

Abstract
Glucose-stimulated insulin secretion from pancreatic β-cells is controlled by ATP-regulated potassium (KATP) channels composed of Kir6.2 and sulfonylurea receptor 1 (SUR1) subunits. The KATP channel-opener diazoxide is FDA-approved for treating hyperinsulinism and hypoglycemia but suffers from off-target effects on vascular KATP channels and other ion channels. The development of more specific openers would provide critically needed tool compounds for probing the therapeutic potential of Kir6.2/SUR1 activation. Here, we characterize a novel scaffold activator of Kir6.2/SUR1 that our group recently discovered in a high-throughput screen. Optimization efforts with medicinal chemistry identified key structural elements that are essential for VU0071063-dependent opening of Kir6.2/SUR1. VU0071063 has no effects on heterologously expressed Kir6.1/SUR2B channels or ductus arteriole tone, indicating it does not open vascular KATP channels. VU0071063 induces hyperpolarization of β-cell membrane potential and inhibits insulin secretion more potently than diazoxide. VU0071063 exhibits metabolic and pharmacokinetic properties that are favorable for an in vivo probe and is brain penetrant. Administration of VU0071063 inhibits glucose-stimulated insulin secretion and glucose-lowering in mice. Taken together, these studies indicate that VU0071063 is a more potent and specific opener of Kir6.2/SUR1 than diazoxide and should be useful as an in vitro and in vivo tool compound for investigating the therapeutic potential of Kir6.2/SUR1 expressed in the pancreas and brain.
Introduction
ATP-regulated potassium (KATP) channels are key determinants of pancreatic β-cell electrophysiology, insulin secretion, and blood glucose homeostasis (Nichols, 2006). Elevations in blood glucose following a meal lead to the uptake and metabolism of glucose in pancreatic β-cells, an increase in the ratio of intracellular ATP/ADP (Nilsson et al., 1996), and ATP-mediated inhibition of KATP channel activity. KATP channel inhibition, in turn, leads to membrane potential (Vm) depolarization, action potential generation, opening of voltage-gated calcium (Ca2+) channels and influx of Ca2+, and fusion of insulin-containing granules with the plasma membrane (Nichols et al., 2007). The regulated secretion of insulin leads to the uptake, utilization, and storage of glucose by insulin-responsive target organs and return of blood glucose to preprandial levels.
Beta-cell KATP channels are hetero-octameric assemblies of four pore-forming inward rectifier potassium (Kir) channel Kir6.2 (KCNJ11) subunits and four regulatory sulfonylurea receptor 1 (SUR1; ABBC8) subunits (Aguilar-Bryan et al., 1992, 1995; Inagaki et al., 1995; Shyng and Nichols, 1997; Martin et al., 2017). Kir6.2/SUR1 channels are also expressed in various brain regions (Karschin et al., 1997; Thomzig et al., 2005), where their physiologic roles are not as well understood. The SUR2 (ABBC9) splice variants SUR2A and SUR2B assemble with Kir6.1 (and Kir6.2) in different types of muscle cells. Whereas vascular smooth muscle cells express Kir6.1/SUR2B channels predominantly, cardiac myocytes express Kir6.2/SUR2A (atria) and Kir6.1/SUR2B (ventricles) (Quayle et al., 1997; Flagg et al., 2010; Li et al., 2013; Aziz et al., 2014). The assembly of unique KATP channel subtypes enables them to carry out specific functions in diverse cell types.
Different KATP channel subtypes exhibit unique pharmacological properties that are imbued by the SUR subunit (Inagaki et al., 1995, 1996; Schwanstecher et al., 1998; D’Hahan et al., 1999a; Moreau et al., 2000). This differential pharmacology creates therapeutic opportunities for modulating the activity of KATP channel activity in specific tissues. Beta-cell Kir6.2/SUR1 channels have been targeted clinically for the treatment of type 2 diabetes since World War II, when the first generation of inhibitory sulfonylurea drugs were discovered to stimulate insulin secretion and lower blood glucose (Loubatières-Mariani, 2007). Sulfonylurea and glinide drugs with improved potency and selectivity for Kir6.2/SUR1 have been developed in subsequent years and are still in clinical use today (Kharade et al., 2016).
The KATP channel opener diazoxide (Proglycem) was approved in 1976 by the U.S. Food and Drug Administration (FDA) for treatment of disorders of excessive insulin secretion and associated hypoglycemia. Diazoxide’s hyperglycemic actions are mediated through opening of pancreatic Kir6.2/SUR1 channels, hyperpolarization of β-cell Vm, inhibition of Ca2+ entry, and suppression of insulin secretion. Diazoxide also lowers blood pressure owing to its promiscuous activity on vascular smooth muscle KATP channels (Standen et al., 1989). Reports of pulmonary hypertension led the FDA to issue a warning against its use in infants for treating hypoglycemia. In addition, diazoxide can cause shortness of breath, tachycardia, chest pain, blurred vision, and swelling, among other side effects. These are probably mediated through the known pleiotropic effects of diazoxide on voltage-gated ion channels, mitochondria, endothelial cells, and cardiac function (Coetzee, 2013). Considerable efforts have been made to develop diazoxide analogs that are more specific for Kir6.2/SUR1 to avoid these off-target effects (Rasmussen et al., 2000; Carr et al., 2003; Dabrowski et al., 2003; Alemzadeh et al., 2004; Zdravkovic et al., 2005; Hansen, 2006).
We previously reported the discovery of VU0071063, a Kir6.2/SUR1 opener that is structurally unrelated to diazoxide (Raphemot et al., 2014). It was discovered in selectivity screens of inhibitors of Aedes aegypti (mosquito) Kir1 channels. VU0071063 activates heterologously expressed Kir6.2/SUR1 channels with a half-maximal efficacy concentration (EC50) of 7 μM, acts directly on Kir6.2/SUR1 channels in excised membrane patches, and is selective for SUR1-containing KATP channels over Kir6.1/SUR2A, Kir6.2/SUR2A, Kv2.1, and other K+ channels. In contrast to diazoxide, VU0071063 does not inhibit mitochondrial complex II. VU0071063 inhibits high glucose–stimulated Ca2+ influx in mouse pancreatic β-cells, consistent with activation of native Kir6.2/SUR1 channels. Here, we expanded the characterization of VU0071063 by studying its structure-activity relationships (SAR), pharmacokinetics, and pharmacodynamics. We report that VU0071063 is more potent than diazoxide in vitro, does not cause vasodilation, and inhibits glucose-stimulated insulin secretion in vitro and in vivo.
Materials and Methods
Reagents.
Initially, small quantities of VU0071063 was purchased from Calbiochem, and larger quantities were prepared following the experimental procedure below. Reagents and solvents were purchased from Sigma-Aldrich or other commercial sources.
Chemical Synthesis.
Synthesis of VU0071063 analogs was carried about by alkylation of commercially available theophylline (1) with various benzyl bromides (2)(see Supplemental Table S1). Benzyl bromides not commercially available were synthesized by reduction of the benzaldehyde to the alcohol then conversion to the bromide via Appel reaction or by radical bromination of the methylbenzene. Generally, one equivalent of theophylline and benzyl bromide were dissolved in 1 ml of dimethylformamide (DMF) in a 2-dram screw cap vial at room temperature. Two equivalents of potassium carbonate were then added and the reactions were stirred overnight. The product was precipitated by addition of 2 ml of water and an additional hour of stirring, then isolated via vacuum filtration. Impure products were purified by normal-phase column chromatography. Products were isolated in 5%–85% yield. Guanine-based compounds were synthesized from guanosine using the method above, except dimethyl sulfoxide (DMSO) was used in place of DMF. After overnight stirring, the sugar was cleaved by addition of concentrated HCl and an hour of stirring, then isolated via vacuum filtration. For VU0071063, theophylline (50 mg, 0.278 mmol) and 4-tert-butylbenzyl bromide (63.038 mg, 0.278 mmol) were mixed in a 2-g vial. To this mixture was added DMF (1 ml, 0.278 M) and then potassium carbonate (77.831 mg, 0.555 mmol). The reaction was stirred overnight, forming a white precipitate, to which 1 ml water was added to induce further precipitation. The white solid was collected via vacuum filtration, washed with water, and dried under vacuum to yield VU0071063 (60 mg, 66%) as a white solid. Heteronuclear multiple bond coherence analysis confirmed N7 alkylation. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.55 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 5.47 (s, 2H), 3.58 (s, 3H), 3.41 (s, 3H), 1.29 (s, 9H); 13C NMR (100.6 MHz, CDCl3) δ (ppm): 155.4, 151.8, 151.7, 148.9, 140.9, 132.5, 127.9, 126.1, 107.1, 50.1, 34.7, 31.3, 29.8, 28.1; high-resolution mass spectrometry (TOF, ES+): calculated for C18H22N4O2, 326.1743; found, 326.1746.
Quantitative Thallium Flux Assays of Kir6.2/SUR1 and Kir6.1/SUR2B Activity.
Thallium flux assays of KATP channel activity were performed essentially as described previously (Raphemot et al., 2014). Briefly, stably transfected T-REx-HEK-293 cells expressing Kir6.2/SUR1 or Kir6.1/SUR2B were cultured in 384-well plates (20,000 cells/20 μl per well; black-walled, clear-bottomed PureCoat amine-coated plates; BD bioscience, Bedford, MA) with a plating media containing Dulbecco’s modified Eagle’s medium containing 10% dialyzed fetal bovine serum, 1 μg/ml G-418, 50 IU/ml penicillin, 50 μg/ml streptomycin, 5 μg/ml blasticidin S, and 250 μg/ml hygromycin, and were induced overnight with 1 μg/ml tetracycline to express Kir6.2/SUR1 or Kir6.1/SUR2B channels. On the day of the experiment, the cell culture medium was replaced with dye-loading solution containing assay buffer (Hanks’ balanced salt solution with 20 mM HEPES, pH 7.3), 0.01% (v/v) Pluronic F-127 (Life Technologies), and 1.2 μM thallium-sensitive dye Thallos-AM (WaveFront Biosciences, Franklin, TN). Following a 1-hour incubation at room temperature, the dye-loading solution was washed from the plates and replaced with 20 μl/well of assay buffer. The plates were transferred to a Panoptic kinetic imaging plate reader (Wavefront Bioscience) into which 20 μl/well of test compounds dissolved in assay buffer was added to wells. After an 8-minute incubation period, a baseline recording was collected at 1 Hz for 10 seconds (excitation 470 ± 20 nm, emission 540 ± 30 nm) followed by addition of 10 μl/well thallium stimulus buffer (125 mM NaHCO3, 1.8 mM CaSO4, 1 mM MgSO4, 5 mM glucose, 1.8 mM Tl2SO4, and 10 mM HEPES, pH 7.4) and data collection for 4 minutes. All compounds were tested at concentrations to obtain 11-point 3-fold dilution concentration-response curves. The data acquisition and analysis were performed using Waveguide (VU-HTS center) and Microsoft Excel. EC50 values were determined by fitting the Hill equation using variable-slope nonlinear regression analyses performed with GraphPad Prism version 5.01 (GraphPad Software, San Diego, CA).
Animal Studies.
The mouse studies performed at the Jaume I University, the animal housing, and all protocols were approved and conducted in accordance with the institutional animal care guidelines, Spanish animal protection law, and conformed to Directive 2010/63 EU of the European Parliament. All animal procedures performed at Vanderbilt were approved by the Vanderbilt Animal Care and Use Committee.
Pressure Myography.
Ductus arteriosus (DA) vessels, which are enriched in Kir6.1/SUR2B channels (Shelton et al., 2014; Yarboro et al., 2018), were isolated from term-gestation fetal mice and mounted on glass pipette tips submerged in microvessel perfusion chambers as previously reported (Reese et al., 2009; Pfaltzgraff et al., 2014; Hooper et al., 2016). Chambers were placed on inverted microscopes equipped with IonOptix digital image capture and dimensioning software. Vessels were pressurized in a stepwise manner to fetal mouse mean arterial pressure (20 mm Hg) using a column of Krebs buffer. Vessels were then challenged with 50 mM KCl to test for reactivity, and noncontractile vessels were excluded from further study. For dose-response studies, vessels were treated with increasing concentrations (1 nM–1 mM) of VU0071063 or pinacidil (Sigma-Aldrich). Lumen diameter measurements were allowed to plateau (20–60 minutes) before addition of the next concentration of drug. At the conclusion of each experiment, vessels were challenged with 50 mM KCl to confirm viability. An N of eight vessels from multiple litters was used for each experimental protocol. Changes in vessel diameter were expressed as a percent change in lumen diameter compared with baseline diameter at resting tone. Dose-response relationships were illustrated using point-fit curves. Myography studies are expressed as means + S.E.M. GraphPad Prism version 6.0 was used to analyze the data. Dose-response curves were compared using two-way analysis of variance (ANOVA) followed by a Tukey’s multiple comparisons test. A multiplicity-adjusted P value <0.05 was taken to be indicative of statistical significance.
Mouse Islet Electrophysiology.
The Vm of β-cells was recorded using sharp microelectrodes from dissected islets of Langerhans obtained from albino mice, killed by cervical dislocation, as previously described (Sánchez-Andrés et al., 1988). The modified Krebs solution used for the isolation had the following composition (in millimolars): 120 NaCl, 25 NaHCO3, 5 KCl, 2.6 CaCl2, and 1 MgCl2, and was equilibrated with a gas mixture containing 95% O2–5% CO2 at 37°C (pH 7 4). An Axoprobe microelectrode amplifier (Axon Instruments, Foster City, CA) was used for performing electrophysiological recordings. Borosilicate microelectrodes (o.d., 2.0 mm; i.d., 1.0 mm; Sutter Instruments, Novato, CA,) were pulled with a Narishige PE2 puller (Narishige, Japan) and filled with 3 M potassium citrate and 50 mM KCl. Data were acquired at 1-kHz frequency sampling using Clampex software (v10.6; Molecular Devices, Sunnyvale, CA) through an acquisition card (Digidata 1500; Molecular Devices), and stored on computer hard disk for further analysis using ClampFit (v10.6; Molecular Devices).
Islet cells were impaled in the presence of 10 mM glucose in modified Krebs solution to identify cells exhibiting robust oscillatory electrical behavior typical of β-cells. Beta-cells were further identified by the characteristic membrane hyperpolarization and cessation of electrical activity upon perfusion with modified Krebs solution without glucose. The minimal criteria for cell viability and quality were: at least −40 mV membrane potential during the silent phases of oscillations, 10 mV of depolarization in response to 20 mM glucose addition, and 10 mV action potential amplitude. Current clamp were performed as follows. Islets were first exposed to 20 mM glucose and then 0 mM glucose to establish maximal (100%) and minimal (0%) KATP channel inhibition, respectively (Göpel et al., 1999). Increasing concentrations of VU0071063 and diazoxide were then added to 20 mM glucose-containing buffer to induce membrane hyperpolarization. Recordings (Supplemental Fig. S1) were made for 2–3 minutes after compound addition to allow the cells to reach a steady state. The Vm recorded in the absence of glucose was subtracted from that recorded in each drug dose, and this difference was normalized to membrane potential recorded in 20 mM glucose. A similar subtraction procedure was used to determine if the KATP channel inhibitor tolbutamide (50 μM) could reverse the membrane potential hyperpolarization induced by 20 μM VU0071063. The tolbutamide effect was calculated as the percentage of the Vm recorded in the presence of 20 mM glucose. Statistically significant differences were determined by two-way ANOVA with Tukey’s or Sidak’s multiple comparison post-hoc test.
Drug Metabolism and Pharmacokinetics.
Detailed methods for determining VU0071063 drug metabolism and pharmacokinetic (DMPK) properties are described in Supplemental Methods.
In Vitro Insulin Secretion Assays.
Islets were isolated from pancreata of 8- to 10-week-old C57BL/6 mice by digesting the pancreas with collagenase P (Roche) and performing density gradient centrifugation as previously described (Roe et al., 1994). Islets were cultured for 16 hours in RPMI 1640 (11875; Gibco) medium supplemented with 15% fetal bovine serum, 100-IU ml−1 penicillin, and 100-mg ml−1 streptomycin before secretion studies. Islets were maintained in a humidified incubator at 37°C under an atmosphere of 95% air–5% CO2. After a 16-hour recovery, mouse islets were incubated with Dulbecco’s modified Eagle’s medium (DMEM) containing 5.6 mM glucose for 1 hour followed by secretion. For insulin secretion measurements, 15 islets were incubated in tubes at 37°C containing either 2 mM (low) or 14 mM (high) glucose in DMEM with or without the test compound for 1 hour. The supernatant was collected, and insulin content was analyzed using a radioimmunoassay-based detection by the Vanderbilt Hormone Core.
In Vivo Glucose Tolerance Tests.
All procedures were performed in the Vanderbilt Mouse Metabolic Phenotyping Center (MMPC; http://www.vmmpc.org) and were approved by the Vanderbilt Animal Care and Use Committee. Male C57BL/6 mice were purchased from Envigo at 8 weeks of age and studied at 10–12 weeks of age. Housing was temperature- (23°C) and humidity-controlled on a 12-hour light/dark schedule with mice given free access to food (Harlan Teklad LM-485, no. 7912) and water. Mice were injected intraperitoneally (at t = −15) with either vehicle (0.5% methyl cellulose + 10% Tween 80 in water), indicated dose of diazoxide, or VU0071063 followed by 1 g/kg of glucose (at t = 0, 15 minutes after drug dose). Blood samples (5 μl for glucose or 50 μl for insulin) were collected from the tail vein. Blood glucose was measured at = −15, 0, 5, 10, 15, 20, 30, 45, 60, 90, and 120 minutes. Samples to measure plasma insulin were taken at 0, 10, 30, 60, and 120 minutes.
Results
VU0071063 SAR.
Optimization efforts for the VU0071063 scaffold were initially aimed at improving potency toward Kir6.2/SUR1 channels heterologously expressed in HEK-293 cells (Fig. 1; Supplemental Table S1). Thallium flux assays were used to quantitate channel activation by synthetic analogs. Parallel synthesis with a varied set of benzyl bromides afforded 29 compounds, none of which improved upon VU0071063. Moving the tert-butyl group to the 3 position (3a) (Supplemental Table S1)resulted in a 3-fold loss in potency and 2-fold loss in efficacy, and the two substitution (3b) lost activity entirely. Modest activity was observed in analogs with trifluoromethoxy substitution at the 3 position (3u), and moving the trifluoromethoxy group to the 4 position (3t) was 1.5-fold less potent than VU0071063. The most potent alkyl-substituted analog was the 4-isopropyl (3c), indicating that the steric bulk of tert-butyl was necessary for optimal interaction with the channel protein. Owing to the steep SAR encountered on the eastern portion of the molecule, efforts were next focused on changes to the western ring. Removal of the N1 methyl group while keeping the 4-tert-butyl benzyl (3ae) did not prove efficacious. Shifting back to the eastern portion, substitution of a tertiary amine or hydroxyl (3ac-ad) on the benzyl ring was introduced to increase hydrogen-bond interactions. These were synthesized by action of methyl magnesium bromide on the nitrile and ketone, respectively, but these analogs were inactive. Changing the scaffold to guanine (3 AM-3an) in the hope of increasing the polarity of the ring system showed that the theophylline backbone was necessary for activity.
Fig. 1.

Overview of VU0071063 structure-activity relationships. Dose-dependent inhibition of Kir6.2/SUR1-mediated thallium flux in HEK-293 cells was evaluated for each compound to determine activity (Supplemental Table S1).
VU0071063 Does Not Activate Heterologously Expressed Kir6.1/SUR2B or Relax Ductus Arteriosus Vessel Tone.
We evaluated the activity of VU0071063 on Kir6.2/SUR1 and Kir6.1/SUR2B channels heterologously expressed in HEK-293 cells, as well as on native Kir6.1/SUR2B expressed in ductus arteriosus vessels. They were chosen for these studies because they are enriched in vascular KATP channels composed of Kir6.1 and SUR2B (Shelton et al., 2014; Yarboro et al., 2018). As shown in Fig. 2A, VU0071063 and diazoxide dose dependently opened Kir6.2/SUR1 with EC50 values of 7.44 μM [95% confidence interval (CI), 6.98–7.90 μM] and 78.42 μM (95% CI, 74.13–83.20 μM), whereas DMSO (solvent control) and pinacidil did not. Both pinacidil (EC50 = 6.50 μM; 95% CI, 5.781–7.33 μM) and diazoxide (no fit) activated Kir6.1/SUR2B channels, whereas VU0071063 (or DMSO) did not (Fig. 2B). Thus, VU0071063 is selective for Kir6.2/SUR1 over Kir6.1/SUR2B channels. Term-gestation mouse ductus arteriosus vessels were challenged next with increasing concentrations of the SUR2-specific opener pinacidil (positive control), diazoxide, or VU0071063 (1 nM–1 mM). As expected, exposure of DA vessels to pinacidil led to a dose-dependent vasodilation by activating vascular KATP channels. Diazoxide also induced relaxation at higher concentrations. However, VU0071063 had no significant effect on DA tone (Fig. 2C). Taken together, these data point to the ability of VU0071063 to discriminate between Kir6.2/SUR1 and Kir6.1/SUR2B channels.
Fig. 2.

Selectivity of VU0071063 for Kir6.2/SUR1 over Kir6.1/SUR2B. Concentration-response curves of the indicated drugs against (A) Kir6.2/SUR1 or (B) Kir6.1/SUR2B expressed in HEK-293 cells determined in thallium flux assays. (C) Concentration-response curves determined in fetal mouse ductus arteriosus vessels. Changes in lumen diameter were measured and are expressed as a percent change in lumen diameter compared with baseline diameter at resting tone. Pinacidil (green, n = 8 vessels) and diazoxide (blue, n = 8 vessels) induced dose-dependent vasodilation, whereas VU0071063 (red, n = 8 vessels) did not. *P < 0.01, pinacidil statistically significantly different from VU0071063. **P < 0.04, diazoxide statistically significantly different from VU0071063.
VU0071063 Inhibits β-Cell Excitability in Mouse Islets.
We next determined if VU0071063 modulates native KATP channel activity in a manner consistent with its pharmacological properties defined using heterologously expressed Kir6.2/SUR1 (Raphemot et al., 2014). In those studies, the EC50 for activation of Kir6.2/SUR1 expressed in HEK-293 cells was approximately 7 μM. In the present study, β-cell Vm was used as a proxy for KATP channel activity, since Vm is largely determined by the activity of resident KATP channels in these cells (Göpel et al., 1999; Nichols, 2006). Sharp microelectrodes were used to measure the Vm from excised mouse islets. Beta-cells were distinguished from other cell types by their responses to glucose (see Materials and Methods). As shown in Fig. 3, A and B, increasing bath glucose from 0 to 20 mM led to a mean ± S.E.M. depolarization of 27.7 ± 1.7 mV (n = 12) mV and action potential generation. In the continuous presence of 20 mM glucose, both diazoxide and VU0071063 led to a dose-dependent hyperpolarization of Vm and cessation of action potentials. Importantly, however, VU0071063 inhibited β-cell excitability at lower doses than did diazoxide. To ensure that the effects of VU0071063 on Vm were mediated through KATP, we confirmed that tolbutamide could reverse the effects of VU0071063 (Fig. 3D).
Fig. 3.

(A) Dose response of VU0071063 and diazoxide on the pancreatic β-cell membrane potential (Vm). After impalement the islet was perifused in the absence of glucose (0 mM glucose) to obtain maximal activation and in the presence of 20 mM glucose to obtain maximal blockade of Kir6.2/SUR1. In 20 mM glucose, increasing (A) diazoxide or (B) VU0071063 concentrations were added and Vm measured at steady state responses. (C) Accumulated dose responses of the VU0071063 and diazoxide effects on the β-cell Vm. The Vm in the presence of 0 and 20 mM glucose was considered 0% and 100%, respectively. *P < 0.001 statistically significant difference between VU0071063 and diazoxide at same dose by two-way ANOVA. (D) Summary of data from three experiments showing β-cell response to VU0071063 and tolbutamide treatment. *P < 0.0001, statistically significant difference between VU0071063 and 20 mM glucose or 20 μM VU0071063 + 50 μM Tolbutamide.
VU0071063 Inhibits Glucose-Stimulated Insulin Secretion In Vitro.
We previously reported that VU0071063 inhibits glucose-induced Ca2+ influx in isolated mouse β-cells, prompting us to examine in the present study if VU0071063 can inhibit insulin secretion in a KATP channel-dependent manner. Isolated mouse islets were treated with either 2 mM (low) or 14 mM (high) glucose alone or together with 10 μM VU0071063 or 10 μM diazoxide for 60 minutes. The inactive VU0071063 analog 34MT (7-(4-(tert-butyl)benzyl)-3-methyl-3,7-dihydro-1H-purine-2,6-dione) (Fig. 4A) was used as a negative control to confirm that any effects of VU0071063 on insulin secretion are mediated through KATP channels. As shown in Fig. 4B, high glucose significantly stimulated insulin secretion (35.6 ± 8.1 ng/ml) above that observed under low-glucose conditions (1.6 ± 0.3 ng/ml). As expected, 10 μM diazoxide significantly reduced the glucose-stimulated insulin secretion (21.14 ± 1.6 ng/ml). Importantly, the same concentration of VU0071063 virtually abolished the glucose-stimulated insulin secretion (4.01 ± 0.84 ng/ml). Consistent with VU0071063-dependent inhibition of insulin secretion being mediated through activation of KATP, 34MT had no effect on glucose-stimulated insulin secretion.
Fig. 4.

Effects of VU0071063 on glucose-induced insulin secretion from mouse islets. (A) Thallium flux concentration-response curves showing activation of Kir6.2/SUR1 channels in response to VU0071063 but not 34MT (structure shown in inset) or DMSO. (B) VU0071063 inhibits glucose-induced insulin secretion in isolated mouse islets. Mouse islets were treated with 10 μM VU0071063, 34MT, or diazoxide in the presence of low (2 mM) or high (14 mM) glucose. VU0071063 inhibited glucose-induced insulin secretion to a greater extent than the equimolar diazoxide, whereas the 34MT did not have any effect on the insulin secretion. The experiment was performed on islets isolated from three different mice. *P ≤ 0.03 statistically significantly different from low glucose, #P ≤ 0.001 statistically significantly different from high glucose + 10 μM VU0071063, @P < 0.04 statistically significantly different from high glucose +10 μM 34MT.
VU0071063 Exhibits Favorable Drug Metabolism and Pharmacokinetic Properties in Mice.
VU0071063 possesses favorable physiochemical properties and displays an attractive drug metabolism and pharmacokinetic profile in both rat and mouse (Table 1). VU0071063 is a low-molecular-weight compound (mw = 326 g/mol) with low lipophilicity (log P = 2.64) and is Lipinski compliant. In vitro, VU0071063 shows good free fraction in both rat and mouse plasma [rat fraction unbound ( fu) = 0.048, mouse fu = 0.133], good fu in rat and mouse brain homogenate (rat fu = 0.023, mouse fu = 0.024), and moderate predicted hepatic clearance in rat (CLhep = 49.6 ml/min per kilogram) and moderate-to-high in mouse (CLhep = 73.2 ml/min per kilogram). VU0071063 achieved high brain exposure in both rat (Kp = 8.06, Kp,uu = 3.86) and mouse (Kp = 2.22, Kp,uu = 0.4). In the case of the rat, it is possible that there is active uptake into the brain via a transporter mechanism. When dosed at 30 mg/kg i.p. in mice, VU0071063 achieved a time of maximal plasma concentration (Tmax) at 0.25 hours, a maximal plasma concentration exposure of 6.8 μM, and a plasma area under the curve for 0–4 hours of 11.4 μM, providing sufficient exposure to support in vivo studies (Fig. 5).
TABLE 1.
In vitro and in vivo pharmacokinetic properties of VU0071063
| Parameter | Value |
|---|---|
| VU0071063 | |
| Molecular weight | 326 |
| cLogP | 2.64 |
| In vitro pharmacokinetics | |
| Rat fu plasma | 0.048 |
| Mouse fu plasma | 0.133 |
| Rat fu brain | 0.023 |
| Mouse fu brain | 0.024 |
| Rat CLHEP (ml/min per kilogram) | 49.6 |
| Mouse CLHEP (ml/min per kilogram) | 73.2 |
| Mouse and rat plasma brain levels | |
| Rat Kp | 8.06 |
| Rat Kp,uu | 3.86 |
| Mouse Kp | 2.22 |
| Mouse Kp,uu | 0.4 |
| In vivo pharmacokinetics (mouse, IP, 30 mg/kg) | |
| Tmax | 0.25 h |
| Cmax | 6.8 μM |
| AUC(0–4 h) | 11.4 μM*h |
CLHEP, predicted hepatic clearance; Kp - ratio of total plasma AUC to total Brain AUC; Kp,uu -ratio of unbound plasma AUC to unbound Brain AUC; Cmax, maximal plasma concentration.
Fig. 5.

VU0071063 pharmacokinetic studies. Plasma and brain concentration of VU0071063 measured over time after 30 mg/kg i.p. dose.
VU0071063 Inhibits Glucose-Stimulated Insulin Secretion In Vivo.
The drug metabolism and pharmacokinetic properties of VU0071063 suggested that it could be a useful tool compound for activating Kir6.2/SUR1 channels in vivo. To determine if VU0071063 could engage β-cell KATP channels in vivo, we measured its effects on blood glucose and insulin levels in glucose tolerance tests in mice. On the basis of the Tmax of VU0071063 (Fig. 5), mice were injected intraperitoneally at t = −15 minutes with either vehicle (0.5% methyl cellulose + 10% Tween 80 in water), 50 mg/kg diazoxide, or 50 or 100 mg/kg VU0071063, followed by intraperitoneal administration of glucose (1 g/kg) 15 minutes later (t = 0 minutes). Blood samples were collected periodically from the tail vein for determination of blood glucose and insulin concentrations. As expected, intraperitoneal administration of glucose led to a significant increase of both blood glucose (Fig. 6A2) and insulin (Fig. 6B2) levels in vehicle-treated animals at the 60-minute time point. Blood glucose in vehicle-treated animals exhibited a biphasic response over the 120-minute collection period and began to wane toward the end of the experiment owing to pancreatic secretion of insulin (Fig. 6A3). Both diazoxide and VU0071063 when administered at a dose of 50 mg/kg led to a significant increase in blood glucose at 60 minutes, but only the diazoxide effect was sustained at 120 minutes. The relatively short pharmacodynamic response of VU0071063 probably reflects its rapid clearance from the plasma following administration (Fig. 5). Diazoxide or VU0071063 (50 mg/kg) significantly reduced plasma insulin concentration at the 60- and 120-minute time points; however, insulin levels began to rebound at 120 minutes compared with the 60-minute time point in diazoxide-treated mice. The effect of 50 mg/kg VU0071063 on insulin levels was sustained through 120 minutes. Increasing the dose of VU0071063 to 100 mg/kg significantly lowered insulin and increased glucose compared with 50 mg/kg diazoxide.
Fig. 6.

Intraperitoneal glucose tolerance test and insulin measurement. Mice were injected intraperitoneally with either vehicle, the indicated dose of diazoxide or VU0071063, followed by 1 g/kg of glucose (after 15 minutes of drug dose). Blood samples were collected from the tail vein once before and at several time intervals after the drug administration for up to 120 minutes for the measurement of glucose and insulin concentration. Time-course showing blood (A1) glucose and (B1) insulin concentration in response to drug treatments. Bar graph showing blood (A2) glucose and (B2) insulin concentrations at 60 minutes, and blood (A3) glucose and (B3) insulin concentrations at 120-minute time points. *P < 0.05 statistically significantly different than vehicle, #P < 0.05 statistically significantly different than 50 mg/kg diazoxide and 100 mg/kg VU0071063, @P < 0.05 statistically significantly different than 50 mg/kg diazoxide.
Discussion
The major findings of this study are: 1) There is a very steep SAR around the VU0071063 chemotype; 2) VU0071063 does not cause vasodilation of DA vessels, indicating a lack of activity on Kir6.1/SUR2B channels; 3) VU0071063 inhibits insulin secretion from isolated β-cells by hyperpolarizing Vm and does so with greater potency than diazoxide; 4) VU0071063 exhibits favorable exposure in the plasma and brain following intraperitoneal administration in mice; and 5) intraperitoneal administration of VU0071063 inhibits glucose-stimulated insulin secretion in a manner that is similar to diazoxide. Taken together with our previous study (Raphemot et al., 2014), this work indicates that VU0071063 is superior in its potency and selectivity to diazoxide and should be a useful tool compound for teasing out specific roles of Kir6.2/SUR1 channels in vitro and in vivo.
To our knowledge, VU0071063 is the only Kir6.2/SUR1-specific opener that is not a synthetic derivative of diazoxide. Novo Nordisk reported on several diazoxide derivatives that exhibit improved selectivity for Kir6.2/SUR1 over vascular KATP channels. They developed these compounds with the goal of improving glucose responsiveness in type 1 and 2 diabetic patients by inducing so-called “β-cell rest,” a concept that inhibiting the electrical activity of β-cells will protect β-cell mass and improve glucose-stimulated insulin secretion. Two of the more widely studied compounds from this work are NNC-55-0118 (6-chloro-1,1-dioxo-N-propan-2-yl-4H-thieno[3,2-e][1,2,4]thiadiazin-3-amine) and NN414 (6-chloro-N-(1-methylcyclopropyl)-1,1-dioxo-4H-thieno[3,2-e][1,2,4]thiadiazin-3-amine) (Table 2). Both compounds open Kir6.2/SUR1 channels heterologously expressed in HEK-293 cells with submicromolar potency and exhibit no activity toward Kir6.2/SUR2A or Kir6.2/SUR2B expressed in Xenopus oocytes (Dabrowski et al., 2003). Diazoxide and NNC-55-0118 (both at 100 mg/kg per day by oral gavage) showed partial and comparable (i.e., approx. 30%) efficacy in protecting β-cell mass in diabetic rats (Rasmussen et al., 2000). NN414 also showed favorable in vitro and in vivo efficacy in protecting β-cell mass and function (Carr et al., 2003; Alemzadeh et al., 2004; Maedler et al., 2004; Ritzel et al., 2004; Skak et al., 2004) and eventually progressed to human trials. In a limited study of 24 patients with type 2 diabetes treated with NN414 for 7 days, no improvements in glycemic control were observed (Zdravkovic et al., 2007).
TABLE 2.
Kir6.2/SUR1-specific opener structures and potencies
| Compound | Structure | Kir6.2/SUR1 EC50 (μM) | Reference |
|---|---|---|---|
| VU0071063 | 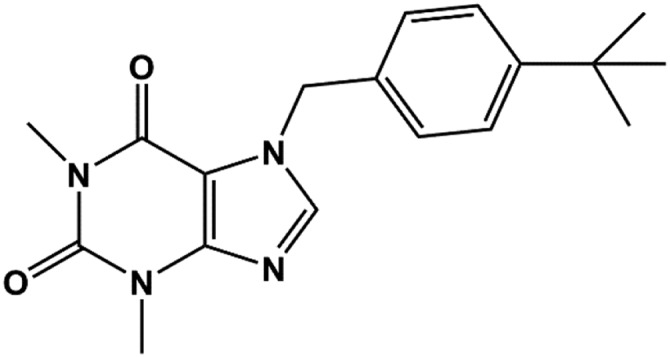 |
7 | Raphemot et al., 2014 |
| Diazoxide | 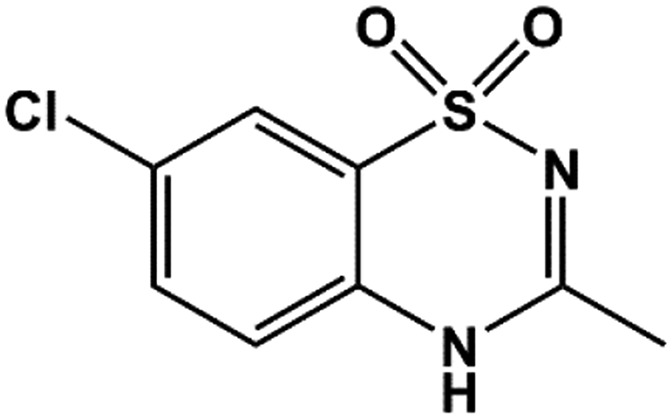 |
30 | Schwanstecher et al., 1998 |
| NN414 | 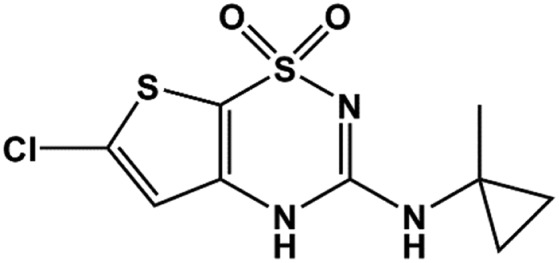 |
0.45 | Dabrowski et al., 2003 |
| NNC 55-0118 | 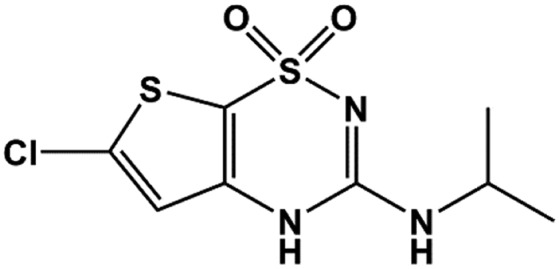 |
0.33 | Dabrowski et al., 2003 |
As noted earlier, VU0071063 is structurally distinct from NN414 and NNC-55-0118, prompting us to expand on the SAR reported previously by our group (Raphemot et al., 2014). The bicyclic system of VU0071063 is identical to that of theophylline, which prompted us originally to test if theophylline itself could activate Kir6.2/SUR1. However, theophylline at a concentration of 250 μM had no effect on Kir6.2/SUR1 activity, indicating the eastern arm of VU0071063 (Table 2) attached to theophylline is essential for opening the channel. Here, we found that the SAR of the eastern arm was very steep, as migrating the tert-butyl group from the 4-position around the ring led to a stepwise loss of potency toward Kir6.2/SUR1. Replacing the 4′-tert butyl group with several other moieties also led to a loss of activity, suggesting that the steric bulk of tert-butyl is important for optimal activity.
We previously reported that VU0071063 is selective for SUR1 over SUR2A (Raphemot et al., 2014). Thus, VU0071063 can open either Kir6.1 or Kir6.2 pores when expressed with SUR1 but not when they are expressed with SUR2A. This indicates that the VU0071063 binding site is probably localized in SUR1. This pharmacological profile predicted that VU0071063 should be able to modulate pancreas and brain (see below) KATP-channel activity without having untoward effects on vascular physiology mediated through Kir6.1/SUR2B channels. Indeed, in the present study, we showed that whereas the SUR2-selective opener pinacidil causes vasodilation of DA vessels, VU0071063 had no significant effect at a concentration that fully inhibits glucose-stimulated insulin secretion in isolated β-cells. As noted earlier, diazoxide can induce vasodilation and lower blood pressure via off-target effects on vascular KATP channels. Opening of vascular KATP channels can be mediated through direct actions on the channel or through inhibition of mitochondrial complex II and oxidative phosphorylation (Grimmsmann and Rustenbeck, 1998). The subsequent reduction of cytosolic ATP concentrations could lead to disinhibition of KATP channel activity and enhancement of diazoxide sensitivity (D’Hahan et al., 1999b). The lack of activity of VU0071063 on mitochondrial complex II (Raphemot et al., 2014) probably increases its selectivity toward pancreatic and brain over vascular KATP channels.
With emerging data implicating Kir6.2/SUR1 as a therapeutic target for the treatment of various neurologic diseases, there is a growing need for the development of brain-penetrant openers for exploring the integrative physiology and druggability of Kir6.2/SUR1 in the brain. There is surprisingly little data on the broader expression patterns of different KATP-channel subunit proteins in the central nervous system. Kir6.2 and SUR1 mRNA expression appears to be widespread and overlapping in the rodent brain, with higher levels of expression in the hippocampus, neocortex, olfactory bulb, cerebellum, midbrain, and brainstem (Karschin et al., 1997). In contrast, Kir6.1 appears to be weakly expressed in the brain (Thomzig et al., 2005). The SUR2 splice variants SUR2A and SUR2B exhibit distinct expression patterns, with SUR2A mRNA expression highest in neurons and SUR2B mRNA expression highest in certain neuronal populations, astrocytes, and oligodendrocytes (Zhou et al., 2012). With few exceptions, the myriad functions of molecularly defined KATP channels in the brain are poorly understood.
In comparison, KATP channels expressed in the hippocampus have been studied extensively and contribute to neuronal protection during ischemia (Sun et al., 2006) and memory processes (Betourne et al., 2009). They are composed primarily of Kir6.2 and SUR1 subunits (Sun et al., 2006; Betourne et al., 2009; Tanner et al., 2011), are opened by diazoxide, and are blocked by the SUR1-preferring inhibitor tolbutamide (Ohno-Shosaku and Yamamoto, 1992; Zawar and Neumcke, 2000; Matsumoto et al., 2002). Hippocampal KATP channels may provide a mechanistic link between hyperglycemia in type 2 diabetes and the development of Alzheimer disease (Ott et al., 1999; Crane et al., 2013; Huang et al., 2014). Holtzman and colleagues reported that elevation of peripheral blood glucose in a mouse model of AD led to a corresponding increase in hippocampal interstitial fluid glucose, lactate (a proxy of neuronal activity), and amyloid-β concentrations (Macauley et al., 2015). Importantly, microdialysis of glibenclamide or diazoxide into the hippocampus enhanced and inhibited, respectively, hyperglycemia-induced increase in amyloid-β. These and other data (Salgado-Puga et al., 2017; Moriguchi et al., 2018) suggest that hippocampal Kir6.2/SUR1 channels may represent a novel therapeutic target for AD. To our knowledge, the ability of NNC-55-0118 and NN414 to cross the blood-brain-barrier has not been reported. However, VU0071063 readily crosses the blood-brain-barrier following peripheral administration and could be useful for evaluating the therapeutic potential of Kir6.2/SUR1 channels for treating Alzheimer disease.
The data presented here and previously (Raphemot et al., 2014) show that VU0071063 can be used to manipulate β-cell physiology both in vitro and in vivo. In isolated cells, VU0071063 dose dependently and reversibly hyperpolarizes the β-cell membrane potential, which, in turn, inhibits glucose-stimulated Ca2+ entry and insulin secretion. The actions of VU0071063 on the β-cell membrane potential are reversed by tolbutamide, and glucose-stimulated insulin secretion is unaffected by the inactive analog 34MT, indicating that the effects are mediated through Kir6.2/SUR1. VU0071063 is more potent (this study) and faster acting (Raphemot et al., 2014) than diazoxide and might therefore represent a better in vitro tool compound for opening Kir6.2/SUR1. VU0071063 exhibits favorable drug metabolism and pharmacokinetic properties that enable it to engage KATP channels in vivo. Following intraperitoneal administration at a dose of 30 mg/kg, VU0071063 reaches peak plasma concentration of over 2000 ng/ml at 15 minutes and declines over the following 4 hours. Brain VU0071063 levels reach concentrations twice that of plasma VU0071063 at 15 minutes post-administration and then decline with a similar time-course. Diazoxide and VU0071063 were likewise efficacious at inhibiting glucose-stimulated insulin secretion and raising blood glucose over a 2-hour period. Opening of hypothalamic KATP channels with diazoxide has been reported to inhibit peripheral gluconeogenesis in rodents (Pocai et al., 2005) and humans (Kishore et al., 2011). It will be of interest in future studies to determine if activation of brain KATP channels with VU0071063 contributes to glucose homeostasis in vivo.
In conclusion, VU0071063 is a new tool compound for opening Kir6.2/SUR1 channels in vitro and in vivo. VU0071063 is specific for SUR1-containing KATP channels, opens native pancreatic KATP channels in vitro and in vivo, and inhibits glucose-stimulated calcium signaling and insulin secretion from β-cells. To our knowledge, VU0071063 is the only Kir6.2/SUR1-specific opener to be discovered in a molecular target-based high-throughput screening campaign. Unlike the other Kir6.2/SUR1-specific channel activators, NN414 and NNC-55-0118, VU0071063 is structurally unrelated to diazoxide. The discovery of VU0071063 suggests that structurally diverse activators of Kir6.2/SUR1 channels can be discovered with high-throughput screening in the future.
Acknowledgments
We acknowledge the assistance of the Vanderbilt Mouse Metabolic Phenotyping Center (DK059637) and the Vanderbilt Hormone Assay and Analytical Core (DK059637, DK020593). We thank Dr. Colin Nichols (Washington University) for the SUR2B cDNA.
Abbreviations
- ANOVA
analysis of variance
- Ca2+
voltage-gated calcium
- DA
ductus arteriosus
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EC50
half-maximal efficacy concentration
- fu
fraction unbound
- G418
(2R,3S,4R,5R,6S)-5-amino-6-[(1R,2S,3S,4R,6S)-4,6-diamino-3-[(2R,3R,4R,5R)-3,5-dihydroxy-5-methyl-4-methylaminooxan-2-yl]oxy-2-hydroxycyclohexyl]oxy- 2-(1-hydroxyethyl)oxane-3,4-dio
- KATP
ATP-regulated potassium channel
- Kir
inward rectifier potassium channel
- SUR
sulfonylurea receptor
- Tmax
maximal plasma concentration
- Vm
membrane potential
- VU0071063
7-(4-(tert-butyl)benzyl)-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione
Authorship Contributions
Participated in research design: Kharade, Sanchez-Andres, Fulton, Shelton, Engers, Hofmann, Dadi, Lantier, Jacobson, Lindsley, Denton.
Conducted experiments: Kharade, Sanchez-Andres, Fulton, Shelton, Engers, Hofmann, Dadi, Blobaum.
Contributed new reagents or analytic tools: Fulton, Engers, Lindsley.
Performed data analysis: Kharade, Sanchez-Andres, Fulton, Lantier, Shelton, Hofmann, Blobaum.
Wrote or contributed to the writing of the manuscript: Kharade, Sanchez-Andres, Shelton, Lantier, Lindsley, Denton.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants R01 DK082884; R01 DK115620]; National Heart, Lung, and Blood Institute [Grant R21 HL132805]; American Diabetes Association [Grant 1-17-IBS-024]; and American Heart Association [Grant AHA15SDG25280015].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Aguilar-Bryan L, Nichols CG, Rajan AS, Parker C, Bryan J. (1992) Co-expression of sulfonylurea receptors and KATP channels in hamster insulinoma tumor (HIT) cells. Evidence for direct association of the receptor with the channel. J Biol Chem 267:14934–14940. [PubMed] [Google Scholar]
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. (1995) Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268:423–426. [DOI] [PubMed] [Google Scholar]
- Alemzadeh R, Fledelius C, Bodvarsdottir T, Sturis J. (2004) Attenuation of hyperinsulinemia by NN414, a SUR1/Kir6.2 selective K-adenosine triphosphate channel opener, improves glucose tolerance and lipid profile in obese Zucker rats. Metabolism 53:441–447. [DOI] [PubMed] [Google Scholar]
- Aziz Q, Thomas AM, Gomes J, Ang R, Sones WR, Li Y, Ng KE, Gee L, Tinker A. (2014) The ATP-sensitive potassium channel subunit, Kir6.1, in vascular smooth muscle plays a major role in blood pressure control. Hypertension 64:523–529. [DOI] [PubMed] [Google Scholar]
- Betourne A, Bertholet AM, Labroue E, Halley H, Sun HS, Lorsignol A, Feng ZP, French RJ, Penicaud L, Lassalle JM, et al. (2009) Involvement of hippocampal CA3 K(ATP) channels in contextual memory. Neuropharmacology 56:615–625. [DOI] [PubMed] [Google Scholar]
- Carr RD, Brand CL, Bodvarsdottir TB, Hansen JB, Sturis J. (2003) NN414, a SUR1/Kir6.2-selective potassium channel opener, reduces blood glucose and improves glucose tolerance in the VDF Zucker rat. Diabetes 52:2513–2518. [DOI] [PubMed] [Google Scholar]
- Coetzee WA. (2013) Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol Ther 140:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, Haneuse S, Craft S, Montine TJ, Kahn SE, et al. (2013) Glucose levels and risk of dementia. N Engl J Med 369:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski M, Larsen T, Ashcroft FM, Bondo Hansen J, Wahl P. (2003) Potent and selective activation of the pancreatic beta-cell type K(ATP) channel by two novel diazoxide analogues. Diabetologia 46:1375–1382. [DOI] [PubMed] [Google Scholar]
- D’hahan N, Jacquet H, Moreau C, Catty P, Vivaudou M. (1999a) A transmembrane domain of the sulfonylurea receptor mediates activation of ATP-sensitive K(+) channels by K(+) channel openers. Mol Pharmacol 56:308–315. [DOI] [PubMed] [Google Scholar]
- D’hahan N, Moreau C, Prost AL, Jacquet H, Alekseev AE, Terzic A, Vivaudou M. (1999b) Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc Natl Acad Sci USA 96:12162–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. (2010) Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev 90:799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. (1999) Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol 521:717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmsmann T, Rustenbeck I. (1998) Direct effects of diazoxide on mitochondria in pancreatic B-cells and on isolated liver mitochondria. Br J Pharmacol 123:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JB. (2006) Towards selective Kir6.2/SUR1 potassium channel openers, medicinal chemistry and therapeutic perspectives. Curr Med Chem 13:361–376. [DOI] [PubMed] [Google Scholar]
- Hooper CW, Delaney C, Streeter T, Yarboro MT, Poole S, Brown N, Slaughter JC, Cotton RB, Reese J, Shelton EL. (2016) Selective serotonin reuptake inhibitor exposure constricts the mouse ductus arteriosus in utero. Am J Physiol Heart Circ Physiol 311:H572–H581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Chung CM, Leu HB, Lin LY, Chiu CC, Hsu CY, Chiang CH, Huang PH, Chen TJ, Lin SJ, et al. (2014) Diabetes mellitus and the risk of Alzheimer’s disease: a nationwide population-based study. PLoS One 9:e87095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. (1996) A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 16:1011–1017. [DOI] [PubMed] [Google Scholar]
- Karschin C, Ecke C, Ashcroft FM, Karschin A. (1997) Overlapping distribution of K(ATP) channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett 401:59–64. [DOI] [PubMed] [Google Scholar]
- Kharade SV, Nichols C, Denton JS. (2016) The shifting landscape of KATP channelopathies and the need for “sharper” therapeutics. Future Med Chem 8:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore P, Boucai L, Zhang K, Li W, Koppaka S, Kehlenbrink S, Schiwek A, Esterson YB, Mehta D, Bursheh S, et al. (2011) Activation of K(ATP) channels suppresses glucose production in humans. J Clin Invest 121:4916–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Knutsen RH, Zhang H, Osei-Owusu P, Moreno-Dominguez A, Harter TM, Uchida K, Remedi MS, Dietrich HH, Bernal-Mizrachi C, et al. (2013) Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J Am Heart Assoc 2:e000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loubatières-Mariani MM. (2007) The discovery of hypoglycemic sulfonamides. J Soc Biol 201:121–125. [DOI] [PubMed] [Google Scholar]
- Macauley SL, Stanley M, Caesar EE, Yamada SA, Raichle ME, Perez R, Mahan TE, Sutphen CL, Holtzman DM. (2015) Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J Clin Invest 125:2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Størling J, Sturis J, Zuellig RA, Spinas GA, Arkhammar PO, Mandrup-Poulsen T, Donath MY. (2004) Glucose- and interleukin-1beta-induced beta-cell apoptosis requires Ca2+ influx and extracellular signal-regulated kinase (ERK) 1/2 activation and is prevented by a sulfonylurea receptor 1/inwardly rectifying K+ channel 6.2 (SUR/Kir6.2) selective potassium channel opener in human islets. Diabetes 53:1706–1713. [DOI] [PubMed] [Google Scholar]
- Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, Chen JZ, Shyng SL. (2017) Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Komiyama S, Akaike N. (2002) Pre- and postsynaptic ATP-sensitive potassium channels during metabolic inhibition of rat hippocampal CA1 neurons. J Physiol 541:511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau C, Jacquet H, Prost AL, D’hahan N, Vivaudou M. (2000) The molecular basis of the specificity of action of K(ATP) channel openers. EMBO J 19:6644–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi S, Ishizuka T, Yabuki Y, Shioda N, Sasaki Y, Tagashira H, Yawo H, Yeh JZ, Sakagami H, Narahashi T, et al. (2018) Blockade of the KATP channel Kir6.2 by memantine represents a novel mechanism relevant to Alzheimer’s disease therapy. Mol Psychiatry 23:211–221. [DOI] [PubMed] [Google Scholar]
- Nichols CG. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440:470–476. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Koster JC, Remedi MS. (2007) beta-cell hyperexcitability: from hyperinsulinism to diabetes. Diabetes Obes Metab 9 (Suppl 2):81–88. [DOI] [PubMed] [Google Scholar]
- Nilsson T, Schultz V, Berggren PO, Corkey BE, Tornheim K. (1996) Temporal patterns of changes in ATP/ADP ratio, glucose 6-phosphate and cytoplasmic free Ca2+ in glucose-stimulated pancreatic beta-cells. Biochem J 314:91–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Yamamoto C. (1992) Identification of an ATP-sensitive K+ channel in rat cultured cortical neurons. Pflugers Arch 422:260–266. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. (1999) Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 53:1937–1942. [DOI] [PubMed] [Google Scholar]
- Pfaltzgraff ER, Shelton EL, Galindo CL, Nelms BL, Hooper CW, Poole SD, Labosky PA, Bader DM, Reese J. (2014) Embryonic domains of the aorta derived from diverse origins exhibit distinct properties that converge into a common phenotype in the adult. J Mol Cell Cardiol 69:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L. (2005) Hypothalamic K(ATP) channels control hepatic glucose production. Nature 434:1026–1031. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. (1997) ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev 77:1165–1232. [DOI] [PubMed] [Google Scholar]
- Raphemot R, Swale DR, Dadi PK, Jacobson DA, Cooper P, Wojtovich AP, Banerjee S, Nichols CG, Denton JS. (2014) Direct activation of β-cell KATP channels with a novel xanthine derivative. Mol Pharmacol 85:858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SB, Sørensen TS, Hansen JB, Mandrup-Poulsen T, Hornum L, Markholst H. (2000) Functional rest through intensive treatment with insulin and potassium channel openers preserves residual beta-cell function and mass in acutely diabetic BB rats. Horm Metab Res 32:294–300. [DOI] [PubMed] [Google Scholar]
- Reese J, O’Mara PW, Poole SD, Brown N, Tolentino C, Eckman DM, Aschner JL. (2009) Regulation of the fetal mouse ductus arteriosus is dependent on interaction of nitric oxide and COX enzymes in the ductal wall. Prostaglandins Other Lipid Mediat 88:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RA, Hansen JB, Veldhuis JD, Butler PC. (2004) Induction of beta-cell rest by a Kir6.2/SUR1-selective K(ATP)-channel opener preserves beta-cell insulin stores and insulin secretion in human islets cultured at high (11 mM) glucose. J Clin Endocrinol Metab 89:795–805. [DOI] [PubMed] [Google Scholar]
- Roe MW, Philipson LH, Frangakis CJ, Kuznetsov A, Mertz RJ, Lancaster ME, Spencer B, Worley JF, III, Dukes ID. (1994) Defective glucose-dependent endoplasmic reticulum Ca2+ sequestration in diabetic mouse islets of Langerhans. J Biol Chem 269:18279–18282. [PubMed] [Google Scholar]
- Salgado-Puga K, Rodríguez-Colorado J, Prado-Alcalá RA, Peña-Ortega F. (2017) Subclinical doses of ATP-sensitive potassium channel modulators prevent alterations in memory and synaptic plasticity induced by amyloid-β. J Alzheimers Dis 57:205–226. [DOI] [PubMed] [Google Scholar]
- Sánchez-Andrés JV, Ripoll C, Soria B. (1988) Evidence that muscarinic potentiation of insulin release is initiated by an early transient calcium entry. FEBS Lett 231:143–147. [DOI] [PubMed] [Google Scholar]
- Schwanstecher M, Sieverding C, Dörschner H, Gross I, Aguilar-Bryan L, Schwanstecher C, Bryan J. (1998) Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J 17:5529–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton EL, Ector G, Galindo CL, Hooper CW, Brown N, Wilkerson I, Pfaltzgraff ER, Paria BC, Cotton RB, Stoller JZ, et al. (2014) Transcriptional profiling reveals ductus arteriosus-specific genes that regulate vascular tone. Physiol Genomics 46:457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S, Nichols CG. (1997) Octameric stoichiometry of the KATP channel complex. J Gen Physiol 110:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skak K, Gotfredsen CF, Lundsgaard D, Hansen JB, Sturis J, Markholst H. (2004) Improved beta-cell survival and reduced insulitis in a type 1 diabetic rat model after treatment with a beta-cell-selective K(ATP) channel opener. Diabetes 53:1089–1095. [DOI] [PubMed] [Google Scholar]
- Standen NB, Quayle JM, Davies NW, Brayden JE, Huang Y, Nelson MT. (1989) Hyperpolarizing vasodilators activate ATP-sensitive K+ channels in arterial smooth muscle. Science 245:177–180. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Miki T, Seino S, French RJ. (2006) Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol 95:2590–2601. [DOI] [PubMed] [Google Scholar]
- Tanner GR, Lutas A, Martínez-François JR, Yellen G. (2011) Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J Neurosci 31:8689–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomzig A, Laube G, Prüss H, Veh RW. (2005) Pore-forming subunits of K-ATP channels, Kir6.1 and Kir6.2, display prominent differences in regional and cellular distribution in the rat brain. J Comp Neurol 484:313–330. [DOI] [PubMed] [Google Scholar]
- Yarboro MT, Durbin MD, Herington JL, Shelton EL, Zhang T, Ebby CG, Stoller JZ, Clyman RI, Reese J. (2018) Transcriptional profiling of the ductus arteriosus: comparison of rodent microarrays and human RNA sequencing. Semin Perinatol 42:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawar C, Neumcke B. (2000) Differential activation of ATP-sensitive potassium channels during energy depletion in CA1 pyramidal cells and interneurones of rat hippocampus. Pflugers Arch 439:256–262. [DOI] [PubMed] [Google Scholar]
- Zdravkovic M, Kruse M, Rost KL, Møss J, Kecskes A. (2007) The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener in subjects with type 2 diabetes. Exp Clin Endocrinol Diabetes 115:405–406. [DOI] [PubMed] [Google Scholar]
- Zdravkovic M, Kruse M, Rost KL, Møss J, Kecskes A, Dyrberg T. (2005) The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener, in healthy male subjects. J Clin Pharmacol 45:763–772. [DOI] [PubMed] [Google Scholar]
- Zhou M, He HJ, Tanaka O, Sekiguchi M, Kawahara K, Abe H. (2012) Localization of the ATP-sensitive K(+) channel regulatory subunits SUR2A and SUR2B in the rat brain. Neurosci Res 74:91–105. [DOI] [PubMed] [Google Scholar]