

Abstract
Changes in the methylation levels of tumor suppressor genes or proto-oncogenes are involved in the pathogenesis of hepatitis C virus (HCV) infection-induced hepatocellular carcinoma (HCC). The aim of the present study was to identify novel aberrantly methylated differentially expressed genes by integrating mRNA expression profile (GSE19665 and GSE62232) and methylation profile (GSE60753) microarrays downloaded from the Gene Expression Omnibus database. Functional enrichment analysis of screened genes was performed using the DAVID software and BinGO database. Protein-protein interaction (PPI) networks were constructed using the STRING database, followed by module analysis with MCODE software. The transcriptional and translational expression levels of crucial genes were confirmed using The Cancer Genome Atlas (TCGA) datasets and Human Protein Atlas database (HPA). A total of 122 downregulated/hypermethylated genes and 63 upregulated/hypomethylated genes were identified. These genes were enriched in the Gene Ontology biological processes terms of ‘inflammatory response’ [Fos proto-oncogene, AP-1 transcription factor subunit (FOS)] and ‘cell cycle process’ [aurora kinase A (AURKA), cyclin dependent kinase inhibitor 3 (CDKN3) and ubiquitin conjugating enzyme E2 C (UBE2C)]. PPI network and module analysis indicated that human oncogenes FOS, AURKA, CDKN3 and UBE2C may be hub genes. mRNA, protein expression and methylation levels of AURKA and FOS were validated by TCGA and HPA data. In conclusion, aberrantly methylated AURKA and FOS may be potential therapeutic targets for HCV-positive HCC.
Keywords: hepatocellular carcinoma, hepatitis C virus, methylation, differentially expressed genes, bioinformatics, aurora kinase A
Introduction
Hepatocellular carcinoma (HCC) is a common malignancy and the leading cause of cancer-related mortality, with an estimated 42,220 new cases diagnosed and 30,200 mortalities in the United States in 2018 (1). Despite curative surgical resection and recent advances in adjuvant chemotherapy, radiotherapy and liver transplantation, recurrence and metastasis occur frequently, leading to the overall 5-year survival rate <20% (2). Although multiple factors have been linked to the development and progression of HCC, hepatitis virus infection is considered to be the predominant underlying cause. It has been reported that the burden of HCC parallels the prevalence of hepatitis C virus (HCV) (3). The 10-year survival rate was reported to be approximately 35% in HCC patients with HCV (4). Thus, it is necessary to explore the molecular mechanisms of HCV-associated hepatocarcinogenesis to screen novel prognostic biomarkers and to develop effective therapeutic strategies.
Although the mechanism of the pathogenesis by which HCV induces HCC is currently unclear, epigenetic changes (such as DNA methylation) have been demonstrated to serve fundamental roles. For example, aberrant hypermethylation of tumor suppressor genes or hypomethylation of proto-oncogenes may result in the decrease or increase in their expression levels and induce excessive proliferation, migration and invasion of hepatocytes (5). Methylation of several genes has been reported in HCV-associated HCC (6). For example, Ramadan et al demonstrated that the frequency of aberrant methylation in the promoter region of serine protease inhibitor kunitz-type 2 gene was significantly higher in HCV-positive HCC cases compared with HCV-positive cirrhosis and normal control patients (7). Takagi et al reported that CpG islands in zygote arrest 1 exon 1 had a higher methylation level in HCV-positive HCC compared with non-tumorous tissues (8). Tsunedomi et al not only demonstrated a correlation between DNA methylation and mRNA expression levels of ATP-binding cassette subfamily B member 6 (ABCB6), but also revealed that aberrant mRNA and DNA methylation levels of ABCB6 may serve as predictive biomarkers for early intrahepatic recurrence of HCV-positive HCC (9). In vitro studies by Mileo et al (10) and Quan et al (11) demonstrated that HCV may promote the progression of HCC cells by downregulating the protein and mRNA levels of proline rich protein BstNI subfamily 2/p130 and secreted frizzled-related protein, a Wnt antagonist, by inducing promoter hypermethylation. However, genes with aberrant DNA methylation for HCV-positive HCC remain largely under-investigated.
The aim of the present study was to identify novel genes to explain the development of HCV-positive HCC by combining mRNA expression profile and methylation profile microarrays, and to confirm their transcriptional and translational expression using The Cancer Genome Atlas (TCGA) datasets and Human Protein Atlas database (HPA). The results of the present study may provide novel therapeutic targets for HCV-positive HCC.
Materials and methods
Microarray data collection
Three microarray datasets: GSE19665 (12), GSE62232 (13) and GSE60753 (14) were downloaded from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) on July 25, 2018. The GSE19665 dataset [platform, GPL570 Affymetrix Human Genome U133 Plus 2.0 Array (HG-U133_Plus_2)] was used to analyze the gene expression profile in 20 HCC and 20 matched non-cancerous tissues, among which 5 pairs were HCV-positive; the GSE62232 dataset (platform, HG-U133_Plus_2) was used to detect the gene expression profile in 81 HCC (including 9 HCV-positive) and 10 normal liver tissues; and the GSE60753 dataset (platform, GPL13534, Illumina HumanMethylation450 BeadChip) was used to determine the DNA methylation profile in 156 HCC (including 12 HCV-positive), 34 normal liver tissues and 1 HCC cell line. Normal liver tissues were not matched with HCC in GSE62232 and GSE60753, but only collected from patients without HCC (such as benign cysts). Only the HCV-positive HCC and normal control samples were used for our following analyses.
Microarray data preprocessing
For the GSE19665 and GSE62232 datasets, the raw data were preprocessed using the oligo package (version 1.42.0; http://www.bioconductor.org/packages/release/bioc/html/oligo.html) in Bioconductor R package (version 3.4.1; http://www.R-project.org), including data transformation, missing value imputation with median, background correction with microarray analysis suite method and quantile normalization. For the GSE60753 dataset, the DNA methylation β values were downloaded and the genes were annotated according to the annotation information from the corresponding platform.
Differential gene expression and methylation analysis
Differentially expressed genes (DEGs) and differentially methylated genes (DMGs) between HCV-positive HCC and control samples were identified using the Linear Models for Microarray Data method (version 3.34.0; http://bioconductor.org/packages/release/bioc/html/limma.html) (15) in the Bioconductor R package. False discovery rate (FDR) <0.05 and |logFC| >1 were defined as the statistical threshold value; where FC is fold change. Hierarchical clustering (16) was performed for the DEGs and DMGs using pheatmap R package (version 1.0.8; http://cran.r-project.org/web/packages/pheatmap) based on Euclidean distance and the results were displayed as a heat map. The upregulated and downregulated shared DEGs in GSE19665 and GSE62232 datasets were then overlapped with the hypomethylated and hypermethylated DMGs, respectively, to identify the methylated-mediated genes. Additionally, the methylated-mediated genes were compared with the human oncogenes downloaded from the ONGene database (http://ongene.bioinfo-minzhao.org) (17) to screen for HCC-related oncogenes.
Protein-protein interaction (PPI) network construction and module analysis
The STRING database (version 10.0; http://string-db.org) (18) was used to predict the interactions between DEGs, and a PPI network was constructed using the obtained interaction pairs using the Cytoscape software (version 3.6.1; http://www.cytoscape.org) (19). The topological characteristics of the nodes (proteins) in the PPI network were computed using the CytoNCA plugin in the Cytoscape software (http://apps.cytoscape.org/apps/cytonca) (20) to determine the hub genes, including ‘degree’ [the number of edges (interactions) of a node (protein)], ‘betweenness’ (the number of shortest paths that run through a node), ‘closeness centrality’ (CC; the average length of the shortest paths to access all other proteins in the network) and ‘average path length’ (APL; the average distance between all pairs of nodes). Functionally related modules with well-interconnected genes were further identified in the PPI network using the Molecular Complex Detection (MCODE; version 1.4.2; http://apps.cytoscape.org/apps/mcode) algorithm (21) with the following scoring options: Degree cutoff=2; node score cutoff=0.2; K-core=2. Modules with MCODE score (Density*Nodes) >3 and node number >6 were considered to be significant.
Function enrichment analysis
Gene Ontology (GO) biological process terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed for the methylated-mediated DEGs using the Database for Annotation, Visualization and Integrated Discovery (DAVID) online tool (version 6.8; http://david.abcc.ncifcrf.gov) (22) and BinGO (23) plugin in Cytoscape to predict their underlying functions. Statistical significance was defined as P<0.05 or FDR <0.05.
Validation of the selected methylation-mediated DEGs
The mRNA and methylation sequencing data of HCC tissues and normal liver tissues from patients without HCC were extracted from TCGA database (https://portal.gdc.cancer.gov) prior to July 25, 2018, which were measured on the Illumina HiSeq 2000 RNA Sequencing platform. Only the HCV-positive HCC samples were included in the present study to confirm the expression consistency of the methylated-mediated DEGs. The expression difference between HCC and controls was determined by Student's independent t-test using the TCGA data. P<0.05 was considered to indicate a statistically significant difference.
In addition, protein expression levels of the methylated-mediated DEGs were also validated by the HPA database (version 18; http://www.proteinatlas.org) (24), which was used to evaluate the translational levels of the DEGs by immunohistochemistry. Results are presented as the sum of scores of staining intensity (negative, weak, moderate or strong) and the percentage of stained cells (<25, 25–75 or >75%): Negative-not detected; weak + <25%-not detected; weak + 25–75 or 75%-low; moderate <25%-low; moderate + 25–75 or 75%-medium; strong <25%-medium, strong + 25–75 or 75%-high.
Results
Differential gene expression and methylation
A flowchart depicting the analytical process is presented as Fig. 1. Following preprocessing, a total of 1,306 (735 downregulated and 571 upregulated) and 1,249 (330 downregulated and 919 upregulated) DEGs were identified between HCV-positive HCC and control tissues in GSE19665 and GSE62232 datasets, respectively, using the cut-off criteria FDR <0.05 and |logFC| >1. The hierarchical-clustering heat map (Fig. 2A and B) indicated that DEGs may be used to distinguish HCV-positive HCC from control samples.
Figure 1.

Analysis plan. The key genes were determined by integrating the methylation and mRNA expression profile microarray datasets and then confirmed using TCGA and HPA data. DAVID, Database for Annotation, Visualization and Integrated Discovery; DEGs, differentially expressed genes; DMGs, differentially methylated genes; GEO, Gene Expression Omnibus; GO, Gene Ontology; HPA, Human Protein Atlas; KEGG, Kyoto Encyclopedia of Genes and Genomes; limma, Linear Models for Microarray Data; PPI, protein-protein interaction; TCGA, The Cancer Genome Atlas.
Figure 2.

Differentially expressed and methylated genes in HCV-positive HCC. (A-C) Hierarchical clustering and heat map analysis of differentially expressed genes in (A) GSE19665 and (B) GSE62232 and of (C) differentially methylated genes in GSE60753. Red, high expression (or hypermethylation); green, low expression (or hypomethylation). HCC hepatocellular carcinoma; HCV, hepatitis C virus.
A total of 23,408 methylated probes were annotated to genes in the GSE60753 dataset. By comparing with normal samples, 1,448 DMGs (903 hypomethylated and 545 hypermethylated) were also obtained in HCV-positive HCC tissues. The hierarchical-clustering heat map (Fig. 2C) revealed that DMGs were different between HCV-positive HCC and control samples.
Following comparison of the DEGs identified in GSE19665 and GSE62232 datasets, 173 downregulated and 278 upregulated DEGs were revealed to be common and their expression trends were consistent in the two datasets (Fig. 3A). Further integration with the DMGs found 122 DEGs were downregulated by DNA hypermethylation and 63 DEGs were upregulated by DNA hypomethylation (Fig. 3B). Among the methylated DEGs, nine were suggested as human oncogenes according to the prediction by ONGene database; five were downregulated: Inhibitor of DNA binding 1, HLH protein (ID1), epithelial cell adhesion molecule (EPCAM), Fos proto-oncogene, AP-1 transcription factor subunit (FOS), ID2 and placenta specific 8 (PLAC8), whereas four were upregulated: Aurora kinase A (AURKA), ubiquitin conjugating enzyme E2 C (UBE2C), erb-b2 receptor tyrosine kinase 3 (ERBB3) and cyclin-dependent kinase inhibitor 3 (CDKN3).
Figure 3.

Shared DEGs in GSE19665 and GSE62232 hepatitis C virus-positive hepatocellular carcinoma datasets. (A and B) Venn diagrams demonstrate (A) shared DEGs in GSE19665 and GSE62232 and (B) their association with DMGs in GSE60753. DEGs, differentially expressed genes; DMGs, differentially methylated genes.
Functional enrichment for the DEGs
The 122 downregulated/hypermethylated and 63 upregulated/hypomethylated DEGs were respectively uploaded to DAVID to predict their functions. Using the threshold value of FDR <0.05, 18 GO biological process terms were obtained for the downregulated/hypermethylated DEGs, including ‘response to wounding’ (FOS) and ‘inflammatory response’ (FOS), whereas 14 GO biological process terms were enriched for the upregulated/hypomethylated DEGs, including ‘cell cycle process’ (AURKA, CDKN3 and UBE2C) and ‘cell cycle’ (CDKN3 and MCM6) (Fig. 4A; Table I). Furthermore, KEGG pathway enrichment analysis was also performed, which resulted in 11 KEGG pathways identified as enriched for downregulated/hypermethylated and 4 enriched for upregulated/hypomethylated DEGs, using the threshold value of P<0.05 (FDR >0.05 for all pathways) (Fig. 4B; Table II). The KEGG pathway enrichment results were consistent with GO biological process term analysis, in which inflammatory-related ‘cytokine-cytokine receptor interaction’ pathway was enriched for downregulated/hypermethylated DEGs, and ‘DNA replication’ and ‘cell cycle’ were enriched for upregulated/hypomethylated DEGs.
Figure 4.

GO and KEGG enrichment analyses of the methylation-related DEGs. (A) GO biological process term analysis. Triangles indicate the significance level (P-value adjusted to FDR). (B) KEGG pathway enrichment. Left, downregulated/hypermethylated DEGs; right, upregulated/hypomethylated DEGs. DEGs, differentially expressed genes; FDR, false discovery rate; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Table I.
GO enrichment for methylation-related differentially expressed genes.
| A, Downregulated/hypermethylated genes | |||
|---|---|---|---|
| GO ID | GO term | FDR | Genes |
| 0051605 | Protein maturation by peptide bond cleavage | 2.87×10−2 | CFP, C8B, C7, FCN3, KLKB1, C1R |
| 0048545 | Response to steroid hormone stimulus | 1.47×10−2 | PRSS8, FOS, GOT1, CCL2, ACADS, WFDC1, CA2, NPY1R, GHR |
| 0046395 | Carboxylic acid catabolic process | 2.18×10−2 | ASPA, GOT1, ACADS, IDO2, KMO, UROC1, PON3 |
| 0045087 | Innate immune response | 2.26×10−3 | CFP, C8B, C7, FCN3, IL1RAP, VNN1, C1R, CD1D, GCH1 |
| 0031960 | Response to corticosteroid stimulus | 2.93×10−2 | PRSS8, FOS, GOT1, CCL2, ACADS, GHR |
| 0019439 | Aromatic compound catabolic process | 2.70×10−2 | EPHX2, IDO2, KMO, PON3 |
| 0016054 | Organic acid catabolic process | 2.18×10−2 | ASPA, GOT1, ACADS, IDO2, KMO, UROC1, PON3 |
| 0010817 | Regulation of hormone levels | 4.64×10−2 | ALDH8A1, SHBG, LY6E, CRHBP, CYP26A1, SRD5A1, BCO2 |
| 0009725 | Response to hormone stimulus | 2.54×10−2 | PRSS8, FOS, GOT1, CCL2, HMGCS2, ACADS, FBP1, WFDC1, CA2, NPY1R, GHR |
| 0009719 | Response to endogenous stimulus | 4.10×10−2 | PRSS8, FOS, GOT1, CCL2, HMGCS2, ACADS, FBP1, WFDC1, CA2, NPY1R, GHR |
| 0009611 | Response to wounding | 1.02×10−3 | C7, CCL2, HPS5, EPHX2, CHST4, C1R, CFP, C8B, FOS, LPA, PLSCR4, FCN3, KLKB1, IL1RAP, PROZ, VNN1, NGFR |
| 0006959 | Humoral immune response | 2.46×10−2 | CFP, C8B, C7, CCL2, FCN3, C1R |
| 0006956 | Complement activation | 2.70×10−2 | CFP, C8B, C7, FCN3, C1R |
| 0006955 | Immune response | 6.96×10−3 | C7, CCL2, CHST4, C1R, VIPR1, CXCL12, CD1D, GCH1, CFP, C8B, FCN3, HAMP, IL1RAP, VNN1 |
| 0006954 | Inflammatory response | 5.26×10−3 | CFP, C8B, FOS, C7, CCL2, FCN3, KLKB1, IL1RAP, EPHX2, VNN1, C1R, CHST4 |
| 0006952 | Defense response | 1.98×10−2 | C7, CCL2, EPHX2, CHST4, C1R, CD1D, GCH1, CFP, C8B, FOS, FCN3, HAMP, KLKB1, IL1RAP, VNN1 |
| 0006575 | Cellular amino acid derivative metabolic process | 2.47×10−2 | GGT5, LY6E, IDO2, VNN1, KMO, BBOX1, GHR, GCH1 |
| 0002526 | Acute inflammatory response | 3.37×10−3 | CFP, C8B, C7, FCN3, KLKB1, EPHX2, VNN1, C1R |
| B, Upregulated/hypomethylated genes | |||
| GO ID | GO term | FDR | Genes |
| 0051726 | Regulation of cell cycle | 3.52×10−2 | TP53BP2, NUSAP1, SFN, CDKN3, UBE2C |
| 0051301 | Cell division | 4.37×10−2 | RAD21, NUSAP1, NDC80, PARD3B, CEP55, UBE2C, CDCA3 |
| 0022616 | DNA strand elongation | 4.65×10−2 | RFC4, FEN1 |
| 0022403 | Cell cycle phase | 3.99×10−2 | RAD21, NUSAP1, NDC80, AURKA, CEP55, CDKN3, UBE2C, CDCA3 |
| 0022402 | Cell cycle process | 2.27×10−2 | RAD21, NUSAP1, NDC80, AURKA, CEP55, CDKN3, UBE2C, CDCA3 |
| 0007067 | Mitosis | 3.63×10−2 | RAD21, NUSAP1, NDC80, AURKA, CEP55, UBE2C, CDCA3 |
| 0007049 | Cell cycle | 3.98×10−2 | RAD21, TP53BP2, E2F8, NUSAP1, NDC80, AURKA, PARD3B, CEP55, CDKN3, UBE2C, CDCA3, MCM6 |
| 0006271 | DNA strand elongation during DNA replication | 4.22×10−2 | RFC4, FEN1 |
| 0006261 | DNA-dependent DNA replication | 4.56×10−2 | RFC4, MCM4, FEN1, MCM6 |
| 0006260 | DNA replication | 4.15×10−2 | RFC4, RNASEH2A, MCM4, FEN1, MCM6 |
| B, Upregulated/hypomethylated genes | |||
| GO ID | GO term | FDR | Genes |
| 000028 | Nuclear division | 3.63×10−2 | RAD21, NUSAP1, NDC80, AURKA, CEP55, UBE2C, CDCA3 |
| 0000279 | M phase | 4.61×10−2 | RAD21, NUSAP1, NDC80, AURKA, CEP55, UBE2C, CDCA3 |
| 0000278 | Mitotic cell cycle | 3.06×10−2 | RAD21, NUSAP1, NDC80, AURKA, CEP55, CDKN3, UBE2C, CDCA3 |
| 0000087 | M phase of mitotic cell cycle | 2.68×10−2 | RAD21, NUSAP1, NDC80, AURKA, CEP55, UBE2C, CDCA3 |
FDR, false discovery rate; GO, Gene Ontology.
Table II.
KEGG pathway enrichment for methylation-related differentially expressed genes.
| A, Downregulated/hypermethylated genes | |||
|---|---|---|---|
| KEGG ID | KEGG pathway | P-value | Genes |
| hsa04610 | Complement and coagulation cascades | 3.13×10−3 | C8B, C7, KLKB1, C1R |
| hsa00250 | Alanine, aspartate and glutamate metabolism | 3.81×10−3 | ASPA, GOT1, ASS1 |
| hsa00380 | Tryptophan metabolism | 6.04×10−3 | AADAT, IDO2, KMO |
| hsa00460 | Cyanoamino acid metabolism | 6.82×10−3 | GBA3, GGT5 |
| hsa00830 | Retinol metabolism | 1.02×10−2 | CYP4A11, CYP26A1, RDH16 |
| hsa04060 | Cytokine-cytokine receptor interaction | 2.67×10−2 | CCL2, IL1RAP, NGFR, CXCL12, GHR |
| hsa00270 | Cysteine and methionine metabolism | 2.91×10−2 | GOT1, BHMT |
| hsa00620 | Pyruvate metabolism | 3.33×10−2 | LDHD, ACOT12 |
| hsa00071 | Fatty acid metabolism | 3.33×10−2 | CYP4A11, ACADS |
| hsa00983 | Drug metabolism | 3.53×10−2 | NAT2, UPP2 |
| hsa04115 | p53 signaling pathway | 4.98×10−2 | GADD45B, IGFBP3 |
| B, Upregulated/hypomethylated genes | |||
| hsa03030 | DNA replication | 1.70×10−5 | RFC4, RNASEH2A, MCM4, FEN1, MCM6 |
| hsa04110 | Cell cycle | 1.79×10−2 | RAD21, SFN, MCM4, MCM6 |
| hsa04120 | Ubiquitin mediated proteolysis | 4.67×10−2 | UBE2C, UBE2Q1 |
| hsa05200 | Pathways in cancer | 4.79×10−2 | LAMC1, CTNNA1 |
KEGG, Kyoto Encyclopedia of Genes and Genomes.
PPI network
The STRING database identified interaction relationships in 105 out of the 185 methylation-related DEGs (68 downregulated and 37 upregulated). The 211 interaction relationship pairs among the DEGs were used to construct a PPI network (Fig. 5A); seven of the previously identified human oncogenes were included (downregulated, ID1, FOS and EPCAM; upregulated, AURKA, CDKN3, UBE2C and ERBB3), as no interactions with other DEGs were identified for ID2 and PLAC8.
Figure 5.

PPI network of the methylation-related differentially expressed genes. (A) An overall PPI network constructed using the protein interaction data from the STRING 10.0 database. (B) Functional highly connected sub-modules extracted from the PPI network using the Molecular Complex Detection plugin of Cytoscape software. Red, upregulated genes; green, downregulated genes; circled genes are known human oncogenes. AURKA, aurora kinase A; CDKN3, cyclin-dependent kinase inhibitor 3; EPCAM, epithelial cell adhesion molecule; ERBB3, erb-b2 receptor tyrosine kinase 3; FOS, Fos proto-oncogene, AP-1 transcription factor subunit; ID1, inhibitor of DNA binding 1, HLH protein; M1, module 1; M2, module 2; M3, module 3; M4, module 4; PPI, protein-protein interaction; UBE2C, ubiquitin conjugating enzyme E2 C.
Two human oncogenes, FOS and CDKN3, were indicated as hub genes of the PPI network as they were shared and ranked in the top 15 for 4 topological characteristics (Table III). In addition, AURKA and UBE2C ranked top 5 in ‘degree’. ID1 was one of the top 10 genes in ‘CC’ and ‘APL’.
Table III.
Top 15 genes based on each topological characteristic.
| Node | Degree | Node | Closeness centrality | Node | Average path length | Node | Betweenness Centrality |
|---|---|---|---|---|---|---|---|
| NDC80 | 18 | FAT1 | 1.00 | FAT1 | 1.00 | FAT1 | 1.00 |
| CDKN3a | 16 | SMAD5 | 1.00 | SMAD5 | 1.00 | FOS | 0.52 |
| AURKAa | 16 | CBFA2T3 | 1.00 | CBFA2T3 | 1.00 | CDKN3a | 0.40 |
| UBE2Ca | 15 | SPP2 | 1.00 | SPP2 | 1.00 | FEN1 | 0.36 |
| NUSAP1 | 15 | FNIP1 | 1.00 | FNIP1 | 1.00 | MTR | 0.35 |
| RFC4 | 14 | WFDC1 | 1.00 | WFDC1 | 1.00 | LPL | 0.25 |
| KIF4A | 14 | ECM1 | 1.00 | ECM1 | 1.00 | LPA | 0.18 |
| CEP55 | 14 | SLC22A1 | 1.00 | SLC22A1 | 1.00 | IGFBP3 | 0.17 |
| FEN1 | 13 | SLC10A1 | 1.00 | SLC10A1 | 1.00 | TXNRD1 | 0.16 |
| ATAD2 | 13 | ID1 | 1.00 | ID1 | 1.00 | SHBG | 0.16 |
| MCM4 | 13 | RRAGD | 1.00 | RRAGD | 1.00 | ASS1 | 0.16 |
| FOS | 11 | CTNNA1 | 0.67 | CTNNA1 | 1.50 | CCL2 | 0.15 |
| MCM6 | 11 | LAMC1 | 0.67 | LAMC1 | 1.50 | ACADS | 0.15 |
| RNASEH2A | 11 | CDKN3a | 0.28 | FOS | 3.54 | APOF | 0.14 |
| DEPDC1 | 8 | FOS | 0.28 | CDKN3a | 3.54 | CANX | 0.14 |
Potential hub gene.
Subsequently, four highly connected PPI sub-modules (Fig. 5B) were extracted from the overall PPI network using MCODE. BinGO enrichment analysis demonstrated that the genes in module 1 (MCODE score=12.81) were involved in mitotic cell cycle (AURKA, CDKN3 and UBE2C); the genes in module 2 (MCODE score=5.067) were associated with detoxification of copper ions; the genes in module 3 (MCODE score=3.771) participated in the regulation of cell migration; and the genes in module 4 (MCODE score=3.41) were associated with carboxylic acid metabolic process (Table IV).
Table IV.
GO enrichment for genes in modules.
| A, Module 1 | |||
|---|---|---|---|
| GO ID | Pcorr | GO term | Genes in test set |
| 48015 | 1.86×10−6 | Phosphoinositide-mediated signaling | FEN1, RFC4, UBE2C, NDC80, AURKA |
| 278 | 3.01×10−5 | Mitotic cell cycle | UBE2C, NUSAP1, NDC80, CEP55, AURKA, CDKN3 |
| 6260 | 3.01×10−5 | DNA replication | FEN1, RNASEH2A, RFC4, MCM4, MCM6 |
| 22403 | 3.65×10−5 | Cell cycle phase | UBE2C, NUSAP1, NDC80, CEP55, AURKA, CDKN3 |
| 280 | 3.65×10−5 | Nuclear division | UBE2C, NUSAP1, NDC80, CEP55, AURKA |
| 7067 | 3.65×10−5 | mitosis | UBE2C, NUSAP1, NDC80, CEP55, AURKA |
| 87 | 3.65×10−5 | M phase of mitotic cell cycle | UBE2C, NUSAP1, NDC80, CEP55, AURKA |
| 7049 | 4.09×10−5 | Cell cycle | UBE2C, NUSAP1, MCM6, NDC80, CEP55, AURKA, CDKN3 |
| 51301 | 1.03×10−4 | Cell division | UBE2C, NUSAP1, NDC80, CEP55, AURKA |
| 22402 | 1.03×10−4 | Cell cycle process | UBE2C, NUSAP1, NDC8, CEP55, AURKA, CDKN3 |
| 279 | 1.35×10−4 | M phase | UBE2C, NUSAP1, NDC80, CEP55, AURKA |
| 35556 | 4.36×10−3 | Intracellular signal transduction | FEN1, RFC4, UBE2C, NDC80, AURKA |
| 6996 | 5.42×10−3 | Organelle organization | UBE2C, KIF4A, NUSAP1, NDC80, CEP55, AURKA |
| 34645 | 1.06×10−2 | Cellular macromolecule biosynthetic process | FEN1, RNASEH2A, RFC4, MCM4, MCM6 |
| 9059 | 1.08×10−2 | Macromolecule biosynthetic process | FEN1, RNASEH2A, RFC4, MCM4, MCM6 |
| 23034 | 1.41×10−2 | Intracellular signaling pathway | FEN1, RFC4, UBE2C, NDC80, AURKA |
| 90304 | 2.84×10−2 | Nucleic acid metabolic process | FEN1, RNASEH2A, RFC4, MCM4, MCM6 |
| 44249 | 4.55×10−2 | Cellular biosynthetic process | FEN1, RNASEH2A, RFC4, MCM4, MCM6 |
| B, Module 2 | |||
| GO ID | Pcorr | GO term | Genes in test set |
| 10273 | 9.41×10−3 | Detoxification of copper ion | MT2A |
| 10038 | 9.41×10−3 | Response to metal ion | MT2A, MT1X |
| 10035 | 1.12×10−2 | Response to inorganic substance | MT2A, MT1X |
| 6882 | 1.12×10−2 | Cellular zinc ion homeostasis | MT2A |
| 55069 | 1.12×10−2 | Zinc ion homeostasis | MT2A |
| 6878 | 1.12×10−2 | Cellular copper ion homeostasis | MT2A |
| 55070 | 1.12×10−2 | Copper ion homeostasis | MT2A |
| 7263 | 1.37×10−2 | Nitric oxide mediated signal transduction | MT2A |
| 46688 | 1.48×10−2 | Response to copper ion | MT2A |
| C, Module 3 | |||
| GO ID | Pcorr | GO term | Genes in test set |
| 30334 | 1.28×10−2 | Regulation of cell migration | CXCL12, IGFBP3, THY1 |
| 51270 | 1.28×10−2 | Regulation of cellular component movement | CXCL12, IGFBP3, THY1 |
| 40012 | 1.28×10−2 | Regulation of locomotion | CXCL12, IGFBP3, THY1 |
| 42325 | 3.08×10−2 | Regulation of phosphorylation | GHR, IGFBP3, THY1 |
| 19220 | 3.08×10−2 | Regulation of phosphate metabolic process | GHR, IGFBP3, THY1 |
| 51174 | 3.08×10−2 | Regulation of phosphorus metabolic process | GHR, IGFBP3, THY1 |
| 45595 | 3.08×10−2 | Regulation of cell differentiation | GHR, IGFBP3, THY1 |
| 7155 | 3.56×10−2 | Cell adhesion | CXCL12, THY1, IGFALS |
| 22610 | 3.56×10−2 | Biological adhesion | CXCL12, THY1, IGFALS |
| 32879 | 3.64×10−2 | Regulation of localization | CXCL12, IGFBP3, THY1 |
| 50793 | 3.69×10−2 | Regulation of developmental process | GHR, IGFBP3, THY1 |
| 48522 | 3.81×10−2 | Positive regulation of cellular process | GHR, CXCL12, IGFBP3, THY1 |
| 48518 | 4.28×10−2 | Positive regulation of biological process | GHR, CXCL12, IGFBP3, THY1 |
| 51239 | 4.29×10−2 | Regulation of multicellular organismal process | GHR, IGFBP3, THY1 |
| 10646 | 4.53×10−2 | Regulation of cell communication | GHR, IGFBP3, THY1 |
| 7166 | 4.93×10−2 | Cell surface receptor linked signaling pathway | GHR, CXCL12, THY1 |
| D, Module 4 | |||
| GO ID | Pcorr | GO term | Genes in test set |
| 19752 | 7.81×10−8 | Carboxylic acid metabolic process | BHMT, GOT1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
| 43436 | 7.81×10−8 | Oxoacid metabolic process | BHMT, GOT1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
| 6082 | 7.81×10−8 | Organic acid metabolic process | BHMT, GOT1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
| 42180 | 7.81×10−8 | Cellular ketone metabolic process | BHMT, GOT1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
| 6520 | 2.23×10−6 | Cellular amino acid metabolic process | BHMT, GOT1, MTR, ASPA, ASS1 |
| 44106 | 6.48×10−6 | Cellular amine metabolic process | BHMT, GOT1, MTR, ASPA, ASS1 |
| 44281 | 1.21×10−5 | Small molecule metabolic process | BHMT, GOT1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
| 6519 | 1.21×10−5 | Cellular amino acid and derivative metabolic process | BHMT, GOT1, MTR, ASPA, ASS1 |
| 9308 | 1.89×10−5 | Amine metabolic process | BHMT, GOT1, MTR, ASPA, ASS1 |
| 44283 | 2.33×10−5 | Small molecule biosynthetic process | BHMT, GOT1, MTR, ALDH8A1, ASS1 |
| 44237 | 1.82×10−3 | Cellular metabolic process | BHMT, GOT1, TXNRD1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
| 8152 | 5.73×10−3 | Metabolic process | BHMT, GOT1, TXNRD1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
| 44249 | 5.73×10−3 | Cellular biosynthetic process | BHMT, GOT1, MTR, ALDH8A1, ASS1 |
| 9058 | 6.30×10−3 | Biosynthetic process | BHMT, GOT1, MTR, ALDH8A1, ASS1 |
| 34641 | 9.55×10−3 | Cellular nitrogen compound metabolic process | BHMT, GOT1, MTR, ASPA, ASS1 |
| 6807 | 1.19×10−2 | Nitrogen compound metabolic process | BHMT, GOT1, MTR, ASPA, ASS1 |
| 44238 | 1.71×10−2 | Primary metabolic process | BHMT, GOT1, MTR, ALDH8A1, ACADS, ASPA, ASS1 |
GO, Gene Ontology; Pcorr, corrected P-value.
Validation of the selected methylation-mediated DEGs
Based on the enrichment and PPI analyses, it was hypothesized that downregulated/hypermethylated FOS and ID1 and upregulated/hypomethylated CDKN3, AURKA and UBE2C may be important human oncogenes for HCV-positive HCC. To further confirm their expression and methylation levels, the mRNA and methylation sequencing data of 58 HCV-HCC tissues and 50 normal controls were obtained from the TCGA database. The results demonstrated that the transcriptional expression and methylation levels of FOS, CDKN3 and AURKA in TCGA sequencing data (Fig. 6B) were consistent with the microarray data (Fig. 6A). However, the methylation level of UBE2C was not significantly different between HCV-positive HCC and normal control TCGA samples, although its expression level was consistent between TCGA sequencing data and our used microarray data (Fig. 6). The methylation level of ID1 had a detection value of 0 in the TCGA and thus comparison was not performed.
Figure 6.

Validation of the hub genes in the samples obtained from TCGA database. (A) Gene expression and methylation levels in samples of microarray datasets GSE19665 (HCV-positive HCC tissues, n=5; normal controls, n=5), GSE62232 (HCV-positive HCC tissues, n=9; normal controls, n=10) and GSE60753 (HCV-positive HCC tissues, n=29; normal controls, n=34). (B) Gene expression and methylation levels in TCGA data (HCV-positive HCC tissues, n=58; normal controls, n=50). Student's independent t-test was used to analyze the differences between HCV-positive HCC and normal controls. *P<0.05, **P<0.01 and ***P<0.001 vs. control. AURKA, aurora kinase A; CDKN3, and cyclin-dependent kinase inhibitor 3; FOS, Fos proto-oncogene, AP-1 transcription factor subunit; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; ID1, inhibitor of DNA binding 1, HLH protein; UBE2C, ubiquitin conjugating enzyme E2 C; n.s., not significant; TCGA, The Cancer Genome Atlas.
In addition, the HPA database was used to confirm the protein expression level of the genes in HCC by immunohistochemistry. Protein expression levels of AURKA in HCC tissues were higher, whereas protein expression levels of FOS in HCC tissues were lower compared with normal hepatocytes (Fig. 7). There was no immunohistochemical result for CDKN3 in the HPA database and no difference was observed in UBE2C and ID1 protein expression levels between HCC and normal control tissues.
Figure 7.
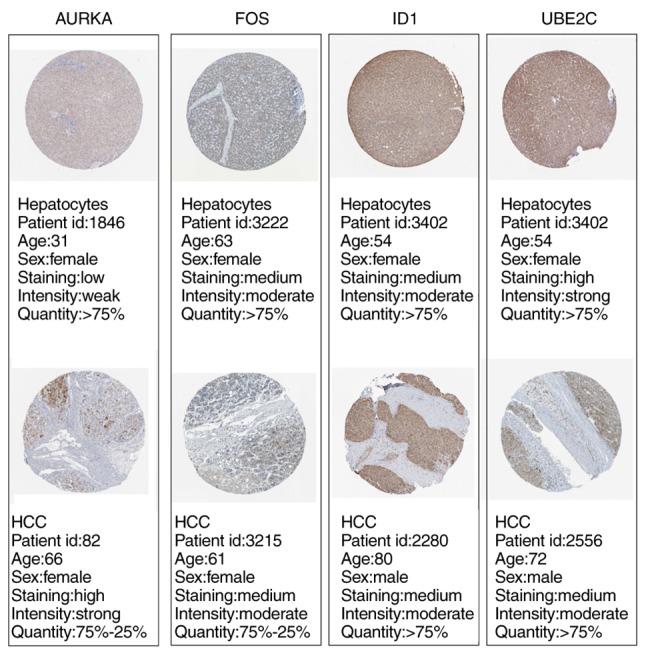
Validation of the hub genes at a translational level using the Human Protein Atlas database. AURKA, aurora kinase A; FOS, Fos proto-oncogene, AP-1 transcription factor subunit; HCC, hepatocellular carcinoma; ID1, inhibitor of DNA binding 1, HLH protein; UBE2C, ubiquitin conjugating enzyme E2 C.
Discussion
Through comprehensive analysis and validation, results from the present study indicated that AURKA and FOS may be crucial genes involved in HCV-positive HCC by participating in the cell cycle process and inflammatory response. HCV may upregulate the expression of AURKA and downregulate FOS by changes in DNA methylation.
HCV stimulates excessive cell proliferation in hepatocytes by dysregulating the cell cycle, which induces the development of HCC (25,26). Several positive cell cycle regulators (such as cyclin D1, cyclin E and Rb/p105) have been identified to be upregulated, whereas negative regulators [such as cyclin-dependent kinase 4 (CDK4), CDK6, p21Cip1, p27Kip1 and p57Kip2) are downregulated in patients with HCV-positive HCC compared with patients with chronic hepatitis C with or without liver cirrhosis (27). AURKA, which is located on chromosome 20q13.2, encodes a serine/threonine kinase involved in the assembly and maintenance of the mitotic spindle (28). Thus, AURKA is speculated to be a crucial gene for the regulation of cell cycle and carcinogenesis in several cancer types, including HCC (29). Yang et al demonstrated that knockdown of AURKA suppressed the growth of ovarian cancer cells by reducing centrosome amplification, malformation of mitotic spindles, and chromosome aberration (30). Additionally, restoring the expression of p21 and pRb attenuated the effects of AURKA silencing on cell cycle progression (30). Using RNA microarray and reverse transcriptase-quantitative PCR analysis, Zhou et al reported that AURKA was significantly upregulated in human urothelial carcinoma compared with normal urothelium (31) and demonstrated that AURKA inhibitor MLN8237 induced cell-cycle arrest, aneuploidy, mitotic spindle failure and apoptosis in human bladder cancer cells, which arrested tumor growth (31). Li et al also used the MLN8237 to demonstrate that AURKA regulated cell cycle in breast cancer cells by modulating the p53/p21/cell division control 2/cyclin B1 pathway (32). Similarly, the verification experiments demonstrated that AURKA inhibitor alisertib arrested HCC cells in G2/M phase and induced an accumulation of aneuploidy by regulating the expression of key cell cycle regulators such as cyclin B1 (33,34). The present study demonstrated that AURKA was highly expressed at the mRNA and protein levels in HCV-positive HCC. In addition to the cell cycle, a recent study has suggested that AURKA contributes to tumor migration, invasion, epithelial mesenchymal transition and cancer stem cell behaviors, which also have been preliminarily validated in HCC (35), but not HCV-related HCC. Thus, further investigation of the roles of AURKA in HCV-related HCC remains necessary.
DNA methylation is an important mechanism for regulating gene expression epigenetically. Hypermethylation of genes is associated with reduced expression, whereas hypomethylation is associated with increased expression. Thus, high expression of AURKA in HCV-positive HCC was predicted to be due to hypomethylation, which was validated by the microarray and TCGA data. This conclusion agreed with a previous study on esophageal cancer, in which AURKA methylation and human papillomavirus infection was higher in precancer, esophagitis and normal tissues compared with cancer tissues (36). However, further experiments are needed to confirm the effects of HCV on the methylation of AURKA and the development of HCC.
FOS is a member of the fos family of transcription factors, which has been extensively demonstrated to be a pro-oncogenic gene and promote proliferation, invasion and metastasis of cancer through AP-1-related mechanisms, including HCV-positive HCC (37). However, in the present study, FOS was downregulated and hypermethylated. This may indicate that FOS may be a dual-function gene, which has been identified in other cancers, such as ovarian cancer (38) and pancreatic cancer (39). Alternatively, the results may be negative due to the small sample size. Further studies with larger sample sizes are needed to confirm role of FOS in HCV-positive HCC.
There were certain limitations to the present study. First, although the known microarray and TCGA sequencing data were included to confirm the expression and methylation levels of crucial genes, the sample size associated with HCV-positive HCC was small. Therefore, more clinical samples need to be collected to further confirm their expression levels. Second, although the present study has suggested that the expression levels of AURKA and FOS may be regulated by methylation, additional in vitro and in vivo experiments using a methylation inhibitor, such as 5-azacytidine, are essential to verify these results. Third, HCV infection-related in vitro and in vivo experiments (accompanied with overexpression or knockdown of genes) are also needed to demonstrate the functional roles of AURKA and FOS in cell proliferation, apoptosis, migration and invasion.
The results of the present study preliminarily indicate that aberrantly methylated AURKA and FOS may be potential therapeutic targets for treatment of HCV-positive HCC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The sequencing datasets GSE19665 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19665), GSE62232 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62232) and GSE60753 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60753) were downloaded from the GEO database in NCBI. The mRNA and microRNA sequencing data were obtained from The Cancer Genome Atlas (https://tcga-data.nci.nih.gov).
Authors' contributions
ZM and XH conceived and designed the study; YL and ZH performed the acquisition of data; ZM and YL conducted the statistical analysis. WL and ZH were involved in the interpretation of the data. ZM drafted the manuscript. XH revised the manuscript. all authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Jing L, Huang L, Yan J, Qiu M, Yan Y. Liver resection for hepatocellular carcinoma: Personal experiences in a series of 1330 consecutive cases in China. ANZ J Surg. 2018;88:E713–E717. doi: 10.1111/ans.14381. [DOI] [PubMed] [Google Scholar]
- 3.Petruzziello A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) related hepatocellular carcinoma. Open Virol J. 2018;12:26–32. doi: 10.2174/1874357901812010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore MS, Bocour A, Tran OC, Qiao B, Schymura MJ, Laraque F, Winters A. Effect of hepatocellular carcinoma on mortality among individuals with Hepatitis B or Hepatitis C infection in New York City, 2001–2012. Open Forum Infect Dis. 2018;5:ofy144. doi: 10.1093/ofid/ofy144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiran M, Chawla YK, Kaur J. Methylation profiling of tumor suppressor genes and oncogenes in hepatitis virus-related hepatocellular carcinoma in northern India. Cancer Genet Cytogenet. 2009;195:112–119. doi: 10.1016/j.cancergencyto.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Zekri AR, Bahnasy AA, Shoeab FE, Mohamed WS, El-Dahshan DH, Ali FT, Sabry GM, Dasgupta N, Daoud SS. Methylation of multiple genes in hepatitis C virus associated hepatocellular carcinoma. J Adv Res. 2014;5:27–40. doi: 10.1016/j.jare.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramadan RA, Zaki MA, Awad AM, El-Ghalid LA. Aberrant methylation of promoter region of SPINT2/HAI-2 gene: An epigenetic mechanism in hepatitis C virus-induced hepatocarcinogenesis. Genet Test Mol Biomarkers. 2015;19:399–404. doi: 10.1089/gtmb.2015.0025. [DOI] [PubMed] [Google Scholar]
- 8.Takagi K, Fujiwara K, Takayama T, Mamiya T, Soma M, Nagase H. DNA hypermethylation of zygote arrest 1 (ZAR1) in hepatitis C virus positive related hepatocellular carcinoma. Springerplus. 2013;2:150. doi: 10.1186/2193-1801-2-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsunedomi R, Iizuka N, Yoshimura K, Iida M, Tsutsui M, Hashimoto N, Kanekiyo S, Sakamoto K, Tamesa T, Oka M. ABCB6 mRNA and DNA methylation levels serve as useful biomarkers for prediction of early intrahepatic recurrence of hepatitis C virus-related hepatocellular carcinoma. Int J Oncol. 2013;42:1551–1559. doi: 10.3892/ijo.2013.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mileo AM, Mattarocci S, Matarrese P, Anticoli S, Abbruzzese C, Catone S, Sacco R, Paggi MG, Ruggieri A. Hepatitis C virus core protein modulates pRb2/p130 expression in human hepatocellular carcinoma cell lines through promoter methylation. J Exp Clin Cancer Res. 2015;34:140. doi: 10.1186/s13046-015-0255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quan H, Zhou F, Nie D, Chen Q, Cai X, Shan X, Zhou Z, Chen K, Huang A, Li S, Tang N. Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial-mesenchymal transition. Oncogene. 2014;33:2826–2835. doi: 10.1038/onc.2013.225. [DOI] [PubMed] [Google Scholar]
- 12.Deng YB, Nagae G, Midorikawa Y, Yagi K, Tsutsumi S, Yamamoto S, Hasegawa K, Kokudo N, Aburatani H, Kaneda A. Identification of genes preferentially methylated in hepatitis C virus-related hepatocellular carcinoma. Cancer Sci. 2010;101:1501–1510. doi: 10.1111/j.1349-7006.2010.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Chen L, Gu J, Zhang H, Yuan J, Lian Q, Lv G, Wang S, Wu Y, Yang YC, et al. Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nat Commun. 2017;8:14421. doi: 10.1038/ncomms14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hlady RA, Tiedemann RL, Puszyk W, Zendejas I, Roberts LR, Choi JH, Liu C, Robertson KD. Epigenetic signatures of alcohol abuse and hepatitis infection during human hepatocarcinogenesis. Oncotarget. 2014;5:9425–9443. doi: 10.18632/oncotarget.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szekely GJ, Rizzo ML. Hierarchical clustering via Joint between-within distances: Extending Ward's minimum variance method. J Classification. 2005;22:151–183. doi: 10.1007/s00357-005-0012-9. [DOI] [Google Scholar]
- 17.Liu Y, Sun J, Zhao M. ONGene: A literature-based database for human oncogenes. J Genet Genomics. 2017;44:119–121. doi: 10.1016/j.jgg.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43 (Database Issue) 2015:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohl M, Wiese S, Warscheid B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 20.Tang Y, Li M, Wang J, Pan Y, Wu FX. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2015;127:67–72. doi: 10.1016/j.biosystems.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Maere S, Heymans K, Kuiper M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 24.Pontén F, Jirström K, Uhlen M. The human protein atlas-a tool for pathology. J Pathol. 2008;216:387–393. doi: 10.1002/path.2440. [DOI] [PubMed] [Google Scholar]
- 25.Irshad M, Gupta P, Irshad K. Molecular basis of hepatocellular carcinoma induced by hepatitis C virus infection. World J Hepatol. 2017;9:1305–1314. doi: 10.4254/wjh.v9.i36.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moustafa S, Karakasiliotis I, Mavromara P. Hepatitis C virus core+1/ARF protein modulates Cyclin D1/pRb pathway and promotes carcinogenesis. J Virol. 2018;92(pii):e02036–17. doi: 10.1128/JVI.02036-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bassiouny AEE, Nosseir MM, Zoheiry MK, Ameen NA, Abdelhadi AM, Ibrahim IM, Zada S, El-Deen AH, El-Bassiouni NE. Differential expression of cell cycle regulators in HCV-infection and related hepatocellular carcinoma. World J Hepatol. 2010;2:32–41. doi: 10.4254/wjh.v2.i1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kufer TA, Silljé HH, Körner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158:617–623. doi: 10.1083/jcb.200204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benten D, Keller G, Quaas A, Schrader J, Gontarewicz A, Balabanov S, Braig M, Wege H, Moll J, Lohse AW, Brummendorf TH. Aurora kinase inhibitor PHA-739358 suppresses growth of hepatocellular carcinoma in vitro and in a xenograft mouse model. Neoplasia. 2009;11:934–944. doi: 10.1593/neo.09664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang G, Chang B, Yang F, Guo X, Cai KQ, Xiao X, Wang H, Sen S, Hung MC, Mills GB, et al. Aurora Kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res. 2010;16:3171–3181. doi: 10.1158/1078-0432.CCR-09-3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou N, Singh K, Mir MC, Parker Y, Lindner D, Dreicer R, Ecsedy JA, Zhang Z, The BT, Almasan A, Hansel DE. The investigational Aurora kinase A inhibitor MLN8237 induces defects in cell viability and cell cycle progression in malignant bladder cancer cells in vitro and in vivo. Clin Cancer Res. 2013;19:1717–1728. doi: 10.1158/1078-0432.CCR-12-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li JP, Yang YX, Liu QL, Pan ST, He ZX, Zhang X, Yang T, Chen XW, Dong W, Qiu JX, Zhou SF. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des Devel Ther. 2015;9:1627–1652. doi: 10.2147/DDDT.S75378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Zhu Q, Yu X, Zhou ZW, Zhou C, Chen XW, Zhou SF. Inhibition of Aurora A Kinase by alisertib induces autophagy and cell cycle arrest and increases chemosensitivity in human hepatocellular carcinoma HepG2 cells. Curr Cancer Drug Targets. 2017;17:386–401. doi: 10.2174/1568009616666160630182344. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Q, Yu X, Zhou ZW, Luo M, Zhou C, He ZX, Chen Y, Zhou SF. A quantitative proteomic response of hepatocellular carcinoma Hep3B cells to danusertib, a pan-Aurora kinase inhibitor. J Cancer. 2018;9:2061–2071. doi: 10.7150/jca.20822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Song G, Xiang J, Zhang H, Zhao S, Zhan Y. AURKA promotes cancer metastasis by regulating epithelial-mesenchymal transition and cancer stem cell properties in hepatocellular carcinoma. Biochem Biophys Res Commun. 2017;486:514–520. doi: 10.1016/j.bbrc.2017.03.075. [DOI] [PubMed] [Google Scholar]
- 36.Mohiuddin MK, Chava S, Upendrum P, Latha M, Zubeda S, Kumar A, Ahuja YR, Hasan Q, Mohan V. Role of Human papilloma virus infection and altered methylation of specific genes in esophageal cancer. Asian Pac J Cancer Prev. 2013;14:4187–4193. doi: 10.7314/APJCP.2013.14.7.4187. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe T, Hiasa Y, Tokumoto Y, Hirooka M, Abe M, Ikeda Y, Matsuura B, Chung RT, Onji M. Protein Kinase R modulates c-Fos and c-Jun signaling to promote proliferation of hepatocellular carcinoma with Hepatitis C virus infection. PLoS One. 2013;8:e67750. doi: 10.1371/journal.pone.0067750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveiraferrer L, Rößler K, Haustein V, Schröder C, Wicklein D, Maltseva D, Khaustova N, Samatov T, Tonevitsky A, Mahner S, et al. c-FOS suppresses ovarian cancer progression by changing adhesion. Br J Cancer. 2013;110:753–763. doi: 10.1038/bjc.2013.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo JC, Li J, Zhao YP, Zhou L, Cui QC, Zhou WX, Zhang TP, You L. Expression of c-fos was associated with clinicopathologic characteristics and prognosis in pancreatic cancer. PLoS One. 2015;10:e0120332. doi: 10.1371/journal.pone.0120332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequencing datasets GSE19665 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19665), GSE62232 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62232) and GSE60753 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60753) were downloaded from the GEO database in NCBI. The mRNA and microRNA sequencing data were obtained from The Cancer Genome Atlas (https://tcga-data.nci.nih.gov).