growth-inhibition assays compared to GDC0941 while still hitting its targets because of IPI145 stability or turnover.
The 2 most effective combinations from our synthetic lethal screen and validation experiments were combinations of the PI3K inhibitor GDC0941 with either the AMP kinase (AMPK) inhibitor dorsomorphin or the microtubule assembly inhibitor vincristine (VCR). Recent work has implied that dorsomorphin inhibits more targets than AMPK alone (Liu et al., 2014). For this reason and the fact that VCR is a known chemotherapeutic agent frequently used in the clinic and part of the standard therapy regimen for T-ALL treatment, we focused on the GDC0941/VCR and IPI145/VCR combinations for the remaining part of our study.
We analyzed the effects of GDC0941/VCR on primary murine T cells. We established the growth-inhibitory and apoptosis effects on VCR alone on primary T cells (Figure S5D). Very similar to results with JURKAT and MOLT3 T-ALL, the GDC0941/VCR combination yielded strong synergistic growth-inhibitory effects in normal T cells (Figure 4I). By contrast, IPI145/VCR did not lead to strong synergistic growth inhibition in normal T cells (Figure 4J), suggesting that this IPI145/VCR combination may be a very attractive combination therapy for T-ALL that does not affect normal cells.
Mechanisms of GDC0941 and VCR Synergy
PI3K inhibition and VCR have been explored in several solid cancers (Badinloo and Esmaeili-Mahani, 2014; Opel et al., 2008; Shingu et al., 2003). VCR is known to be cytotoxic at higher concentrations (Jackson and Bender, 1979; Kobayashi et al., 1998).
To rule out the possibility that our results were specific for GDC0941 or IPI145, we tested 2 other PI3K inhibitors that are also in clinical development, BKM120 (buparlisib) and XL147 (pilaralisib). When combined with VCR, both inhibitors showed strong synergistic effects, similar to GDC0941 (Figure 5A). When treating JURKAT cells with GDC0941 and VCR alone or in combination over an extended dose range, we observed a large therapeutic window in which each drug had minor effects as single agents while being strongly growth inhibitory when combined (Figure 5B). Examining cell growth overtime showed similar results: both drugs could be administered at a concentration that produced almost no apparent effect when given alone, yet yielded highly synergistic inhibition when the same concentration of each inhibitor was applied jointly (Figure 5B). Only the GDC0941/VCR combination strongly increased the frequency of apoptotic and dead JURKAT cells when analyzed after 16 and 48 h, especially at the later time point, which showed similar efficiency as DXR treatment (Figure 5C). These results were gratifying, as the goal of our synthetic lethal screen was to identify inhibitor pairs that could transform the solely cytostatic effects of PI3K inhibition on T-ALL (Figure 1F) into cytotoxic effects.
Figure 5. Mechanistic Effects of Observed GDC0941 and Vincristine Synergy.
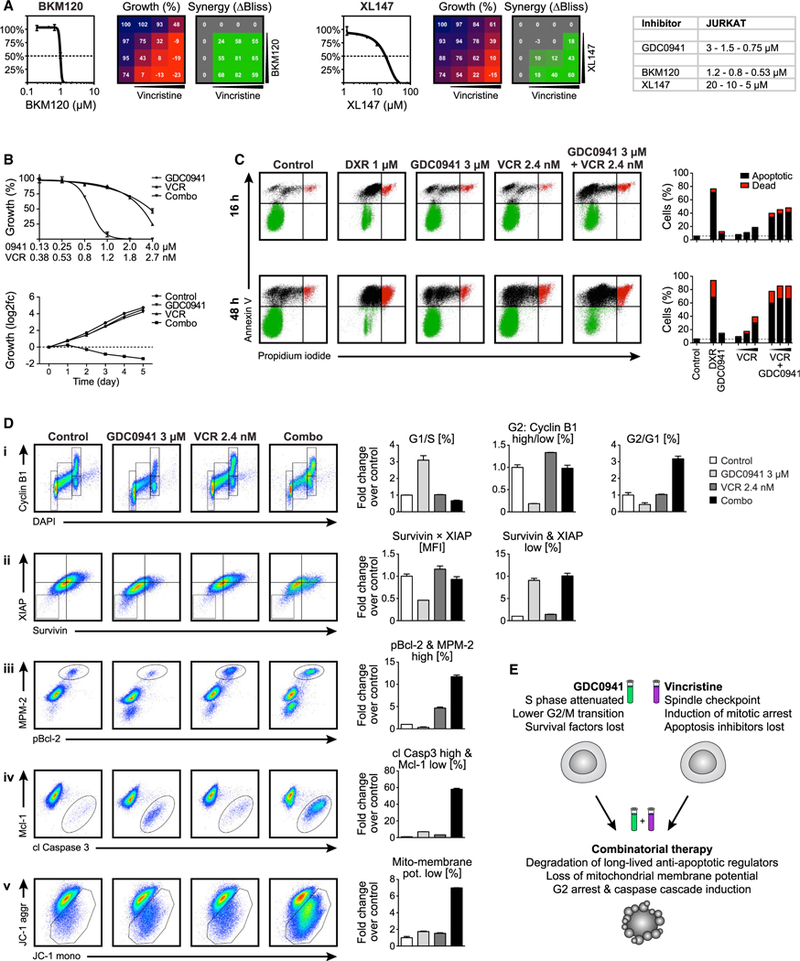
(A) 4 × 4 grids with combinatorial inhibitor titrations to test for the synergy of PI3K inhibition by BKM120 (left) or XL147 (right) with vincristine (VCR) in JURKAT cells.
(B) Single and combinatorial titration of GDC0941 and VCR in 3-day growth assay (top), and cell growth overtime with 1 μM GDC0941 and 1.2 nM VCR alone or in combination (bottom).
(C) Flow cytometric analysis of JURKAT treated with 1 μM DXR alone or 3 μM GDC0941 and a titration of VCR at 1.6, 2.4, and 3.6 nM, alone or in combination; stained for apoptotic cells with annexin V and dead cells with propidium iodide.
(D) Flow cytometric analysis of JURKAT cells treated with 3 μM GDC0941 and 2.4 nM VCR, alone or in combination; stained for (i) markers of cell proliferation, (ii) anti-apoptotic proteins, (iii) mitotic arrest indicators, (iv) apoptosis induction cascade, and (v) mitochondrial membrane potential.
(E) Schematic representation of mechanisms for the combinatorial efficacy of PI3K and tubulin inhibition.
(A)–(D) are representative examples of ≥3 independent experiments.
To mechanistically dissect the combinatorial effects of GDC0941 and relatively low doses of VCR, we treated cells for 24 h at the EC50 of these drugs; quantitatively analyzed the cell cycle, protein translation, apoptosis, and mitochondrial membrane potential; and applied FACS barcoding to ensure identical staining of cells with specific antibodies and dyes (Figure S6A). PI3K signaling has been reported to be required to drive protein synthesis during G1 phase, but is also necessary to allow entry into S phase (Gille and Downward, 1999). GDC0941 treatment caused a significant reduction of cells in S phase, with a corresponding increase in cells arrested in G1 (Figure 5D, i). Cells already in G2 entered mitosis at a much lower rate, as seen by a strong reduction in cyclin B1-high cells. In contrast, VCR alone did not induce any gross changes in cell-cycle progression, whereas combinatorial treatment induced a G2 arrest (Figure 5D, i). To assess the effects of protein translation attenuation by PI3K inhibition, the anti-apoptotic proteins survivin and XIAP were quantified, which are known to feature a very short half-life (only ~30 min) and thus need constant replenishment to efficiently interfere with apoptosis induction (Dan et al., 2016; White-Gilbertson et al., 2009; Zhao et al., 2010). As expected, GDC0941 treatment led to reduced levels of these short-lived regulators, thus leading to a marked increase in the percentage of survivin- and XIAP-low cells, whereas VCR had no effect (Figure 5D, ii). At high concentrations, VCR affects the global cellular tubulin network. However, at the low concentrations used here, it is thought to interfere mostly with the tubulin dynamics of the spindle apparatus (Jordan and Wilson, 2004). Accordingly, VCR induced a 5-fold increase in cells that are positive for the mitosis-specific antibody mouse monoclonal mitotic (MPM-2), which recognizes mitotic phosphoproteins and marks cells in mitotic arrest (Tapia et al., 2006), along with the inactivation of the anti-apoptotic protein Bcl-2, which is known to be phosphorylated by agents damaging microtubules (Ling et al., 2002; Srivastava et al., 1999); this effect was further exacerbated by the addition of GDC0941 (Figure 5D, iii).
The hallmarks of true apoptosis induction include the activation of the caspase cascade and the loss of mitochondrial membrane potential. When cells were analyzed for cleaved caspase 3 along with the loss of the apoptosis inhibitor Mcl-1 and for staining patterns with the membrane-permeable JC-1 dye to monitor mitochondrial charge, the combined action of GDC0941 and VCR revealed very strong synergy (Figures 5D, iv and 5D, v). In conclusion, treatment with GDC0941 or VCR at low concentrations primes T-ALL cells for apoptosis, and these effects become highly detrimental for cell survival upon combined treatment (Figure 5E).
GDC0941 and VCR Combination Therapy
With the promising results of GDC0941/VCR in JURKAT cells, we next assessed the efficacy of this combination in a panel of cell lines from other cancers (Figure 6A). We extended our analysis to the entire panel of 10 T-ALL cell lines, for which the efficacy of GDC0941 alone had already been determined (Figure S1D) in 8 × 8 grids of 7 inhibitor concentrations prepared using a robotic liquid handling station with a 96-well pin tool (see Figure S6B for inhibitor concentration ranges). Cell densities were determined on a plate reader using a luminescence-based viability assay and then used to obtain cell counts and ΔBliss values as before (Figures 6B and S7). To allow for easy comparison between the different cell lines tested, we plotted the 9 highest ΔBliss values for each cell line (Figure S6C). All 10 T-ALL cell lines exhibited a relatively high ΔBliss value—on average ~40—indicating that ~40% more growth inhibition was observed compared to the sum of the inhibitors’ effects in a single treatment (Figure 6B). The extensive 8 × 8 grid setup allowed us to evaluate whether synergy is observed over large inhibitor ranges or only when specific concentrations of inhibitors are administered (Figures 6B and S7).
Figure 6. GDC0941 and VCR Treatment Synergy in 28 Cell Lines of 5 Different Cancer Types.
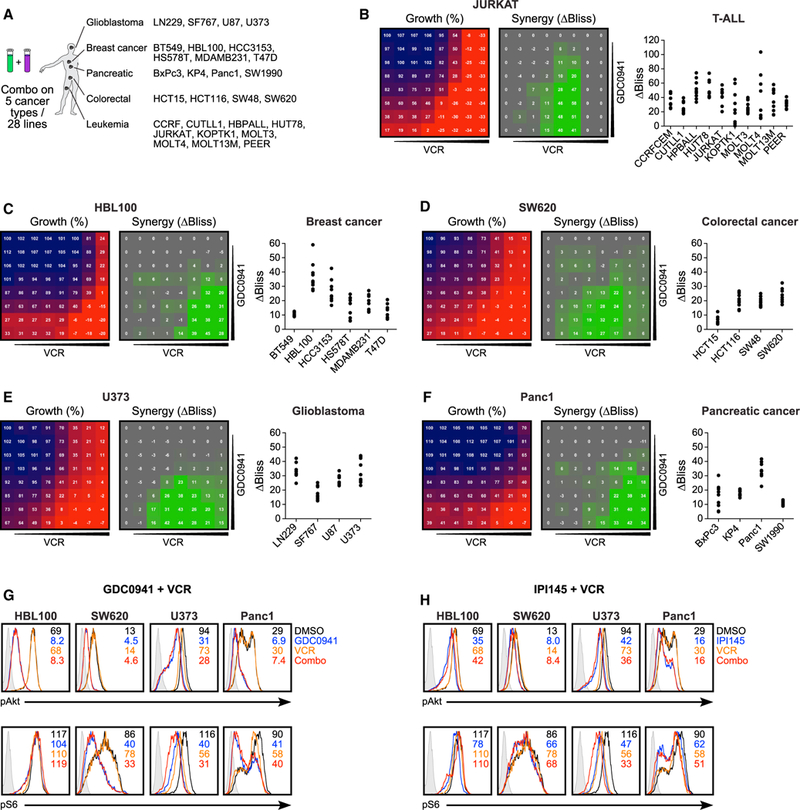
(A) Selection of 28 cell lines from 5 different cancers to be tested for GDC0941 and vincristine synergy.
(B) Robotics-generated 8 × 8 grids allowed for the analysis of synthetic lethality byGDC0941 and vincristine (VCR) on 10humanT-ALLcell lines; grids for growth and synergy shown for JURKAT cells (left), along with summary for the 10 best ΔBliss values for all of the lines tested (right).
(C–F) Same as (B), but for breast cancer (C), colorectal cancer (D), glioblastoma (E), and pancreatic cancer (F), each with 1 selected cell line highlighted and summary plots for the other lines tested.
(G) Phospho-flow analysis of baseline (black) phospho-Akt (top) and phospho-S6 (bottom) levels, and treatment with GDC0941 (blue), second inhibitor alone (orange), or a combination of both (red) (n = 2).
(H) Same as (G), but with treatment with IPI145 (blue).
(G) and (H) are representative examples of ≥3 independent experiments (n = 2).
We then expanded to 18 breast cancer, colorectal cancer, glioblastoma, and pancreatic ductal adenocarcinoma cell lines. Glioblastoma cell lines revealed a similarly robust and uniform sensitivity to the GDC0941/VCR combination treatment, as was observed in T-ALL, whereas responses in the other cancer types were more heterogeneous (Figures 6C–6F, S8, and S9). In all 18 solid tumor cell types and all 10 T-ALL cell lines we observed some degree of synergy with the GDC0941/VCR combination treatment, arguing that our synthetic lethal screen leukemia platform using JURKAT and MOLT3 T-ALL cell lines yielded results that are, in principle, applicable to other cancer types. Exposure of HBL110 (breast), SW620 (colorectal), U373 (glioblastoma), and Panc1 (pancreatic) cancer cell lines to GDC0941 reduced Akt phosphorylation and to a lesser extent S6 phosphorylation, whereas VCR did not affect the phosphorylation levels of these two kinases (Figure 6G). Many solid cancers signal through the PI3K-α and -β isoforms (LoRusso, 2016), and in agreement with this notion, the IPI145 inhibitor had only modest effects on Akt and S6 phosphorylation in the 4 cell lines (Figure 6H). For this reason, we did not explore the growth inhibition assays with this PI3K-α-sparing compound.
GDC0941 and VCR in Mouse Leukemia Preclinical Trials
Mouse cancer models generated using insertional mutagenesis (IM) recapitulate the multi-step pathogenesis and inter- and in- tratumoral genetic heterogeneity that is a hallmark of advanced human cancers (Uren and Toretsky, 2005). Injecting neonatal mice with the MOL4070LTR retrovirus induced aggressive T-ALLs that can be transplanted into cohorts of recipients to perform controlled preclinical trials of chemical inhibitors and drug combinations (Lauchle et al., 2009; Dail et al., 2010, 2014; Burgess et al., 2017). T-ALL JW81 is an aggressive leukemia that causes lethality after ~15 days in transplant recipients characterized by high blood leukocyte counts and extensive proliferation of leukemic blasts in the bone marrow that also invade the CNS (Figure 7A) (Dail et al., 2014). We first determined the maximum tolerated doses (MTDs) of GDC0941 and VCR alone and in combination. Treatment with GDC0941 (100 mg/kg/day) and the highest dose of VCR tested (0.3 mg/kg twice weekly) resulted in a transient reduction in body weight after 1 week, but all of the mice recovered and did not exhibit other toxicities (Figure 7B; data not shown). For the preclinical trial, cryopreserved JW81 cells were expanded in a sublethally irradiated recipient mouse, and bone marrow from this animal was harvested and transplanted into 20 congenic secondary recipients (Figure 7C). These mice were randomly assigned to 1 of 4 treatment groups: (1) control vehicle, (2) VCR alone, (3) GDC0941 alone, and (4) GDC0941/VCR combination. GDC0941 was administered daily and VCR twice per week until the mice developed progressive disease and required euthanasia (Figure 7C). Whereas treatment with VCR failed to extend survival, GDC0941 exhibited modest efficacy as a single agent, which was consistent with prior data (Dail et al., 2014). In contrast, the GDC0941/VCR combination was synergistic with a ~1.5-fold increase in survival compared to mice treated with the control vehicle (median time to euthanasia of 15 versus 23 days; p = 0.0015; Figures 7D and S6D). We confirmed the beneficial effects of the GDC0941/VCR combination in an independent cohort of recipients transplanted with T-ALL JW81 with slightly altered drug dosing (Figure 7E).
Figure7. GDC0941 and Vincristine Combinatorial Treatment in Preclinical Trials.
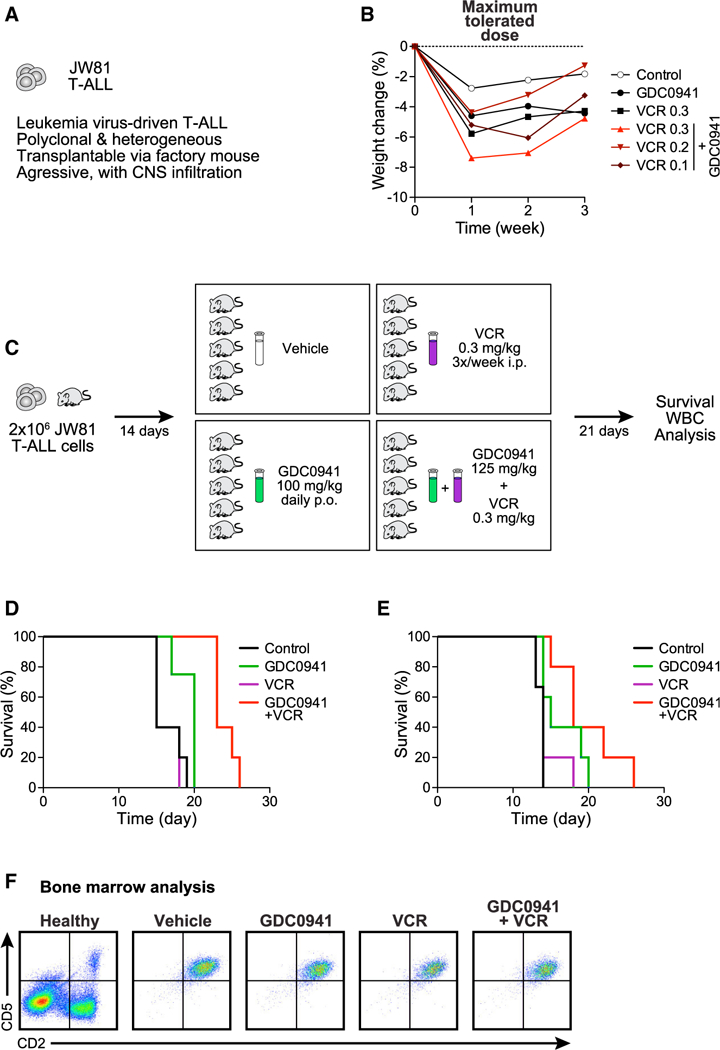
(A) Characteristics of murine T-ALL JW81.
(B) Body weight measurements over time to determine the maximum tolerated dose of GDC0941 (100 mg/kg body weight) and vincristine (VCR) (0.3 mg/kg body weight) alone, or for GDC0941 in combination with 3 different doses of VCR (mg/kg body weight).
(C) Dosing and application scheme for GDC0941 and VCR in preclinical trials.
(D) Survival curves for mice treated with vehicle control, GDC0941 alone, VCR alone, or the GDC0941/VCR combination therapy. GDC0941 at 125 mg/kg/day starting 4 days post-transplant and VCR at 0.3 mg/kg twice weekly starting 6 days post-transplant. Mantel-Cox tests comparing treatments provided the following p values: control versus VCR p = 0.6015, control versus GDC0941 p = 0.0323, VCR versus GDC0941 p = 0.0392, VCR versus combination p = 0.0023, GDC0941 versus combination p = 0.0050, and control versus combination p = 0.0015. For additional statistical analysis, see Figure S6.
(E) As in (D), but GDC0941 at 100 mg/kg/day and VCR at 0.2 mg/kg twice weekly, both starting 4 days post-transplant. Mantel-Cox test p values: control versus VCR p = 0.1690, control versus GDC0941 p = 0.0645, VCR versus GDC0941 p = 0.1721, VCR versus combination p = 0.0290, GDC0941 versus combination p = 0.1762, and control versus combination p = 0.0067.
(F) Analysis of bone marrow cells harvested at the time of euthanasia from representative recipients of the indicated therapy.
In addition to modulating survival, VCR treatment modified the pattern of leukemic involvement at the time of death. Whereas all of the recipient mice assigned to receive control vehicle or GDC0941 alone exhibited diffuse leukemic involvement of hematologic tissues at euthanasia, several moribund mice that were treated with VCR alone or in combination with GDC0941 had substantially lower blood leukocyte counts, smaller spleens, and reduced total cell yield from harvested bone marrow (data not shown). Despite the beneficial effect of the GDC0941/VCR combination in recipients transplanted with the primary T-ALL JW81, all of the animals in the trial eventually succumbed from progressive leukemia, which can be identified by the CD2/CD5 staining of JW81 blast cells in the bone marrow (Figure 7F). Furthermore, the animals exhibited neurological symptoms that are consistent with the infiltration of the CNS by leukemia cells at euthanasia. These observations suggest that treatment with VCR attenuated leukemic growth in the bone marrow and other hematologic tissues, but that it was ineffective in the CNS because it does not cross the blood-brain barrier. This interpretation is fully consistent with clinical trials of children with ALL performed in the 1960s in which CNS disease only emerged as a major sanctuary site for leukemia cells and an important cause of relapse and death after combination regimens that included VCR, glucocorticoids, and other drugs induced prolonged hematologic remissions in many patients (George et al., 1968). This realization, in turn, resulted in changes in front line ALL therapeutic trials to incorporate prophylactic treatment of the CNS (Aur et al., 1971; Liu et al., 2017).
DISCUSSION
This resource provides methods for high-throughput screening to find novel combination therapies, along with a large dataset for potential combinations with PI3K inhibition. In total, we were able to confirm 9 of 10 predicted synthetic lethal interactions with GDC0941, combining the results from the JURKAT and MOLT3 screens. Such unbiased and robust screening technologies are essential to find good drug candidates, and using high-complexity libraries increases the statistical power of such screens by masking off-target effects. This in turn reduces the rates of false-positive and false-negative results, which was a major limitation of many older shRNA screens featuring only a limited number of shRNAs per target.
When comparing the 2 cell lines tested, JURKAT and MOLT3, the majority of genes depleted or enriched were shared. This especially holds true when focusing on genes involved in PI3K signaling, which was surprising to us, considering the different baseline pAkt levels of these 2 cell lines. Similarly, the strong presence of candidates from the cell-cycle and cell-division machinery in both cell lines also indicates shared nodes, which can be effectively targeted using inhibitor combinations. In a recent publication on a positive-selection genome-scale shRNA screen, further synergistic interactions with PI3K inhibition have been found using a breast cancer cell line (Zwang et al., 2017). While Zwang et al. (2017) used the power of targeted positive selection with apoptosis-specific cell sorting, we used a high-complexity shRNA library along with next-generation sequencing to find dropout candidates.
From our screens, interference with tubulin assembly turned out to be the best candidate synthetic lethal interaction. Administering VCR at a low dose can lead to elevated levels of mitotic arrest, yet it does not trigger apoptosis induction. These are early signs of spindle assembly checkpoint signaling due to interference with the chromosome segregation machinery by tubulin inhibition (Kothari et al., 2016). In combination with PI3K inhibition, this seems to have fatal effects. GDC0941 alone induces strong growth retardation along with the loss of short-lived anti-apoptotic proteins, most probably due to the reported translation attenuation following PI3K inhibition (Bader et al., 2005). In combination, the effects of each single agent act synergistically and induce full-blown apoptosis. This synthetic lethality could also be verified in all of the other T-ALL cell lines and most solid cancer cell lines tested, which points to a more universal combinatorial effect of PI3K and tubulin inhibition. The synergistic growth inhibition that we uncovered with the PI3K inhibitor IPI145 and VCR in T-ALL was not apparent in normal primary T cells. These findings suggest that the IPI145/ VCR combination may be a very attractive therapy that inhibits leukemia but does not affect normal cells. Using a very potent but also generally toxic pan-PI3K inhibitor to identify synthetic lethality with high confidence in whole-genome screens followed by specific tweaking of the specific small molecules may be a productive strategy. The fact that IPI15/VCR combination is less toxic for normal T cells opens the door to explore new avenues for therapy in the clinic that effectively inhibit the leukemia but leave the patient’s immune system intact and functional.
We also demonstrated the in vivo efficacy of combination treatment with GDC0941 and VCR using an established retroviral insertional mutagenesis (RIM)-induced mouse model of T-ALL that recapitulates the highly aggressive and heterogeneous characteristics of human T-ALL. These results suggest PI3K inhibition as a potential therapeutic strategy for augmenting the efficacy of modern T-ALL treatment regimens, which include VCR and other chemotherapeutic agents, particularly in patients with refractory or relapsed disease. A recent comprehensive genome-wide analysis of ~260 T-ALL cases showing that PTEN and AKT mutations are correlated with treatment failure provide further support for targeting aberrant PI3K signaling (Liu et al., 2017).
With VCR being part of the standard of care therapy for leukemia and GDC0941 being far advanced in clinical development, this combination therapy could be beneficial for cancer patients, yielding improved efficacy and reduced side effects. While we are still on the verge of fully understanding the signaling pathways involved in cancer growth, more in-depth screening but also sophisticated modeling of signaling networks will help to combat the complexity that cancer research is facing (Saez-Rodriguez et al., 2015). While still in the more distant future, more advanced screening technologies may become sufficiently fast and powerful to allow for a fully personalized analysis of the best combination of the most effective inhibitors for each individual patient.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies and Dyes | ||
| pAkt (Ser473), h | Cell Signaling Tech. | CST #4058 |
| pS6 (Ser235/236), h | Cell Signaling Tech. | CST #2211 |
| Annexin V-APC | eBioscience | BMS306APC-20 |
| Cyclin B1 (V152), h | Cell Signaling Tech. | CST #4135 |
| XIAP, h | BD Biosciences | 610762 |
| Survivin, h | Cell Signaling Tech. | CST # 2808 |
| MPM-2, h | Millipore | 05–368 |
| pBcl-2 (Ser70), h | Cell Signaling Tech. | CST #2827 |
| Mcl-1, h | R&D Systems | MAB828 |
| cl. Caspase 3 (Asp175), h | Cell Signaling Tech. | CST #9661 |
| JC-1 | Enzo Life Sciences | ENZ-52304 |
| FITC anti-mouse CD5 Antibody | BioLegend | 100606 |
| PE anti-mouse CD2 Antibody | BioLegend | 100107 |
| PE Donkey Anti-Rabbit IgG | Jackson Immuno | 711–116-152 |
| APC Goat Anti-Mouse IgG | Jackson Immuno | 115–135-164 |
| BV421 Goat Anti-Rabbit IgG | BD Biosciences | 565014 |
| DAPI | Molecular Probes | D1306 |
| Propidium Iodide | Molecular Probes | P3566 |
| Fixable Viability Dye eFluor 780 | eBioscience | 65–0865-14 |
| Alexa Fluor 488 NHS Ester | Molecular Probes | A20000 |
| Alexa Fluor 546 NHS Ester | Molecular Probes | A20002 |
| Acridine Orange | Sigma-Aldrich | 235474 |
| Biological Samples | ||
| Primary murine acute myeloid leukemia sample (JW81),female | Lab of Kevin Shannon | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GDC-0941 (Pictilisib) | Selleckchem | S1065 |
| IPI-145 (Duvelisib) | Selleckchem | S7028 |
| DXR | Selleckchem | S1208 |
| BKM120 (Buparlisib) | Selleckchem | S2247 |
| XL147 (Pilaralisib) | Selleckchem | S7645 |
| AS-605240 | Selleckchem | S1410 |
| EHop-016 | Selleckchem | S7319 |
| CTA 056 | Tocris | 4726/10 |
| VCR | Selleckchem | S1241 |
| Centrinone B | Tocris | 5690/10 |
| BX-795 | Selleckchem | S1274 |
| Dorsomorphin | Selleckchem | S7840 |
| Defactinib | Selleckchem | S7654 |
| ARRY-520 | Tocris | 4676/10 |
| Mps1-IN-5 | MedChem Express | HY-12858 |
| GDC-0941 | Genentech | N/A |
| Critical Commercial Assays | ||
| QIAamp DNA Blood Maxi Kit | QIAGEN | 51192 |
| PstI | New England Biolabs | R3140T |
| Silica column | Denville Scientific | CM-0600–20 |
| HiFi Phusion polymerase | New England Biolabs | M0530L |
| Midi GeBAflex tubes | Gene Bio-Application | T010 |
| Experimental Models: Cell Lines | ||
| JURKAT cells (human, male) | Lab of Jeroen Roose | N/A |
| MOLT3 cells (human, male) | Lab of Jeroen Roose | N/A |
| CCRFCEM cells (human, female) | Lab of Jeroen Roose | N/A |
| CUTLL1 cells (human, male) | Lab of Jeroen Roose | N/A |
| HPBALL cells (human, male) | Lab of Jeroen Roose | N/A |
| HUT78 cells (human, male) | Lab of Jeroen Roose | N/A |
| KOPTK1 cells (human, male) | Lab of Jeroen Roose | N/A |
| MOLT4 cells (human, male) | Lab of Jeroen Roose | N/A |
| MOLT13M cells (human, female) | Lab of Jeroen Roose | N/A |
| PEER cells (human, female) | Lab of Jeroen Roose | N/A |
| LN229 cells (human, female) | Lab of William Weiss | N/A |
| SF767 cells (human, female) | Lab of William Weiss | N/A |
| U87 cells (human, female) | Lab of William Weiss | N/A |
| U373 cells (human, male) | Lab of William Weiss | N/A |
| BT549 cells (human, female) | Lab of Zena Werb | N/A |
| HBL100 cells (human, female) | Lab of Zena Werb | N/A |
| HCC3153 cells (human, female) | Lab of Zena Werb | N/A |
| HS578T cells (human, female) | Lab of Zena Werb | N/A |
| MDAMB231 cells (human, female) | Lab of Zena Werb | N/A |
| T47D cells (human, female) | Lab of Zena Werb | N/A |
| BxPc3 cells (human, female) | Lab of Rushika Perera | N/A |
| KP4 cells (human, male) | Lab of Rushika Perera | N/A |
| Panc1 cells (human, male) | Lab of Rushika Perera | N/A |
| SW1990 cells (human, male) | Lab of Rushika Perera | N/A |
| HCT15 cells (human, male) | Lab of Jeroen Roose | N/A |
| HCT116 cells (human, male) | Lab of Jeroen Roose | N/A |
| SW48 cells (human, female) | Lab of Jeroen Roose | N/A |
| SW620 cells (human, male) | Lab of Jeroen Roose | N/A |
| Experimental Models: Organisms/Strains | ||
| F1 C57BL/6 × 129Sv/Jae mice | UCSF Laboratory Animal Resource Center, Breeding Core | N/A |
| C57BL/6 mice | Lab of Jeroen Roose | N/A |
| Oligonucleotides | ||
| TruSeq Index Primers (The barcoding primers are proprietary to Illumina and were shared with the UCSF Genomics Core with the constraint not to publish the individual sequences.) | Illumina | N/A |
| Recombinant DNA | ||
| UCSF EXPANDed RNAi library | Lab of Michael McManus, UCSF ViraCore | Bassik et al., 2009 |
| Software and Algorithms | ||
| Galaxy | usegalaxy.org | |
| Bowtie | Langmead et al., 2009 | bowtie-bio.sourceforge.net |
| DESeq2 | Love et al., 2014 | (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) |
| MAGecK | Li et al., 2014 | (http://sourceforge.net/projects/mageck) |
| Ingenuity Pathway Analysis | QIAGEN Bioinformatics | N/A |
| RStudio | RStudio | v1.0.136 |
| R | The R Project | v3.3.2 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeroen P. Roose (jeroen.roose@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Cell lines
T-ALL cell lines (JURKAT, MOLT3, CCRFCEM, CUTLL1, HPBALL, HUT78, KOPTK1, MOLT4, MOLT13M) were grown in RPM11640 medium containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml L-glutamine, 55 μM b-mer-captoethanol, and 10 mM HEPES; for growth of PEER T-ALL cells, medium was further supplemented with non-essential amino acids (all GIBCO). Solid cancer cell lines (Breast: BT549, HBL100, HCC3153, HS578T, MDAMB231, T47D; Colorectal: HCT15, HCT116, SW48, SW620; Glioblastoma: LN229, SF767, U87, U373; and Pancreatic: BXPC3, KP4, PANC1, SW1990) were grown in DMEM containing FBS, penicillin, streptomycin, L-glutamine, and HEPES at the above concentrations. All cell lines were incubated at 37°C, with cells in RPMI at 5% CO2 and cells in DMEM at 10% CO2. Origin and sex of all cell lines are stated in the Key Resources Table.
Primary mouse T cells
Primary mouse T cells were isolated from cervical, brachial, axillary, and inguinal lymph nodes of C57BL6 male mice, 6–10 weeks old. CD4+T cells were isolated by MACS negative isolation (Miltenyi). Cells were counted and plated at 4E6 cells/ml on 24 well plates that had been pre-coated with 10μg/ml anti-CD3 (UCSF Cell Culture Facility; Clone 145–2C11). T cells were cultured in RPMI supplemented with 10% FBS, 1% sodium pyruvate, 1% non-essential amino acids, 1% HEPES, 1% penicillin/streptomycin/glutamine, and 0.1% beta-mercaptoethanol (all GIBCO), with 5 μg/ml anti-CD28 (UCSF Cell Culture Facility; Clone 37.51) for 24h, at 37°C with 5% CO2. Cells were then washed with RPMI and seeded with 50 U/ml of IL-2 for drug titration, proliferation, and apoptosis analysis.
Mouse preclinical trials
All animal experiments conformed to national regulatory standards and were approved by the University of California, San Francisco Committee on Animal Research. All mice were healthy, immune competent, and drug and test naive. Mice were housed in micro-isolater cages and provided chow and water ad libitum. Therapeutic studies were carried out in male recipient mice to exclude the possibility of sex-linked variation in drug metabolism. Animals were randomly assigned to experimental treatment groups, then drug or control vehicle was administered without blinding. To test a maximum tolerated dose for GDC0941 and VCR, 8–12 weeks old F1 C57BL/6 × 129Sv/Jae mice were sublethally irradiated and treatment was started 4 days later with 3 mice per group. Hydroxypropyl methylcellulose vehicle (0.5% hydroxypropyl methylcellulose and 0.2% Tween 80) and PBS were administered to control mice. GDC0941 was administered daily by oral gavage and VCR 2x/week by intraperitoneal injection. Mice were weighed weekly to adjust dosing. After 4 weeks of treatment, mice were euthanized and general examination of organs performed; a complete blood count (CBC) was run for one mouse from each group. No obvious signs of toxicity were observed. For the preclinical trials, freshly thawed JW81 T-ALL cells were transplanted into sublethally irradiated recipient 8–12 weeks old F1 C57BL/6 × 129Sv/Jae mice via the tail vein (Origin and sex of JW81 cells are stated in the Key Resources Table). When these animals became moribund, they were sacrificed and 2×106 freshly harvested bone marrow cells were transplanted into sublethally irradiated 8–12 weeks old F1 C57BL/6 × 129Sv/Jae recipient mice via the tail vein. Treatment was started 4 days (GDC0941) or 6 days (VCR) post-transplant and continued until mice became moribund and were euthanized. The trial was performed with GDC0941 at 125 mg/kg, VCR at 0.3 mg/kg, or a combination of both at the same concentrations, with administration as above and 5 mice per treatment group. Bone marrow harvested from mice enrolled on the preclinical trial at euthanasia was subjected to RBC lysis and incubated with PE- or FITC-conjugated antibodies against CD2 (BioLegend, 100107) and CD5 (BioLegend, 100606), respectively. Whole bone marrow harvested from a wild-type F1 C57BL/6 × 129Sv/Jae mouse was analyzed as a control.
METHOD DETAILS
Inhibitor titrations
Inhibitors were prepared in DMSO or water, according to the instructions of the manufacturer. Using a serial dilution series, inhibitor concentration ranges inducing growth attenuation were determined for JURKAT and MOLT3 cells. For that, cells were seeded in duplicates at a density of 200,000 cells/ml in 24 well plates and cultured for 3 days, before final cell concentrations were determined.
Apoptosis/dead cell staining and counting
To quantify cell counts and proportions of live, apoptotic, and dead cells by flow cytometry, cell samples were mixed with 123count eBeads (eBioscience), 20 nM acridine orange (Aldrich), 200 nM propidium iodide (Molecular Probes), 2 mM calcium chloride (Sigma), and Annexin V-APC (1/500, eBioscience) in duplicates. To exclude debris from nucleated cells, only acridine orange-positive events were considered, before plotting Annexin V versus propidium iodide to distinguish live, apoptotic, and dead cells. Cell counts were determined as follows: Cell concentration = bead concentration × (live cell count × bead volume) / (bead count × total cell volume).
Intracellular staining for flow cytometry
Antibody staining was all completed in duplicates, using FACS buffer, consisting of PBS with 2% FBS, 2 mM EDTA, and 0.1% sodium azide. Prior to fixation, cells were centrifuged at 500 rcf. for 5 minutes at 4°C, after fixation, cells were centrifuged at 2.500 rcf. For 2 minutes at RT. To label dead cells, samples were incubated with Fixable Viability Dye eFluor 780 (½000, eBioscience) for 30 minutes on ice. To determine the mitochondrial membrane potential, cells were incubated in 5 mg/ml JC-1 (Enzo Life Sciences) in growth medium for 10 minutes at 37°C, before cells were washed once with FACS buffer and kept on ice until acquisition. For intracellular staining, cells were washed once in PBS, fixed in 2% PFA for 5 minutes at RT, washed again in PBS, and then permeabilized in 90% methanol overnight at –20°C. Prior to subsequent staining, cells were rehydrated in FACS buffer for 5 minutes at RT, before washing cells twice in FACS buffer to remove remaining methanol. Phosphorylated Akt and ribosomal protein S6 was stained using rabbit primary antibodies: pAkt (Ser473) and pS6 (Ser235/236) (both Cell Signaling Technologies), followed by staining with donkey anti- rabbit-PE (Jackson). For barcoding, cells were incubated with dilutions of the succinimidyl esters of Alexa Fluor 488 (150, 10, and 0.2 ng/ml) and Alexa Fluor 546 (200, 22, and 1.0 ng/ml, both Molecular Probes) in 0.5× FACS buffer for 15 minutes at RT. Before pooling barcoded cells, samples were incubated and washed repeatedly in FACS buffer at RT to eliminate any reactive dye leaking from the cells. Staining was performed with mouse primary antibodies: Cyclin B1 (Cell Signaling Technologies), Mcl-1 (R&D Systems), MPM-2 (Millipore), XIAP (BD Biosciences), and rabbit primary antibodies: cleaved Caspase 3, phospho-Bcl-2, Survivin (all Cell Signaling Technologies), followed by staining with goat anti-rabbit-BV421 (BD Biosciences) and goat anti-mouse-APC (Jackson Immuno Research). For cell cycle analysis, cells were resuspended in 1 μg/ml DAPI (Molecular Probes).
Lentivirus shRNA synthetic lethal screen
JURKAT and MOLT3 cells were transduced in duplicates with lentivirus shRNA libraries in roller bottles, at a multiplicity of infection of 0.7 to reduce the likelihood of multiple integrations per cell. Two days later expression of the lentivirus marker mCherry was verified by flow cytometry and transduced cells were selected with puromycin (Sigma) for another 3 days. Cultures were split and further incubated with either DMSO control or GDC0941 at concentrations inducing approx. 20% growth retardation (JURKAT: 1.4 μM, MOLT3: 0.3 μM). Cells were kept under these conditions and subcultured every 2 or 3 days. On day 22 aliquots of 120×106 cells were spun down in replicates and snap-frozen on dry ice.
shRNA processing from screen samples
Genomic DNA was extracted from frozen cell pellets using the QIAamp DNA Blood Maxi Kit (QIAGEN) following the instructions of the manufacturer. To release the proviral DNA, genomic DNA was digested with PstI over night and then run on a preparative 0.6% agarose gel to separate the 2.2 kb proviral insert. DNA was released from the gel fragment using sequential freeze-thaw cycles and then purified on a silica column (Denville Scientific). shRNAs were amplified from the proviral DNA using HiFi Phusion polymerase (NEB) and barcoded TrueSeqIndex forward primers and a common reverse primer. PCR products were purified on silica columns and separated on a preparative 7.5% polyacrylamide gel to isolate the 270 bp PCR amplicon. DNA was release from the polyacrylamide gel fragments using electroelution in Midi GeBAflex tubes (Gene Bio-Application) and purified on silica columns. Final DNA concentrations of all samples were estimated by BioAnalyzer (Agilent Technologies) measurement, and adjusted before submitting to 50 bp single end deep sequencing on a HiSeq 2500 sequencer (Illumina).
Combinatorial inhibitor grids
To determine synergy, inhibitors were combined and ΔBliss values determined (Bliss, 1939). A Bliss expectation (representing the inhibition expected when inhibitors act only additively) was calculated from single inhibitor treatments using the formula (A + B) – (A × B) for every concentration used. The ΔBliss is the difference between the Bliss expectation and the actual inhibition, with positive values depicting synergistic effects. For 4×4 combinatorial inhibitor grids, cells were seeded in duplicates at a density of 200,000 cells/ml in 24 well plates and incubated with combinations of GDC0941 and a second inhibitor at serial dilutions for 3 days. Final cell concentrations were determined using apoptosis/dead cell staining and counting. For 8×8 combinatorial inhibitor grids, 96 well plates with combinations of serial dilutions of GDC0941 and VCR were prepared using a robotic liquid handling station with a 96 well pin tool dispensing 200 nL from a 1000x stock compound plate, frozen down, and thawed when needed. For suspension cells 300,000 cells/ml were added to each well, for adherent cells 30,000 cells/ml in each well, sealed with AeraSeal (Excel Scientific), and incubated for 3 days. Cell densities were read out using CellTiter-Glo (Promega) according to the instructions of the manufacturer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Processing of deep sequencing reads in Galaxy
Sequencing raw reads were processed using Galaxy. Individual files from different lanes with the same barcode were concatenated tail to head before reads were trimmed to 34 bp to contain the shRNA sense arm and the hairloop sequence. All sequences not containing the common hairloop sequence were discarded to only contain amplicons from proviral DNA. After removing the hairpin loop sequence all sequences shorter than 18 bp were discarded to contain only true shRNA reads. In addition, all low-quality reads were discarded as well. Using Bowtie (Langmeadet al., 2009), reads were aligned to the shRNA library and individual reads for each shRNA identifier were summed up.
Statistical processing of shRNA counts in R
We performed data processing in R using DESeq2 (Love et al., 2014) to assign weight and statistical significance to each of the shRNA-targeted genes. To compare replicates with each other and control with treated samples, individual shRNA reads of each identifier were plotted against each other, with a narrow distribution of counts indicating a high reproducibility between samples while broader distributions show differences between control and treated samples. To calculate changes in shRNA counts between samples and to evaluate statistical significance, shRNA read counts were analyzed with DESeq2, using standard settings. shRNA identifiers were matched with gene names and all shRNAs targeting the same gene consolidated and plotted. To determine single values for each gene from the joint information of all shRNAs targeting the same gene, the count difference between significantly depleted and enriched shRNAs (D) was calculated, and also the mean log2 fold change of all significant shRNAs (m). Those two values were plotted for each gene to estimate the overall distribution of all genes represented in the screens. For a more focused view on the depleted genes, only genes that were represented by at least 4 significant shRNAs and with a D of at least −3 were considered. See figure legends 2 and 3.
Ingenuity pathway analysis- IPA
Prior to IPA, shRNA counts were re-calculated with MAGecK (Li et al., 2014) in R to estimate p values for each gene along with log2 fold changes. IPA was done with all genes represented in the screen and custom and integrated pathway maps were generated to determine common patterns in signaling pathways. See Figure S2.
Statistical evaluation of preclinical trials
A total of n = 3–5 mice were randomly assigned to each of the control vehicle or experimental arms for MTD and preclinical trial studies as described. Survival analysis was calculated from the day of transplant, and mice were euthanized when they appeared moribund. Statistical significance was calculated by comparing independent treatment arms using the log-rank test. Rarely, mice were excluded from analysis due to failure of leukemia cell engraftment after transplantation. Data analysis from previous studies has verified the statistical power of the cohort size used in these trials to reliably detect significant differences (Burgess et al., 2017; Dail et al., 2010, 2014; Lauchle et al., 2009).
Supplementary Material
Highlights.
A leukemia platform allows for saturated shRNA-based synthetic lethality screens
High-complexity shRNA libraries enable lethality identification with high fidelity
Small molecule inhibitors and cancer cell lines confirm predicted lethality
PI3K inhibitors and vincristine synergize in in vivo preclinical mouse trials
ACKNOWLEDGMENTS
We thank the members of the Roose lab and the Heme-Onc community at UCSF for useful suggestions and comments, and Byron Hahn and the Preclinical Therapeutics Core at the UCSF Helen Diller Family Comprehensive Cancer Center for help with the preclinical trials. Research support for this study came from a Gabrielle’s Angel Foundation grant, an Alex’s Lemonade Stand Foundation Innovator Award, the NIH/NCI (R01-CA187318), and the NIH/NHLBI (R01-HL120724) (all to J.P.R.). Further support came from a Leukemia & Lymphoma Society grant (to M.M.) and NIH grants R01CA212767 and U01CA217882 (to M.T.M.). A.W. is supported by a postdoctoral fellowship, PF-14–070-01-TBG, from the American Cancer Society, including a supplement from the Hillcrest Committee. This work was also supported, in part, by NIH grants R37 72614 and R01 CA193994 (to K.S.). K.S. is an American Cancer Society Research Professor.
Footnotes
DECLARATION OF INTERESTS
J.R. is a co-founder of and scientific advisor to Seal Biosciences and serves on the scientific advisory committee for the Mark Foundation for Cancer Research. A portion of this work has been submitted for patent application.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.045.
DATA AND SOFTWARE AVAILABILITY
All individual targets can be explored in our searchable database of our synthetic lethal screen with GDC0941 (web page: https://mmues.shinyapps.io/K7screen/).
REFERENCES
- Al-Lazikani B, Banerji U, and Workman P (2012). Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 30, 679–692. [DOI] [PubMed] [Google Scholar]
- Aur RJA, Simone J, Hustu HO, Walters T, Borella L, Pratt C, and Pinkel D (1971). Central nervous system therapy and combination chemotherapy of childhood lymphocytic leukemia. Blood 37, 272–281. [PubMed] [Google Scholar]
- Babij C, Zhang Y, Kurzeja RJ, Munzli A, Shehabeldin A, Fernando M, Quon K, Kassner PD, Ruefli-Brasse AA, Watson VJ, et al. (2011). STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res. 71, 5818–5826. [DOI] [PubMed] [Google Scholar]
- Bader AG, Kang S, Zhao L, and Vogt PK (2005). Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 5, 921–929. [DOI] [PubMed] [Google Scholar]
- Badinloo M, and Esmaeili-Mahani S (2014). Phosphatidylinositol 3-kinases inhibitor LY294002 potentiates the cytotoxic effects of doxorubicin, vincristine, and etoposide in a panel of cancer cell lines. Fundam. Clin. Pharmacol. 28, 414–422. [DOI] [PubMed] [Google Scholar]
- Bassik MC, Lebbink RJ, Churchman LS, Ingolia NT, Patena W, LeProust EM, Schuldiner M, Weissman JS, and McManus MT (2009). Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nat. Methods 6, 443–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss CI (1939). The Toxicity of Poisons Applied Jointly. Ann. Appl. Biol. 26, 585–615. [Google Scholar]
- Boettcher M, and McManus MT (2015). Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 58, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL (1989). ras oncogenes in human cancer: a review. Cancer Res. 49, 4682–4689. [PubMed] [Google Scholar]
- Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT, et al. (2013). Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2, e00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KK, and Toker A (2015). The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess MR, Hwang E, Mroue R, Bielski CM, Wandler AM, Huang BJ, Firestone AJ, Young A, Lacap JA, Crocker L, et al. (2017). KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 168, 817–829.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, and Varmus H (2015). A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et al. (2013). Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 23, 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, and Der CJ (2014). Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dail M, Li Q, McDaniel A, Wong J, Akagi K, Huang B, Kang HC, Kogan SC, Shokat K, Wolff L, et al. (2010). Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc. Natl. Acad. Sci. USA 107, 5106–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dail M, Wong J, Lawrence J, O’Connor D, Nakitandwe J, Chen S-C, Xu J, Lee LB, Akagi K, Li Q, et al. (2014). Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature 513, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan HC, Sun M, Kaneko S, Feldman RI, Nicosia SV, Wang H-G, Tsang BK, and Cheng JQ (2016). Retraction. J. Biol. Chem. 291, 22846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Rodon J, Serra V, and Tabernero J (2014). Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 13, 1021–1031. [DOI] [PubMed] [Google Scholar]
- Downward J (2006). Cancer biology: signatures guide drug choice. Nature 439, 274–275. [DOI] [PubMed] [Google Scholar]
- Ehrhardt M, Craveiro RB, Holst MI, Pietsch T, and Dilloo D (2015). The PI3K inhibitorGDC-0941 displays promising in vitro and in vivo efficacy for targeted medulloblastoma therapy. Oncotarget 6, 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinn IW, O’Brien S, Kahl B, Patel M, Oki Y, Foss FF, Porcu P, Jones J, Burger JA, Jain N, et al. (2017). Duvelisib, a novel oral dual inhibitor of PI3K-δ,γ, is clinically active in advanced hematologic malignancies. Blood 131 , 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris G, Wozniak A, Sciot R, Li H, Friedman L, Van Looy T, Wellens J, Vermaelen P, Deroose CM, Fletcher JA, et al. (2013). A potent combination of the novel PI3K inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin. Cancer Res. 19, 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-yl-methyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Fransecky L, Mochmann LH, and Baldus CD (2015). Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol. Cell. Ther. 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez JM, Wullschleger S, Preston G, Guichard S, Fleming S, Alessi DR, and Duce SL (2011). Effect of PI3K- and mTOR-specific inhibitors on spontaneous B-cell follicular lymphomas in PTEN/LKB1-deficient mice. Br. J. Cancer 104, 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam P, Karhinen L, Szwajda A, Jha SK, Yadav B, Aittokallio T, and Wennerberg K (2016). Identification ofselective cytotoxic and synthetic lethal drug responses in triple negative breast cancer cells. Mol. Cancer 15, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George P, Hernandez K, Hustu O, Borella L, Holton C, and Pinkel D (1968). A study of “total therapy” of acute lymphocytic leukemia in children. J. Pediatr. 72, 399–408. [DOI] [PubMed] [Google Scholar]
- Giancotti FG (2014). Deregulation ofcell signaling in cancer. FEBS Lett. 588, 2558–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille H, and Downward J (1999). Multiple ras effector pathways contribute to G(1) cell cycle progression. J. Biol. Chem. 274, 22033–22040. [DOI] [PubMed] [Google Scholar]
- Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D, Moreau LA, Winter SS, et al. (2009). High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 114, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Haritunians T, Mori A, O’Kelly J, Luong QT, Giles FJ, and Koeffler HP (2007). Antiproliferative activity of RAD001 (everolimus) as a single agent and combined with other agents in mantle cell lymphoma. Leukemia 21, 333–339. [DOI] [PubMed] [Google Scholar]
- Hartman JL 4th, Garvik B, and Hartwell L (2001). Principles for the buffering of genetic variation. Science 291, 1001–1004. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, and Friend SH (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Hartzell C, Ksionda O, Lemmens E, Coakley K, Yang M, Dail M, Harvey RC, Govern C, Bakker J, Lenstra TL, et al. (2013). Dysregulated RasGRP1 responds to cytokine receptor inputinT cell leukemogenesis. Sci. Signal. 6, ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes D (2011). PI3K pathway inhibitors approach junction. Nat. Rev. Drug Discov. 10, 563–564. [DOI] [PubMed] [Google Scholar]
- Jackson DV Jr., and Bender RA (1979). Cytotoxic thresholds of vincristine in a murineand a human leukemia cell line in vitro. Cancer Res. 39,4346–4349. [PubMed] [Google Scholar]
- Jordan MA, and Wilson L (2004). Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Takemura Y, Holland JF, and Ohnuma T (1998). Vincristine saturation of cellular binding sites and its cytotoxic activity in human lymphoblastic leukemia cells: mechanism of inoculum effect. Biochem. Pharmacol. 55, 1229–1234. [DOI] [PubMed] [Google Scholar]
- Kothari A, Hittelman WN, and Chambers TC (2016). Cell Cycle-Dependent Mechanisms Underlie Vincristine-Induced Death of Primary Acute Lymphoblastic Leukemia Cells. Cancer Res. 76, 3553–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksionda O, Limnander A, and Roose JP (2013). RasGRP Ras guanine nucleotide exchange factors in cancer. Front. Biol. (Beijing) 8, 508–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksionda O, Mues M, Wandler AM, Donker L, Tenhagen M, Jun J, Ducker GS, Matlawska-Wasowska K, Shannon K, Shokat KM, and Roose JP (2018). Comprehensive analysis of T cell leukemia signals reveals heterogeneity in the PI3 kinase-Akt pathway and limitations of PI3 kinase inhibitors as monotherapy. PLoS One 13, e0193849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, Zweibel J, Collins J, and Doroshow JH (2010). Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat. Rev. Drug Discov. 9, 843–856. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauchle JO, Kim D, Le DT,Akagi K, Crone M, Krisman K, Warner K, Bonifas JM, Li Q, Coakley KM, et al. (2009). Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature 461, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y-H, Liebes L, Ng B, Buckley M, Elliott PJ, Adams J, Jiang J-D, Muggia FM, and Perez-Soler R (2002). PS-341, a novel proteasome inhibitor, induces Bcl-2 phosphorylation and cleavage in association with G2-M phase arrest and apoptosis. Mol. Cancer Ther. 1, 841–849. [PubMed] [Google Scholar]
- Liu X, Chhipa RR, Nakano I, and Dasgupta B (2014). The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol. Cancer Ther. 13, 596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, McCastlain K, Edmonson M, Pounds SB, Shi L, et al. (2017). The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetti A, Cappellini A, Sparta AM, Chiarini F, Buontempo F, Evangelisti C, Evangelisti C, Orsini E, McCubrey JA, and Martelli AM (2015). PI3K pan-inhibition impairs more efficiently proliferation and survival of T-cell acute lymphoblastic leukemia cell lines when compared to isoform-selective PI3K inhibitors. Oncotarget 6, 10399–10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso PM (2016). Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors. J. Clin. Oncol. 34, 3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, and Zhang J (2016). Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 116, 6607–6665. [DOI] [PubMed] [Google Scholar]
- Luo T, Masson K, Jaffe JD, Silkworth W, Ross NT, Scherer CA, Scholl C, Frohling S, Carr SA, Stern AM, et al. (2012). STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proc. Natl. Acad. Sci. USA 109, 2860–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini M, Ciraolo E, Gulluni F, and Hirsch E (2013). Targeting PI3K in cancer: any good news? Front. Oncol. 3, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MS, and Miller LD (2012). RAS mutations and oncogenesis: not all RAS mutations are created equally. Front. Genet. 2, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munugalavadla V, Mariathasan S, Slaga D, Du C, Berry L, Del Rosario G, Yan Y, Boe M, Sun L, Friedman LS, et al. (2014). The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene 33, 316–325. [DOI] [PubMed] [Google Scholar]
- O’Brien C, Wallin JJ, Sampath D, GuhaThakurta D, Savage H, Pun-noose EA, Guan J, Berry L, Prior WW, Amler LC, et al. (2010). Predictive biomarkers of sensitivity to the phosphatidylinositol 3′ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin. Cancer Res. 16, 3670–3683. [DOI] [PubMed] [Google Scholar]
- Opel D, Westhoff M-A, Bender A, Braun V, Debatin K-M, and Fulda S (2008). Phosphatidylinositol 3-kinase inhibition broadly sensitizes glioblastoma cells to death receptor- and drug-induced apoptosis. Cancer Res. 68, 6271–6280. [DOI] [PubMed] [Google Scholar]
- Ostrem JML, and Shokat KM (2016). Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov. 15, 771–785. [DOI] [PubMed] [Google Scholar]
- Proctor J, Yang Y, Gao X, Zhang W, Huang S, Changelian P, Kutok JL, McGovern K, and You MJ (2013). The Potent PI3K-δ,γ Inhibitor, IPI-145, Exhibits Preclinical Activity In Murine and Human T-Cell Acute Lympho-blastic Leukemia. Blood 122, 1438. [Google Scholar]
- Pui C-H, and Evans WE (2006). Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354, 166–178. [DOI] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, and Bar-Sagi D (2011). RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RL, McPherson HR, Kettlewell L, Shnyder SD, Hurst CD, Alder O, and Knowles MA (2016). PIK3CA dependence and sensitivity to therapeutic targeting in urothelial carcinoma. BMC Cancer 16, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Rodriguez J, MacNamara A, and Cook S (2015). Modeling Signaling Networks to Advance New Cancer Therapies. Annu. Rev. Biomed. Eng. 17, 143–163. [DOI] [PubMed] [Google Scholar]
- Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, et al. (2009). Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137, 821–834. [DOI] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, and Bollag G (2007). Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308. [DOI] [PubMed] [Google Scholar]
- Shingu T, Yamada K, Hara N, Moritake K, Osago H, Terashima M, Ue- mura T, Yamasaki T, and Tsuchiya M (2003). Synergistic augmentation of antimicrotubule agent-induced cytotoxicity by aphosphoinositide 3-kinase inhibitor in human malignant glioma cells. Cancer Res. 63, 4044–4047. [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, and McCormick F (2017). RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims D, Mendes-Pereira AM, Frankum J, Burgess D, Cerone M-A, Lombardelli C, Mitsopoulos C, Hakas J, Murugaesu N, Isacke CM, et al. (2011). High-throughput RNA interference screening using pooled shRNA libraries and next generation sequencing. Genome Biol. 12, R104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava RK, Sasaki CY, Hardwick JM, and Longo DL (1999). Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J. Exp. Med. 190, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam PS, Whye DW, Efimenko E, Chen J, Tosello V, De Keers-maecker K, Kashishian A, Thompson MA, Castillo M, Cordon-Cardo C, et al. (2012). Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell 21,459–472. [DOI] [PubMed] [Google Scholar]
- Tapia C, Kutzner H, Mentzel T, Savic S, Baumhoer D, and Glatz K (2006). Two mitosis-specific antibodies, MPM-2 and phospho-histone H3 (Ser28), allow rapid and precise determination of mitotic activity. Am. J. Surg. Pathol. 30, 83–89. [DOI] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, and Zhao JJ (2015). PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 15, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren A, and Toretsky JA (2005). Pediatric malignancies provide unique cancer therapy targets. Curr. Opin. Pediatr. 17, 14–19. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Stephens L, and Hawkins P (2012). PI3Ksignalling: the path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 13, 195–203. [DOI] [PubMed] [Google Scholar]
- Vivanco I, and Sawyers CL (2002). The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501. [DOI] [PubMed] [Google Scholar]
- von Lintig FC, Huvar I, Law P, Diccianni MB, Yu AL, and Boss GR (2000). Ras activation in normal white blood cells and childhood acute lympho-blastic leukemia. Clin. Cancer Res. 6, 1804–1810. [PubMed] [Google Scholar]
- Wallin JJ, Guan J, Prior WW, Lee LB, Berry L, Belmont LD, Koeppen H, Belvin M, Friedman LS, and Sampath D (2012). GDC-0941, a novel class I selective PI3K inhibitor, enhances the efficacy of docetaxel in human breast cancer models by increasing cell death in vitro and in vivo. Clin. Cancer Res. 18, 3901–3911. [DOI] [PubMed] [Google Scholar]
- Weigelt B, and Downward J (2012). Genomic Determinants of PI3K Pathway Inhibitor Response in Cancer. Front. Oncol. 2, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weїwer M, Spoonamore J, Wei J, Guichard B, Ross NT, Masson K, Silkworth W, Dandapani S, Palmer M, Scherer CA, et al. (2012). A Potent and Selective Quinoxalinone-Based STK33 Inhibitor Does Not Show Synthetic Lethality in KRAS-Dependent Cells. ACS Med. Chem. Lett. 3, 1034–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White-Gilbertson S, Kurtz DT, and Voelkel-Johnson C (2009). The role of protein synthesis in cell cycling and cancer. Mol. Oncol. 3, 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemels JL, Zhang Y, Chang J, Zheng S, Metayer C, Zhang L, Smith MT, Ma X, Selvin S, Buffler PA, and Wiencke JK (2005). RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia 19, 415–419. [DOI] [PubMed] [Google Scholar]
- Wilson RC, and Doudna JA (2013). Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 42, 217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DG, Faia KL, DiNitto JP, Ali JA, White KF, Brophy EE, Pink MM, Proctor JL, Lussier J, Martin CM, et al. (2013). PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem. Biol. 20, 1364–1374. [DOI] [PubMed] [Google Scholar]
- Xie L, Gazin C, Park SM, Zhu LJ, Debily MA, Kittler ELW, Zapp ML, Lapointe D, Gobeil S, Virbasius C-M, and Green MR (2012).A synthetic interaction screen identifies factors selectively required for proliferation and TERT transcription in p53-deficient human cancer cells. PLoS Genet. 8, e1003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. (2012). The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Meng Q, Liu L-Z, You Y-P, Liu N, and Jiang B-H (2010). Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem. Biophys. Res. Commun. 395, 219–224. [DOI] [PubMed] [Google Scholar]
- Zwang Y, Jonas O, Chen C, Rinne ML, Doench JG, Piccioni F, Tan L, Huang H-T, Wang J, Ham YJ, et al. (2017). Synergistic interactions with PI3K inhibition that induce apoptosis. eLife 6, e24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.