

Abstract
A series of 1,2,3-triazole analogs of the amino acids L-histidine and L-tryptophan were modeled, synthesized and tested for L-type amino acid transporter 1 (LAT1; SLC7A5) activity to guide the design of amino acid-drug conjugates (prodrugs). These triazoles were conveniently prepared by the highly convergent Huisgen 1,3-dipolar cycloaddition (Click Chemistry). Despite comparable predicted binding modes, triazoles generally demonstrated reduced cell uptake and LAT1 binding potency relative to their natural amino acid counterparts. The structure-activity relationship (SAR) data for these triazoles has important ramifications for treating cancer and brain disorders using amino acid prodrugs or LAT1 inhibitors.
Keywords: membrane transporter, solute carrier family, triazole, click chemistry, amino acid, cancer, blood-brain barrier
Graphical Abstract
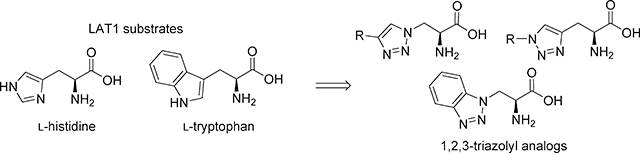
Conjugation of drugs with amino acids (“amino acid prodrugs”) has become an increasingly common strategy for targeted drug delivery across the blood-brain barrier (BBB) and into cancer cells by way of the L-type amino acid transporter 1 (LAT1; SLC7A5). LAT1 is a sodium-independent membrane solute carrier protein1–3 found in several tissues throughout the body, with high levels of expression in brain,4, 5 spleen, testis, placenta, thymus, skeletal muscle and various cancer cells.6–10 LAT1 transports lipophilic amino acids such as phenylalanine, leucine, isoleucine, methionine, tryptophan as well as thyroid hormones including thyroxine. Additionally, LAT1 transports the hydrophilic amino acid histidine. We and others have shown that LAT1 can also transport aryl-substituted phenylalanine analogs containing various functional groups,2, 11–13 as well as amino acid bioisosteres such as hydroxamic acids and esters.14 Given these findings that LAT1 is relatively tolerant of structural changes to the amino acid promoiety, we sought an amino acid scaffold that would be amenable to the rapid assembly of prodrugs while maintaining substrate activity.
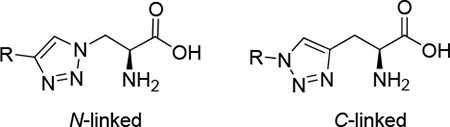
The Huisgen 1,3-dipolar cycloaddition, also known as “Click Chemistry”,15–20 provides a synthetic strategy for connecting an amino acid moiety to a drug molecule via a 1,2,3-triazole ring. Specifically, we envisioned using Click Chemistry to attach an alkyne- or azide-substituted amino acid to a drug molecule possessing a biodegradable functional group that is linked to an azide or alkyne moiety (Figure 1). These 1,2,3-triazoles (hereafter referred to as “triazoles”) are structurally similar to the amino acids L-histidine and L-tryptophan. Thus, we hypothesized that triazoles would be LAT1 substrates based on the similarity of their predicted binding modes to these endogenous ligands (supplementary data). Moreover, L-histidine is one of the most potent LAT1 substrates,12, 21 so we thought triazole analogs would also have good potency. Click Chemistry offers the additional advantage of obtaining a single triazole regioisomer. Prior to using this strategy for designing amino acid prodrugs, we wanted to test its feasibility by determining the structure-activity relationship (SAR) for a series of triazole structural analogs that we have designated as N-linked or C-linked based on which triazole atom is connected to the amino acid beta carbon (Figure 1). We varied both the size and polarity of the triazole substituent R to compare with the SAR of structurally-related histidine analogs and tryptophan. Compounds were evaluated in trans-stimulation and cis-inhibition cell assays (as described below).
Figure 1.

Click Chemistry design strategy for N- and C-linked β−1,2,3-triazolyl amino acid prodrugs.
Scheme 1 shows the synthesis of azide intermediate 4 used to prepare N-linked triazoles. Starting with commercially available Boc-protected serine 1, we prepared tert-butyl ester 2 using tert-butyl (Z)-N,N’-diisopropylcarbamimidate.22 We were unable to prepare a tosylate of alcohol 2 by reaction with tosyl chloride presumably due to steric crowding of the Boc and tert-butyl ester groups. Instead we prepared benzyl sulfonate 3 as previously described.23 Subsequent nucleophilic substitution with sodium azide using tetrabutylammonium bromide as a phasetransfer agent provided the desired azide 423 in moderate yield (60%).
Scheme 1.

Synthesis of azide intermediate 4.
Using methodology similar to what was previously published for preparing triazoles 7ac,24 cycloaddition of azide 4 with various substituted alkynes (5a-e) was catalyzed by copper(I) ion generated in situ from sodium ascorbate reduction of copper(II) acetate to give protected triazole analogs 6a-e in 35–100% yields (Scheme 2). Both the tert-butyl ester and Boc protecting groups were conveniently removed in one step using trifluoroacetic acid (TFA) in the presence of carbocation scavenger triethylsilane (Et3SiH). Additionally, Et3SiH has been shown to increase the rate of Boc deprotection.25 These deprotection conditions were also used in the synthesis of C-linked triazole 11d (Scheme 3) and benzotriazole 13 (Scheme 4).
Scheme 2.

Synthesis of N-linked triazoles 7a-e.
Scheme 3.

Synthesis of C-linked triazoles 11d-e.
Scheme 4.

Synthesis of benzotriazole 13.
Scheme 3 illustrates the synthesis of the two C-linked triazoles 11d and 11e. Benzyl triazole 11e was prepared from benzyl azide 8e and unprotected, commercially available (S)-2-aminopent-4-ynoic acid using Click Chemistry conditions “A” as had been applied in the synthesis of N-linked triazoles of Scheme 2. As 11e was chronologically the first triazole analog we synthesized in this project, we had hoped to avoid using protecting groups. However, we found that separation of the amino acid 11e from the other reactants (i.e. diacetoxycopper and sodium ascorbate) proved challenging; thus, we modified our synthesis to use protected amino acids in the Click Chemistry reactions for all other analogs of Schemes 2–4. Hence, alkyne 926 was prepared by Boc and tert-butyl ester protection of (S)-2-aminopent-4-ynoic acid. For unknown reasons, we found that methyl azide 8d (prepared in situ from sodium azide and iodomethane) did not react with alkyne 9 using the same conditions as for N-linked triazoles of Scheme 2. Alternative conditions27 “B” employing CuI and triethylamine (Et3N) were found to give desired 10d in 60% yield. Methyl triazole 11d was obtained following deprotection of 10d using TFA and Et3SiH.
Benzotriazole 12 was prepared by a [3+2] cycloaddition of azide 4 and benzyne generated in situ by a cesium fluoride mediated elimination of 2-(trimethylsilyl)phenyl triflate (Scheme 4; 72% yield).28 Desired amino acid 13 was obtained by deprotection of 12 with TFA and Et3SiH, as performed for triazoles of Scheme 2.
Triazoles of Schemes 2–4 were tested for LAT1 activity as previously described (24-well poly-D plate configuration)12, 29 using human embryonic kidney cells containing inducible LAT1 (HEK-hLAT1)30 in two different assays: cis-inhibition and trans-stimulation, to determine ligand potency and relative transport rates, respectively. In the cis-inhibition assay, test compounds were incubated with cells in the presence of a known substrate, [3H]-gabapentin (6 nM). Compounds were screened at 200 μM to determine %inhibition of [3H]-gabapentin uptake relative to a LAT1 inhibitor BCH (2 mM),31 set to 100% inhibition. Select compounds were then tested at various concentrations to determine the half maximal inhibitory concentration (IC50). cis-Inhibition indicates ligand potency, however, it does not distinguish between LAT1 inhibitors and substrates. To identify whether a compound was a substrate, we performed a transstimulation assay. This experiment is possible due to the alternating access mechanism of LAT1,32 in which an exchange occurs between intra- and extra-cellular amino acids in a 1:1 mole ratio. The assay is performed by loading cells with [3H]-gabapentin prior to exposure to test compounds. The efflux rate of [3H]-gabapentin (fmol/min) was calculated at 3 minutes after adding test compound. % Efflux was normalized relative to L-phenylalanine 18, which had an efflux rate of 2.7 ± 0.3 fmol/min, from an average of seven experiments.
Positive controls included natural amino acid substrates L-histidine 15, L-phenylalanine 18 and L-tryptophan 22. Non-substrate L-arginine 14 was used as a negative control, which had a relative efflux rate of 28% in our trans-stimulation assay. Non-substrates generally show 20−35% relative efflux due to background levels of exported [3H]-gabapentin. Low levels of endogenous amino acids present in the cell assay may cause efflux via LAT1 or an alternate transporter.
Unfortunately, triazole analogs of Table 1 (7a-e, 11d-e) were considerably less potent (lower %inhibition and greater IC50 values) in our cis-inhibition assay than related histidine analogs (15–17) of Table 2. Unsubstituted N-linked triazole 7a (R = H) showed the highest relative efflux rate and greatest potency of the triazole series. However, both the potency (160 μM vs. 110 μM) and relative %efflux (82% vs. 140%) for 7a were inferior to L-histidine 15. In contrast to histidine analogs 16–17, substitution in the triazole series was less well tolerated. Only a methyl substituent in compound 7d displayed comparable %efflux to its related histidine analog 16, but showed a 2-fold decrease in potency (440 μM vs. 190 μM). Moreover, benzyl substitution in triazoles 7e and 11e resulted in loss of potency relative to analog 17. Though one could interpret the negative %inhibition for 7e and 11e as due to LAT1 activation for [3H]gabapentin uptake, we think it is possible that a change in plasma membrane integrity could cause an uptake signal increase compared with control in the absence of any test compounds. Nonetheless, these negative %inhibition values do not change the interpretation of our data, that triazoles 7e and 11e have poor potency. However, triazoles of Table 1 (7a-e, 11d-e) demonstrated relative %efflux greater than our negative control L-arginine 14 (37–82% vs. 28%), suggesting that they are being transported into cells by LAT1.
Table1.
LAT1 trans-stimulation and cis-inhibition cell assay activity for N- and C-linked triazoles 7a-e and 11d-e.
 | |||||
|---|---|---|---|---|---|
| Compounda | R | Regioisomer | Relative %Effluxb | % Inhibitionc | IC50 (μM)d |
| 7a | H | N | 82 | 58 | 160 ± 27 |
| 7b | Ph | N | 37 | 13 | >1000 |
| 7c | CH2OH | N | 57 | 14 | >1000 |
| 7d | Me | N | 70 | 38 | 440 ± 190 |
| 11d | Me | C | 63 | 20 | >1000 |
| 7e | Bn | N | 49 | −6 | NDe |
| 11e | Bn | C | 43 | −19 | NDe |
Cell assay data was obtained at least in triplicate (24-well poly-D plate configuration) in each condition. All compounds above are single enantiomers of L configuration.
Compounds were tested at 200 μM for their ability to cause efflux (fmol/min) of [3H]-gabapentin from pre-loaded HEK-hLAT1 cells. Efflux of [3H]-gabapentin was calculated at 3 min after adding test compound. %Efflux was normalized relative to L-Phe (18), which had an efflux rate of 2.7 ± 0.3 fmol/min, from an average of seven experiments.
Compounds were tested at 200 μM for their ability to inhibit uptake of [3H]-gabapentin into HEK-hLAT1 cells. Data are presented as % inhibition relative to background signal in the absence of a test compound.
For IC50 determinations, varying concentrations of each compound were added, from 0.1 μM to 500 μM. IC50 and standard deviation of each compound was calculated by GraphPad Prism version 5.0. %[3H]-Gabapentin uptake at each concentration was normalized relative to %inhibition by BCH31 at 2 mM, which was set to 100% inhibition.
Not determined.
Table 2.
LAT1 trans-stimulation and cis-inhibition cell assay activity for some naturally occurring amino acids and structural analogs including benzotriazole 13.a
 | ||||
|---|---|---|---|---|
| Compound | R | Relative %Efflux | % Inhibition | IC50 (μM) |
| 14 (L-Arg ) | NH2(NH)CNH(CH2)2− | 28 | 49 | |
| 15 (L-His) | 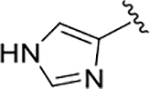 |
140 | 92 | 110 ± 93 |
| 16 | 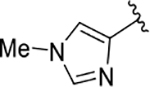 |
85 | 70 | 190 ± 25 |
| 17 | 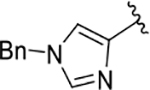 |
81 | 74 | 170 ± 53 |
| 18 (L-Phe) | 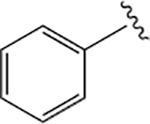 |
100 | 85 | 69 ± 29 |
| 19 | 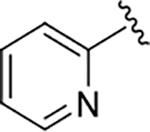 |
140 | 84 | 160 ± 56 |
| 20 | 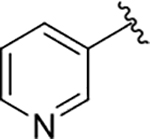 |
130 | 98 | 69 ± 17 |
| 21 | 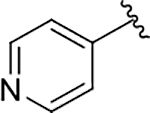 |
89 | 99 | 100 ± 53 |
| 22 (L-Trp) | 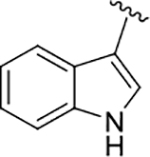 |
59 | 79 | 160 ± 87 |
| 13 (L-Trp) | 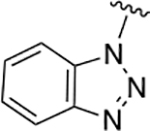 |
100 | 59 | 340 ± 104 |
Cell assay conditions were as described for Table 1.
It should be noted that we were unable to prepare the C-linked analog of triazole 7a by reaction of TMS-azide with alkyne 9 using either conditions A or B of Scheme 3. An alternate, multi-step synthesis (not involving Click Chemistry) of this unsubstituted C-linked triazole in racemic form has previously been described;33 however, given the lack of potency for C-linked triazoles 11d and 11e, we decided not to pursue this compound.
Relative to triazoles of Table 1, benzotriazole 13 (Table 2) demonstrated improved %efflux and potency. Additionally, it appeared to be a better substrate in terms of %efflux compared with the natural substrate tryptophan 22 (100% vs. 59%), albeit with a 2-fold decrease in potency. Thus, all of the triazole analogs of Tables 1 and 2 had diminished potency relative to their corresponding histidine and tryptophan analogs.
Based on our models, both triazoles and the corresponding histidine analogs dock within a model of the LAT1 binding site in a comparable manner (supplementary data). Therefore, we cannot explain the difference in activity based on their predicted binding modes. Furthermore, comparing cLogP values for triazoles with their histidine congeners did not provide any additional insight into why triazoles have decreased potency. For example, N-linked triazoles (7a-7e) exhibit an almost order of magnitude higher cLogP34 than their histidine counterparts (e.g. cLogP: 7a = −2.9 vs. 15 = −3.7). Whereas, C-linked triazoles, which were even less potent, had more negative cLogP values (e.g. 11d = −4.2 vs. 16 = −3.7). Moreover, pyridyl analogs 19–21, which we previously reported as potent LAT1 substrates,12 had a comparable cLogP to triazole 7a (−3.1 vs. −2.9, respectively). The only apparent difference between these triazoles and their related histidine and pyridyl analogs is heterocycle basicity. A 1,2,3-triazole ring is non-basic with a reported pKa of 1.15 for its conjugate acid.35 However, our models, representing snapshots with the highly dynamic transport cycle do not indicate the presence of acidic side chains in the vicinity of the ligand binding site. Thus, we cannot determine the potential functional role of a basic heterocycle (i.e. imidazole and pyridine). Furthermore, imidazole and pyridine rings would be primarily neutral under the conditions of our cell assay (pH 7.4), even though their protonation states inside the binding site is unknown. Given the fact that most natural LAT1 substrates do not contain a basic heterocycle (e.g. phenylalanine 18, tryptophan 22, leucine, methionine, tyrosine, etc.), it is unclear to us why triazoles have decreased potency compared with structurally-related histidine and pyridyl analogs.
A series of substituted 1,2,3-triazolyl amino acids were synthesized and tested for LAT1 substrate activity. The synthesis of these triazole analogs is convenient, potentially serving as a template for conjugation of drug molecules with amino acids (i.e. hypothetical prodrugs of Figure 1) via the highly convergent Huisgen 1,3-dipolar cycloaddition. However, only a benzotriazole analog 13 exhibited a better uptake rate than the corresponding natural amino acid analog L-tryptophan 22, and all of the triazoles of the current work were less potent than their corresponding histidine and tryptophan equivalents. Despite having poor potency for inhibiting LAT1 uptake of [3H]-gabapentin, triazoles may be weak binding substrates, as evidenced by their activity in a trans-stimulation assay relative to non-substrate L-arginine 14. Though we have no evidence to explain why a basic heterocycle (e.g. imidazole and pyridine) provides greater potency than these non-basic triazoles, our results suggest that the binding site is sensitive to relatively subtle changes in ligand properties. Moreover, it was previously noted that LAT1 is selective for aromatic amino acids relative to aliphatic ones, and this preference does not correlate well with lipophilicity.36, 37 Our data expand upon that observation by showing that aromaticity does not guarantee good ligand binding. Thus, for reasons that are not obvious to us, amino acids containing a 1,2,3-triazole ring appear to be inferior LAT1 substrates compared with histidine and tryptophan structural analogs. These findings could influence the design of amino acid containing prodrugs as well as LAT1 inhibitors for treating brain disorders and cancer.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health’s National Institute of Neurological Disorders and Stroke grant R15 NS099981 (to A.A.T), the National Institutes of Health’s National Institute of General Medical Sciences grant R01 GM117163 (to H.C.C. and K.M.G.) and R01 GM108911 (to A.S. and C.C.). We appreciate OpenEye Scientific Software Inc. for granting us access to its high-performance molecular modeling applications through its academic license program.
Footnotes
Conflicts of interest
The authors confirm no conflict of interest.
Appendix A. Supplementary data
Full experimental details, compound characterization data, including 1H and 13C NMR spectra for newly synthesized compounds 7d and 11d, and ligand docking description along with models of compounds 7e, 11e and 17 docked in the LAT1 active site can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Kanai Y; Segawa H; Miyamoto K; Uchino H; Takeda E; Endou H J. Biol. Chem 1998, 273, 23629. [DOI] [PubMed] [Google Scholar]
- 2.Uchino H; Kanai Y; Kim DK; Wempe MF; Chairoungdua A; Morimoto E; Anders MW; Endou H Mol. Pharmacol 2002, 61, 729. [DOI] [PubMed] [Google Scholar]
- 3.del Amo EM; Urtti A; Yliperttula M Eur. J. Pharm. Sci 2008, 35, 161. [DOI] [PubMed] [Google Scholar]
- 4.Boado RJ; Li JY; Nagaya M; Zhang C; Pardridge WM Proc. Natl. Acad. Sci. USA 1999, 96, 12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kido Y; Tamai I; Uchino H; Suzuki F; Sai Y; Tsuji A J. Pharm. Pharmacol 2001, 53, 497. [DOI] [PubMed] [Google Scholar]
- 6.Yanagisawa N; Satoh T; Hana K; Ichinoe M; Nakada N; Endou H; Okayasu I; Murakumo Y Cancer Biomark. 2015, 15, 365. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi H; Ishii Y; Takayama T J. Surg. Oncol 2005, 90, 233. [DOI] [PubMed] [Google Scholar]
- 8.Ebara T; Kaira K; Saito J; Shioya M; Asao T; Takahashi T; Sakurai H; Kanai Y; Kuwano H; Nakano T Anticancer Res. 2010, 30, 4223. [PubMed] [Google Scholar]
- 9.Ichinoe M; Mikami T; Yoshida T; Igawa I; Tsuruta T; Nakada N; Anzai N; Suzuki Y; Endou H; Okayasu I Pathol. Int 2011, 61, 281. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi K; Ogata S; Nakanishi K; Ozeki Y; Hiroi S; Tominaga S; Aida S; Matsuo H; Sakata T; Kawai T Lung Cancer 2010, 68, 58. [DOI] [PubMed] [Google Scholar]
- 11.Ylikangas H; Malmioja K; Peura L; Gynther M; Nwachukwu EO; Leppanen J; Laine K; Rautio J; Lahtela-Kakkonen M; Huttunen KM; Poso A ChemMedChem 2014, 9, 2699. [DOI] [PubMed] [Google Scholar]
- 12.Chien HC; Colas C; Finke K; Springer S; Stoner L; Zur AA; Venteicher B; Campbell J; Hall C; Flint A; Augustyn E; Hernandez C; Heeren N; Hansen L; Anthony A; Bauer J; Fotiadis D; Schlessinger A; Giacomini KM; Thomas AA J. Med. Chem 2018, 61, 7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Augustyn E; Finke K; Zur AA; Hansen L; Heeren N; Chien H-C; Lin L; Giacomini KM; Colas C; Schlessinger A; Thomas AA Bioorg. Med. Chem. Lett 2016, 26, 2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zur AA; Chien H-C; Augustyn E; Flint A; Heeren N; Finke K; Hernandez C; Hansen L; Miller S; Lin L; Giacomini KM; Colas C; Schlessinger A; Thomas AA Bioorg. Med. Chem. Lett 2016, 26, 5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Z; Yao Z; Xu X Curr. Org. Chem 2017, 21, 2240. [Google Scholar]
- 16.Singh MS; Chowdhury S; Koley S Tetrahedron 2016, 72, 5257. [Google Scholar]
- 17.Wang C; Ikhlef D; Kahlal S; Saillard J-Y; Astruc D Coord. Chem. Rev 2016, 316, 1. [Google Scholar]
- 18.Schoffelen S; Meldal M Alkyne-azide reactions, 1st ed; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014. [Google Scholar]
- 19.Heravi MM; Tamimi M; Yahyavi H; Hosseinnejad T Curr. Org. Chem 2016, 20, 1591. [Google Scholar]
- 20.Kolb HC; Finn MG; Sharpless KB Angew. Chem. Int. Ed. Engl 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Yanagida O; Kanai Y; Chairoungdua A; Kim DK; Segawa H; Nii T; Cha SH; Matsuo H; Fukushima J; Fukasawa Y; Tani Y; Taketani Y; Uchino H; Kim JY; Inatomi J; Okayasu I; Miyamoto K; Takeda E; Goya T; Endou H Biochim. Biophys. Acta 2001, 1514, 291. [DOI] [PubMed] [Google Scholar]
- 22.West KR; Bake KD; Otto S Org. Lett 2005, 7, 2615. [DOI] [PubMed] [Google Scholar]
- 23.Zou Y; Fahmi NE; Vialas C; Miller GM; Hecht SM J. Am. Chem. Soc 2002, 124, 9476. [DOI] [PubMed] [Google Scholar]
- 24.Gajewski M; Seaver B; Esslinger CS Bioorg. Med. Chem. Lett 2007, 17, 4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta A; Jaouhari R; Benson TJ; Douglas KT Tetrahedron Lett. 1992, 33, 5441. [Google Scholar]
- 26.McConathy J; Zhou D; Shockley SE; Jones LA; Griffin EA; Lee H; Adams SJ; Mach RH Mol. Imaging 2010, 9, 329. [PubMed] [Google Scholar]
- 27.Kaur J; Bhardwaj A; Sharma SK; Wuest F Bioorg. Med. Chem 2013, 21, 4288. [DOI] [PubMed] [Google Scholar]
- 28.Shi F; Waldo JP; Chen Y; Larock RC Org. Lett 2008, 10, 2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geier EG; Schlessinger A; Fan H; Gable JE; Irwin JJ; Sali A; Giacomini KM Proc. Natl. Acad. Sci. USA 2013, 110, 5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerangue NUS Patent 7,462,459 B2, 2008.
- 31.Segawa H; Fukasawa Y; Miyamoto K; Takeda E; Endou H; Kanai Y J. Biol. Chem 1999, 274, 19745. [DOI] [PubMed] [Google Scholar]
- 32.Fotiadis D; Kanai Y; Palacin M Mol. Aspects Med 2013, 34, 139. [DOI] [PubMed] [Google Scholar]
- 33.Ikeda Y; Kawahara S.-i.; Taki M; Kuno A; Hasegawa T; Taira K Protein Eng. 2003, 16, 699. [DOI] [PubMed] [Google Scholar]
- 34.cLogP was calculated using ChemDraw v.17.1.
- 35.Catalan J; Abboud JLM; Elguero J Adv. Heterocycl. Chem 1987, 41, 187. [Google Scholar]
- 36.Ylikangas H; Peura L; Malmioja K; Leppanen J; Laine K; Poso A; Lahtela-Kakkonen M; Rautio J Eur. J. Pharm. Sci 2013, 48, 523. [DOI] [PubMed] [Google Scholar]
- 37.Puris E; Gynther M; Huttunen J; Petsalo A; Huttunen KM J. Controlled Release 2017, 261, 93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.