

Abstract
Our early efforts to find a covalent inhibitor of mortalin, a member of the 70kD heat shock protein (Hsp70) family, led us to solve the structure of mortalin nucleotide binding domain (NBD) in complex with N6-propargyladenosine-5’-diphosphate. The acquired structure emphasized the ability of the nucleotide binding pocket to accommodate modified ADP compounds. A library of ADP analogues modified at either the 2- or N6-positions of adenosine were screened against mortalin-NBD. Competitive inhibition and binding assays of the analogues demonstrate that modifications at the 2- or N6-positions have potential to bind and inhibit mortalin uniquely compared to other Hsp70 homologs, and that modifications at the 2-position confer the greatest selectivity in binding and inhibition of mortalin-NBD.
Introduction
Heat shock protein 70s, Hsp70s, are molecular chaperones of approximately 70 kDa molecular weight that are essential for cellular homeostasis, protein maintenance, and stress responses.1 Hsp70 family members are structurally conserved, ubiquitously expressed in multiple cellular compartments, and participate in a wide variety of functions.2 All Hsp70s perform these functions by undergoing a cycle of conformational changes controlled by the reversible binding of adenosine-5’-triphosphate (ATP) and subsequent hydrolysis to alter the affinity for a protein substrate. In eukaryotic cells, the Hsc70 and Hsp70 family members are localized to the cytosol, Grp78 to the endoplasmic reticulum, and mortalin is primarily localized to the mitochondria.3
Mortalin, the mitochondrial Hsp70, was first discovered in mice as a cellular mortality factor.4 In humans one isoform has been identified as a single gene product that exhibits high sequence homology to the prokaryotic Hsp70, DnaK.5 Although classified as a heat shock protein mortalin is not inducible by heat, rather it is upregulated by other forms of cellular stress such as elevated levels of reactive oxygen species (ROS). In the mitochondria, mortalin associates with the TIM (translocase of the inner membrane) protein complex to assist with mitochondrial import of proteins and the folding of proteins in the mitochondria.6 Mortalin is involved in iron-sulfur (Fe-S) cluster biogenesis by stabilizing the disordered form of the iron-sulfur cluster protein scaffold necessary for the assembly of 2Fe-2S and insertion into Fe-S apoproteins.7 Mortalin also plays a cytoprotective role by preventing cell lysis following immune attack by the membrane attack complex by disrupting formation of the complex.8 Mortalin is also able to modulate mitochondrial homeostasis by assisting the tumor necrosis factor receptor associated protein 1 and the voltage-dependent anion channel, and regulates ATP levels, membrane potential and permeability, and response to elevated levels of ROS.9–11
The overall structure of mortalin is conserved among the Hsp70 family members. Similar to other Hsp70s mortalin is composed of two main domains, the ~42 kD nucleotide binding domain (NBD) and a ~25 kD substrate binding domain (SBD). The four NBD subdomains IA, IB, IIA, and IIB fold into a pair of lobes to form the nucleotide-binding pocket located at the central cleft of the NBD. The SBD can be partitioned into a twisted β-sandwich domain (SBDβ) and a ~12 kD α-helical lid domain (SBDα). The interdomain linker connects the NBD and SBD and allows for communication of allosteric changes upon the hydrolysis of ATP by the NBD. Prior crystal structures of mortalin include the SBD (PDB ID 3N8E, unpublished), apo NBD (PDB ID 4KBO), and the ADP bound state of the NBD (PDB ID 6NHK).12,13
Mortalin is overexpressed in cancer cells and plays multiple roles in survival of the cancerous cell.14,15 Drug resistance in some cancer cells has been attributed to elevated levels of mortalin.16 In fact, aberrant localization of mortalin to the cytoplasm has been cited as an anti-apoptotic mechanism in cancerous cells.17 Under this mechanism, mortalin is translocated to the cytoplasm where mortalin binds to the apoptotic signaling protein p53 thereby sequestering and preventing p53 from entering the nucleus and performing its apoptotic role.18,19,20 Once sequestered by mortalin, p53 is degraded by the MDM2-mediated ubiquitin proteasome pathway.21 This mechanism inactivates p53 and allows for the immortalization of the cancer cell. The mortalin targeting compound MKT-077, a cationic rhodacyanine dye analogue, was shown to disrupt the binding of p53 to mortalin-NBD.22,23 This prevents the sequestering of p53 and results in reactivation of p53 function and sensitization of the cancer cell to apoptosis.24,25 While MKT-077 is effective at sensitizing several cancer cell lines to apoptosis, it was aborted from clinical trials due to high renal toxicity.26,27 However, interest in MKT-077 has remained strong and multiple derivatives have been synthesized and studied.28 Cancer cells use mortalin’s normally beneficial functions to continue to resist immune attack and survive the stressors associated with growing in aberrant locations in the body. The upregulation of mortalin is an essential process for the survival of cancer cells and the development mortalin-specific targeting compounds could be beneficial in sensitizing cancer cells to therapeutics with fewer toxicity issues than MKT-077.
The mortalin-NBD is a potential target for nucleotide-competitive inhibitors and allosteric inhibitors. Elucidation of the mortalin-NBD structure has allowed the identification of potential sites that are not conserved in other mammalian Hsp70s for binding functional groups that could potentially increase the specificity of inhibitors for mortalin. The broad-spectrum inhibitors of mortalin, including MKT-077 and withanone, have many off-target binding partners resulting in severe side effects such as renal toxicity.26,29 For example, the ATP competitive compound VER-155008 binds to the nucleotide binding pocket of human Hsp70 holding the Hsp70-NBD in a half-open conformation and acting as an ATP-competitive inhibitor that prevents allosteric control between the NBD and SBD.30 Increasing the specificity of inhibitors for mortalin-NBD may result in fewer off-target interactions and reduce toxicity in the treatment of cancers. In this study, we present the crystal structure of human mortalin-NBD in complex with the N6-propargyladenosine-5’-diphosphate (N6-propargyl ADP) at a resolution of 2.00 Å. This structure, of a modified nucleotide bound to the mortalin-NBD highlights the potential of nucleotide-competitive inhibitors for binding to mortalin-NBD. Subsequently we identified multiple modified ADP analogues that are accommodated by the mortalin nucleotide binding pocket and inhibit ATPase activity of the NBD. Our results further suggest how additional modifications to these nucleotide-competitive inhibitors could further the development of a mortalin-specific inhibitor.
Methods
Cloning
Mortalin-NBD (52–431) was cloned into a pHis//2 vector as previously described.12 The pHis//2 Mortalin-NBD (52–431) expression vector encodes for a His6-tag, followed by the tobacco etch virus (TEV) protease cleavage sequence, and the coding sequence for the wild-type human mortalin-NBD residues 52–431. Hsp70-NBD and Hsc70-NBD were cloned as previously described to produce pET15b-Hsp70-NBD and pET15b-Hsc70-NBD expression vectors encoding for a His6-tag, followed by the tobacco etch virus (TEV) cleavage sequence, and the corresponding sequence coding for wild-type human Hsp70-NBD residues 1–381 or Hsc70-NBD residues 1–385.13
Protein expression
Expression constructs were transformed into Escherichia coli Rosetta2 (DE3) cells (EMD Chemicals) after DNA sequence verification. Multiple colonies of transformed cells were used to inoculate starter cultures of lysogenic broth, containing 100 µg/ml carbenicillin and 34 µg/ml chloramphenicol, then incubated overnight at 37 °C. The following morning, starter cultures were then transferred to terrific broth expression culture (Research Products International) and grown at 37 °C until the OD600 reached 1.0. Protein expression was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 400μM into the culture flasks. Cell cultures were harvested by centrifugation at 4000 rpm at 4 °C for 20 min to pellet the induced cells. The cells were subsequently resuspended in a buffer containing 180 mM Tris (pH 8.0), 450 mM NaCl, 10% (v/v) Glycerol. For storage the cells were transferred to 50 ml centrifuge tubes with the addition of 20 µg/ml DNaseI (MP Biomedicals), 1 mg/ml lysozyme (Sigma), and100 µg/ml 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF; Gold Biotechnology). The resuspended cells were then stored at −80 °C until purification after being flash-frozen in liquid nitrogen.
Protein purification
Frozen cells stocks were thawed overnight at 4 °C on a slow rotator and subsequently sonicated. The lysed cells were then centrifuged at 12,500 rpm for 45 min at 4 °C and the supernatant was filtered using a 0.45 µm (VWR) syringe filter into a new 50 mL centrifuge tube. An ÄKTA pure fast protein liquid chromatography (FPLC) system (GE Healthcare) was used to load the supernatant with the expressed His6-tagged construct onto a 5 mL HisTrap HP column (GE Healthcare). Buffer A (25 mM HEPES/NaOH pH 7.8, 150 mM NaCl) was used as to equilibrate the HisTrap column, which was subsequently loaded with lysate and washed with 15 column volumes (CV) of buffer A to remove non-specifically bound proteins. 5 CVs of 100% buffer B (25 mM HEPES/NaOH, pH 7.8, 150 mMs NaCl, 500 mM imidazole) was used to elute the His6-tagged sample. A 1 mg aliquot of His6-tagged tobacco etch virus (TEV) protease was added to the pooled fractions containing the His6-tagged construct and dialyzed overnight at 4 °C to remove the His6-tags and to remove excess imidazole. The cleaved sample was then reloaded onto a 5 mL HisTrap HP column, the flow-through containing the cleaved sample was collected and then washed with 5 CV of buffer A. The HisTrap column was washed with 5 CV of buffer B to remove TEV and any uncleaved His6-tagged construct. The sample was further purified using a HiLoad 16/60 Superdex 75 (S75) size exclusion column (GE Healthcare) equilibrated with buffer A. Samples were concentrated by centrifugation prior to application to the size exclusion column and elution fractions were tested for purity by SDS-PAGE electrophoresis. Fractions containing the desired construct were concentrated and applied to 5 mL HiTrap Desalting columns (GE Healthcare) washed with 5 CV of 25 mM HEPES/NaOH pH 7.8. The sample was then applied to HiTrap Blue HP column (GE Healthcare) and washed with 5 CV of 25 mM HEPES/NaOH pH 7.8 to remove any unbound sample. The protein was eluted with 10 CV gradient of 2 M NaCl, 25 mM HEPES/NaOH pH 7.8. The peak fractions containing the desired eluate were pooled and dialyzed into 25 mM HEPES/NaOH, pH 7.8, 50 mM NaCl for analysis.
Crystallization, structure determination and refinement
Human mortalin-NBD was concentrated to 20 mg/mL and co-crystalized with N6-propargyl ATP using sitting drop vapor diffusion method screened with the sparse matrix crystallization screens. Human mortalin-NBD was incubated with N6-propargyladenosine-5’-triphosphate (N6-propargyl ATP) at 20 mM and placed in a clear glass tube under UV light (350 nm) in a UV reactor for 60 minutes on ice. The N6-propargyl ATP/mortalin-NBD sample was then aliquoted into matrix crystallization screens MCSG 1–4 (Microlytic), JBScreen PACT++ (Qiagen), and JCSG++ (Qiagen). Using the mixture of N6-propargyl ATP and mortalin-NBD crystals were grown in 0.1M sodium acetate, pH 4.5, 0.2M lithium sulfate, and 30% (w/v) polyethylene glycol 8000. Harvested crystals were cryoprotected by brief transfer through LV CryoOil (MiTeGen) and immediately frozen in liquid nitrogen prior to X-ray diffraction. X-ray diffraction data was collected at 1.00 Å on beamline 4.2.2 at the Advanced Light Source in Lawrence Berkeley National Laboratory and processed using XDS.31 Phases were calculated by molecular replacement using PHASER with the structure of human mortalin-NBD in the apo state (PDB ID 4KBO) as the search model in PHASER.32 Structure refinement and model building were carried out using PHENIX and Coot.33,34 Restraints for refining the N6-propargyl ADP ligand were generated using eLBOW35 Briefly, the two-dimensional structure of N6-propargyl ADP was prepared in ChemDraw Prime 16.0 (PerkinElmer) and converted to SMILES format. N6-propargyl ADP in SMILES format was used as an input into eLBOW and a simple force-field optimization was carried out in which the three-dimensional structure of N6-propargyl ADP, including hydrogens, was produced. The three-dimensional structure from the simple optimization was then used as input into eLBOW for an AM1 geometry optimization. The output three-dimensional coordinate as a .pdb file and restraints as a .cif file were then examined using the restraints editor especially ligands (REEL)36 application within PHENIX prior to use in subsequent structure refinements and model building for the mortalin-NBD/N6-propargyl ADP complex in PHENIX and Coot. Hydrogen atoms for N6-propargyl ADP were not used for refinement of the mortalin-NBD/N6-propargyl ADP complex. Stereochemical and geometric analyses of the mortalin-NBD structure were conducted with MolProbity.37 All molecular structure figures were prepared with PyMOL.38 The structure of the ADP-bound mortalin-NBD, structure factors, and stereochemical restraints for the N6-propargyl ADP ligand were deposited in the PDB with accession code 6P2U.
ITC ADP Binding
The interaction of NBD of human mortalin with modified ADP analogues was measured by isothermal titration calorimetry (ITC) using a Nano ITC isothermal titration calorimeter (TA Instruments) at 25 °C. A library of commercially available ADP analogues with varied substituents at the N6- or 2-position were purchased (Biolog). Concentrations were determined using a 660 nm Protein Assay (Pierce). Nucleotide free mortalin-NBD and modified ADP analogues, at 50 μM and 500 μM respectively, were prepared in binding buffer containing 20 mM HEPES/NaOH (pH 7.8), 50 mM NaCl, 5 mM KCl, 5 mM MgCl2, and 5 mM Na2HPO4. The enthalpy change for each injection was calculated by integrating the area under the peaks of the recorded time course of the power change. The data were analyzed with NanoAnalyze v3.8.0 software using the nonlinear least-squares fitting to a single-site binding model to calculate the dissociation constants (Kd), binding stoichiometry (n), the binding enthalpy change (ΔH), and binding entropy change (ΔS).
Competitive Inhibition Kinetics
ATPase activity of mortalin-NBD ATPase activity was measured spectrophotometrically using the EnzChek Phosphate Assay kit (Invitrogen) in an effort to determine inhibition of mortalin by modified ADP analogues. This method quantifies inorganic phosphate (Pi) released from ATP hydrolysis by the enzyme. ATP hydrolysis activity was collected from triplicate 50 μL reaction mixtures in 384 well optical bottom plates (Thermo Scientific Nunc™) using a BioTek Synergy II plate reader reading absorbance at 360 nm. Raw data were converted to micromolar phosphate concentrations using a standard curve. The negative control for the assay was all components assembled in the absence of mortalin. Samples containing the Pi freed by ATP hydrolysis were incubated with 0.2 U of purine nucleoside phosphorylase (PNP) and 0.2 μM MESG, a chromogenic substrate, for 10 min at 25 °C. Mortalin-NBD, prepared in binding buffer, was mixed with the samples and ADP or modified ADP analogue inhibitors with concentrations ranging from (0–3 mM). ATP (Sigma) was added to the samples to a final concentration of 50 μM and the absorbance was immediately measured at 360 nm once every minute for 60 min at 25 °C. For data analysis, the values were determined from the experimental reaction values minus the corresponding zero-enzyme control values in Excel. The fractional activity was determined by dividing varying concentrations of the corresponding inhibitor by the uninhibited ATPase activity defined as the amount of Pi released per minute (V0, μM/min). The fractional activity was plotted against the log10 of inhibitor concentration and a one-site logIC50 curve fit for competitive binding (Prism 6, GraphPad) was used to obtain the 50% inhibitory concentration (IC50). The apparent inhibition constant, Kiapp, was calculated in Prism 6. The amount of Pi released per minute (V0, μM/min) was plotted against the concentration of the inhibitor μM and using Prism 6, a Morrison equation curve fit was used to determine the apparent inhibition constant, Kiapp. Et the concentration of enzyme catalytic sites (1:1 in mortalin-NBD) was 1 μM, the concentration of substrate (S) ATP was 50 μM, and the Michaelis-Menten constant (KM) determined in previous experiments without competitor were used as constraints in the Morrison equation curve fitting to determine the apparent inhibition constant, KIapp.
Results and Discussion
Selection of a suitable ADP analogue
To take advantage of the non-conserved residue C317 in the NBD of human mortalin, N6-propargyl ATP was tested as a possible suicide inhibitor. We co-crystallized and solved the structure of the human mortalin-NBD with N6-propargyl ADP in the NBP to a 2.00 Å resolution by molecular replacement, using the NBD of apo human mortalin (PDB ID 4KBO) as a search model. The data collection and refinement statistics are shown in Table 1. The overall structure is similar to the closed conformation NBD exhibited in another crystal structure of human mortalin with ADP bound (PDB ID 6NHK).13 Prior to crystallization, the mixture of N6-propargyl ATP and mortalin-NBD were exposed to UV light with the goal of initiating a thiol-alkyne click reaction between the thiol of C317 and the N6-propargyl alkyne of the nucleotide analogue. This covalent modification would then occlude the NBP and inhibit mortalin ATPase activity. This inhibition of the mortalin chaperone cycle would be beneficial for hindering the growth of cancer cells that are dependent on the overexpression of mortalin for survival. Unfortunately, the crystal structure revealed that while N6-propargyl ATP bound to mortalin-NBD and was hydrolyzed to form N6-propargyl ADP, the N6-propargyl alkyne is held at a geometry that does not allow for the formation of a bond to C317 (Figure 1A). The propargyl group is facing in the opposing direction, while C317 is rotated to form a sulfur-π interaction with the adenosine moiety of N6-propargyl ADP. While the desired interaction did not form between the NBD of human mortalin and N6-propargyl ADP, the crystal structure did imply that the mortalin-NBD can accommodate modified nucleotides. Our analysis of the structure suggests room for a potential modification at the N6-position of the adenosine moiety for interactions expanding beyond the NBP at this position (Figure 1B). Additionally, we found that the 2-position on the adenosine moiety lies adjacent to a small pocket, which we hereafter refer to as the 2C pocket, with the potential for accommodating small polar modifications at the 2-position of the adenosine moiety (Figure 1C). As mortalin has been previously determined to be essential for the growth of fast-growing cancers, the development of new inhibitors is important. ADP-based inhibitors modified at the N6- and 2-positions could yield information beneficial for the development of futured inhibitors of mortalin-NBD. Furthermore, development of a mortalin-specific inhibitor that does not target other Hsp70 family members could be an avenue for cancer drug development with decreased toxicity.
Table 1.
Data collection and refinement statistics for N6-propargyl ADP bound Mortalin-NBD
| Data collection | |
|---|---|
| X-ray source | Beamline ALS 4.2.2 |
| Wavelength (Å) | 1.00 |
| Resolution range (Å) | 47.04 – 2.00 (2.07 – 2.00) |
| Space group | P 212121 |
| a, b, c (Å) | 51.04, 68.25, 121.22 |
| α, β, γ (°) | 90, 90, 90 |
| Reflections | 200,934 (19,593) |
| Unique reflections | 29,169 (2,847) |
| Multiplicity | 6.9 (6.9) |
| Completeness (%) | 99 (98) |
| Mean I/sigma (I) | 10.62 (1.95) |
| Wilson B-factor (Å2) | 21.71 |
| CC½ | 0.994 (0.59) |
| Refinement | |
| PDB ID | 6P2U |
| Number of reflections | 29,165 (2,845) |
| Reflections used for R-free | 1,996 (195) |
| Rwork / Rfree | 0.216 / 0.238 |
| Number of atoms Protein/water/ligands) | 2,806 / 271 / 36 |
| Average B factors (protein/water/ligands) | 24.60 / 29.33 / 27.96 |
| Model Quality | |
| Bond length rmsd (Å) | 0.007 |
| Bond angle rmsd (°) | 0.91 |
| Ramachandran favored (%) | 99 |
| Ramachandran allowed (%) | 1.0 |
| Ramachandran outliers (%) | 0 |
| Poor rotamers (%)b | 0 |
| Clashscore b | 4.28 |
| Clash percentileb | 99th percentile (N=715; 2.00 Å ± 0.25 Å) |
| MolProbity scoreb | 1.21 |
| MolProbity score percentileb | 100th percentile (N=12,522; 2.00 Å ± 0.25 Å) |
Data for the highest resolution shell is represented by the values in parentheses.
Values calculated using MolProbity v4.4.37
Figure 1.

(A) Close-up view of the mortalin nucleotide binding pocket with N6-propargyl ADP bound. Throughout the figure mortalin is shown with each subdomain backbone individually colored (subdomain IA pink; subdomain IB magenta; subdomain IIA white; subdomain IIB cyan), N6-propargyl ADP-Pi (yellow) is shown as sticks, the Mg2+ ion (green) is represented as a sphere and electron density (grey) is displayed as a transparent surface overlay. (B) Close-up of the surface density (orange) view showing possible interaction pockets for N6 modified ADP analogues. (C) Close-up of a transparent surface overlay (pink) showing a pocket unique to mortalin that indicates a possible site for C2 ADP modifications.
We hypothesize that modified ADP analogues that further increase affinity and specificity for mortalin will likely correlate to an improvement in the aptness to inhibit the NBD. Compared to the Hsp70 NBP, mortalin NBD features the 2C pocket and targeting this pocket could increase affinity for mortalin-NBD. The 2C pocket may also afford increased specificity as the structure of Hsp70-NBD (PDB ID 3ATU) indicates that this pocket is occluded by a phenylalanine in Hsp70. The corresponding residue in mortalin is an isoleucine, and the decreased bulk provides the potential for targeting the 2C pocket. Similarly, in comparison to Hsp70, the region of mortalin-NBD near the N6 of the adenosine moiety harbors aliphatic residues, not present in Hsp70, which could be potential targets for increasing inhibitor specificity. For example, relatively bulky hydrophobic groups could potentially serve as N6-substituents capable of binding mortalin-NBD with increased specificity over Hsp70.
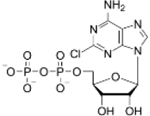
In order to determine if a particular ADP analogue would be a suitable candidate for inhibition of mortalin we tested whether the mortalin-NBD would have a tighter affinity for an ADP analogue versus ADP, with a previously reported as Kd of 5.30 ± 0.106 μM.12,13 ITC was utilized to screen a library of commercially available ADP analogues to determine suitability of modifications at either the N6- or 2-position. Out of the twelve modified ADP candidates, eight were found to bind to the mortalin-NBD (Figure S1) and the binding data (Table 2) shows that the mortalin-NBD can accommodate various groups at the N6- and 2-positions. Analogues with higher affinity for mortalin-NBD than ADP dissociation constants (Kd) of 4.19 ± 1.06 μM for 6-PhEt ADP, 3.52 ± 0.224 μM for 6-Bn ADP, and 1.77 ± 0.192 μM for 2-Cl ADP which exhibited the highest affinity of any tested analogue. The affinity of mortalin-NBD for 2-Cl ADP is approximately ~3-fold tighter than ADP. In contrast to 2-Cl ADP, 2-methylthio-adenosine-5’-diphosphate (2-MeS ADP) resulted in a nearly 2-fold decrease in affinity compared to ADP, suggest that in comparison to the bulkier 2-MeS substituent, the 2-Cl group is in an optimal size for matching the small 2C pocket adjacent to the NBP. Contrary to the clear steric preference for a less bulky group at the 2-position, results for modifications at the N6-position strike a different balance. The affinity for N6-benzyladenosine-5’-diphosphate (6-Bn ADP) is ~1.5-fold tighter than ADP and the affinity for N6-(2-phenylethyl)adenosine-5’-diphosphate (6-PhEt ADP) is ~1.25-fold tighter than ADP. Although neither modification is as strong an improvement in affinity as 2-Cl ADP, the 6-PhEt and 6-Bn modifications enable increased affinity not afforded by any of the other tested analogues with modifications at the N6-position.
Table 2.
Dissociation constants, change in enthalpy, and the change in entropy of mortalin-NBD for modified ADP analogues
| Ligand | ADP | 2-Cl ADP | 6-Bn ADP | 6-PheEt ADP | 6-Fu ADP | 2-MeS ADP | 6-(3-MeBn) ADP | 6-cHe ADP | |
|---|---|---|---|---|---|---|---|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
||
| Mortalin-NBD | Kd (M) | 5.30E-06 ± 1.26E-07 | 1.77E-06 ± 1.92E-07 | 3.52E-06 ± 2.24E-07 | 4.19E-06 ± 1.06E-06 | 5.84E-06 ± 2.42E-06 | 9.81E-06 ± 4.84E-06 | 1.14E-05 ± 3.45E-06 | 1.41E-05 ± 1.67E-06 |
| ΔΗ (kJ/mol) | −27.3 ± 1.29 | −19.0 ± 0.721 | −12.5 ± 3.51 | −33.9 ± 2.07 | −21.1 ± 1.16 | −12.4 ± 3.62 | −13.1 ± 0.228 | −22.09 ± 0.29 | |
| ΔS (J/mol·K) | 9.36 ± 4.48 | 45.4 ± 3.04 | 61.8 ± 12.1 | −10.4 ± 5.17 | 28.7 ± 6.85 | 54.5 ± 16.5 | 50.2 ± 1.73 | 18.84 ± 0.87 | |
| Hsc70-NBD | Kd (M) | 6.65E-08 ± 1.02E-08 | ~ | 1.59E-07 ± 3.69E-08 | 2.37E-07 ± 3.85E-08 | 8.70E-06 ± 2.64E-06 | ~ | 3.35E-06 ± 7.66E-08 | 3.57E-07 ± 3.21E-08 |
| ΔΗ (kJ/mol) | −30.79 ± 0.09 | ~ | −17.06 ± 0.27 | −18.06 ± 0.09 | −22.83 ± 2.15 | ~ | −42.78 ± 3.79 | −25.13 ± 2.35 | |
| ΔS (J/mol·K) | 34.19 ± 1.55 | ~ | 72.13 ± 2.77 | 65.31 ± 1.22 | 19.22 ± 5.06 | ~ | −41.10 ± 12.94 | 37.74 ± 7.36 | |
| Hsp70-NBD | Kd (M) | 4.35E-08 ± 4.27E-09 | ~ | 2.16E-06 ± 4.25E-07 | 1.43E-07 ± 3.83E-08 | 4.27E-06 ± 9.38E-07 | ~ | 2.52E-07 ± 3.39E-08 | 1.99E-07 ± 5.56E-08 |
| ΔΗ (kJ/mol) | −23.98 ± 0.85 | ~ | −16.49 ± 4.25 | −45.20 ± 0.87 | −10.02 ± 2.35 | ~ | −20.03 ± 0.94 | −20.78 ± 0.61 | |
| ΔS (J/mol·K) | 60.54 ± 3.65 | ~ | 53.04 ± 14.20 | −22.97 ± 3.25 | 68.77 ± 8.28 | ~ | 58.44 ± 3.96 | 57.67 ± 0.64 | |
~ denotes ambiguous data
Beyond binding to mortalin-NBD, we sought to determine whether binding of ADP analogues would be specific to mortalin, or common among the cytosolic homologs. Results from ITC experiments with Hsc70-NBD and Hsp70-NBD (Table 2), in comparison to mortalin-NBD, indicates that modifications at the 2-position preclude binding of the ADP analogues to Hsc70-NBD and Hsp70-NBD. ITC data for both homologs towards the 2-Cl ADP and 2-MeS ADP analogues (Figures S2 and S3) exhibited variable low intensity injection peaks indicative of heat of dissolution without subsequent binding and did not yield data that could be fitted for obtaining Kd, ΔH, or ΔS. In contrast, the cytosolic homologs did have demonstrate capability to bind the N6-modified ADP analogues, though all were found to have diminished affinity relative to ADP (Table 2). In comparison to analogues modified at the N6-position, previously reported adenosine-derived inhibitors with aromatic modifications at the 8-position take advantage R272 residue of cytosolic homologs to allow for additional π-stacking interactions, but restrict the possible orientations of the adenosine moiety.39 These prior results with analogues modified at the 8-position, and our results here for N6-modified ADP analogues, are consistent with the role of R272 in contributing to nucleotide and inhibitor binding in the cytosolic homologs.40 However, mortalin lacks R272 and instead features a cysteine residue at the corresponding position, which may place fewer or weaker restrictions on binding geometry of the adenosine moiety. The more accommodating NBP of mortalin therefore may allow for more favorable placement of ADP analogues functionalized at the N6- or 2-positions compared to Hsc70 and Hsp70.
Inhibition by ADP analogues modified at the N6- or 2-position
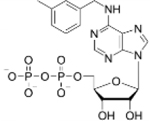
Following analyses of binding of modified ADP analogues to mortalin-NBD, Hsc70-NBD, and Hsp70-NBD we measured the ability of the tested analogues to inhibit ATP hydrolysis. 2-Cl ADP, 6-Bn ADP, and 6-PheEt ADP were determined to inhibit ATP hydrolysis of mortalin-NBD more effectively compared to ADP (Figure 2A). The Kiapp for ADP indicates that ADP must be present at a concentration approximately seven times higher than ATP in order to inhibit 50% of the ATPase activity (Table 3). Although 6-PheEt ADP had a slight increase in affinity determined by ITC, this slight increase translated into a large improvement in capability to inhibit mortalin-NBD compared to unmodified ADP. Similarly, 6-Bn ADP exhibited a larger increase in affinity, as measured by ITC, and correspondingly large improvement was observed for inhibition of mortalin-NBD. The greatest inhibition of mortalin-NBD inhibition was demonstrated by 2-Cl ADP with a KIapp of 45.05 μM. Mortalin-NBD also had the greatest affinity for 2-Cl ADP, consistent with increased affinity of modified nucleotides corresponding to greater improvement in inhibition of mortalin-NBD. The logIC50 values are also in agreement with the apparent inhibition constants (Table 3 and Figure S4). All other tested ADP analogues were able to inhibit ATPase activity of mortalin-NBD, however inhibition was weaker than that for unmodified ADP. For example, 2-MeS ADP demonstrated a larger Kiapp compared to ADP, and in comparison to results for 2-Cl ADP, this data suggests that placement of the 2-methylthio functional group is sterically hindered in the 2C pocket of mortalin and that the smaller 2-chloro group of 2-Cl ADP is better accommodated in the 2C pocket. The increased Kiapp of N6-furfuryladenosine-5’-diphosphate (6-Fu ADP) suggests that the five membered ring of the furfuryl functional group has a less favorable geometry relative to N6-benzyl modification of 6-Bn ADP. Inhibition data for N6-(3-methylbenzyl)adenosine-5’-diphosphate (6–3MeBn ADP) suggests that the addition of methyl group at the 3-position of the N6-benzyl group disrupts the ability of 6–3MeBn ADP to inhibit the mortalin-NBD, possibly due to unfavorable steric hindrance. The weakest inhibition of all tested commercially available ADP analogues, as judged by the compound with the largest Kiapp for mortalin-NBD, was N6-cyclohexyladenosine-5’-diphosphate (6-cHe ADP), an analogue which lacks aromaticity for the N6-substituent and has no alkyl linker to between the substituent and the N6 of the adenosine moiety, in contrast to 6-Bn ADP, 6-PheEt ADP, 6-Fu ADP, and 6–3MeBn ADP. This suggests either that additional π-stacking interactions may occur between mortalin-NBD and ADP analogues modified with aromatic substituents at the N6-position, or that mortalin-NBD is better able to accommodate planar aromatic substituents at the N6-position compared to the nonplanar cyclohexyl group of 6-cHe ADP.
Figure 2.

Competitive inhibition of phosphate release assays used to derive the apparent inhibition constants of (A) mortalin-NBD, (B) Hsc70-NBD, and (C) Hsp70-NBD ATP hydrolysis by modified ADP analogues. Figures are shown with nonlinear regression fits and error bars indicating the standard deviation of data collected in triplicate. Error bars for some data points are smaller than the symbol drawn.
Table 3.
Apparent inhibition constants (KIapp) and log IC50 values for Mortalin-NBD, Hsc70-NBD, Hsp70-NBD by various modified ADP analogues
| Ligand | ADP | 2-Cl ADP | 6-Bn ADP | 6-PheEt ADP | 6-Fu ADP | 2-MeS ADP | 6-(3-MeBn) ADP | 6-cHe ADP | |
|---|---|---|---|---|---|---|---|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
||
| Mortalin-NBD | KIapp (µM) | 352.90 ± 36.21 | 45.05 ± 2.42 | 86.51 ± 10.26 | 97.29 ± 9.96 | 564.60 ± 49.63 | 523.20 ± 98.35 | 675.30 ± 99.35 | 733.10 ± 82.07 |
| log IC50 | 2.613 ± 0.065 | 1.752 ± 0.037 | 2.074 ± 0.084 | 2.238 ± 0.041 | 2.747 ± 0.071 | 2.955 ± 0.169 | 3.154 ± 0.124 | 3.203 ± 0.082 | |
| Hsc70-NBD | KIapp (µM) | 239.10 ± 32.21 | ~7642 ± ~2173 | 294.50 ± 67.24 | 444.50 ± 101.4 | 723.3 ± 178.5 | ~13558 ± ~6528 | 681.8 ± 179.2 | 622.2 ± 182.8 |
| log IC50 | 2.734 ± 0.117 | ~ | 3.168 ± 0.179 | 3.323 ± 0.237 | 3.831 ± 0.446 | ~ | 3.905 ± 0.360 | 3.349 ± 0.368 | |
| Hsp70-NBD | KIapp (µM) | 440.3 ± 47.8 | ~9907 ± ~3125 | 1612 ± 136.7 | 1161 ± 189.2 | 1857 ± 171.7 | ~41786 ± ~11896 | 1302 ± 148.5 | 1239 ± 164.2 |
| log IC50 | 2.697 ± 0.091 | ~ | 3.368 ± 0.105 | 3.494 ± 0.132 | 3.511 ± 0.113 | ~ | 3.273 ± 0.129 | 3.396 ± 0.129 | |
~ denotes ambiguous data
In comparison to the performance of 2-Cl ADP, 6-Bn ADP, and 6-PhEt ADP as inhibitors of mortalin-NBD, all of the tested ADP analogues exhibited either poor, or no inhibitory activity against the cytosolic homologs Hsc70-NBD and Hsp70-NBD and all the tested analogues were less effective than ADP (Table 3). The role of R272 in π-stacking interactions and restricting the conformation of the adenosine moiety may prevent N6-modified ADP analogues from adopting favorable conformations with either of the cytosolic homologs. Most notably, 2-Cl ADP and 2-MeS ADP demonstrate no reliably demonstrable inhibition of Hsc70-NBD (Figure 2B) or Hsp70-NBD (Figure 2C). It is likely that these functional groups are sterically unfavorable modifications at the 2-position of adenosine due to the absence of the 2C pocket which is occluded by I343 and F310 in Hsc70 and Hsp70. In addition to steric clashes preventing placement of the 2-Cl or 2-MeS in similar binding modes as used for mortalin, modifications at the 2-position would thereby alter the adenosine binding mode and potentially disrupt the π-stacking interaction with R272 making the 2-Cl and 2-MeS ADP analogues less able to compete with ATP and inhibit enzymatic activity. Therefore, functional groups at the 2-position of the ADP analogues exhibit selectivity for inhibiting mortalin ATPase activity versus the cytosolic homologs due to the unique 2C pocket within the mortalin-NBD. It is possible that N6-modified ADP analogues could exhibit further improvements in mortalin binding selectivity and inhibition with the incorporation of a 2-Cl modification.
In closing, this study demonstrates that modified ADP analogues are capable of acting as competitive inhibitors of ATPase activity. Analogues functionalized at the 2-position of the adenosine moiety demonstrated selectivity for binding and inhibiting the mortalin-NBD compared to Hsc70-NBD and Hsp70-NBD. Although the inhibitory activities of 2- and N6-modified ADP analogs studied here are unlikely to be high enough to enable use as therapeutics, these results set the stage for further development of mortalin inhibitors based on our observations that particular modifications at the 2- and N6-positions enable increased binding selectivity and improved inhibition of mortalin. Selective inhibition of the activity of Hsp70 chaperones, such as mortalin, offers an avenue to influence protein homeostasis and cell survival in an effort to develop anti-cancer therapeutics.
Supplementary Material
Figure S1. ITC measurements and model fits to derive the binding affinities of the mortalin-NBD towards the modified ADP analogues.
Figure S2. ITC measurements and model fits to derive the binding affinities of the Hsc70-NBD towards the modified ADP analogues.
Figure S3. ITC measurements and model fits to derive the binding affinities of the Hsp70-NBD towards the modified ADP analogues.
Figure S4. Competitive inhibition curves used to determine log IC50 values of ATP hydrolysis for mortalin-NBD, Hsc70-NBD, and Hsp70-NBD by modified ADP analogues.
Figure S5. Data of Mortalin-NBD ATP hydrolysis inhibition by the indicated modified ADP analogue that were measured by an EnzChek phosphate assay kit (Invitrogen).
Figure S6. Data of Hsc70-NBD ATP hydrolysis inhibition by the indicated modified ADP analogue that were measured by an EnzChek phosphate assay kit (Invitrogen).
Figure S7. Data of Hsp70-NBD ATP hydrolysis inhibition by the indicated modified ADP analogue that were measured by an EnzChek phosphate assay kit (Invitrogen).
Acknowledgments
The authors acknowledge support from National Science Foundation grant MCB 15–52113 (RCP) and National Institutes of Health grant R35 GM128595 (RCP). RCP also acknowledges institutional support from Miami University through the Robert H. and Nancy J. Blayney Professorship.
Footnotes
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
The authors declare no competing financial interest.
References
- (1).Hartl FU; Bracher A; Hayer-Hartl M Molecular Chaperones in Protein Folding and Proteostasis. Nat 2011, 475, 324–332. [DOI] [PubMed] [Google Scholar]
- (2).Mayer MP; Bukau B Hsp70 Chaperones: Cellular Functions and Molecular Mechanism. Cell. Mol. Life Sci 2005, 62, 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wadhwa R; Taira K; Kaul SC An Hsp70 Family Chaperone, Mortalin/Mthsp70/PBP74/Grp75: What, When, and Where? Cell Stress and Chaperones 2009, 7, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kumazaki T Expression of Endothelin, Fibronectin, and Mortalin as Aging and Mortality Markers. Exp. Gerontol 1997, 32, 95–103. [DOI] [PubMed] [Google Scholar]
- (5).Flachbartova Z; Kovacech B Mortalin - a Multipotent Chaperone Regulating Cellular Processes Ranging from Viral Infection to Neurodegeneration. Acta Virol 2013, 57, 3–15. [DOI] [PubMed] [Google Scholar]
- (6).D’Silva P; Liu Q; Walter W; Craig EA Regulated Interactions of mtHsp70 with Tim44 at the Translocon in the Mitochondrial Inner Membrane. Nat. Struct. Biol 2004, 11, 1084–1091. [DOI] [PubMed] [Google Scholar]
- (7).Luo W-I; Cowan JA The Role of Mortalin in Iron Homeostasis. In Mortalin Biology: Life, Stress and Death; Springer Netherlands: Dordrecht, 2012; Vol. 2, pp 31–54. [Google Scholar]
- (8).Pilzer D; Fishelson Z Mortalin/GRP75 Promotes Release of Membrane Vesicles from Immune Attacked Cells and Protection from Complement-Mediated Lysis. Int Immunol 2005, 17, 1239–1248. [DOI] [PubMed] [Google Scholar]
- (9).Gesualdi NM; Chirico G; Pirozzi G; Costantino E; Landriscina M; Esposito F Tumor Necrosis Factor-Associated Protein 1 (TRAP-1) Protects Cells from Oxidative Stress and Apoptosis. Stress 2009, 10 (4), 342–350. [DOI] [PubMed] [Google Scholar]
- (10).Szabadkai G; Bianchi K; Várnai P; De Stefani D; Wieckowski MR; Cavagna D; Nagy AI; Balla T; Rizzuto R Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol 2006, 175, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Voos W Chaperone–Protease Networks in Mitochondrial Protein Homeostasis. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2013, 1833 (2), 388–399. [DOI] [PubMed] [Google Scholar]
- (12).Amick J; Schlanger SE; Wachnowsky C; Moseng MA; Emerson CC; Dare M; Luo WI; Ithychanda SS; Nix JC; Cowan JA; et al. Crystal Structure of the Nucleotide-Binding Domain of Mortalin, the Mitochondrial Hsp70 Chaperone. Protein Sci 2014, 23, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Moseng MA; Nix JC; Page RC Biophysical Consequences of EVEN-PLUS Syndrome Mutations for the Function of Mortalin. J. Phys. Chem. B 2019, 123, 3383–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wadhwa R; Takano S; Kaur K; Deocaris CC; Pereira-Smith OM; Reddel RR; Kaul SC Upregulation of Mortalin/Mthsp70/Grp75 Contributes to Human Carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [DOI] [PubMed] [Google Scholar]
- (15).Nomikos A; Dundas SR; Murray GI Mortalin Expression in Normal and Neoplastic Tissues. In Mortalin Biology: Life, Stress and Death; Springer Netherlands: Dordrecht, 2012; Vol. 2, pp 257–265. [Google Scholar]
- (16).Yun C-O; Bhargava P; Na Y; Lee J-S; Ryu J; Kaul SC; Wadhwa R Relevance of Mortalin to Cancer Cell Stemness and Cancer Therapy. Nature Publishing Group 2017, 7, 42016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kaul SC; Wadhwa R; Komatsu Y; Sugimoto Y; Mitsui Y On the Cytosolic and Perinuclear Mortalin: An Insight by Heat Shock. Biochem. Biophys. Res. Commun 1993, 193, 348–355. [DOI] [PubMed] [Google Scholar]
- (18).Wadhwa R; Takano S; Robert M Inactivation of Tumor Suppressor P53 by Mot-2, a Hsp70 Family Member. J. Biol. Chem 1998, 273, 29586–29591. [DOI] [PubMed] [Google Scholar]
- (19).Walker C; Böttger S; Low Ben. Mortalin-Based Cytoplasmic Sequestration of P53 in a Nonmammalian Cancer Model. Am. J. Pathol 2010, 168, 1526–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wadhwa R; Takano S; Mitsui Y; Kaul SC NIH 3T3 Cells Malignantly Transformed by Mot-2 Show Inactivation and Cytoplasmic Sequestration of the P53 Protein. Cell Res 1999, 9, 261–269. [DOI] [PubMed] [Google Scholar]
- (21).Kaul SC; Aida S; Yaguchi T; Kaur K; Wadhwa R Activation of Wild Type P53 Function by Its Mortalin-Binding, Cytoplasmically Localizing Carboxyl Terminus Peptides. J. Biol. Chem 2005, 280, 39373–39379. [DOI] [PubMed] [Google Scholar]
- (22).Wadhwa R; Sugihara T; Yoshida A; Nomura H; Reddel RR; Simpson R; Maruta H; Kaul SC Selective Toxicity of MKT-077 to Cancer Cells Is Mediated by Its Binding to the Hsp70 Family Protein Mot-2 and Reactivation of P53 Function. Cancer Res 2000, 60, 6818–6821. [PubMed] [Google Scholar]
- (23).Pilzer D; Saar M; Koya K; Fishelson Z Mortalin Inhibitors Sensitize K562 Leukemia Cells to Complement-Dependent Cytotoxicity. Int. J. Cancer 2009, 126, 1428–1435. [DOI] [PubMed] [Google Scholar]
- (24).Deocaris CC; Widodo N; Shrestha BG; Kaur K; Ohtaka M; Yamasaki K; Kaul SC; Wadhwa R Mortalin Sensitizes Human Cancer Cells to MKT-077-Induced Senescence. Cancer Lett 2007, 252, 259–269. [DOI] [PubMed] [Google Scholar]
- (25).Pilzer D; Saar M; Koya K; Fishelson Z Mortalin Inhibitors Sensitize K562 Leukemia Cells to Complement-Dependent Cytotoxicity. Int. J. Cancer 2009, 126, 1428–1435. [DOI] [PubMed] [Google Scholar]
- (26).Britten CD; Rowinsky EK; Baker SD; Weiss GR; Smith L; Stephenson J; Rothenberg M; Smetzer L; Cramer J; Collins W; et al. A Phase I and Pharmacokinetic Study of the Mitochondrial-Specific Rhodacyanine Dye Analog MKT 077. Clin. Cancer Res 2000, 6, 42–49. [PubMed] [Google Scholar]
- (27).Propper DJ; Braybrooke JP; Taylor DJ; Lodi R; Styles P; Cramer JA; Collins WCJ; Levitt NC; Talbot DC; Ganesan TS; et al. Phase I Trial of the Selective Mitochondrial Toxin MKT 077 in Chemo-Resistant Solid Tumours. Ann. Oncol 1999, 10, 923–927. [DOI] [PubMed] [Google Scholar]
- (28).Rousaki A; Miyata Y; Jinwal UK; Dickey CA; Gestwicki JE; Zuiderweg ERP Allosteric Drugs: The Interaction of Antitumor Compound MKT-077 with Human Hsp70 Chaperones. J. Mol. Biol 2011, 411, 614–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Grover A; Priyandoko D; Gao R; Shandilya A; Widodo N; Bisaria VS; Kaul SC; Wadhwa R; Sundar D Withanone Binds to Mortalin and Abrogates Mortalin–P53 Complex: Computational and Experimental Evidence. Int. J. Biochem. Cell Biol 2012, 44, 496–504. [DOI] [PubMed] [Google Scholar]
- (30).Wen W; Liu W; Shao Y; Chen L VER-155008, a Small Molecule Inhibitor of HSP70 with Potent Anti-Cancer Activity on Lung Cancer Cell Lines. Exp. Biol. Med 2014, 239, 638–645. [DOI] [PubMed] [Google Scholar]
- (31).Kabsch W Integration, Scaling, Space-Group Assignment and Post-Refinement. Acta Cryst 2010, D66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; et al. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Cryst 2010, D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Cryst 2010, D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Moriarty NW; Grosse-Kunstleve RW; Adams PD Electronic Ligand Builder and Optimization Workbench (eLBOW): a Tool for Ligand Coordinate and Restraint Generation. Acta Cryst 2009, D65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Moriarty NW; Draizen EJ; Adams PD An Editor for the Generation and Customization of Geometry Restraints. Acta Cryst 2017, D73, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Cryst 2009, D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).The PyMOL Molecular Graphics System, version 1.6; Schrodinger; LLC: Portland, OR, 2010. [Google Scholar]
- (39).Massey AJ; Williamson DS; Browne H; Murray JB; Dokurno P; Shaw T; Macias AT; Daniels Z; Geoffroy S; Dopson M; et al. A Novel, Small Molecule Inhibitor of Hsc70/Hsp70 Potentiates Hsp90 Inhibitor Induced Apoptosis in HCT116 Colon Carcinoma Cells. Cancer Chemother. Pharmacol 2009, 66, 535–545. [DOI] [PubMed] [Google Scholar]
- (40).Schlecht R; Scholz SR; Dahmen H; Wegener A; Sirrenberg C; Musil D; Bomke J; Eggenweiler H-M; Mayer MP; Bukau B Functional Analysis of Hsp70 Inhibitors. PLoS ONE 2013, 8, e78443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. ITC measurements and model fits to derive the binding affinities of the mortalin-NBD towards the modified ADP analogues.
Figure S2. ITC measurements and model fits to derive the binding affinities of the Hsc70-NBD towards the modified ADP analogues.
Figure S3. ITC measurements and model fits to derive the binding affinities of the Hsp70-NBD towards the modified ADP analogues.
Figure S4. Competitive inhibition curves used to determine log IC50 values of ATP hydrolysis for mortalin-NBD, Hsc70-NBD, and Hsp70-NBD by modified ADP analogues.
Figure S5. Data of Mortalin-NBD ATP hydrolysis inhibition by the indicated modified ADP analogue that were measured by an EnzChek phosphate assay kit (Invitrogen).
Figure S6. Data of Hsc70-NBD ATP hydrolysis inhibition by the indicated modified ADP analogue that were measured by an EnzChek phosphate assay kit (Invitrogen).
Figure S7. Data of Hsp70-NBD ATP hydrolysis inhibition by the indicated modified ADP analogue that were measured by an EnzChek phosphate assay kit (Invitrogen).