

Abstract
Motivation
Phosphorylation is the most studied post-translational modification, which is crucial for multiple biological processes. Recently, many efforts have been taken to develop computational predictors for phosphorylation site prediction, but most of them are based on feature selection and discriminative classification. Thus, it is useful to develop a novel and highly accurate predictor that can unveil intricate patterns automatically for protein phosphorylation sites.
Results
In this study we present DeepPhos, a novel deep learning architecture for prediction of protein phosphorylation. Unlike multi-layer convolutional neural networks, DeepPhos consists of densely connected convolutional neuron network blocks which can capture multiple representations of sequences to make final phosphorylation prediction by intra block concatenation layers and inter block concatenation layers. DeepPhos can also be used for kinase-specific prediction varying from group, family, subfamily and individual kinase level. The experimental results demonstrated that DeepPhos outperforms competitive predictors in general and kinase-specific phosphorylation site prediction.
Availability and implementation
The source code of DeepPhos is publicly deposited at https://github.com/USTCHIlab/DeepPhos.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Post-translational modification (PTM) is a pivotal mechanism of regulating cellular functions by the covalent and generally enzymatic modification, which plays vital roles in regulating various biological processes, e.g. gene expression, cell division and cell signaling (Walsh, 2006). As one of the most well studied PTMs, protein phosphorylation usually phosphorylated on serine (S), threonine (T) and tyrosine (Y) residues, and it is fundamental for regulating cellular process such as DNA repair, growth, motility, metabolism and cell cycle control (Li et al., 2008; Matthews, 1995; Trost and Kusalik, 2011). There are evidences showing that more than 30% of eukaryotic proteins can be phosphorylated, and half of them closely relate to different kinds of diseases, especially cancer (Walsh, 2006).
Due to the significance of phosphorylation in understanding biological systems of proteins and guidance to basic biomedical drug design, researches on phosphorylation were booming in the last decades. And numerous trials including experimental methods and computational prediction strategies have been made to identify phosphorylation sites (Huang et al., 2015; Qin et al., 2016; Trost et al., 2013; Trost et al., 2016). Conventional biological experimental identification methods including low-throughput 32P-labeling (Aponte et al., 2009; Beausoleil et al., 2006) and high throughput Mass Spectrometry techniques can annotate phosphorylation sites accurately, therefore accumulating a large number of phosphorylation examples. However, traditional experimental methods are labor-intensive and time-consuming especially applied in verifying huge amounts of candidate phosphorylation sites (Liu et al., 2018; Wang et al., 2017; Wen et al., 2016). Alternately, computational approaches are becoming popular to deal with the difficulties of experimental strategies.
To date, there are more than 40 computational methods for identifying phosphorylation sites, and a considerable number of them are based on machine learning algorithms including Support Vector Machine (Huang et al., 2015), Bayesian decision theory (Xue et al., 2006), logistic regression (Li et al., 2018) and Random forest (Fan et al., 2014). For example, Gao et al. (2010) proposed an algorithm called Musite, which uses protein disorder scores as well as local amino acid sequences frequencies and k-nearest neighbor features to further improve the prediction accuracy. Xue et al. (2008) proposed a Markov cluster algorithm based method, Group-based Prediction System (GPS), which uses an amino acid substitution matrix to predict kinase-specific phosphorylation sites.
These computational methods and tools promote the understanding on phosphorylation and have efficient improvement on performance. However, the majority of them perform a three-stage classification using multiple sequence based features, such as protein disorder, physicochemical properties or extra domain knowledge (Gao et al., 2010; Xue et al., 2008). Generally, the first stage is to generate all the features using additional tools, but not all of these features would be used in final prediction for the large number of features and the redundancy among features (Fan et al., 2014; Wei et al., 2017). So these predictors choose some crucial and effective features in the second stage. Finally, applying the selected features to the machine learning algorithm for the discriminative classification. Although these methods have achieved good performance, they rely on the ranking of important features to identify the phosphorylation sites, and this situation may bring over optimization and cause bias in evaluation (Wang et al., 2017). Recently, deep learning has made breakthrough on image recognition (Cireşan et al., 2011), natural language understanding and sentiment analysis (Collobert et al., 2011). A distinctive advantage of deep learning lies on the fact that instead of choosing features manually, it can automatically discover complex patterns and capture the high level abstraction adaptively from the training data, which seems to be an attractive solution for the challenge of phosphorylation site prediction.
So far, a number of deep learning-based explorations have been successfully undertaken in different bioinformatics areas (Alipanahi et al., 2015; Sun et al., 2017). For example, to predict DNA and RNA targets of binding proteins, Babak et al. proposed a novel method called DeepBind (Alipanahi et al., 2015) that is established upon convolution neural network (CNN), and the results suggest that CNN has stable ability for abstract sequence representation and motif discovery. Indeed, deep learning is attractive for PTM site prediction. For example, Muscadel (Chen et al., 2018) successfully applies long short-term memory (LSTM) recurrent neural networks (RNNs) to predict eight types of lysine PTMs, and DeepNitro (Xie et al., 2018) uses multi-layer deep neural network to predict nitration and nitrosylation sites. As a pioneer approach for phosphorylation site prediction, Wang et al. lately presented Musitedeep (Wang et al., 2017) that adopts a multi-layer CNN architecture to discover complex sequential representation automatically. By comprehensive analysis of various results obtained by MusiteDeep and other competitive methods, the CNN architecture is shown to be superior to the traditional methods, which inspires further comprehensive exploration and more carefully designed CNN architectures that hold the promise of performance improvement in predicting phosphorylation sites.
In this work, we present a novel multi-layer CNN architecture, DeepPhos, to accurately predict phosphorylation sites with protein sequential information. Different from aforementioned deep learning methods, DeepPhos consists of so-called densely connected CNN (DC-CNN) blocks in which convolutional layers are connected to each other simultaneously via intra block concatenation layers (Intra-BCL), to not only efficiently enhance the flow of phosphorylation information but also integrate different levels of representations extracted by convolutional layers. Meanwhile, multiple DC-CNN blocks with distinct window and filter sizes are adopted to capture the vital sequence representations of protein phosphorylation sites automatically, which are further integrated by an inter block concatenation layer (Inter-BCL) to make final prediction. To evaluate the performance of DeepPhos, we collected plenty of verified phosphorylation examples from several databases, which are used to train and valuate the models. The evaluation results reveal that DeepPhos outperforms existing methods in general phosphorylation prediction. In addition, our architecture can be successfully applied to a series of kinase-specific phosphorylation site prediction tasks varying from kinase group level to individual kinase level by layer transfer from a base general DeepPhos model. Further evaluation also demonstrates DeepPhos has better performance for kinase-specific phosphorylation site prediction.
2 Materials and methods
2.1 Overview
DeepPhos is a novel deep learning architecture for phosphorylation site prediction, and the working flow of DeepPhos is described in Supplementary Figure S1 and Figure 1a. First, all verified data from multiple databases was used to construct the dataset for deep learning models. The dataset construction including data collection and pre-processing is illustrated in Section 2.2. The following procedures consists of training of DeepPhos for general and kinase-specific prediction at group, family, subfamily and individual kinase level, which were evaluated on independent test data for performance assessment. The details of architecture and training process for general and kinase-specific prediction are introduced in Subsection 2.3. Subsection 2.4 is the performance assessment adopted in this study.
Fig. 1.
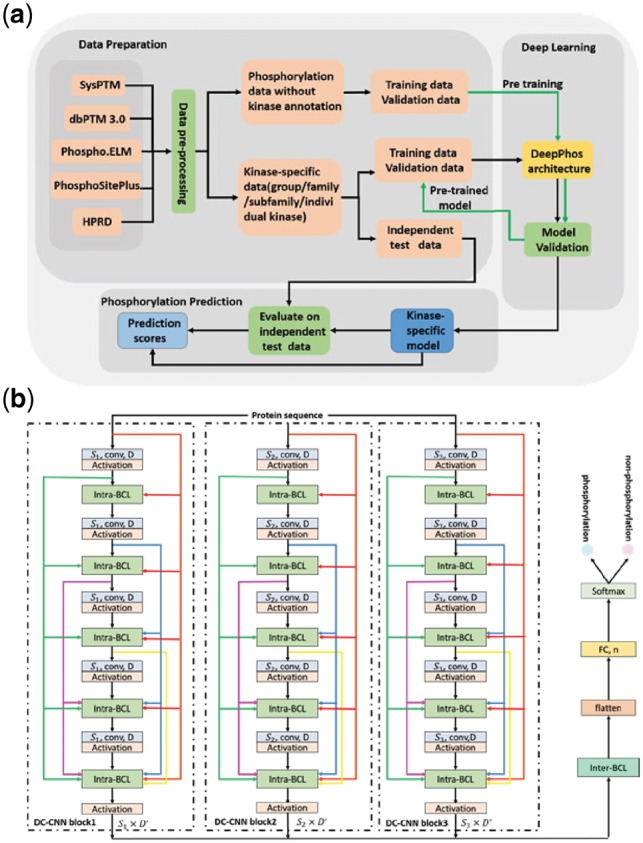
DeepPhos overall framework. (a) The working flow of kinase-specific phosphorylation site prediction in DeepPhoos. (b) The network layout of the DeepPhos approach. The raw sequences are transformed into a set of sequence features by one-hot codings. Intra-BCLs between two convolutional layers in each DC-CNN block are designed for connecting previous and current feature maps as shown by the different colorful arrows, in which feature maps are concatenated in the feature dimension. in the convolutional layers refers to the width of the filters used in convolutional operation, D represents the number of filters used in convolutional layers, and refers to the size of output feature maps. The representations generated by multi-blocks are further integrated by inter-BCL and transformed to FC layer (n represents the number of neurons), and finally generate the phosphorylation prediction by softmax function (Color version of this figure is available at Bioinformatics online.)
2.2 Data collection and pre-processing
To ensure the high quality of data, we collected the experimentally verified phosphorylation sites of human proteins from several databases including Phospho.ELM (Diella et al., 2004), PhosphositePlus (Hornbeck et al., 2012), HPRD (Peri et al., 2004), dbPTM (version 3.0) (Lu et al., 2013) and SysPTM (Li et al., 2009). For general site prediction, we removed all repetitive items from different databases, then the CD-HIT tool (Huang et al., 2010) with similarity threshold of 40% (Pan et al., 2014) was applied to phosphorylation proteins to reduce the sequence redundancy of phosphorylation proteins and avoid model overfitting. Finally 12 810 protein sequences were reserved for further general phosphorylation site prediction in this work. Next, we extracted all experimentally verified phosphorylation sites for S/T sites and Y sites from those filtered proteins sequences as positive examples, the number of S/T sites and Y sites is 140 120 and 27 691, respectively. As for negative examples, we randomly selected a subset of the other S/T and Y sites to match the number of positive examples (Gnad et al., 2010). For kinase-specific prediction, similar pre-processing procedure was used and we then cluster all the 8130 phosphorylation sites with kinase annotations into group, family, subfamily and individual kinase levels as described in previous study (Xue et al., 2008). Meanwhile, a common performance evaluation strategy used in deep learning methods for sequence analysis was adopted in this study, which separates the dataset into strictly non-overlapping training, validation and independent test randomly (Min et al., 2017; Zhou and Troyanskaya, 2015). In this way, the training dataset is used to adjust the weights of the model, and the validation dataset is used to avoid overfitting (Min et al., 2017). The independent test data (∼10% for general and ∼20% for kinase-specific sites) is used to assess the performance of DeepPhos and compare with other phosphorylation predictors (Ismail et al., 2016; Li et al., 2015). The detailed description of training dataset and independent dataset can be found in Supplementary Materials.
2.3 Training of DeepPhos model
DeepPhos is a novel CNN architecture, which can map the local protein sequence to a high dimensional continuous representation by a series of non-liner transformation and finally generate the classification results of phosphorylation sites. Instead of using common multi-layer CNN directly, DeepPhos utilizes different DC-CNN blocks (Fig. 1b) that could efficiently ensure the critical protein sequence information for phosphorylation prediction. More details of the architecture are described as below:
For a local protein sequence x, the input of DeepPhos with totally K DC-CNN blocks is a set of sequence features for DC-CNN block k), with I and Lk being the size of the amino acid symbol dictionary and the corresponding local window size of phosphorylation sites, respectively. In this study, we code protein sequences by one-hot encoding scheme, and therefore I is set to 21 (Khurana et al., 2018). We carefully explored various configurations of DC-CNN blocks with distinct window sizes in the task of phosphorylation site prediction and finally develop an efficient network architecture with K = 3 and window sizes of 15, 33 and 51, which have been previously proposed for phosphorylation site (Blom et al., 2004; Wei et al., 2017; Xue et al., 2006) for DC-CNN block 1, 2 and 3, respectively.
The convolutional layers in each DC-CNN block perform one-dimension convolution operation along the protein sequence length (Khurana et al., 2018) (Supplementary Fig. S2) and generate corresponding values, which are then applied to activation function (here we use ReLU), in order to activate the neurons and realize the non-linear transformation. For DC-CNN block k, the feature maps generated by the first convolutional layer are defined as:
| (1) |
where represents the weight matrix with the size of , is the size of filters and =3, 7, 13 for k = 1, 2, 3, D is the number of filters and refers to the bias item. In order to reduce the risk of overfitting in training, dropout is used in each convolutional layer, which abandons some neurons randomly after convolution layer.
To enhance the flow of phosphorylation information in the DC-CNN blocks of DeepPhos, we introduce Intra-BCLs that connect all previous convolutional layers with subsequent convolutional layers (Fig. 1b). Consequently, the input of the ith convolutional layer receives the feature maps as the concatenation of sequence feature and the output of all previous layers, which can help to transfer the abstraction of previous layers with different levels to current layer as the network becomes deep (Huang et al., 2017). Accordingly, the output feature maps of the ith convolutional layer in DC-CNN block k are concatenated along the feature dimension, which can be calculated as follows:
| (2) |
where refers to the feature maps produced in the (i−1)th convolutional layer, with is the total number of filters for convolutional layer 1 to i and C is the number of convolutional layers in each DC-CNN block and is set to 5 in this study.
After generating the sequence representations of protein phosphorylation sites using different DC-CNN blocks, they are further integrated by an Inter-BCL in DeepPhos (Fig. 1b) that performs concatenation along the first dimension as follows:
| (3) |
where represents the feature maps produced by convolutional layer C in the kth DC-CNN block. In this way, multiple feature maps are concatenated and then transformed to one-dimensional tensor by a flatten layer. After that fully connect neural network is applied to generate the input of final softmax function, , and n is the number of neurons. Finally, the prediction score of phosphorylation is calculated as follows:
| (4) |
where , q represents the number of categories to be predicted, and is between 0 and 1. In this study, the phosphorylation prediction task is a binary classification problem, so the scores of non-phosphorylation can be formulated as:
| (5) |
The standard cross-entropy for binary classification problem is adopted as cost function to minimize the training error:
| (6) |
where N refers to the total number of training examples, refers to the jth input local sequence and refers to corresponding phosphorylation status label of the jth input sequence. In addition, to relieve the overfitting, L2 regularization is adopted in training, thus the final objective function of DeepPhos is defined as:
| (7) |
where λ is the regularization coefficient, and means the L2 norm of weight matrix. In this study, mini batch strategy is used during training process, which divides the training dataset into several parts according to the mini-batches for each epoch stochastically. We choose Adam optimizer, a widely used optimizer that can adjust learning rate automatically.
DeepPhos can be applied to phosphorylation site prediction including general and kinase-specific prediction at group, family, subfamily or individual kinase level. To this end, for general phosphorylation site prediction, all available S/T and Y phosphorylation sites data are used to train deep learning models. On the other hand, training of deep learning models for kinase-specific phosphorylation site prediction is more challenging as currently most of the verified phosphorylation sites lack corresponding kinase annotation (Wang et al., 2017). To address this issue, we first trained and validated a base deep learning model by phosphorylation data without kinase annotation. Afterwards, we further fine-tuned to obtain final deep learning model using kinase-specific training and validation data (Fig. 1b). In this study, we adopted a transfer learning fine-tuning strategy called layer transfer (Yosinski et al., 2014), to transfer the network in including convolutional layers for all DC-CNN blocks, Intra-CBLs/Inter-CBLs, as well as the learned weight matrices and bias items associated with the convolutional layers.
2.4 Performance evaluation
To assess the performance of phosphorylation site prediction, several commonly used statistical measurements are employed in this study, including sensitivity (Sn), specificity (Sp), overall accuracy (Acc), precision (Pre), Matthew’s correlation coefficient (MCC) and F1 scores. The detailed definitions are:
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
Here, TP is the number of positive samples correct classified in prediction, and TN represents the number of negative samples correct classified by predictors. FP and FN represent the numbers of positive or negative samples that classified by mistake, respectively. Therefore, Sn refers to the percentage of true positive samples correct classified by predictors, and similarly, Sp is the percentage of true negative samples. Pre represents the ratio of true positive samples that generated by predictors, MCC indicates the balance quality of positive and negative data, F1 score is a metric that comprehensively considers precision and recall. Furthermore, we also used receiver-operating characteristic (ROC) curve as well as the area under ROC curve (AUC) to assess the overall performance, the closer ROC curve to the left corner, the closer the AUC value to 1, which demonstrates that the overall performance is better.
3 Results
3.1 Evaluation of DeepPhos for general phosphorylation site prediction
In this section, we first compared DeepPhos with different deep learning network architectures including CNN (LeCun et al., 1998), RNN, fully connected neural network (FCNN) and LSTM on the independent test data as described in Section 2.2. The AUC values of these methods on residues S/T and Y were listed in Supplementary Table S1. In general, DeepPhos obtained higher AUC values than other deep learning architectures, showing that DeepPhos had better overall performance. For example, on Y sites, the AUC value of our architecture is 71.58%, which has 3.14, 2.81, 3.66 and 3.82% improvement over CNN, RNN, LSTM and FCNN, respectively. In addition to AUC values, Sn, Sp, Acc, MCC and F1 score were also calculated in this study to evaluate the performance of DeepPhos. Here, we followed the study of Liu et al. (2018) to set the Sp threshold at the high and medium stringency level =95 and 90%, respectively. Details of these measurements about S/T and Y sites were listed in Table 1 and Supplementary Table S2. It is obvious that DeepPhos consistently achieved higher performance on all the measurements than other deep learning architectures. For S/T sites, Sn, Acc, MCC, Pre values and F1 score of the prediction performance of DeepPhos at the high-stringency level are 33.86, 64.43, 36.48, 87.13 and 48.77%, respectively. Among the other methods, the performance of LSTM is better than FCNN, RNN and CNN at the medium and high-stringency level. For example, the Sn value of LSTM is 31.44%, while the values of FCNN, RNN and CNN are 23.77, 26.98 and 30.19%, respectively. However, the performance of LSTM is not as good as other deep learning architectures on Y sites, indicating that LSTM may not be an ideal architecture for phosphorylation site prediction. In comparison with other architectures, DeepPhos achieves prediction performance with Sn of 18.59%, Acc of 56.79%, MCC of 21.05%, Pre of 78.76% and F1 score of 30.08% on Y sites at the high-stringency level. In short, DeepPhos achieved better overall performance than other deep learning architectures on S/T and Y sites, suggesting that DeepPhos is an efficient deep learning framework that suitable for phosphorylation site prediction. In addition, Supplementary Figure S3 shows that the performance of the independent test data is similar to that obtained by 10-fold cross-validation as following previous study (Dou et al., 2014).
Table 1.
Performance of different architectures on S/T sites at the medium and high-stringency levels
| Stringency | Measure | DeepPhos | FCNN | RNN | LSTM | CNN |
|---|---|---|---|---|---|---|
| Sp=90% | Sn(%) | 47.80 | 37.95 | 40.75 | 45.21 | 43.77 |
| Acc(%) | 68.90 | 63.98 | 65.37 | 67.60 | 66.89 | |
| MCC(%) | 41.69 | 32.74 | 35.32 | 39.37 | 38.09 | |
| Pre(%) | 82.70 | 79.15 | 80.28 | 81.88 | 81.40 | |
| F1(%) | 60.58 | 51.30 | 54.06 | 58.25 | 56.93 | |
| Sp=95% | Sn(%) | 33.86 | 23.77 | 26.98 | 31.44 | 30.19 |
| Acc(%) | 64.43 | 59.39 | 60.99 | 63.22 | 62.59 | |
| MCC(%) | 36.48 | 26.75 | 29.98 | 34.25 | 33.06 | |
| Pre(%) | 87.13 | 82.62 | 84.36 | 86.28 | 85.77 | |
| F1(%) | 48.77 | 36.92 | 40.88 | 46.09 | 44.66 |
Note: Best performing method in bold.
To further assess the performance of DeepPhos, we compared DeepPhos with several existing tools for prediction of general phosphorylation sites using independent test data. For general phosphorylation prediction on S/T and Y sites, several well-known predictors including NetPhos3.0 (Blom et al., 2004), PPSP (Xue et al., 2006), Musite (Gao et al., 2010) and MusiteDeep (Wang et al., 2017) were used to make comparison. In general, DeepPhos achieved higher performance than other four predictors. For Y sites, the AUC value of DeepPhos is 71.58%, which is 15.51, 7.49, 17.58 and 4.98% higher than NetPhos3.0, PPSP, Musite and MusiteDeep, respectively. As for S/T sites, NetPhos3.0, PPSP, Musite and MusiteDeep predictors achieve AUC value of 63.18, 74.14, 57.94 and 77.58%, respectively, while DeepPhos obtains a better performance than these predictors with AUC value of 80.43%. Furthermore, we also calculated the Sn, Pre, Acc, MCC and F1 score of all compared predictors, and the bar graphs of these predictors are displayed in Figure 2. We find that Musite has satisfied Pre values, but other measurements are not as good as other predictors when predicting Y sites. PPSP, NetPhos3.0 and MusiteDeep have a good balance of prediction performance. Consistent with previous study (Wang et al., 2017), deep learning-based MusiteDeep shows better performance compared with other predictors. Take Y site as an example, the performance of MusiteDeep at medium level are Sn of 24.95%, Acc of 57.48%, MCC of 19.70%, Pre of 71.41% and F1 score of 36.98%. In comparison, DeepPhos further improves the prediction performance and the corresponding Sn, Acc, MCC, Pre and F1 score reach 30.11, 60.06, 25.13, 75.09 and 42.99%, respectively. These results show that with the novel deep learning architecture, DeepPhos compared favorably with existing predictors in general phosphorylation prediction.
Fig. 2.
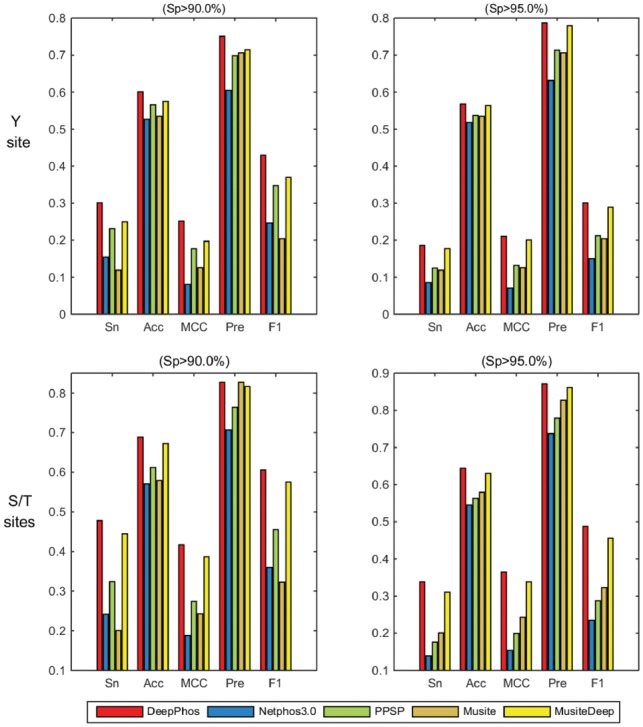
Performance comparison of different predictors for general phosphorylation S/T and Y sites prediction at the high and medium stringency levels. The left field represents the Sp values of 90%, the right part represents the Sp of 95%. The upper rows refer to the performance of Y sites, the rows under refer to the S/T sites. The Sn, Acc, MCC, Pre and F1 represent the different measurements (Color version of this figure is available at Bioinformatics online.)
3.2 Comparison with existing tools for kinase-specific phosphorylation site prediction
In this session, we compared DeepPhos with some existing predictors for kinase-specific prediction including PPSP, GPS and MusiteDeep based on independent test data. In consistent with previous studies (Wang et al., 2017; Xu et al., 2018), we selected some kinase groups, families, subfamilies and individual kinases on S/T and Y sites with the largest sample sizes for performance assessment. We compared with MusiteDeep only in family level on S/T sites for MusiteDeep can be applied to predict kinase families (Wang et al., 2017). Figure 3 and Supplementary Figure S4 display the ROC curves with the corresponding AUC values of different predictors. From the results, DeepPhos achieved comparable or better performance than other predictors. Take group CMGC as an example, the AUC value of DeepPhos is 91.85%, while PPSP and GPS have the AUC value of 82.12 and 83.23%, respectively. As for group CAMK, DeepPhos obtains the AUC value of 90.92%, while the corresponding AUC of PPSP and GPS are 71.26 and 70.57%, respectively. Also, as shown in Supplementary Figure S4, the AUC value of group AGC, Atypical and TK of DeepPhos are 88.43, 83.20 and 82.04%, respectively, which are higher than other methods. Take family CDK as an example, DeepPhos achieves the AUC value of 95.98%, which has an improvement of 3.01, 9.88 and 5.53% compared with MusiteDeep, PPSP and GPS. As for subfamily CDC2, the performance of both GPS and DeepPhos are significantly better than PPSP, which achieve the AUC values of 91.08 and 93.60%, respectively. For prediction of subfamily and individual kinase level, GPS also achieved good performance with high AUC values. For example, the performance of subfamily CDC2, subfamily ERK1, kinase CK2a1 and kinase CDK1 are AUC of 91.08, 92.36, 84.43 and 91.8%, respectively. In comparison, DeepPhos achieved comparable or better AUC values than GPS.
Fig. 3.

ROC curves of DeepPhos and other predictors for group CMGC, group CAMK, family PKC, family CDK, subfamily PKCa, subfamily CDC2, kinase SRC and kinase PRKCA. Here, DeepPhos (red lines) is compared with PPSP (green lines), GPS (the yellow lines) and MusiteDeep (light orange lines) (Color version of this figure is available at Bioinformatics online.)
In addition to AUC values, we listed the metrics of Sn, Acc, MCC, Pre and F1 scores with the high specificity stringency of different predictors in Table 2 and Supplementary Table S3. From these results, we find that DeepPhos offered a balanced performance with not only good Sn and Pre, but also higher MCC and F1 scores. For example, in prediction of group CMGC, DeepPhos achieves the Sn of 67.24%, Acc of 81.07%, MCC of 64.75%, Pre of 93.104% and F1 score of 78.10%, while other predictors have lower values. In conclusion, aforementioned analysis shows that DeepPhos obtained better performance in prediction of kinase-specific phosphorylation sites varying from group, family to subfamily and individual kinase level.
Table 2.
Performance of different predictors for kinase-specific phosphorylation site prediction at the high stringency level
| Kinase | Measure | DeepPhos | GPS | PPSP |
|---|---|---|---|---|
| Group | Sn(%) | 57.80 | 9.83 | 20.81 |
| CAMK | Acc(%) | 74.68 | 48.42 | 54.43 |
| MCC(%) | 55.65 | 9.27 | 23.10 | |
| Pre(%) | 93.46 | 70.83 | 83.72 | |
| F1(%) | 71.43 | 17.26 | 33.33 | |
| Group | Sn(%) | 67.24 | 50.10 | 29.90 |
| CMGC | Acc(%) | 81.07 | 72.47 | 62.33 |
| MCC(%) | 64.75 | 50.43 | 32.79 | |
| Pre(%) | 93.14 | 91.00 | 85.79 | |
| F1(%) | 78.10 | 64.62 | 44.35 |
Note: Best performing method in bold.
To further analyze the significance of layer transfer on kinase-specific prediction, we also compared the results of DeepPhos with the model without layer transfer at group level. Take CMAK group as an example, the AUC value of DeepPhos is 90.9%, while the AUC value of model without layer transfer is 84.0%, more details were displayed in Supplementary Table S4. The results show that the application of layer transfer in DeepPhos was significant in kinase-specific phosphorylation prediction for solving the small number of examples with kinase annotation.
3.3 Visualization of learned features
In order to distinguish the abstractions generated by our deep learning architecture, we visualized the features extracted by DeepPhos and original one-hot coding of protein sequences in this section. To observe the difference between phosphorylation and non-phosphorylation intuitively, a popular visualization algorithm t-Distributed Stochastic Neighbor Embedding (Maaten and Hinton, 2008) was used here to visualize the results, by which we squeezed the high-dimensional features into 2D space and normalized the value to −1 to 1.Take group CMGC as an example, the representation of abstract features extracted by our architecture and the original sequence features were plotted in Figure 4. It is difficult to separate the phosphorylation sites from non-phosphorylation sites by original sequences one-hot coding, while it is much clearer for us to identify these two classes after the abstract representation of DeepPhos. The comparison for some other kinase groups was also displayed in Supplementary Figure S4. Through the visualization of t-Distributed Stochastic Neighbor Embedding, we demonstrated that sequences of raw proteins can be mapped into meaningful representation through the non-linear transformation generated by DeepPhos, which can be helpful for further analysis of phosphorylation sites.
Fig. 4.
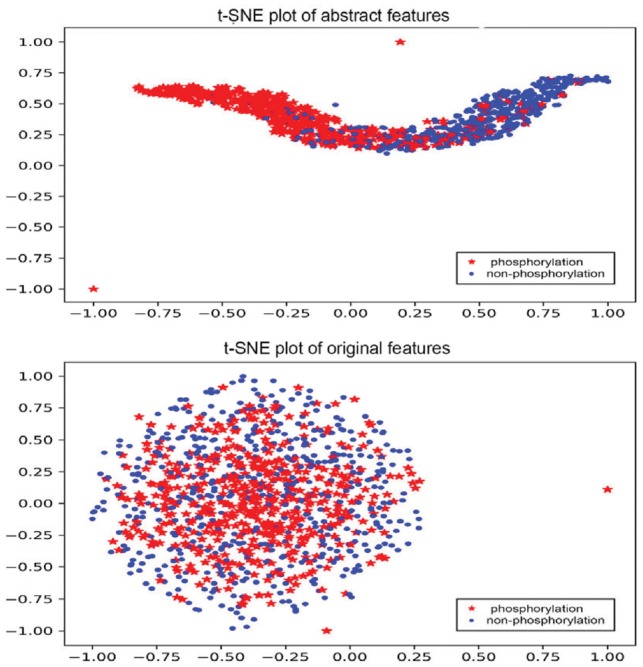
Visualization of abstract features extracted by DeepPhos and original sequence features by one-hot codings. The red star represents the phosphorylation sites with kinase annotation belonging to group CMGC, the blue dot represents the non-phosphorylation sites (Color version of this figure is available at Bioinformatics online.)
3.4 Analysis of potential phosphorylation sites
To evaluate the ability of DeepPhos in discovery of unknown phosphorylation sites, by following previous study (Wang et al., 2016) we checked the top 20 candidate phosphorylation sites manually from the high-quality protein database Uniport. The candidate phosphorylation sites were extracted from our independent test that are not verified by experiment but likely to be phosphorylation sites according to DeepPhos. The top 20 candidate sites of S/T and Y were listed in Table 3, Supplementary Table S5, respectively. In Table 3, we find three sites, i.e. Ser788 of protein Q9H987 that ranks four with score of 0.9637, Ser476 of protein O43526 with score of 0.9393 and Ser783 of protein Q9UPX0 with score of 0.9358, seem to be phosphorylated according to the evidences in Uniport database (bold in Table 3). These results demonstrated that DeepPhos can be applied to detect unknown phosphorylation sites practically, which can be helpful for discovering the mechanisms of related biological processes.
Table 3.
The top 20 candidate phosphorylation sites on S/T of independent test
| Rank | Protein | Position | Score | Rank | Protein | Position | Score |
|---|---|---|---|---|---|---|---|
| 1 | Q8N5F7 | 217 | 0.9916 | 11 | Q9HBD4 | 1458 | 0.9400 |
| 2 | Q9Y438 | 857 | 0.9786 | 12 | O43526 | 476 | 0.9393 |
| 3 | Q9NSI6 | 1822 | 0.9698 | 13 | O14529 | 418 | 0.9391 |
| 4 | Q9H987 | 788 | 0.9637 | 14 | Q5CZC0 | 6583 | 0.9380 |
| 5 | Q8N3T6 | 807 | 0.9598 | 15 | Q8N3T6 | 557 | 0.9367 |
| 6 | Q9H4Q3 | 239 | 0.9549 | 16 | Q15648 | 1447 | 0.9362 |
| 7 | Q9C0D6 | 525 | 0.9546 | 17 | Q9UPX0 | 783 | 0.9359 |
| 8 | Q09MP3 | 21 | 0.9545 | 18 | Q6ZQQ6 | 2242 | 0.9358 |
| 9 | O60393 | 444 | 0.9495 | 19 | Q9H0D2 | 1268 | 0.9353 |
| 10 | O14529 | 986 | 0.9475 | 20 | A1L0S7 | 953 | 0.9343 |
Note: The sites seem to be phosphorylated according to the evidences in Uniport database in bold.
4 Discussion
Phosphorylation is of significance in biological process, which relates to various diseases. Due to the limitations of experimental verifying sites that cost time and money, it is very useful to develop effective computational methods for phosphorylation prediction. Hence, in this study, we propose DeepPhos, a novel deep learning architecture, which can predict potential general phosphorylation sites and kinase-specific sites including group, family, subfamily and individual kinase levels. DeepPhos has a better performance than existing phosphorylation predictors evaluated by independent test. In addition to performance metrics, we visualized the features extracted by DeepPhos, the visualization results show that the proposed architecture can transform protein sequences to meaningful representations. Furthermore, there are evidences in Uniport database corresponding to the highly ranked results, which can be helpful for further biology research.
The major contributions of our work can be summarized as follows. Firstly, different from existing deep learning architectures such as deep learning architectures such as CNN (Wang et al., 2017), LSTM (Chen et al., 2018), deep neuron networks (Xie et al., 2018) in PTM site prediction, the Intra-BCL in each DC-CNN block could assign different weights to input automatically and enhance the flow of phosphorylation information, in this way, useful information can pass to help obtain final decision. Another difference in the architecture of DeepPhos is that application of distinct filter sizes according to different windows can integrate multiple abstracts of sequence features by Inter-BCL, and finally catch the non-linear relationships between original raw protein sequences and the phosphorylation prediction results. Secondly, by applying layer transfer to a pre-trained model using the large amount of phosphorylation data without kinase annotation, the proposed framework can efficiently deal with kinase-specific phosphorylation site prediction including group, family, subfamily and individual kinase level. Meanwhile, the dropout and L2 regularization were also used here to prevent overfitting, leading to better fine-tune and generalization performance. Finally, as an efficient deep learning architecture for protein sequence representation, DeepPhos can be further modified and extended for the tasks of PTM site prediction using the training data of different type of PTMs.
Although our architecture has shown promising performance of phosphorylation site prediction, there are still some limitations that can be further improved. Since the deep learning method is still a black-box (Ma et al., 2018), our method cannot be explained well with meaningful biological process. Our future work would concentrate on the considerable biological interpretation and carry on to improve the framework by combing some efficient modules, such as the generative adversarial networks (Goodfellow et al., 2014) and attention mechanisms (Mnih et al., 2014). Although attention mechanisms have been used in PTM site prediction (Wang et al., 2017), there are some improved attention mechanisms such as spatial (Chen et al., 2017) and modular attention (Yu et al., 2018), which can be further explored in future study. In addition, we would use other parameter optimization strategies such as batch normalization (Ioffe and Szegedy, 2015), for optimizing the parameters of deep learning models. Moreover, features based on amino acid properties or protein function are also very important for PTM site prediction (Dou et al., 2014, 2017; Song et al., 2017), which can be further combined with protein sequences in future study. In conclusion, we propose a novel deep learning architecture for phosphorylation site prediction, DeepPhos, which can be applied to reduce the cost and provide clues for further biological research.
Funding
This work was supported by National Natural Science Foundation of China (61871361, 61471331, 61571414, 61772368, 61572363, 91530321, 61602347) and Natural Science Foundation of Shanghai (17ZR1445600).
Conflict of Interest: none declared.
Supplementary Material
References
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831.. [DOI] [PubMed] [Google Scholar]
- Aponte A.M. et al. (2009) 32P labeling of protein phosphorylation and metabolite association in the mitochondria matrix. Methods Enzymol., 457, 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil S.A. et al. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol., 24, 1285.. [DOI] [PubMed] [Google Scholar]
- Blom N. et al. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics, 4, 1633–1649. [DOI] [PubMed] [Google Scholar]
- Chen L. et al. (2017) SCA-CNN: Spatial and channel-wise attention in convolutional networks for image captioning. In: 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR). pp. 6298–6306. IEEE.
- Chen Z. et al. (2018) Large-scale comparative assessment of computational predictors for lysine post-translational modification sites. Brief. Bioinform., 30285084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cireşan D. et al. (2011) A committee of neural networks for traffic sign classification. In: The 2011 International Joint Conference on Neural Networks (IJCNN). pp. 1918–1921. IEEE.
- Collobert R. et al. (2011) Natural language processing (almost) from scratch. J. Mach. Learn. Res., 12, 2493–2537. [Google Scholar]
- Diella F. et al. (2004) Phospho. ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinformatics, 5, 79.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y. et al. (2014) PhosphoSVM: prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino Acids, 46, 1459–1469. [DOI] [PubMed] [Google Scholar]
- Dou Y. et al. (2017) Prediction of protein phosphorylation sites by integrating secondary structure information and other one-dimensional structural properties In: Prediction of Protein Secondary Structure. Springer, New York, pp. 265–274. [DOI] [PubMed] [Google Scholar]
- Fan W. et al. (2014) Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino Acids, 46, 1069–1078. [DOI] [PubMed] [Google Scholar]
- Gao J. et al. (2010) Musite: a tool for global prediction of general and kinase-specific phosphorylation sites. Mol. Cell. Proteomics, 9, 2586–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F. et al. (2010) Predicting post-translational lysine acetylation using support vector machines. Bioinformatics, 26, 1666–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow I. et al. (2014) Generative adversarial nets. In: Advances in Neural Information Processing Systems, pp. 2672–2680.
- Hornbeck P.V. et al. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res., 40, D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G. et al. (2017) Densely Connected Convolutional Networks. In: CVPR. p.3.
- Huang S.-Y. et al. (2015) Using support vector machines to identify protein phosphorylation sites in viruses. J. Mol. Graph. Model., 56, 84–90. [DOI] [PubMed] [Google Scholar]
- Huang Y. et al. (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics, 26, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioffe S., Szegedy C. (2015) Batch normalization: accelerating deep network training by reducing internal covariate shift. In: ICML, p.3.
- Ismail H.D. et al. (2016) RF-Hydroxysite: a random forest based predictor for hydroxylation sites. Mol. BioSyst., 12, 2427–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S. et al. (2018) DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics, 34, 2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y. et al. (1998) Gradient-based learning applied to document recognition. Proc. IEEE, 86, 2278–2324. [Google Scholar]
- Li F. et al. (2018) Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics, 34, 4223–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. et al. (2015) GlycoMine: a machine learning-based approach for predicting N-, C-and O-linked glycosylation in the human proteome. Bioinformatics, 31, 1411–1419. [DOI] [PubMed] [Google Scholar]
- Li H. et al. (2009) SysPTM: a systematic resource for proteomic research on post-translational modifications. Mol. Cell. Proteomics, 8, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T. et al. (2008) Prediction of kinase‐specific phosphorylation sites with sequence features by a log‐odds ratio approach. Proteins, 70, 404–414. [DOI] [PubMed] [Google Scholar]
- Liu Y. et al. (2018) PTM-ssMP: a Web Server for Predicting Different Types of Post-translational Modification Sites Using Novel Site-specific Modification Profile. Int. J. Biol. Sci., 14, 946–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.-T. et al. (2013) DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res., 41, D295–D305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J. et al. (2018) Using deep learning to model the hierarchical structure and function of a cell. Nat. Methods, 15, 290.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, Hinton G. (2008) Visualizing data using t-SNE. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- Matthews H.R. (1995) Protein kinases and phosphatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: a possible regulator of the mitogen-activated protein kinase cascade. Pharmacol. Ther., 67, 323–350. [DOI] [PubMed] [Google Scholar]
- Min X. et al. (2017) Chromatin accessibility prediction via convolutional long short-term memory networks with k-mer embedding. Bioinformatics, 33, i92–i101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnih V. et al. (2014) Recurrent models of visual attention. In Advances in Neural Information Processing Systems, pp. 2204–2212.
- Pan Z. et al. (2014) Systematic analysis of the in situ crosstalk of tyrosine modifications reveals no additional natural selection on multiply modified residues. Sci. Rep., 4, 7331.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri S. et al. (2004) Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res., 32, D497–D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin G.-M. et al. (2016) PhosD: inferring kinase–substrate interactions based on protein domains. Bioinformatics, 33, 1197–1204. [DOI] [PubMed] [Google Scholar]
- Song J. et al. (2017) PhosphoPredict: a bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Sci. Rep., 7, 6862.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D. et al. (2017) Prognosis prediction of human breast cancer by integrating deep neural network and support vector machine: supervised feature extraction and classification for breast cancer prognosis prediction. In: 2017 10th International Congress on Image and Signal Processing, BioMedical Engineering and Informatics (CISP-BMEI). pp. 1–5. IEEE.
- Trost B. et al. (2013) DAPPLE: a pipeline for the homology-based prediction of phosphorylation sites. Bioinformatics, 29, 1693–1695. [DOI] [PubMed] [Google Scholar]
- Trost B., Kusalik A. (2011) Computational prediction of eukaryotic phosphorylation sites. Bioinformatics, 27, 2927–2935. [DOI] [PubMed] [Google Scholar]
- Trost B. et al. (2016) DAPPLE 2: a tool for the homology-based prediction of post-translational modification sites. J. Proteome Res., 15, 2760–2767. [DOI] [PubMed] [Google Scholar]
- Walsh C. (2006) Posttranslational Modification of Proteins: Expanding Nature's Inventory. Roberts and Company Publishers, Greenwood Village, CO. [Google Scholar]
- Wang D. et al. (2017) MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics, 33, 3909–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-R. et al. (2016) ESA-UbiSite: accurate prediction of human ubiquitination sites by identifying a set of effective negatives. Bioinformatics, 33, 661–668. [DOI] [PubMed] [Google Scholar]
- Wang X. et al. (2017) Prediction of phosphorylation sites based on Krawtchouk image moments. Proteins, 85, 2231–2238. [DOI] [PubMed] [Google Scholar]
- Wei L. et al. (2017) PhosPred-RF: a novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans. Nanobioscience, 16, 240–247. [DOI] [PubMed] [Google Scholar]
- Wen P.-P. et al. (2016) Accurate in silico prediction of species-specific methylation sites based on information gain feature optimization. Bioinformatics, 32, 3107–3115. [DOI] [PubMed] [Google Scholar]
- Xie Y. et al. (2018) DeepNitro: prediction of Protein Nitration and Nitrosylation Sites by Deep Learning. Genomics Proteomics Bioinformatics, 16, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. et al. (2018) PhosContext2vec: a distributed representation of residue-level sequence contexts and its application to general and kinase-specific phosphorylation site prediction. Sci. Rep., 8, 8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y. et al. (2006) PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics, 7, 163.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y. et al. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics, 7, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosinski J. et al. (2014) How transferable are features in deep neural networks? In: Advances in Neural Information Processing Systems, pp. 3320–3328.
- Yu L. et al. (2018) Mattnet: Modular attention network for referring expression comprehension. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR) pp. 1307–1315. IEEE. [Google Scholar]
- Zhou J., Troyanskaya O.G. (2015) Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods, 12, 931.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.