

Abstract
We describe the preparation of gold(I)-compounds that are amenable to efficient bioconjugation with monoclonal antibodies via activated ester or maleimide linkers. New Trastuzumab-gold conjugates were synthesized and fully chracterized. These bioconjugates are significantly more cytotoxic (sub-micromolar range) to HER2-positive breast cancer cells than the gold complexes and Trastuzumab.
Antibody-drug conjugates (ADCs) represent a promising therapeutic approach for cancer chemotherapy.1–8 These conjugates combine the antigen-targeting specificity of monoclonal antibodies (mAbs) with the cytotoxic potency of chemotherapeutics. Human tumor cells have unique or overexpressed tumor-specific antigens and mAbs can specifically bind to these antigens. Monoclonal antibodies have been used as anticancer agents as they can induce an immunological response or inhibit cellular signalling pathways. However, therapeutic efficacy is limited by the cell death effect that the mAb may generate and addition of small molecule cytotoxic payloads have shown to increase the efficacy.1
Since the vast majority of cytotoxic drugs do not discriminate tumor and healthy tissues, the use of mAbs as targeting vehicles give rise to more selective chemotherapeutic treatments. Two ADCs were approved by the US Food and Drug Administration (FDA) and the European Medicine Agency (EMA), Adcetris® (brentuximab vedotin) in 2011 and Kadcyla® (Trastuzumab emtansine or T-DM1) in 2013. More recently in 2017, inotuzumab (Besponsa™) and gemtuzumab ozogamicin (Mylotarg™) have also been approved by the FDA. In addition, over 65 ADCs (in 2017) are in the clinical pipeline.1–8 The current research focus is on new generations of ADCs based on site-specific conjugation and a different type of payload which would minimize their high cost-of-goods.
In terms of cytotoxic payloads, very few ADCs based on metal-compounds as cytotoxic loads have been described.9,10 Gold(I) compounds have emerged as potential anticancer chemotherapeutics11–14 with Auranofin([(2R,3R,4S,5R,6S)-3,4,5-tris(acetyloxy)-6-{[(triethyl-λ5-phosphanylidene)aurio]sulfanyl} oxan-2-yl]methyl acetate, a compound containing the [AuPEt3]+ fragment)15 being evaluated in clinical trials.16,17 AF is known for its redox enzymes inhibitory and ROS scavenging properties.15 The first ADC based on a N-heterocyclic carbene gold fragment conjugated to an engineered Trastuzumab antibody, Thiomab, was reported recently by Bernardes et al.18 With an GI50 in the low micromolar range, the anti-proliferative activity of this ADC in HER2 positive breast cancer cell line showed a promising moderate improvement as compared to the gold-complex drug. However, the direct conjugation of the gold atom to the free cysteine of Thiomab (Au-S-cysteine bond) may pose in vivo stability issues.19 Linkers in ADC constructs do not only serve the purpose of bridging the antibody and the cytotoxic drug, but also play a relevant role in the ADC stability during preparation, storage and systemic circulation period.19 They can also facilitate intracellular targeted release by inclusion of appropriate functionalities.19
We describe here the preparation of gold compounds containing the [Au(PPh3)]+ motif and a linker. This study stands as a proof-of-concept for the efficient and versatile bioconjugation of gold complexes to mAbs.
The preparation of the new linkers containing the [AuPPh3]+ fragment (1-4) are depicted in Scheme 1. All linkers employed are commercially available with linker b (PTDA) already described for the preparation of ADCs.20,21 The first strategy (A) is based on a copper-free cycloaddition reaction between a linker terminal alkyne and gold azide complex. This reaction was first described by Gray et al. in 2007,22 and employed in the preparation of peptides containing gold.23 The second strategy (B) is based on amide bond formation between a linker with a terminal amine and a gold thiolate compound containing a free carboxylate group ([Au(mba)(PPh3)] extensively used in our research group as a component of anticancer heterometallic complexes.24,25 The second method is simpler and more environmentally friendly than the cycloaddition reactions as it involves shorter reaction times at room temperature, and less toxic reagents (see Supporting Information, SI).
Scheme 1.
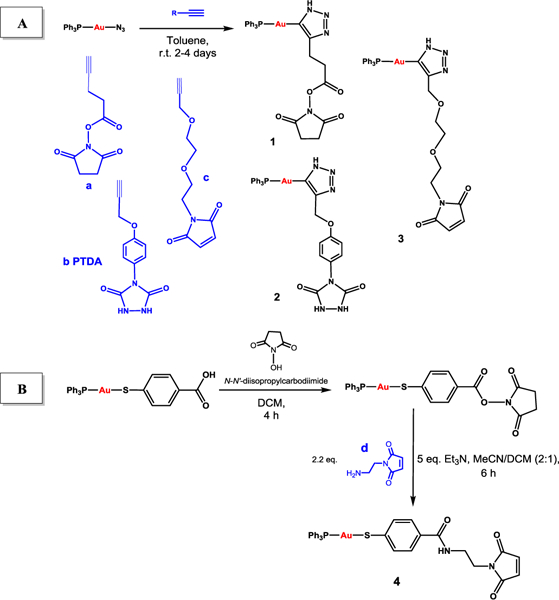
Synthetic strategies (A and B) to generate linkers containing gold(I) compounds (1-4) amenable to bioconjugation to monoclonal antibodies.
Compounds 1-4 were obtained in moderate yields (30–60%) and were fully characterized (see SI, Section 1). Compound 3 could only be characterized by elemental analysis and partially by NMR, as it is insoluble in most organic and inorganic solvents and only slightly soluble in DMSO. Compounds 1-4 stability profile in solution (either in d6-DMSO, or mixtures d6-DMSO:PBS-D2O in a ratio 3:1) was studied by 1H and 31P{1H} NMR spectroscopy. Compounds 2 and 4 remained intact in d6-DMSO for up to 14 and 40 days, respectively. Compound 4 was equally stable in d6-DMSO:PBS-D2O. Compound 1 (containing hygroscopic linker a) displayed the lowest stability, with half-lives of 6 and 5 hours in d6-DMSO and d6-DMSO/PBS-D2O, respectively. However, this stability is sufficient for subsequent bioconjugation reactions.
Two compounds (1 and 4) with two distinct groups amenable for bioconjugation and good solubility were selected for bioconjugation reactions to the anti-HER2 antibody, Trastuzumab. Trastuzumab (Herceptin™, Genentech) is a humanised IgG1 mAb, which has been exploited successfully as a targeting vector for both therapeutic agent and radioactive isotopes. Its main application lies in the treatment of HER2 positive cancers either alone, or in combination with chemotherapy, hormone blockers or tyrosine kinase inhibitors. Overexpression of HER2 correlates with increased tumor aggression and metastatic potential.26
Compound 1 activated NHS ester moiety can react with Trastuzumab available lysine residues27 (Scheme 2) while compound 4 maleimide moiety can react with Trastuzumab cysteine residues available after reduction of Trastuzumab interchain disulfide bonds.28 The selectivity of the conjugation method is of primary importance as non-site selective methods will result in heterogeneous ADC mixtures with a broad range of properties (e.g. binding potency, stability). Due to the high number of lysine accessible in a typical antibody (up to 20), lysine conjugation results in heterogeneous ADC population with a broad range of drug-to-antibody ratio (DAR) and a plethora of potential regioisomers. Methods for site-selective conjugation were evaluated to overcome ADCs’ heterogeneity and include the reduction of antibody interchain disulfide bound to provide free cysteine.28
Scheme 2.
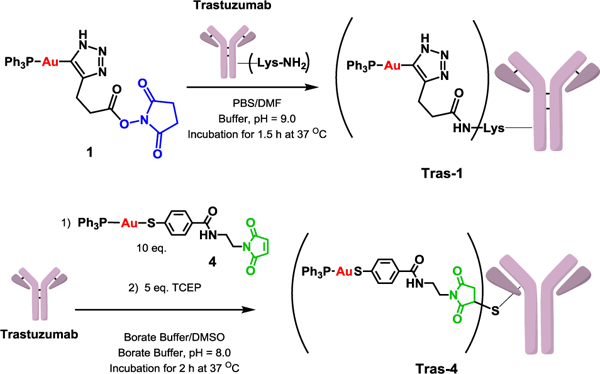
Preparation of novel Trastuzumab-gold conjugates Tras-1 and Tras-4.
Two new antibody gold-based conjugates (AGCs), Tras-1 and Tras-4, were successfully obtained using the lysine and cysteine conjugation strategies with yields of 55% and 70% respectively (see Supporting Information). To our knowledge, they are the first AGCs that conjugate gold complexes to an antibody via a linker. Tras-1 and Tras-4 were characterized by MALDI-TOF and size exclusion HPLC (see Supporting Information). Lysine conjugation resulted in Tras-1 with a DAR of 2.7–3.2 while conjugation through cysteine resulted in Tras-4 with a DAR of 2.7 (see SI). DARs strongly influence the efficacy of ADCs. Low DARs can result in reduced potency of the ADC while high DARs can result in increased pharmacokinetic of the ADC.29 To mitigate such effects, we aimed for DAR comprised between 2 and 4 for our AGCs. Furthermore, Tras-1 and Tras-4 were obtained with purity > 95%. Binding affinity of Tras-1 and Tras-4 for HER2 was evaluated in an ELISA assay (Figure 1). Compared to unmodified Trastuzumab (EC50 = 0.22 ± 0.03 nM), both Tras-1 and Tras-4 showed a slight decrease in binding affinity for HER2 with EC50 of1.13 ± 0.14 and 0.36 ± 0.01 nM, respectively. As expected, Tras-1 showed the lowest affinity for HER2. This slight decrease in affinity is hypothesized to result from the non-site selective modification of the Trastuzumab antibody through lysine conjugation. Modification through cysteine conjugation (Tras-4) resulted indeed in a much more moderate decrease in affinity. Even though, the AGCs demonstrated a decrease affinity for HER2, their binding affinity is still in the same range as unmodified Trastuzumab. The stability of the AGCs was studied in human serum over a 7-day period (see SI). The presence of unconjugated gold was evaluated by ICP-OES. No release of gold was observed up to 7 days, confirming the stability our AGCs.
Fig. 1.
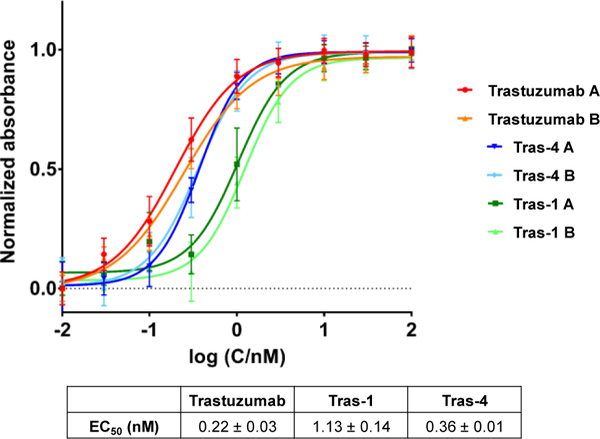
Binding affinity of AGCs Tras-1 and Tras-4 to HER2 as compared to unmodified Trastuzumab.
Cell viability assays were carried out to investigate the antiproliferative effect of our AGCs. We evaluated the cytotoxicity of soluble gold compounds 1, 2 and 4, gold starting materials [AuCl(PPh3)] and [Au(mba)(PPh3)], unmodified Trastuzumab and new AGCs Tras-1 and Tras-4 on HER2-positive breast cancer cells (MCF-7 and BT-474) and on a non-cancerous human breast cell line (MCF-10A). Auranofin was used for comparative purposes. The main results are collected in Table 1 and the results for compound 2 can be found in the SI (Table S2). Cells were incubated with the compounds for 72 hours and viability was assessed with Presto Blue (see Supporting Information including statistical analysis). Starting materials [AuCl(PPh3)] and [Au(mba)(PPh3)], were cytotoxic to the breast cancer cell line MCF-7 in the low micromolar range (EC50 = 3.6 and 2.4 μM, respectively) when compared to the values for the healthy cell line MCF-10A. Compound 4 showed a comparable cytotoxicity value (EC50 = 2.7 μM) while compound 1 was ten-times less toxic (EC50 = 38.8 μM). In general, the modification of the gold starting materials by incorporation of linkers amenable to bioconjugation did not disrupt considerably the cytotoxic potential of our gold-complexes. As mentioned before, linkers bring considerable advantages in terms of ADC stability.19
Table 1.
Cell viability EC50 values (μM) in breast cancer HER2-positive MCF-7 and BT-474 cells, and non-cancerous breast MCF-10A cells for: a) gold starting materials [AuCl(PPh3)] and [Au(mba)(PPh3)], b) new gold-containing linkers 1, and 4, and c) novel antibody gold-based conjugates Tras-1 and Tras-4. Auranofin (AF) was used as control. [a]
| [AuCl(PPh3)] | [Au(mba)(PPh3)] | 1 | 4 | Trastuzumab | Tras-1 | Tras-4 | AF | |
|---|---|---|---|---|---|---|---|---|
| MCF-7 (A) | 3.57 ± 0.33 | 2.36 ± 0.32 | 38.76 ± 4.85 | 2.73 ± 0.86 | ˃ 60 μM | 2.67 ± 0.70 | 0.63 ± 0.05 | 4.09 ± 0.07 |
| BT-474 (B) | 3.32 ± 0.10 | 3.51 ± 0.16 | 23.28 ± 0.31 | 0.81 ± 0.01 | ˃ 60 μM | 1.73 ± 0.17 | 0.32 ± 0.01 | 3.94 ± 0.33 |
| MCF-10A (C) | 18.94 ± 1.30 | 5.52 ±0.36 | 44.57 ± 5.15 | 6.77 ± 0.67 | ˃ 50 μM | 5.69 ± 0.45 | 4.04 ± 0.20 | 3.19 ± 0.45 |
| EC50(C)/EC50(A) | 5.30 ± 0.61 | 2.34 ± 0.35 | 1.15 ± 0.20 | 2.48 ± 0.82 | 0.83 ± 0.13 | 2.13 ± 0.58 | 6.41 ± 0.60 | 0.78 ± 0.11 |
| EC50(C)/EC50 (B) | 5.70 ± 0.43 | 1.57 ± 0.13 | 1.91 ± 0.22 | 8.35 ± 0.83 | 0.83 ± 0.13 | 3.29 ± 0.41 | 12.63 ± 0.74 | 0.81 ± 0.13 |
Compounds were dissolved in 1% of DMSO, DMF or DMSO/triethylene glycol (as described in the Supporting Information) and diluted with media before addition to cell culture medium for a 72 hour incubation period. Trastuzumab, Tras-1 and Tras-4 were directly diluted in media. Compounds performed in duplicate and STDEV is derived from these two separate experiments.
The AGCs, Tras-1 and Tras-4, displayed enhanced cytotoxicity (very low micromolar and sub-micromolar range) compared to the gold linkers (1 and 4) or Trastzumab in the HER2-positive MCF-7 and BT-474 cell lines. Tras-4 was a significantly more cytotoxic than starting material [Au(mba)PPh3] in the BT-474 cell line (P<0.01, unpaired t-test). Conversely, all the compounds and AGCs demonstrated reduced cytotoxicity to the non-cancerous human breast epithelial cell line MCF-10A. This observation is particularly important in the case of our AGCs since it indicates a HER2-mediated toxicity. The selectivity of Tras-4 is higher compared to Tras-1, all gold starting materials and Auranofin.
In conclusion, we have prepared antibody-gold based conjugates that incorporate linkers. Two bioconjugation strategies were investigated to offer site-specific or random functionalization of the antibody. Importantly, the conjugation of gold compounds to Trastuzumab maintains their affinity toward HER2. The two generated AGCs are significantly more cytotoxic to HER2-positive breast cancer cell lines than the gold-containing linkers and Trastuzumab. For ADCs to be efficacious, their cytotoxicity in cells has to be in the low or sub-nanomolar range.1 The very promising EC50 values (low micromolar and sub-micromolar range) obtained for the new AGCs with a non-optimized payload, the fragment [Au(PPh3)]+, warrant the study of new antibody drug conjugates based on gold-cytotoxic payloads displaying low nanomolar EC50 values (such as specific gold-N-heterocyclic carbenes).30 The synthesis described here are much less complex than those described for the incorporation of conventional payloads. Gold payloads may therefore become competitive in terms of the high cost-of-goods of ADCs. The versatility of the strategies developed will allow the synthesis of a library of gold-AGCs with a range of linkers with variable pharmacological profiles or intracellular release functionalities, enabling the preparation of gold-based biological imaging probes.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants 1SC1CA182844 and 2SC1GM127278–05A1 (M.C.). J.S.L. acknowledges the NIH grant P30 CA08748, Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, and The Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center for financial support. N.C. thanks a postdoctoral Fellowship from the Fundación Alfonso Martín Escudero. S.P. acknowledges the fellowship from the François Wallace Monahan Fellowship from the JLM Benevolent Fund. We are grateful to Mike A. Cornejo, Tina B. Elie and Dr. Jan-Philip Meyer for assistance with some of the cell viability studies and the preparation of one of the AGCs. This article is dedicated to Prof. Ernesto Carmona (Universidad de Sevilla-CSIC) on the occasion of his 70th birthday.
Footnotes
Conflicts of interest
“There are no conflicts to declare”.
Electronic Supplementary Information (ESI) available: Experimental section (synthesis and characterization of all compounds and antibody gold conjugates), NMR and MS-ESI spectra for new compounds 1–4 and their stability studies by NMR spectroscopy; HPLC and MALDI-TOF spectra for Tras-1 and Tras-4 as well as ELISA assays for the binding affinity of Tras-1, Tras-4 and Trastuzumab to HER2. It also includes details on the cell viability assays for all compounds and AGCs described. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Ducry L and Stump B, Bioconjugate Chem. 2010, 21, 5. [DOI] [PubMed] [Google Scholar]
- 2.Ducry L in Antibody-drug conjugates, (Ed.: Ducry L), Methods in Molecular Biology 1045, Springer Protocols. Humana Press, New York, 2013. [Google Scholar]
- 3.de Goeij BECG and Lambert JM, Current Opinion in Inmun. 2016, 40, 14. [DOI] [PubMed] [Google Scholar]
- 4.Jerjian TV, Glode AE, Thompson LA and O’Bryant CL, Pharmacotherapy, 2016, 36, 99. [DOI] [PubMed] [Google Scholar]
- 5.Thomas A, Teicher BA and Hassan R, Lancet Oncol. 2016, 17, e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polakis P, Pharmacol. Rev. 2016, 68, 3. [DOI] [PubMed] [Google Scholar]
- 7.Diamantis N and Banerji U, Brit. J. Cancer, 2016, 114, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert JM and Berkenblit A, Annu. Rev. Med., 2018, 69, 191. [DOI] [PubMed] [Google Scholar]
- 9.Falvo E, Treamante E, Fraioli R, Leonetti C, Zamparelli C, Boffi A, Morea V, Ceci P and Giacomini P, Nanoscale, 2013, 5, 12278. [DOI] [PubMed] [Google Scholar]
- 10.McIntosh DP, Cooke RJ, McLachlan AJ, Daley-Yates PT and Rowland M, J. Pharma. Sci. 1997, 86, 1478. [DOI] [PubMed] [Google Scholar]
- 11.Porchia M, Pellei M, Marinelli M, Tisato F, Del Bello F and Santini C, Eur. J. Med. Chem. 2018, 146, 709. [DOI] [PubMed] [Google Scholar]
- 12.Nardon C, Pettenuzzo N and Fregona D, Curr. Med. Chem. 2016, 23, 3374. [DOI] [PubMed] [Google Scholar]
- 13.Zou T, Lum CT, Lok C-N, Zhang J-J and Che CM, Chem. Soc. Rev. 2015, 44, 8786. [DOI] [PubMed] [Google Scholar]
- 14.Bertrand B and Casini A, Dalton Trans. 2014, 43, 4209. [DOI] [PubMed] [Google Scholar]
- 15.Roder C and Thomson MJ, Drugs RD, 2015, 15, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Auranofin and Sirolimus in Treating Participants With Ovarian Cancer. https://clinicaltrials.gov/ct2/show/NCT03456700
- 17.Sirolimus and Auranofin in Treating Patients With Advanced or Recurrent Non-Small Cell Lung Cancer or Small Cell Lung Cancer. https://clinicaltrials.gov/ct2/show/NCT01737502.
- 18.Matos MJ, Labao-Almeida C, Sayers C, Dada O, Tacke M and Bernardes GJI, Chem. A Eur. J., 2018, 24, 12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J, Feng J, Lu A and Zhang G, Int. J. Mol. Sci. 2016, 17, 561.27089329 [Google Scholar]
- 20.Ban H, Gavrilyuk J and Barbas CF III, J. Am. Chem. Soc., 2010, 132, 1523. [DOI] [PubMed] [Google Scholar]
- 21.Ban H, Nagano M, Gavrilyuk J, Hakamata W, Inokuma T and Barbas CF III, Biconjugate. Chem. 2013, 24, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Partyka DV, Updegraff III JB, Zeller M, Hunter AD and Gray TG, Organometallics 2007, 26, 183. [Google Scholar]
- 23.Koster SD, Alborzinia H, Can S, Kiatnovic I, Wolf S, Rubbianni R, Ott I, Rierester P, Prokop A, Merz K and Metzler-Nolte N, Chem. Sci. 2012, 3, 2062. [Google Scholar]
- 24.Fernández-Gallardo J, Elie BT, Sadhukha T, Prabha S, Sanaú M, Rotenberg SA, Ramos JM and Contel M, Chem. Sci. 2015, 6, 5269 and refs therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Contel M, Fernández-Gallardo J, Elie BT, Ramos JW, US Patent 9,315,531 (April/19/2016).
- 26.Baselga J and Swain SM, Nature Rev. Cancer 2009, 9, 463 and refs therein. [DOI] [PubMed] [Google Scholar]
- 27.Wakandar AA, Feeney MB, Rivera J, Chen Y, Kim M, Sharma VK and Wang YJ, Bioconjugate Chem., 2010, 21, 1588. [DOI] [PubMed] [Google Scholar]
- 28.Schumacher FF, Nunes JPM, Maruani A, Chudasama V, Smith MEB, Chester KA, Baker JR and Caddick S, Org. Biomol. Chem., 2014, 12, 7261–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang Y, Tang F, Yang Y, Zhao L, Zhou H, Dong J and Huang W, Sci. Reports, 2017, 7: 7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mora M, Gimeno C, Visbal R et al. , Chem. Soc. Rev. 2018, DOI: 10.1039/C8CS00570B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.