

Abstract
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection. While many individual cells and systems in the body are involved in driving the excessive and sometimes sustained host response, pathogen engagement with endothelial cells and platelets early in sepsis progression, are believed to be key. Significant progress has been made in establishing key molecular interactions between platelets and pathogens and endothelial cells and pathogens. This review will explore the growing number of compensatory connections between bacteria and viruses with platelets and endothelial cells and how a better understanding of these interactions are informing the field of potential novel ways to treat the dysregulated host response during sepsis.
Keywords: sepsis, endothelial cell, platelets, hyper-activation, micro-organisms
Introduction
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection (Sepsis 3) and despite being the primary cause of in-hospital mortality there is little in the drug discovery pipeline for this disease (1). Treatment primarily focuses on the use of antibiotics but with the growing incidence of antibiotic-resistant strains of bacteria and the time it takes to diagnose sepsis there is clearly a need to discover novel approaches to treating sepsis. As the definition indicates that sepsis is a dysregulated host response (2) an obvious novel treatment strategy is to correct this dysregulated host response. Through significant advances in our understanding of the molecular interactions two possible theories have emerged that help explain the nature of the dysregulation. The platelet-pathogen theory suggests that pathogens bind to platelets activating them. These activated platelets bind to both endothelial cells and immune cells activating them which causes damage and disruption to the endothelial layer, leading to loss of barrier integrity, fluid leakage resulting in shock (Figure 1). Alternatively, the endothelial-pathogen theory suggests that pathogens bind to endothelial cells activating them. This leads to a release of granules and pro-inflammatory cytokines and chemokines, that recruit platelets to form a thrombus encasing the pathogens and immune cells and contribute to excessive thrombocytopenia and hyper-inflammatory response. Pathogen binding to endothelial cells also causes apoptosis which results in disruption of the endothelial layer leading to adherens junction disassembly, increased vascular permeability, fluid leakage, and shock (Figure 2). In this review, we will discuss each of these theories outlining the molecular mechanisms leading to each.
Figure 1.

Platelet Theory. An invading pathogen binds to platelets either directly (absence of plasma protein) or indirectly (presence of plasma protein). Binding results in platelet activation via Src kinases which results in prostanoid release, cytokine secretion, granule secretion, and activation of GPIIbIIIa. Release of ADP and thromboxane A2 (TXA2) serves to amplify the platelet response. In conjunction with this, activation of GPIIbIIIa allows fibrinogen binding resulting in platelet aggregation. Secretion of platelet cytokines and chemokines activates the vascular endothelium. For example, secreted TNFa activates the TNFR1 receptor on endothelial cells which triggers the death pathway resulting in apoptosis. This results in endothelial cell shrinkage and loss of barrier integrity leading to increased vascular permeability and shock. Separation of endothelial cells allows for pathogens to escape the bloodstream and infect major organs which eventually leads to multi organ failure.
Figure 2.

Endothelial theory. Similar to platelets invading pathogens bind either directly or indirectly to the vascular endothelium. Binding results in endothelial cell activation that results in apoptosis. Although the pathway to apoptosis has not yet been defined apoptosis leads to endothelial cell shrinkage and loss of barrier integrity leading to increased vascular permeability and shock. Separation of endothelial cells allows for pathogens to escape the bloodstream and infect major organs which eventually leads to multi organ failure. Endothelial cell activation also leads to granule secretion which deposits vonwillebrand factor on the surface of the endothelial cells. This encourages platelet rolling, activation and aggregate formation. In addition, bacteria also roll on the deposited vonwillebrand factor eventually coming to a halt and firmly adhering. Passing platelets bind to the immobilized bacteria on the endothelial cells which leads to inappropriate thrombus formation and exacerbation of the dysregulation.
Platelet Function
After red blood cells platelets are the most numerous cell-like particle in the blood. Their total volume and surface area when combined together is larger than that of all the leukocytes taken together. They are not true cells as they have no nuclei and are in fact fragments of megakaryocytes (3). Their primary role is in hemostasis and they clump together to occlude any breach in the vasculature. They can be considered to have three distinct functions: adhesion, secretion and aggregation and platelet activation links these processes through multiple signaling pathways.
Adhesion
Platelets typically travel close to the endothelium patrolling for breaches in the vasculature. The resting endothelium is inert ensuring platelets do not adhere, however when the endothelial cell layer is damaged exposing sub-endothelial matrix proteins (which as occurs with a cut), or becomes activated through inflammation the area becomes highly thrombogenic and platelets subsequently adhere. Key proteins in the matrix that support platelet adhesion are collagen and von Willebrand Factor (VWF). Adhesion is a highly controlled event and is mediated by platelet receptors such as integrin α2β1 and glycoprotein (GP) VI or collagen and GPIb/IX/V for VWF. The VWF-GPIb/IX/V interaction is shear-dependent and is primarily involved in adhesion under high shear stress such as in coronary arteries. See Nieswandt et al. for a review on platelet function (4).
Activation
While binding to either collagen or VWF facilitates adhesion of platelets to the site of injury it also triggers activation of the platelet leading to platelet secretion and aggregation. The adhesion process is not the only process for activating platelets as soluble mediators can also lead to platelet activation. Substances like adenosine diphosphate (ADP), thrombin, thromboxane A2, and adrenaline can also trigger platelet activation through their respective receptors. To further link all of the platelet functions the primary source of these soluble mediators are platelets in a process known as secretion. Most of these receptors mediate their effects through two different signaling pathways. Some receptors stimulate phospholipase (PL) A2 which ultimately leads to the production of thromboxane A2 while others trigger PLC activation (5).
Secretion
Once activated, platelets secrete the contents of their granules. Platelets contain different types of granules such as alpha and dense granules and lysosomes. The content of these granules plays an important role in haemostasis. Secreted ADP activates the surrounding platelets and this is critical in creating the growing clot. The granules also secrete adhesion molecules such as fibrinogen as well as up-regulating adhesion receptors such as GPIIb/IIIa (fibrinogen receptor) and p-selectin (CD62) to the platelet surface (6).
Aggregation
While platelet activation leads to granule secretion it also leads to activation of GPIIb/IIIa on the platelet surface. GPIIb/IIIa is an integrin (αIIbβ3) fibrinogen receptor and is usually present in a resting, non-binding conformation. Once activated it can bind soluble fibrinogen and as fibrinogen is a large bivalent molecule one fibrinogen molecule can bind to two GPIIb/IIIa molecules. If these GPIIb/IIIa molecules are on different platelets the effect is to link two platelets together. Considering that there are around 50,000 GPIIb/IIIa molecules per platelet this creates a platelet-rich clot cross-linked by fibrinogen. This process is known as aggregation and is critical for sealing a breach in the vasculature (7).
Platelet Signaling
Platelet activation occurs through two processes—outside-in signaling and inside-out signaling and is reviewed in more detail by Stalker et al. (8). The classic example of outside-in signaling is the process by which soluble mediators such as ADP, thrombin, and thromboxane (Tx) A2 activate platelets. These bind to membrane receptors (in most cases G-protein-coupled receptors) triggering downstream events. Agonist binding to these receptors trigger activation of one of two phospholipases (PL)—PLA2 or PLC. PLA2 is the primary PL and acts to release arachidonic acid from the inner membrane of the platelet. This arachidonic acid is a substrate for cyclooxygenase (COX). In platelets, the COX isoform is COX 1 and it converts arachidonic acid into prostaglandin (PG) H2. PGH2 is an intermediate in the signaling process and it is further metabolized to its active product by cell specific enzymes. In platelets the primary enzyme is thromboxane synthase which converts PGH2 to TxA2 which binds to receptors and triggers further platelet activation. This pathway is completely inhibited by COX 1 inhibitors such as aspirin.
The second signaling pathway uses a number of different isoforms of PLC which cleave inositol triphosphate (IP3) and diacylglycerol (DAG) from the membrane. IP3 binds to an intracellular Ca2+ channel which increases intracellular Ca2+ levels through release from the endoplasmic reticulum. DAG ultimately activates protein kinase C which activates further downstream events. This pathway is not inhibited by aspirin. Agonists are often divided into weak agonists which are PLA2-dependent and strong agonists which are PLC-dependent, although this is often concentration-dependent with low concentrations of agonists using PLA2 and high concentrations using PLC.
While soluble agonists activate platelets via PLA2/PLC this is not the only process for activating platelets. Platelet adhesion also leads to platelet activation however, the activation process for each of the adhesion receptors is receptor specific. One example is that of platelet activation in response to adhesion to fibrinogen. The platelet receptor involved is GPIIb/IIIa and initially it was considered that GPIIb/IIIa was merely an adhesion receptor as there were no obvious signaling pathways associated with it. However, it is now clear that GPIIb/IIIa can recruit signaling molecules [Src family kinases, focal adhesion kinase (FAK) etc.] and generate activating signals (9). GPIb/IX/V acts by recruiting 14-3-3ζ, actin binding protein, Src, FAK etc (10). Receptors that contain an ITAM (immunoreceptor, tyrosine-based activation motif) or ITAM-like domain such as Fc receptors and CLEC-2 recruit the tyrosine kinase syk when dimerized (11). Fc receptors can heterodimerise, that is they can dimerize with other receptors such as GPVI and GPIb. GPVI contains an SH3 domain that recruits the src family kinases and when dimerized with FcR-γ it triggers the recruitment of syk and phosphorylation of FcR (12).
The wave of outside-in activation is followed by a wave of inside-out signaling. This primarily involves talin binding to the β3 subunit of GPIIb/IIIa (13). This inside-out signaling is essential for full activation of the platelet.
Platelets also have inhibitory signaling pathways to counter the activating pathways. The primary pathway is mediated by prostacyclin (PGI2). When it binds to is receptor it increases cAMP which in turn activates PKA and inhibits platelet activation. A related mechanism is that of nitric oxide (NO) which directly enters the platelet and activates soluble guanylate cyclase increasing cGMP levels. Both prostacyclin and NO are produced by healthy endothelial cells to prevent clot formation.
In sepsis, thrombocytopenia develops in up to 50% of cases and is associated with poor outcome (14). This thrombocytopenia is likely to play a significant role in the pathogenesis of sepsis leading to development of multiple organ dysfunction syndrome (MOPS), disseminated intravascular coagulation (DIC) and/or massive bleeding as a result of platelet consumption and thrombus formation (15). It is well-established that innate immune cells (IIC) such as macrophages, natural killer cells (NK) cells, neutrophils, dendritic cells etc, release a plethora of pro-inflammatory mediators creating a so-called cytokine storm (16). We now also know that in addition to their hemostatic functions platelets also play a role in inflammation and regulation of inflammatory response by secreting cytokines, interferons, and chemokines. For example, Staphylococcus and Streptococcus spps can trigger platelet aggregation, cytokine release, and thrombocytopenia (17–19).
Platelets and Immunity
The critical role of platelets in the innate immune response is largely mediated by their ability to interact with other immune cells mainly neutrophils (20, 21). Platelets express receptors on their surface that are usually associated with immune cells such as FcγRIIa and Toll-like receptors (TLR) 2 and 4 (22). For example, in 2007 Clark et al., demonstrated that lipopolysaccharide (LPS) binds to platelet TLR4 which mediates attachment to neutrophils. Critical to this interaction is platelet activation which results in granule secretion, P-selectin expression on the surface of the platelet, and crosslinking to its counter receptor P-selectin glycoprotein ligand-1 on the leukocyte surface. Other studies have demonstrated that platelet GPIbα can bind VWF and crosslink the platelet to neutrophils via the β2 integrin (CD18). Neutrophil Extracellular Traps (NETs) are web-like structures composed of a chromatin backbone, histones and anti-microbial proteins and their main function is to trap and kill bacteria, virus, and fungi, avoiding their dissemination. While NET formation is a critical event in innate immunity, uncontrolled formation may exert significant tissue damage which contributes significantly to the already difficult to control host dysregulation (23). Dengue virus has been shown to activate platelets in a CLEC-2-dependent manner producing extracellular vesicles that induce NET formation (24). Regardless of the interaction, platelet attachment to the neutrophil results in rapid activation and most importantly the formation of NETs and together they play an important role in the pathogenesis of sepsis (25).
As platelet activation occurs during inflammation and infections such as sepsis there is also a need to control excessive platelet activation. One controlling factor is that of C-reactive protein (CRP) which is an acute phase protein synthesized in the liver in response to infection. It exists in a monomeric (mCRP) and pentameric (pCRP) forms which have opposing effects. pCRP is known to inhibit platelet aggregation by binding to GPIIb/IIIa and thus will act to reduce thrombus formation (26). Another agent that regulates the platelet response is nitric oxide (NO). NO is produced by the endothelium and is a potent vasodilator but also an inhibitor of platelet activation. During sepsis NO levels increase due to production by immune cells. This increased NO contributes to vasodilation and hypotension as well as inhibiting platelet function (27, 28). Thus, platelet activation status during sepsis depends on the balance between activating and inhibiting factors.
Platelet Immune Receptors
Platelets express immunoreceptor tyrosine-based activation motifs (ITAMs)-containing receptors such as FcγRIIa, GPVI, and C-type lectin-like receptor (CLEC)-2 (29). The presence of FcγRIIa, a receptor for the Fc portion of IgG, on platelets is unusual as it is a receptor involved in phagocytosis and all other FcγRIIa-expressing cells are phagocytic (29). However, while not true phagocytic cells platelets do engulf bacteria in a manner that has some similarities to phagocytosis (30, 31). Platelet FcγRIIa is fully functional and can trigger platelet aggregation. Immune complexes [or even heat-agglutinated immunoglobulin (Ig) G] directly induce platelet aggregation in an FcγRIIa-dependent manner. Furthermore, bacteria that become coated in IgG can also induce platelet aggregation in an FcγRIIa- dependent manner (see below). The functionality of TLRs is more complex. While studies show that TLRs can mediate platelet activation others show that they don't (32). There is evidence to suggest that platelets activated by TLRs can engage with neutrophils and/or monocytes triggering their activation (33). Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), also known as CD209, is a C-type lectin which is usually expressed on macrophages and dendritic cells that is known to be involved in the phagocytosis of HIV and is also expressed on platelets (34). DC-SIGN and FcγRIIa are particularly implicated in platelet activation in Dengue virus infection (DENV) and incubation of platelets with anti-DC-SIGN antibodies prevented DENV-mediated platelet activation (35).
Other receptors shown to facilitate platelet interactions in response to pathogens are sialic-acid-binding immunoglobulin-like lectins (Siglecs)- a type I transmembrane proteins, that play role in regulating the host's immune responses to pathogen (36). In platelets Siglec-7 is most abundantly expressed and its function depends on the P2Y1 platelet receptor and of the GPIIb/IIIa integrin. It is proposed that Siglec-7 down-regulates pathogen-induced platelet activation by inducing apoptosis (37). Along with FcγRIIa, CLEC-2, and GPVI are ITAM receptors found on platelets. The ligands for CLEC-2 is podoplanin and for GPVI it is collagen, fibrinogen, and fibrin (38). These receptors have been found to play a role in the interaction with pathogens. Thus, CLEC-2 has been shown to bind to human immunodeficiency virus (HIV) and GPVI has been shown to bind to Hepatitis C virus (39, 40). GPVI has been shown to be important in Klebsiella pneumoniae sepsis models (41). CLEC-2 has been shown to drive thrombosis following Salmonella infection (42). Furthermore, the platelet CLEC-2-podoplanin interaction has been found to be an important modulator of inflammation during sepsis (43, 44).
Platelet Cytokines
Platelets release cytokines either directly into the bloodstream by de-granulation, or by secreting platelet-derived micro-vesicles (PDMV), which make up between 60 and 90% of extracellular vesicles (EV) in plasma and contribute to hemostatic and immune function of platelets (45–47). These “immuno-parcels” can elicit innate and adaptive immune responses at distant sites by delivering variety of immunomodulatory factors, such as CD154 (also known as soluble CD40 Ligand, sCD40). CD154 from PDMV is enough to activate antigen specific splenic B cell response in CD154−/− mice, in both T cell-dependent and independent manner (48). PDMV also contain a variety of nucleic acids including messenger and micro-RNAs (49). For example, platelets contain mRNA of pro-IL-1β, which upon platelet activation is translated in situ and fully synthesized pro-IL-1β is then released into circulation (50). IL-1β but not IL-1α binds to fibrinogen and it is the bound form of IL-1β that has enhanced action to induce monocyte chemoattractant protein 1 (MCP-1) and nitric oxide (NO) production by endothelial cells via NFκB pathway (51). Among other pro-inflammatory modulators released by platelets are: MCP-1, macrophage inflammatory protein (MIP)-1α, regulated on activation, normal T cell expressed and secreted (RANTES), IL-8, tumor growth factor (TGF)-β, angiogenesis and growth factors, and various immunoglobulins (48, 52–55).
Platelet Bacterial Interactions
There are several platelet receptors that are involved in either direct interactions with pathogens either through direct interactions between microbial adhesins and platelet surface component or indirect associations via a bridging molecule (17, 20, 56, 57). The best studied interaction is that between S. aureus and platelets but the interaction with other Gram-positive and Gram-negative bacteria has also been described (17). The most significant causative agents of sepsis are S. aureus is the major cause (21%), E. coli (16%), Staphylococcus epidermidis (11%), and S. pneumoniae (4%) and these are also the best studied for their interactions with platelets (58).
Staphylococci-Platelet Interactions
S. aureus expresses several cell wall anchored surface proteins that enable binding of the bacteria to platelets (59). During the exponential growth phase S. aureus expresses Clumping factor (Clf) B, fibronectin-binding protein (FnBP) A and B; while ClfA is expressed during the stationary phase. These proteins bind to fibrinogen facilitating its binding to and activation of platelet GPIIb/IIIa (60–62). S. aureus surface protein A (SpA) is known to bind IgG but also binds to domain A1 on VWF which mediates binding to platelet GPIbα (63). However, these interactions are insufficient to activate platelets and all require simultaneous activation of FcγRIIa through engagement with opsonising IgG's on the surface of the bacterium. Engagement of integrins and FcγRIIa lead to platelet activation, aggregation, adenosine triphosphate (ATP) release, and thrombus formation. In the absence of plasma proteins S. aureus can also bind directly to GPIIb/IIIa via its iron-regulated surface determinant protein B (IsdB) inducing platelet adherence and aggregation (64). Four more platelet activating proteins that are secreted by S. aureus have been recently identified by Binsker et al: Extracellular adherence protein (Eap), the chemotaxis inhibitory protein of S. aureus (CHIPS), the formyl peptide receptor-like 1 inhibitory protein (FLIPr), and the major autolysin Atl (AltA) which were all shown to induce P-selecting expression, while Eap, CHIPS, and AltA also induced platelet aggregation (65). Staphylococcus epidermidis expresses serine–aspartate repeat protein (Sdr) G, a member of Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMM), that engage with GPIIb/IIIa either via fibrinogen or directly (66).
Streptococcci-Platelet Interactions
A large number of streptococci have been shown to interact with platelets including Streptococcus gordonii, Streptococcus sanguinis, Streptococcus oralis, Streptococcus agalactiae, and Streptococcus pneumoniae (17). A common mechanism through which the streptococci interact with platelets is via a serine-rich repeat protein (Srrp). This Srrp is expressed on the surface of many streptococci (67–70). Numerous studies have demonstrated that the Srrp binds to platelet GPIbα and induces activation. Under low shear conditions (<500 s−1) platelets roll along immobilized streptococci using a mechanism characteristic of the interaction observed when platelets interact with VWF. Deletion of the Srrp ablates the interaction with platelets under both static and shear conditions (68, 70). S. gordonii have also been shown to bind GPIIb/IIIa. A common feature in this interaction is the presence of an RGD-like sequence in a large cell wall surface protein (3,500 amino acid residues) that mediates the binding called Platelet Adherence protein A (PadA) (71, 72). Binding resulted in src induced rearrangement of the platelet actin cytoskeleton leading to filopodia and lamellipodia formation resulting in platelet spreading over the bacteria (73). Protein analysis and site directed mutagenesis revealed that PadA contains two critical integrin-recognition motifs (383RGT and 484AGD) that mediate interaction with GPIIb/IIIa (73). S. pneumoniae has also been shown to bind to and induce platelet activation resulting in dense granule secretion in a TLR2-dependent manner. Although the bacterial component that interacts with platelet TLR2 was not identified a likely candidate is wall lipoteichoic acid (74).
Platelet-Gram-Negative Bacteria Interactions
Escherichia coli, Helicobacter pylori, Porphyromonas gingivalis, and Brucella abortus all have been shown to bind platelets (18, 75–78). E. coli O157:H7 interact with platelets via platelet TLR4 and P-selectin (CD62) leading to secretion of CD40L, increase in fibrinogen binding on platelets and the formation of aggregates (79). This platelet activation is both FcγRIIa- and GPIIb/IIIa-dependent, and requires opsonisation of bacteria with IgG (18, 32). Platelet activation induced by H. pylori has been shown to be FcγRIIa and GPIbα-dependent (77, 80). B. abortus binds directly to platelets in a dose-dependent manner, although platelet receptors for this interaction remain to be established. This interaction induced enhanced fibrinogen binding and P-selectin expression, and promoted infection of monocytes by delivering bacteria to them (75). P. gingivalis has also been shown to bind to platelets in an IgG-dependent manner. Depletion of IgG or pre-incubation of platelets with an anti-FcγRIIa antibody abolished platelet activation and aggregation (81).
Complement-Dependent Platelet Activation
While some bacteria have surface proteins that can interact with platelets many bacteria can activate platelets despite the absence of such proteins. S. aureus mutants which lack known platelet interacting proteins such as ClfA and ClfB and are thus unable to bind fibrinogen can still induce platelet activation. This is also true of strains of S. sanguinis that do not bind to platelets. However, the aggregation profile of these bacteria is quite different. While wildtype S. aureus induces aggregation within 2–3 min these non-interacting bacteria take more than 15 min to induce aggregation although the aggregation is still FcγRIIa-dependent. Bacterial-induced aggregation is usually mediated by FcγRIIa and a co-receptor such as GPIIb/IIIa and GPIb, in the case of non-binding bacteria the co-receptor is a complement receptor. This slow aggregation requires complement assembly most likely by the alternative pathway. The delay in onset of aggregation probably reflects the time required for complement membrane attack complex formation (82, 83).
Platelet-Virus Interactions
Bacteria are not the only pathogens that affect platelet function during infection. Viral Haemorrhagic Fevers (VHF's) are very contagious zoonotic diseases that occur all over the world although more prevalent in tropical and warm climates (84). As name suggests VHF are associated with thrombocytopenia, hemorrhage, and fever caused by systemic inflammation. VHF viruses cause diseases such as Ebola, Lassa, Marburg, Yellow fever, and Dengue (69). Viruses interact with platelets mainly via FcγRIIa, integrins, DC-SIGN, and complement receptors (85). The best characterized of these interactions is with the Dengue virus which binds to DC-SIGN on platelets, causing their activation, mitochondrial dysfunction, and apoptosis via caspase-9 and 3 engagement thus contributing to systemic inflammation and platelet depletion (Figure 3) (35).
Figure 3.
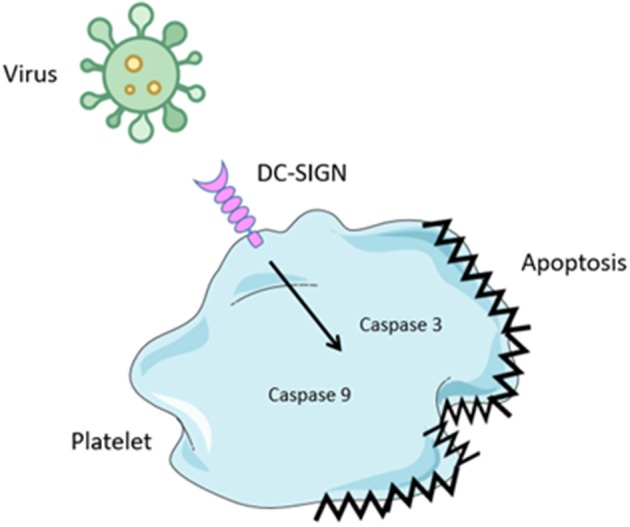
Virus binding to platelets. Several virus have been shown to bind to platelets. Binding is mediated by attachment to the Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), a c-type lectin receptor. Engagement results in activation of caspase 3 and 9 leading to platelet apoptosis. This results in thrombocytopenia.
Endothelial Cell Function
Endothelial cells make up a highly adaptive single cell layer displaying distinct apical and basolateral sides in blood vessels. They appear elongated in the direction of blood flow and form a tight cobblestone pattern. Endothelial cells are highly metabolically active and are constantly sensing alterations in the local extracellular environment (86). The endothelium receives and integrates information from hormones, neurotransmitters, pericytes, smooth muscle cells, leukocytes, platelets, viral or bacterial infection, proinflammatory cytokines, and oxygen tension (87). Endothelial cells also respond to vascular injury and high hydrodynamic shear stress. The primary function of the endothelium is to regulate systemic blood flow and maintain blood vessel wall permeability which selectively controls the movement of fluid, ions, and macromolecules between the circulating blood and the surrounding tissues.
Barrier Integrity
The passage of molecules, cells, and fluid through the endothelial cell layer is a tightly controlled process. In the healthy state endothelial cells are held together to ensure barrier integrity through intracellular junctions called tight junctions and adherens junctions (87). Critical tight junction proteins include occludins and claudins which are localized at the apical area of the intracellular cleft. These tight junction proteins are predominantly responsible for control of permeability of solutes between blood and tissues (88). Adherens junction proteins on the other hand are predominantly responsible for maintaining endothelial cell-endothelial cell connection to ensure vascular integrity. The main adherens junction protein responsible for this is VE-cadherin which is localized at the basal membrane (89, 90).
Anticoagulant and Anti-thrombotic Surface
Under resting conditions the luminal surface of the endothelium is typically both anticoagulant and antithrombotic in order to maintain fluidity within the circulation (91). Healthy endothelial cells express a number of inhibitors to prevent the synthesis and activity of the key penultimate enzyme in the coagulation cascade, thrombin. Anti-coagulation is ensured when thrombin interacts with the endothelial cell integral membrane protein thrombomodulin. Engagement induces activation of protein C which forms a complex with protein S and results in inactivation of factor VIIIa and factor Va, critical co-factors for coagulation (92). Sustained or prolonged release of mediators from healthy endothelial cells inhibit activation and adhesion of platelets to the endothelium. Prostacyclin (PGI2) is released from resting endothelial cells and binds to the Gs-coupled prostacyclin I2 receptor (IP receptor) expressed on platelets (93). Receptor activation results in activation of protein kinase A which increases platelet cAMP, thus preventing platelet activation. Similarly, lipid-soluble nitric oxide is also released from resting endothelial cells and activates protein kinase G which increases cGMP, also preventing platelet activation. Increases in cAMP or cGMP inhibit platelet aggregation, platelet secretion and platelet adherence to the vessel wall (94).
Leukocyte Recruitment
The endothelium responds to tissue invasion by transporting leukocytes from the bloodstream to subendothelial compartments. Circulating leukocytes i.e., neutrophils and monocytes are recruited to the source of infection and release TNFα, a potent endothelial cell activator. TNFα induces the expression of adhesion molecules on the surface of endothelial cells facilitating leukocyte rolling-and-adhesion. MLK is involved in the release of Weibel-Palade bodies (WPBs) stored in the endoplasmic reticulum which harbor P-selectin (95). P-selectin is subsequently transported, by WPBs, and presented on the luminal side of the endothelium (96). The initial interaction between endothelium and leukocyte involves selectins (P- and E-selectins). These selectins recognize sialyl-Lewis-x moieties of leukocyte glycoproteins allowing reversible adherence to the endothelium (97). A second interaction between lymphocyte function-associated antigen 1 (LFA-1) and macrophage-1 antigen (MAC-1) on the surface of leukocytes with intracellular adhesion molecule (ICAM)-1 and ICAM-2 on the endothelium occurs, however, the initial interaction is weak which allows leukocytes to roll along the vasculature (98, 99). Chemokines, such as CXCL8 upregulated by NF-κB and AP-1, bind to receptors on leukocytes inducing a conformational change in LFA-1 and MAC-1 (CD18/CD11b) allowing them to adhere to ligands with high affinity (100). This facilitates firm adhesion of leukocytes to the endothelium arresting rolling movement. In a process known as extravasation, LFA-1, MAC-1 (CD18/CD11b), and platelet endothelial cell adhesion molecule (PECAM, CD31) enable the leukocytes to squeeze between endothelial cell junctions. The leukocytes migrate to the basement membrane (diapedesis) where enzymes break down the extracellular matrix (101). The leukocytes continue migrating through subendothelial tissue following a chemokine (CXCL8; CCL2) concentration gradient, a process known as chemotaxis, to the source of infection where they encounter pathogens.
Endothelial Cell-Bacterial Interactions
Gram Positive Bacteria-Endothelial Cell Interactions
Bacterial interaction with endothelial cells is not well-defined and as a result few interactions have been identified. Lack of progression in this field can be attributed to poor models used to study the interactions. Endothelial cells exist in a dynamic environment surrounded by various circulatory cells (leukocytes, red blood cells, platelets) in plasma. To better reflect the physiological conditions during infection Cheung and Fischetti demonstrated that when endothelial cells are grown in the presence of tumor necrosis factor α (TNFα), significantly more S. aureus bind to the endothelial cells. S. aureus binding was further increased when fibrinogen was added to the endothelial cells (102). These findings suggest that S. aureus binds to fibrinogen and cross-links the bacteria to the “activated” endothelial cell (103). Binding was abolished when S. aureus cells were treated with trypsin, suggesting that the bacterial adhesin was a cell wall protein. Using surface-biotinylated solubilized components of S. aureus revealed a critical role for protein A in binding endothelial cells (104). Protein A is known to bind a number of plasma proteins including IgG and VWF (63, 105). Subsequent studies demonstrated that upon endothelial cell activation, release of intracellular calcium causes mobilization of weibel palade bodies which results in deposition of VWF onto the surface of the cell, thus producing a binding site for protein A on S. aureus. Claes et al. identified a second VWF binding protein expressed on S. aureus called vwb (106). This protein typically acts as a coagulase and activates prothrombin to generate fibrin. Much similar to before deposition of VWF on the surface of endothelial cells following activation or injury provides a binding site for vwb. A S. aureus strain deficient in the vwb protein or an antibody against the A1 domain of vWf significantly reduced S. aureus adhesion to endothelial cells an in vivo model of blood stream infection. Collectively these data suggest that S. aureus use at least two different mechanisms to interact with surface deposited VWF after endothelial cell activation. While these observations are critical in our understanding of how S. aureus interacts with the endothelium it doesn't identify the primary interaction that triggers endothelial cell activation to lead to VWF deposition. Using a shear based model, McDonnell et al., identified a very early interaction that drives vascular dysregulation early in infection (107). Using primary human endothelial cells sheared at physiological shear rates experienced in the vasculature the authors demonstrated that the S. aureus ClfA binds plasma fibrinogen and crosslinks the bacteria to the major endothelial cell receptor αVβ3. Binding via this mechanism resulted in VWF deposition on the surface of the endothelial cells which will allow both bacterial and platelet attachment. Binding also resulted in a loss of barrier integrity as determined by an increase in vascular permeability and loss of VE-cadherin expression. Permeability changes is a common characteristic in sepsis patients and facilitates dissemination of infection to all major organs, thus contributing to organ failure. Blocking S. aureus attachment to αVβ3 prevented VWF deposition and loss of barrier integrity. S. aureus attachment also triggered significant cytokine and chemokine release contributing to hyper-inflammation and immune cell recruitment (108).
While it is evident that S. aureus has evolved to possess various mechanisms to attach to endothelial cells the functional significance of these interactions are still under investigation. One clear functional interaction demonstrates the ability of S. aureus to internalize into endothelial cells (109). Internalization likely occurs to evade immune or anti-microbial attack, as neither immune cells or antibiotics are capable of entering into endothelial cells. Internalization is mediated by Fnbp expressed on S. aureus which binds plasma fibronectin and cross links to endothelial cell receptor α5β1.
Gram Negative Bacteria-Endothelial Cell Interactions
In contrast to S. aureus interactions with endothelial cells which focused primarily on identifying bacteria proteins, research investigating the interaction between Gram negative bacteria interaction and endothelial cells has focused on its major cell wall component LPS and the downstream signaling as a result of this interaction. Possibly the best described interaction involves the Toll-Like Receptors (TLRs). For example, TLR4 recognizes LPS (110). TLR4 signaling begins with the formation of a TLR4/myeloid differentiation 2 (MD2) complex. Upon LPS binding, homodimerization of two TLR4/MD2 receptors occurs, inducing a conformational change that allows the Toll/interleukin-1 receptor-like (TIR) domains of TLR4 to recruit adaptor proteins for the activation of MyD88-dependent pathway at the plasma membrane. These adaptor proteins subsequently activate interleukin (IL)-1R associated kinases (IRAKs) and tumor necrosis factor (TNF) receptor associated factor 6 (TRAF6) (111, 112). This, in turn, activates transforming growth factor β-activated kinase 1 (TAK1) resulting in MAP kinase kinase (MKK) inducement of the MAPK signaling cascade (113). The MAPK signaling cascade activates nuclear transcription factors such as nuclear factor (NF)-κB and activator protein (AP)-1 (114). The activation of NF-κB and AP-1 induces the production of pro-inflammatory cytokines and chemokines driving the acute phase inflammatory response (115). Furthermore, LPS can stimulate a MyD88-independent pathway following internalization of the TLR4-MD2 complex (116). TLR4-MD2 complex utilizes adaptor proteins TIR domain-containing adaptor inducing IFN-β (TRIF), TIR domain-containing adaptor molecule-1 (TICAM-1), and TRIF-related adaptor molecule (TRAM) to activate TNF receptor associated factor 3 (TRAF3) (117–119). TRAF3 activates the kinase TBK1 and IKKε stimulating interferon regulatory factor 3 (IRF3) nuclear translocation, resulting in the production of type-I interferons (116). Type-I interferons are associated with upregulation of anti-inflammatory cytokines IL-10 and IL-27 which inhibit acute phase pro-inflammatory cytokine (TNFα and IL-1) and chemokine (CXCL1 and CXCL2) production (120–122). Nuclear upregulation of both pro- and anti-inflammatory genes corresponds to type-II endothelial activation.
While there is little doubt that LPS plays a key role in driving the inflammatory response during sepsis Gram negative bacteria i.e., E. coli can also bind to endothelial cells. McHale et al., demonstrated that the highly conserved outer membrane protein A (ompA) binds directly to aVb3 on endothelial cells in the absence of plasma proteins (123). In this unique interaction, the ompA protein contains the RGD integrin recognition motif that binds directly to the RGD binding site on αVβ3. Similar to S. aureus binding to αVβ3, E. coli attachment results in loss of barrier integrity causing an increase in permeability and loss of VE-cadherin expression.
Endothelial Cell-Virus Interactions
While it generally accepted that virus are capable of binding to and dysregulating the endothelial cell barrier, the mechanisms through which they interact is not well-characterized. For example, the dengue virus envelope protein has been shown to bind to host cell Fc receptors, DC-SIGN (CD209), ICAM3 (CD-50), CD14, mannose receptor (CD206), HSP70/90, GRP78, and heparan sulfate proteoglycans (HSPGs), all of which are expressed on endothelial cells (124–129). In addition, hantaviruses have been shown to bind to endothelial cell αVβ3 which recruits VEGF receptor 2 to activate Src mediated internalization of VE-cadherin. Internalization causes loss of barrier integrity resulting in localized increases in vascular permeability and oedema (Figure 4) (130–134).
Figure 4.

Virus binding to endothelial cells. Virus can bind to the major endothelial cell receptor aVb3. Binding results in recruitment of the Vascular Endothelial cell Growth Factor Receptor 2 (VEGFR2) which activates Src kinase. This directs VE-cadherin internalization, dissociation of adherens junctions, and an increase in vascular permeability. Separation of endothelial cells allows for pathogens to escape the bloodstream and infect major organs which eventually leads to multi organ failure.
Novel Targets in Sepsis
As our basic understanding of the molecular mechanisms through which bacteria interact with either platelets or endothelial cells develops, key novel targets that drive dysregulation in both of these cells is becoming clear. While it can be argued that both platelets and endothelial cells may be innocent by-standers in these diseases there is strong evidence to support a role for them in driving the early signals that tips sepsis into a state of excessive and sustained host dysregulation. For example, if, as we have discussed above, platelets respond to pathogens by becoming activated they will aggregate forming micro-thrombi. These micro-thrombi can occlude the micro-circulation in many organs such as liver, kidney and brain. These occlusions cause ischemic damage which, as it accumulates, leads to organ failure. Furthermore, serotonin released from these activated platelets causes severe vasodilation leading to shock (135). The combination of organ damage and shock is what defines sepsis. On the other hand, several studies have demonstrated that upon entry to the bloodstream, bacteria bind to the vascular endothelium within minutes. Bacterial binding causes endothelial cell injury that results in loss of barrier integrity which causes the down-regulation of the critical adherens junction protein VE cadherin. This process facilitates bacterial dissemination to all major organs causing secondary infection and therefore contributing to organ failure. In addition, bacteria binding to the endothelium results in a significant cytokine and chemokine release driving the hyper-inflammatory response during sepsis.
As a result of our improved understanding of the molecular interactions that drive dysregulation in the bloodstream, it has led to identification of key novel targets that could control sepsis better. For example, a number of meta-analysis of retrospective studies showed that patients on aspirin or clopidogrel who develop sepsis have better outcomes than those not on an anti-platelet agent (136). This is despite the fact that patients on an anti-platelet agent are generally much older and sicker (usually anti-platelet agents are used post-MI) than those not on an anti-platelet agent. As conventional anti-platelet agents are designed to inhibit the hemostatic properties of platelets what about a new generation of anti-platelet agent that inhibits the interaction of the platelet with the pathogen? For instance a preliminary study has shown benefit of targeting the CLEC-2 ligand podoplanin in mouse models of acute respiratory distress syndrome (44).
There are three approaches to targeting platelets in sepsis. The first is to target platelet activation. This can easily be achieved using existing anti-platelet agents—primarily aspirin and clopidogrel. The advantage is that there is a lot of experience with these agents and both are off patent and thus inexpensive. This is particularly important in treating sepsis in developing countries. There is good evidence to suggest that these agents have benefit in sepsis from retrospective studies and certainly a good quality prospective study is warranted (137, 138). The disadvantage with these agents is the potential for bleeding complications. This is especially true for patients with thrombocytopenia where there is a risk of preserving platelet number at the expense of platelet function. However, the meta-analysis suggests that this risk is more than compensated by the benefit. One factor with the meta-analyses is that patients were already on anti-platelet therapy prior to developing sepsis. This is the ideal situation as it prevents platelets being activated. However, when a patient is diagnosed with sepsis and then given aspirin it might be a bigger challenge as thrombocytopenia will already be established. Thus, they need to be given anti-platelet agents as early as possible to maximize their benefit.
A precision medicine approach can also be used where the platelet receptor that binds to the pathogen is targeted. For instance, GPIIb/IIIa is the receptor for S. aureus and thus GPIIb/IIIa antagonists have the potential to prevent platelet activation in S. aureus-mediate sepsis (139). However, the difficulty here is that the pathogen must be identified prior to treatment. The big challenge in sepsis is identification of the pathogen. Once identified antibiotic therapy is the only effective solution. Another difficulty is that many of these receptors are involved in haemostasis and thus their inhibition will lead to increased bleeding problems.
A third approach is to target platelet-immune receptors. This has the potential to prevent platelet activation induced by pathogens without compromising their hemostatic properties. Furthermore, they are pathogen-independent—or at least involved with many pathogens. One example of such a strategy is to target FcγRIIa on the basis that most bacteria use it as a co-receptor for platelet activation. Small molecules that inhibit FcγRIIa have been discovered and a monoclonal antibody against FcγRIIa is entering PI studies (140, 141). Such an agent could be given to patients prior to confirmation of sepsis. This would slow the progression of sepsis allowing time for appropriate antibiotic therapy to take effect. Not only would this improve survival it may reduce the incidence and severity of post-sepsis syndrome. Furthermore, as it does not impact haemostasis there is no risk of bleeding with patients and the identity of the pathogen is not necessary.
Similarly, given the unique and critical finding that a growing number of pathogens bind directly to the vascular endothelium using the same receptor, αVβ3, inhibition of this receptor may prevent endothelial injury thus preventing the patient from progressing to shock. In addition, by preventing pathogens from internalizing into endothelial cells may also help reduce the incidence of recurrent infection which is common in sepsis, a step is also partly mediated by αVβ3. Currently there are no drugs available to prevent bacterial attachment to the vascular endothelium and therefore endothelial cell dysregulation is difficult to control, however identification of the molecular interactions between bacteria and the endothelial cells makes it an attractive future target.
Clinical Implications—Perspectives
As described before, platelet activation is a key factor in the pathogenesis of sepsis, but what has been crucially lacking in this regard are (i) widespread acceptance and acknowledgment of the fundamental role of platelets in this area, (ii) the understanding that platelet activation can lead to microthrombi from platelet aggregation which can then lead to single or multiple organ failure, (iii) thrombocytopenia in sepsis is at least partly (or predominantly in authors' opinion) due to platelet aggregation in addition to decreased platelet production and destruction by the micro-organisms, and (iv) inadequacy of the trials using antiplatelet agents in conjunction with the standard therapies in the comprehensive management of sepsis. One of the fundamental issues in translational research in this area has been delineating when the platelet activation is protective in the fight against the infections from the destructive role by forming platelet aggregates and microthrombi. Clarification of the timing when the beneficial role changes to a damaging role can aid in targeting the antiplatelet therapy before organ failure has developed. In addition, identifying which specific receptors and molecular mechanisms are involved in the different infections and at different stages would help in selecting appropriate antiplatelet therapies rather than using the conventional antiplatelet drugs in all cases.
An additional area of interest is dealing with the platelet-endothelial interactions and how they may be perturbed in sepsis (142). Although there have been many studies on the topic in the setting of cardiovascular diseases, it is still early days for clinical interventions in sepsis. Two key experimental trials have shown promise by inhibiting histones and neutrophil extracellular trap formation, which are key players in the platelet-endothelial interactions. Esmon's group showed in an animal model of sepsis that they can protect the host from DIC by specifically blocking the protein, histone H4 (143). A more recent paper noted the formation of cell-free DNA and NETS in sepsis (144). They also correlated with sepsis severity. Importantly, the use of recombinant DNAse could cause the degradation of NETs which could attenuate organ damage in combination with antibiotics.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge support from the Higher Education Authority Project grant [grant number (11/BioAT/1377E); and the Science Foundation Ireland (SFI) Career Development Award (grant number 13/CDA/2119) and the Health Research Board [grant number 06/RP/211].
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. (2018) 392:75–87. 10.1016/s0140-6736(18)30696-2 [DOI] [PubMed] [Google Scholar]
- 3.Sugimoto N, Eto K. Platelet production from induced pluripotent stem cells. J Thromb Haemost. (2017) 15:1717–27. 10.1111/jth.13736 [DOI] [PubMed] [Google Scholar]
- 4.Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. (2011) 9(Suppl 1):92–104. 10.1111/j.1538-7836.2011.04361.x [DOI] [PubMed] [Google Scholar]
- 5.Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost. (2016) 14:918–30. 10.1111/jth.13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yadav S, Storrie B. The cellular basis of platelet secretion: Emerging structure/function relationships. Platelets. (2017) 28:108–18. 10.1080/09537104.2016.1257786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holinstat M. Normal platelet function. Cancer Metastasis Rev. (2017) 36:195–8. 10.1007/s10555-017-9677-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stalker TJ, Newman DK, Ma P, Wannemacher KM, Brass LF. Platelet signaling. Handb Exp Pharmacol. (2012) 210:59–85. 10.1007/978-3-642-29423-5_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. (2012) 24:600–6. 10.1016/j.ceb.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canobbio I, Balduini C, Torti M. Signalling through the platelet glycoprotein Ib-V-IX complex. Cell Signal. (2004) 16:1329–44. 10.1016/j.cellsig.2004.05.008 [DOI] [PubMed] [Google Scholar]
- 11.Haining EJ, Cherpokova D, Wolf K, Becker IC, Beck S, Eble JA, et al. CLEC-2 contributes to hemostasis independently of classical hemITAM signaling in mice. Blood. (2017) 130:2224–8. 10.1182/blood-2017-03-771907 [DOI] [PubMed] [Google Scholar]
- 12.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost. (2005) 3:1752–62. 10.1111/j.1538-7836.2005.01429.x [DOI] [PubMed] [Google Scholar]
- 13.Shen B, Zhao X, O'Brien KA, Stojanovic-Terpo A, Delaney MK, Kim K, et al. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. (2013) 503:131–5. 10.1038/nature12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham SM, Liles WC. Platelets in sepsis: beyond hemostasis. Blood. (2016) 127:2947–9. 10.1182/blood-2016-03-706168 [DOI] [PubMed] [Google Scholar]
- 15.Dewitte A, Lepreux S, Villeneuve J, Rigothier C, Combe C, Ouattara A, et al. Blood platelets and sepsis pathophysiology: a new therapeutic prospect in critical ill patients? Ann Intensive Care. (2017) 7:115 10.1186/s13613-017-0337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. (2017) 39:517–28. 10.1007/s00281-017-0639-8 [DOI] [PubMed] [Google Scholar]
- 17.Kerrigan SW. The expanding field of platelet-bacterial interconnections. Platelets. (2015) 26:293–301. 10.3109/09537104.2014.997690 [DOI] [PubMed] [Google Scholar]
- 18.Watson CN, Kerrigan SW, Cox D, Henderson IR, Watson SP, Arman M. Human platelet activation by Escherichia coli: roles for FcgammaRIIA and integrin alphaIIbbeta3. Platelets. (2016) 27:535–40. 10.3109/09537104.2016.1148129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McNicol A, Agpalza A, Jackson EC, Hamzeh-Cognasse H, Garraud O, Cognasse F. Streptococcus sanguinis-induced cytokine release from platelets. J Thromb Haemost. (2011) 9:2038–49. 10.1111/j.1538-7836.2011.04462.x [DOI] [PubMed] [Google Scholar]
- 20.Cox D, Kerrigan SW, Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost. (2011) 9:1097–107. 10.1111/j.1538-7836.2011.04264.x [DOI] [PubMed] [Google Scholar]
- 21.Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. (2018) 122:337–51. 10.1161/CIRCRESAHA.117.310795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamzeh-Cognasse H, Berthelot P, Tardy B, Pozzetto B, Bourlet T, Laradi S, et al. Platelet toll-like receptors are crucial sensors of infectious danger moieties. Platelets. (2018) 29:533–40. 10.1080/09537104.2018.1445842 [DOI] [PubMed] [Google Scholar]
- 23.Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. (2019) 133:2178–85. 10.1182/blood-2018-11-844530 [DOI] [PubMed] [Google Scholar]
- 24.Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402. 10.1038/s41467-019-10360-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67. 10.1182/blood-2016-09-741298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brennan MP, Moriarty RD, Grennan S, Chubb AJ, Cox D. C-reactive protein binds to alphaIIbbeta3. J Thromb Haemost. (2008) 6:1239–41. 10.1111/j.1538-7836.2008.02993.x [DOI] [PubMed] [Google Scholar]
- 27.Spiller F, Oliveira Formiga R, Fernandes da Silva Coimbra J, Alves-Filho JC, Cunha TM, Cunha FQ. Targeting nitric oxide as a key modulator of sepsis, arthritis and pain. Nitric Oxide. (2019) 89:32–40. 10.1016/j.niox.2019.04.011 [DOI] [PubMed] [Google Scholar]
- 28.Gresele P, Momi S, Guglielmini G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem Pharmacol. (2019) 166:300–12. 10.1016/j.bcp.2019.05.030 [DOI] [PubMed] [Google Scholar]
- 29.Kuriri FA, O'Malley CJ, Jackson DE. Molecular mechanisms of immunoreceptors in platelets. Thrombosis Res. (2019) 176:108–14. 10.1016/j.thromres.2019.01.019 [DOI] [PubMed] [Google Scholar]
- 30.White JG. Platelets are covercytes, not phagocytes: uptake of bacteria involves channels of the open canalicular system. Platelets. (2005) 16:121–31. 10.1080/09537100400007390 [DOI] [PubMed] [Google Scholar]
- 31.Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, et al. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell. (2017) 171:1368–82.e23. 10.1016/j.cell.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 32.Moriarty RD, Cox A, McCall M, Smith SG, Cox D. Escherichia coli induces platelet aggregation in an FcgammaRIIa-dependent manner. J Thromb Haemost. (2016) 14:797–806. 10.1111/jth.13226 [DOI] [PubMed] [Google Scholar]
- 33.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. 10.1038/nm1565 [DOI] [PubMed] [Google Scholar]
- 34.Tomo S, Mohan S, Ramachandrappa VS, Samadanam DM, Suresh S, Pillai AB, et al. Dynamic modulation of DC-SIGN and Fc?R2A receptors expression on platelets in dengue. PLOS ONE. (2018) 13:e0206346. 10.1371/journal.pone.0206346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hottz ED, Oliveira MF, Nunes PC, Nogueira RM, Valls-de-Souza R, Da Poian AT, Weyrich AS, et al. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost. (2013) 11:951–62. 10.1111/jth.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, et al. The inflammatory role of platelets via their TLRs and Siglec receptors. Front Immunol. (2015) 6:83. 10.3389/fimmu.2015.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen KA, Hamzeh-Cognasse H, Palle S, Anselme-Bertrand I, Arthaud CA, Chavarin P, et al. Role of Siglec-7 in apoptosis in human platelets. PLoS ONE. (2014) 9:e106239. 10.1371/journal.pone.0106239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. (2019) 129:12–23. 10.1172/jci122955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, et al. DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J Virol. (2006) 80:8951–60. 10.1128/jvi.00136-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zahn A, Jennings N, Ouwehand WH, Allain JP. Hepatitis C virus interacts with human platelet glycoprotein VI. J Gen Virol. (2006) 87:2243–51. 10.1099/vir.0.81826-0 [DOI] [PubMed] [Google Scholar]
- 41.Claushuis TAM, de Vos AF, Nieswandt B, Boon L, Roelofs J, de Boer OJ, et al. Platelet glycoprotein VI aids in local immunity during pneumonia-derived sepsis caused by gram-negative bacteria. Blood. (2018) 131:864–76. 10.1182/blood-2017-06-788067 [DOI] [PubMed] [Google Scholar]
- 42.Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, et al. Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J Clin Invest. (2015) 125:4429–46. 10.1172/jci79070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun. (2017) 8:2239. 10.1038/s41467-017-02402-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lax S, Rayes J, Thickett DR, Watson SP. Effect of anti-podoplanin antibody administration during lipopolysaccharide-induced lung injury in mice. BMJ Open Respir Res. (2017) 4:e000257. 10.1136/bmjresp-2017-000257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brisson AR, Tan S, Linares R, Gounou C, Arraud N. Extracellular vesicles from activated platelets: a semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. (2017) 28:263–71. 10.1080/09537104.2016.1268255 [DOI] [PubMed] [Google Scholar]
- 46.Zaldivia MTK, McFadyen JD, Lim B, Wang X, Peter K. Platelet-derived microvesicles in cardiovascular diseases. Front Cardiovasc Med. (2017) 4:74. 10.3389/fcvm.2017.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Badimon L, Suades R, Fuentes E, Palomo I, Padro T. Role of platelet-derived microvesicles as crosstalk mediators in atherothrombosis and future pharmacology targets: a link between inflammation, atherosclerosis, and thrombosis. Front Pharmacol. (2016) 7:293. 10.3389/fphar.2016.00293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nomura S, Fujita S, Nakanishi T, Yokoi T, Shimamoto K, Miyamoto R, et al. Platelet-derived microparticles cause CD154-dependent activation of dendritic cells. Platelets. (2012) 23:81–2. 10.3109/09537104.2011.595850 [DOI] [PubMed] [Google Scholar]
- 49.Garcia BA, Smalley DM, Cho H, Shabanowitz J, Ley K, Hunt DF. The platelet microparticle proteome. J Proteome Res. (2005) 4:1516–21. 10.1021/pr0500760 [DOI] [PubMed] [Google Scholar]
- 50.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. (2001) 154:485–90. 10.1083/jcb.200105058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sahni A, Guo M, Sahni SK, Francis CW. Interleukin-1beta but not IL-1alpha binds to fibrinogen and fibrin and has enhanced activity in the bound form. Blood. (2004) 104:409–14. 10.1182/blood-2004-01-0126 [DOI] [PubMed] [Google Scholar]
- 52.Cha JK, Jeong MH, Bae HR, Han JY, Jeong SJ, Jin HJ, et al. Activated platelets induce secretion of interleukin-1beta, monocyte chemotactic protein-1, and macrophage inflammatory protein-1alpha and surface expression of intercellular adhesion molecule-1 on cultured endothelial cells. J Korean Med Sci. (2000) 15:273–8. 10.3346/jkms.2000.15.3.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gawaz M, Neumann FJ, Dickfeld T, Koch W, Laugwitz KL, Adelsberger H, et al. Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation. (1998) 98:1164–71. [DOI] [PubMed] [Google Scholar]
- 54.von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. (2001) 103:1772–7. 10.1161/01.cir.103.13.1772 [DOI] [PubMed] [Google Scholar]
- 55.Sut C, Tariket S, Aubron C, Aloui C, Hamzeh-Cognasse H, Berthelot P, et al. The non-hemostatic aspects of transfused platelets. Front Med (Lausanne). (2018) 5:42. 10.3389/fmed.2018.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deppermann C, Kubes P. Platelets and infection. Semin Immunol. (2016) 28:536–45. 10.1016/j.smim.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 57.Arman M, Krauel K, Tilley DO, Weber C, Cox D, Greinacher A, et al. Amplification of bacteria-induced platelet activation is triggered by FcgammaRIIA, integrin alphaIIbbeta3, and platelet factor 4. Blood. (2014) 123:3166–74. 10.1182/blood-2013-11-540526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. (2009) 302:2323–9. 10.1001/jama.2009.1754 [DOI] [PubMed] [Google Scholar]
- 59.Garciarena CD, McHale TM, Watkin RL, Kerrigan SW. Coordinated molecular cross-talk between Staphylococcus aureus, endothelial cells and platelets in bloodstream infection. Pathogens. (2015) 4:869–82. 10.3390/pathogens4040869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McDevitt D, Francois P, Vaudaux P, Foster TJ. Identification of the ligand-binding domain of the surface-located fibrinogen receptor (clumping factor) of Staphylococcus aureus. Mol Microbiol. (1995) 16:895–907. [DOI] [PubMed] [Google Scholar]
- 61.McDevitt D, Nanavaty T, House-Pompeo K, Bell E, Turner N, McIntire L, et al. Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur J Biochem. (1997) 247:416–24. [DOI] [PubMed] [Google Scholar]
- 62.Walsh EJ, Miajlovic H, Gorkun OV, Foster TJ. Identification of the Staphylococcus aureus MSCRAMM clumping factor B (ClfB) binding site in the alphaC-domain of human fibrinogen. Microbiology. (2008) 154:550–8. 10.1099/mic.0.2007/010868-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Seaghdha M, van Schooten CJ, Kerrigan SW, Emsley J, Silverman GJ, Cox D, et al. Staphylococcus aureus protein A binding to von Willebrand factor A1 domain is mediated by conserved IgG binding regions. FEBS J. (2006) 273:4831–41. 10.1111/j.1742-4658.2006.05482.x [DOI] [PubMed] [Google Scholar]
- 64.Miajlovic H, Zapotoczna M, Geoghegan JA, Kerrigan SW, Speziale P, Foster TJ. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology. (2010) 156:920–8. 10.1099/mic.0.036673-0 [DOI] [PubMed] [Google Scholar]
- 65.Binsker U, Palankar R, Wesche J, Kohler TP, Prucha J, Burchhardt G, et al. Secreted immunomodulatory proteins of Staphylococcus aureus activate platelets and induce platelet aggregation. Thromb Haemost. (2018) 118:745–57. 10.1055/s-0038-1637735 [DOI] [PubMed] [Google Scholar]
- 66.Brennan MP, Loughman A, Devocelle M, Arasu S, Chubb AJ, Foster TJ, et al. Elucidating the role of Staphylococcus epidermidis serine-aspartate repeat protein G in platelet activation. J Thromb Haemost. (2009) 7:1364–72. 10.1111/j.1538-7836.2009.03495.x [DOI] [PubMed] [Google Scholar]
- 67.Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, Lopez JA, et al. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibalpha. Mol Microbiol. (2005) 58:380–92. 10.1111/j.1365-2958.2005.04830.x [DOI] [PubMed] [Google Scholar]
- 68.Kerrigan SW, Jakubovics NS, Keane C, Maguire P, Wynne K, Jenkinson HF, et al. Role of Streptococcus gordonii surface proteins SspA/SspB and Hsa in platelet function. Infect Immun. (2007) 75:5740–7. 10.1128/iai.00909-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seo HS, Minasov G, Seepersaud R, Doran KS, Dubrovska I, Shuvalova L, et al. Characterization of fibrinogen binding by glycoproteins Srr1 and Srr2 of Streptococcus agalactiae. J Biol Chem. (2013) 288:35982–96. 10.1074/jbc.M113.513358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol. (2005) 129:101–9. 10.1111/j.1365-2141.2005.05421.x [DOI] [PubMed] [Google Scholar]
- 71.Petersen HJ, Keane C, Jenkinson HF, Vickerman MM, Jesionowski A, Waterhouse JC, et al. Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect Immun. (2010) 78:413–22. 10.1128/IAI.00664-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haworth JA, Jenkinson HF, Petersen HJ, Back CR, Brittan JL, Kerrigan SW, et al. Concerted functions of Streptococcus gordonii surface proteins PadA and Hsa mediate activation of human platelets and interactions with extracellular matrix. Cell Microbiol. (2017) 19:1. 10.1111/cmi.12667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keane C, Petersen HJ, Tilley D, Haworth J, Cox D, Jenkinson HF, et al. Multiple sites on Streptococcus gordonii surface protein PadA bind to platelet GPIIbIIIa. Thromb Haemost. (2013) 110:1278–87. 10.1160/th13-07-0580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keane C, Tilley D, Cunningham A, Smolenski A, Kadioglu A, Cox D, et al. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J Thromb Haemost. (2010) 8:2757–65. 10.1111/j.1538-7836.2010.04093.x [DOI] [PubMed] [Google Scholar]
- 75.Trotta A, Velasquez LN, Milillo MA, Delpino MV, Rodriguez AM, Landoni VI, et al. Platelets promote Brucella abortus monocyte invasion by establishing complexes with monocytes. Front Immunol. (2018) 9:1000. 10.3389/fimmu.2018.01000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fejes AV, Best MG, van der Heijden WA, Vancura A, Verschueren H, de Mast Q, et al. Impact of Escherichia coli K12 and O18:K1 on human platelets: differential effects on platelet activation, RNAs and proteins. Sci Rep. (2018) 8:16145. 10.1038/s41598-018-34473-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yeh JJ, Tsai S, Wu DC, Wu JY, Liu TC, Chen A. P-selectin-dependent platelet aggregation and apoptosis may explain the decrease in platelet count during Helicobacter pylori infection. Blood. (2010) 115:4247–53. 10.1182/blood-2009-09-241166 [DOI] [PubMed] [Google Scholar]
- 78.Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, et al. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology. (2003) 124:1846–54. 10.1016/s0016-5085(03)00397-4 [DOI] [PubMed] [Google Scholar]
- 79.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. (2006) 108:167–76. 10.1182/blood-2005-08-3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Corcoran PA, Atherton JC, Kerrigan SW, Wadstrom T, Murray FE, Peek RM, et al. The effect of different strains of Helicobacter pylori on platelet aggregation. Can J Gastroenterol. (2007) 21:367–70. 10.1155/2007/490852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Naito M, Sakai E, Shi Y, Ideguchi H, Shoji M, Ohara N, et al. Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesin but not Rgp proteinase. Mol Microbiol. (2006) 59:152–67. 10.1111/j.1365-2958.2005.04942.x [DOI] [PubMed] [Google Scholar]
- 82.Miajlovic H, Loughman A, Brennan M, Cox D, Foster TJ. Both complement- and fibrinogen-dependent mechanisms contribute to platelet aggregation mediated by Staphylococcus aureus clumping factor Infect Immun B. (2007) 75:3335–43. 10.1128/IAI.01993-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, et al. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor Mol Microbiol A. (2005) 57:804–18. 10.1111/j.1365-2958.2005.04731.x [DOI] [PubMed] [Google Scholar]
- 84.Zapata JC, Cox D, Salvato MS. The role of platelets in the pathogenesis of viral hemorrhagic fevers. PLoS Negl Trop Dis. (2014) 8:e2858. 10.1371/journal.pntd.0002858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seyoum M, Enawgaw B, Melku M. Human blood platelets and viruses: defense mechanism and role in the removal of viral pathogens. Thromb J. (2018) 16:16. 10.1186/s12959-018-0170-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. 10.1038/nri2171 [DOI] [PubMed] [Google Scholar]
- 87.McCarron JG, Lee MD, Wilson C. The endothelium solves problems that endothelial cells do not know exist. Trends Pharmacol Sci. (2017) 38:322–38. 10.1016/j.tips.2017.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hagen SJ. Non-canonical functions of claudin proteins: beyond the regulation of cell-cell adhesions. Tissue Barriers. (2017) 5:e1327839. 10.1080/21688370.2017.1327839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci. (2013) 126:2545–9. 10.1242/jcs.124529 [DOI] [PubMed] [Google Scholar]
- 90.Dejana E, Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol. (2012) 19:218–23. 10.1097/MOH.0b013e3283523e1c [DOI] [PubMed] [Google Scholar]
- 91.Esmon CT, Esmon NL. The link between vascular features and thrombosis. Annu Rev Physiol. (2011) 73:503–14. 10.1146/annurev-physiol-012110-142300 [DOI] [PubMed] [Google Scholar]
- 92.Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. (2015) 15:130. 10.1186/s12872-015-0124-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. (2010) 4:302–12. 10.2174/1874192401004010302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raslan Z, Naseem KM. The control of blood platelets by cAMP signalling. Biochem Soc Trans. (2014) 42:289–94. 10.1042/bst20130278 [DOI] [PubMed] [Google Scholar]
- 95.Vijayan KV. Myosin IIa signal von Willebrand factor release. Blood. (2018) 131:592–3. 10.1182/blood-2017-12-821579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. (2011) 117:5033–43. 10.1182/blood-2010-09-267492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chantarasrivong C, Ueki A, Ohyama R, Unga J, Nakamura S, Nakanishi I, et al. Synthesis and functional characterization of novel sialyl LewisX mimic-decorated liposomes for E-selectin-mediated targeting to inflamed endothelial cells. Mol Pharm. (2017) 14:1528–37. 10.1021/acs.molpharmaceut.6b00982 [DOI] [PubMed] [Google Scholar]
- 98.de Fougerolles AR, Stacker SA, Schwarting R, Springer TA. Characterization of ICAM-2 and evidence for a third counter-receptor for LFA-1. J Exp Med. (1991) 174:253–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stanley P, Bates PA, Harvey J, Bennett RI, Hogg N. Integrin LFA-1 alpha subunit contains an ICAM-1 binding site in domains V and VI. EMBO J. (1994) 13:1790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Green CE, Schaff UY, Sarantos MR, Lum AF, Staunton DE, Simon SI. Dynamic shifts in LFA-1 affinity regulate neutrophil rolling, arrest, and transmigration on inflamed endothelium. Blood. (2006) 107:2101–11. 10.1182/blood-2005-06-2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ley K, Reutershan J. Leucocyte-endothelial interactions in health and disease. Handb Exp Pharmacol. (2006) 176(Pt 2):97–133. [DOI] [PubMed] [Google Scholar]
- 102.Cheung AL, Koomey JM, Lee S, Jaffe EA, Fischetti VA. Recombinant human tumor necrosis factor alpha promotes adherence of Staphylococcus aureus to cultured human endothelial cells. Infect Immun. (1991) 59:3827–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cheung AL, Krishnan M, Jaffe EA, Fischetti VA. Fibrinogen acts as a bridging molecule in the adherence of Staphylococcus aureus to cultured human endothelial cells. J Clin Invest. (1991) 87:2236–45. 10.1172/jci115259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tompkins DC, Blackwell LJ, Hatcher VB, Elliott DA, O'Hagan-Sotsky C, Lowy FD. Staphylococcus aureus proteins that bind to human endothelial cells. Infect Immun. (1992) 60:965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cedergren L, Andersson R, Jansson B, Uhlen M, Nilsson B. Mutational analysis of the interaction between staphylococcal protein A and human IgG1. Protein Eng. (1993) 6:441–8. [DOI] [PubMed] [Google Scholar]
- 106.Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vandenbriele C, Vanhoorelbeke K, et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood. (2014) 124:1669–76. 10.1182/blood-2014-02-558890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McDonnell CJ, Garciarena CD, Watkin RL, McHale TM, McLoughlin A, Claes J, et al. Inhibition of major integrin alphaV beta3 reduces Staphylococcus aureus attachment to sheared human endothelial cells. J Thromb Haemost. (2016) 14:2536–47. 10.1111/jth.13501 [DOI] [PubMed] [Google Scholar]
- 108.McLoughlin A, Rochfort KD, McDonnell CJ, Kerrigan SW, Cummins PM. Staphylococcus aureus-mediated blood-brain barrier injury: an in vitro human brain microvascular endothelial cell model. Cell Microbiol. (2017) 19:3. 10.1111/cmi.12664 [DOI] [PubMed] [Google Scholar]
- 109.Peacock SJ, Foster TJ, Cameron BJ, Berendt AR. Bacterial fibronectin-binding proteins and endothelial cell surface fibronectin mediate adherence of Staphylococcus aureus to resting human endothelial cells. Microbiology. (1999) 145(Pt 12):3477–86. 10.1099/00221287-145-12-3477 [DOI] [PubMed] [Google Scholar]
- 110.Triantafilou M, Triantafilou K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. (2002) 23:301–4. 10.1016/S1471-4906(02)02233-0 [DOI] [PubMed] [Google Scholar]
- 111.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 112.He W, Qu T, Yu Q, Wang Z, Lv H, Zhang J, et al. LPS induces IL-8 expression through TLR4, MyD88, NF-kappaB and MAPK pathways in human dental pulp stem cells. Int Endod J. (2013) 46:128–36. 10.1111/j.1365-2591.2012.02096.x [DOI] [PubMed] [Google Scholar]
- 113.Molteni M, Gemma S, Rossetti C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm. (2016) 2016:6978936. 10.1155/2016/6978936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mancuso G, Midiri A, Beninati C, Piraino G, Valenti A, Nicocia G, et al. Mitogen-activated protein kinases and NF-kappa B are involved in TNF-alpha responses to group B streptococci. J Immunol. (2002) 169:1401–9. 10.4049/jimmunol.169.3.1401 [DOI] [PubMed] [Google Scholar]
- 115.Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, et al. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol. (2004) 24:7806–19. 10.1128/MCB.24.17.7806-7819.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Premkumar V, Dey M, Dorn R, Raskin I. MyD88-dependent and independent pathways of Toll-Like Receptors are engaged in biological activity of Triptolide in ligand-stimulated macrophages. BMC Chem Biol. (2010) 10:3. 10.1186/1472-6769-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. (2003) 300:1524–5. 10.1126/science.1085536 [DOI] [PubMed] [Google Scholar]
- 118.Rajaiah R, Perkins DJ, Ireland DD, Vogel SN. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc Natl Acad Sci USA. (2015) 112:8391–6. 10.1073/pnas.1424980112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. (2011) 147:868–80. 10.1016/j.cell.2011.09.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe. (2016) 19:760–9. 10.1016/j.chom.2016.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Di Paolo NC, Shafiani S, Day T, Papayannopoulou T, Russell DW, Iwakura Y, et al. Interdependence between interleukin-1 and tumor necrosis factor regulates TNF-dependent control of mycobacterium tuberculosis infection. Immunity. (2015) 43:1125–36. 10.1016/j.immuni.2015.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. (2014) 511:99–103. 10.1038/nature13489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McHale TM, Garciarena CD, Fagan RP, Smith SGJ, Martin-Loches I, Curley G, et al. Inhibition of vascular endothelial cell leak following Escherichia coli attachment in an experimental model of sepsis. Crit Care Med. (2018) 46:e805-e810. 10.1097/ccm.0000000000003219 [DOI] [PubMed] [Google Scholar]
- 124.Chen Y, Maguire T, Marks RM. Demonstration of binding of dengue virus envelope protein to target cells. J Virol. (1996) 70:8765–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miller JL, de Wet BJ, Martinez-Pomares L, Radcliffe CM, Dwek RA, Rudd PM, et al. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. (2008) 4:e17. 10.1371/journal.ppat.0040017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mondotte JA, Lozach PY, Amara A, Gamarnik AV. Essential role of dengue virus envelope protein N glycosylation at asparagine-67 during viral propagation. J Virol. (2007) 81:7136–48. 10.1128/jvi.00116-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. (2003) 197:823–9. 10.1084/jem.20021840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Navarro-Sanchez E, Altmeyer R, Amara A, Schwartz O, Fieschi F, Virelizier JL, et al. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. (2003) 4:723–8. 10.1038/sj.embor.embor866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, Ruigrok RW, et al. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology. (2002) 292:162–8. 10.1006/viro.2001.1232 [DOI] [PubMed] [Google Scholar]
- 130.Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc Natl Acad Sci USA. (1998) 95:7074–9. 10.1073/pnas.95.12.7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J Virol. (1999) 73:3951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gavrilovskaya IN, Peresleni T, Geimonen E, Mackow ER. Pathogenic hantaviruses selectively inhibit beta3 integrin directed endothelial cell migration. Arch Virol. (2002) 147:1913–31. 10.1007/s00705-002-0852-0 [DOI] [PubMed] [Google Scholar]
- 133.Matthys VS, Gorbunova EE, Gavrilovskaya IN, Mackow ER. Andes virus recognition of human and Syrian hamster beta3 integrins is determined by an L33P substitution in the PSI domain. J Virol. (2010) 84:352–60. 10.1128/jvi.01013-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Raymond T, Gorbunova E, Gavrilovskaya IN, Mackow ER. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent alphavbeta3 integrin conformers. Proc Natl Acad Sci USA. (2005) 102:1163–8. 10.1073/pnas.0406743102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci USA. (2018) 115:E1550-e1559. 10.1073/pnas.1720553115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ouyang Y, Wang Y, Liu B, Ma X, Ding R. Effects of antiplatelet therapy on the mortality rate of patients with sepsis: a meta-analysis. J Crit Care. (2019) 50:162–8. 10.1016/j.jcrc.2018.12.004 [DOI] [PubMed] [Google Scholar]
- 137.Du F, Jiang P, He S, Song D, Xu F. Antiplatelet therapy for critically Ill patients: a pairwise and Bayesian network meta-analysis. Shock. (2018) 49:616–24. 10.1097/shk.0000000000001057 [DOI] [PubMed] [Google Scholar]
- 138.Trauer J, Muhi S, McBryde ES, Al Harbi SA, Arabi YM, Boyle AJ, et al. Quantifying the effects of prior acetyl-salicylic acid on sepsis-related deaths: an individual patient data meta-analysis using propensity matching. Crit Care Med. (2017) 45:1871–9. 10.1097/ccm.0000000000002654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.O'Brien L, Kerrigan SW, Kaw G, Hogan M, Penades J, Litt D, et al. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein. Mol Microbiol A. (2002) 44:1033–44. 10.1046/j.1365-2958.2002.02935.x [DOI] [PubMed] [Google Scholar]
- 140.Pietersz GA, Mottram PL, van de Velde NC, Sardjono CT, Esparon S, Ramsland PA, et al. Inhibition of destructive autoimmune arthritis in FcgammaRIIa transgenic mice by small chemical entities. Immunol Cell Biol. (2009) 87:3–12. 10.1038/icb.2008.82 [DOI] [PubMed] [Google Scholar]
- 141.Chen B, Vousden KA, Naiman B, Turman S, Sun H, Wang S, et al. Humanised effector-null FcgammaRIIA antibody inhibits immune complex-mediated proinflammatory responses. Ann Rheum Dis. (2019) 78:228–37. 10.1136/annrheumdis-2018-213523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mangiacapra F, Barbato E. Clinical implications of platelet-vessel interaction. J Cardiovasc Transl Res. (2013) 6:310–5. 10.1007/s12265-012-9441-0 [DOI] [PubMed] [Google Scholar]
- 143.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. (2011) 118:1952–61. 10.1182/blood-2011-03-343061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS ONE. (2016) 11:e0148142. 10.1371/journal.pone.0148142 [DOI] [PMC free article] [PubMed] [Google Scholar]