

Abstract
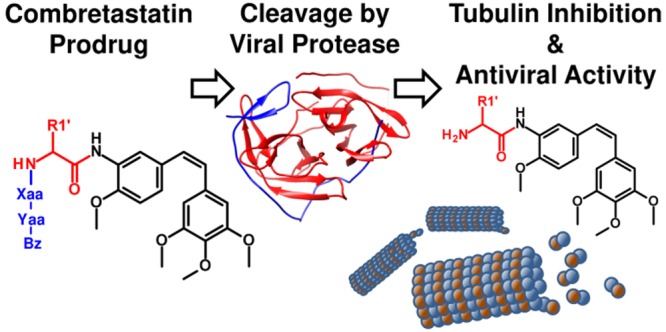
Infections with flaviviruses such as dengue virus (DENV) are prevalent throughout tropical regions worldwide. Replication of these viruses depends on tubulin, a host cell factor that can be targeted to obtain broad-spectrum antiviral agents. Targeting of tubulin does, however, require specific measures to avoid toxic side-effects. Herein, we report the synthesis and biological evaluation of combretastatin peptide hybrids that incorporate the cleavage site of the DENV protease to allow activation of the tubulin ligand within infected cells. The prodrug candidates have no effect on tubulin polymerization in vitro and are 20–2000-fold less toxic than combretastatin A-4. Several of the prodrug candidates were cleaved by the DENV protease in vitro with similar efficiency as the natural viral substrates. Selected compounds were studied in DENV and Zika virus replication assays and had antiviral activity at subcytotoxic concentrations.
Keywords: Dengue virus, Zika virus, tubulin, combretastatin, serine protease
With an estimated number of over 100 million infections per year, dengue is considered the most common vector borne virus infection.1 The dengue virus (DENV), a distinct member of the genus Flavivirus, is transmitted by mosquitoes of the genus Aedes, which are widespread in tropical and subtropical regions around the globe. While in many cases the infection is excruciating but heals without severe consequences, it can also develop into the potentially fatal dengue hemorrhagic fever and shock syndrome. The average annual number of deaths is estimated to be in the five-digit range.2,3 Thus far, no specific antiviral treatment is available, making symptomatic therapy and vector control the only countermeasures. The development of dengue vaccines is highly challenging, on the one hand, due to the occurrence of four different serotypes (DENV-1 to DENV-4) and, on the other hand, due to the mechanism of antibody-dependent enhancement.4 For the only approved vaccine (Dengvaxia), the WHO noted an increased risk of severe dengue for vaccinated individuals who get infected for the first time after vaccination.5 Therefore, developing new antiviral agents and exploring new paths in treatment remains an important challenge.
Besides targeting the viral factors required for productive replication or involved in pathogenesis, host factors are promising and valuable targets to combat viral infections.6 In particular, host factors bear a lower risk of resistance development because they do not experience an evolutionary pressure by antiviral treatment. Furthermore, host targets have the potential to serve as target for broad-spectrum antiviral drugs, which may cover, for example, all four DENV serotypes and other flaviviruses like Zika virus (ZIKV) and West Nile virus.7
One potential host target for the development of novel antivirals is tubulin. Several reports indicate that microtubules are involved in flaviviral replication and that inhibitors of tubulin polymerization have antiviral activity. The cytoskeleton is used by flaviviruses for intracellular transport and virus egress.8 Acosta et al. showed a significant reduction in virus yield caused by inhibitors of microtubule polymerization and suggested that the structural integrity of the microtubule network is a requirement for efficient virus entry.9 In addition, recent work shows a crucial role of microtubules for the replication of ZIKV.10 However, targeting host factors also comes at the cost of side effects and potential systemic toxicity. This is particularly true for tubulin, a prominent anticancer drug target, whose ligands frequently possess considerable systemic toxicity. Therefore, a therapeutic or preventive exposure to tubulin ligands in the context of viral infections can only be considered if a mechanism is in place that prevents unspecific toxicity. We already reported prodrugs based on combretastatin and colchicine that were activated by human carboxylesterase 1, an enzyme highly abundant in monocytes and hepatocytes, the primary target cells of DENV.11 In the present work, we report the concept of tubulin-ligand prodrugs that are specifically activated by the viral protease.
The selective activation of tubulin-ligand prodrugs requires a mechanism, such as enzymatic cleavage, that is exclusively present in cells infected with flaviviruses. The flaviviral RNA genome encodes a polyprotein, which is processed by the viral protease NS2B-NS3.12 In the present study, we evaluated the capability of flaviviral proteases to hydrolyze and thereby activate peptide hybrids of the tubulin ligand combretastatin. Some examples have already been described in which drugs are activated or released by a similar mechanism. Yasobu et al. reported a colchicine prodrug that is released upon hydrolysis by cathepsin B.13 In the field of tumor research, several antitumor drugs are activated by proteases that are overexpressed in tumor cells.14
The substrate recognition properties of the DENV protease are well-defined. Analysis of the cleavage sites in the viral polyprotein recognized by the NS2B-NS3 protease demonstrates a requirement for basic residues (Arg, Lys) particularly in the P1 and P2 positions. Small amino acids (Gly, Ser) are commonly present in P1′ (Figure 1).15 A wider range of residues is tolerated in the other positions near the cleavage site.
Figure 1.

Cleavage site sequence logo for the flaviviral NS2B-NS3 protease, based on the six polyprotein cleavage sites from 50 different flaviviruses.15,16
Since tubulin is an extensively studied target in antitumor therapy, numerous inhibitors of tubulin polymerization have already been synthesized and characterized, and the biochemical properties of tubulin are well-characterized in this context. Thus far, six distinct binding sites on tubulin for different classes of ligands have been identified.17 A large number of ligands is derived from the alkaloid colchicine, which binds into a pocket that has consequently been termed “colchicine binding site”.18 Another ligand that binds into the colchicine binding site is combretastatin A-4 (CA4; see Figure 2), a natural product that was originally isolated from the tree Combretum caffrum.19 Combretastatin analogs are synthetically accessible via the Wittig reaction of two substituted aromatic moieties.20 Different functional groups on both aromatic rings can be employed, resulting in numerous structural variations and well-defined structure–activity relationships.21−23 A major drawback of CA4 and its analogs is their low aqueous solubility, resulting in difficulties to develop pharmaceutical formulations for these drugs.
Figure 2.

Structure of CA4 in the tubulin binding pocket and some of its interactions with surrounding amino acids according to the crystal structure by Gaspari et al. (pdb code: 5LYJ).19 The structural elements essential for binding to tubulin are highlighted in gray.
Figure 2(19) shows the essential structural elements of CA4 analogs that are required for activity: These are the methoxy groups and the cis-configured double bond. Not essential is the hydroxyl group at position B-3, which can therefore be modified to attach the protease recognition sequence. Two widely known examples for this approach are the phosphoric acid ester prodrug fosbretabulin24—developed with the intention to increase aqueous solubility—and the serine ester prodrug ombrabulin.25 Fosbretabulin showed antitumor activity in phase II clinical trials.26 Other modifications in the B-3 position include the design of peptidic prodrugs, such as poly-l-Glu derivatives, to achieve a specific uptake of CA4 into tumor cells.27 Notably, the concept of peptidic CA4 prodrugs that incorporate cationic residues does not only allow site-specific activation by the DENV protease, but it is also expected to modulate the physicochemical properties of the molecules and increase their solubility by introduction of a highly polar moiety.
CA4 was synthesized according to Scheme 1. The cis-configured double bond that connects the two aromatic rings was obtained in a Wittig reaction between the phosphonium salt 1 and isovanillin. The reaction required warming to 60 °C. Under these conditions, the main reaction product was the cis-isomer, possibly due to the lower stability of the intermediate betaine. Despite the slightly acidic phenolic hydroxyl group, no protective groups were required if at least two equivalents of base were used.
Scheme 1. Synthesis of CA4 and “Combretamin” (3).

To attach a peptide sequence that is recognized by the DENV protease, the hydroxyl group at position B-3 was replaced by an amino group. To obtain the derivative 3, hereafter called “combretamin”, a nitro derivative of CA4 was synthesized (2) and then reduced to furnish the amine. Initial attempts with Zn/HOAc28 or H2/Pd resulted either in incomplete reduction or hydrogenation of the double bond.
Eventually, an optimized protocol using five equivalents of each SnCl2 and NaBH4, under absence of light and oxygen, provided the amine 3 in quantitative yield. The oligopeptides were synthesized by solid phase peptide synthesis (Scheme 2). The N-terminus of the peptides was capped with a benzoyl group to protect the otherwise free amino group and to facilitate UV detection of the products. The coupling of the peptides to combretamin was performed in solution, resulting in the compounds 6a to 6h.
Scheme 2. Synthesis of B-3-Combretastatin Peptide Hybrids.

The expected NS2B-NS3 cleavage site is marked with a vertical arrow.
In addition, we synthesized an analog without any prime site residues between the cleavage site and the combretastatin moiety. This Arg-Arg-combretamin was not hydrolyzed by the DENV protease, but acted as an inhibitor in the biochemical protease inhibition assay29 with a remarkably low IC50 of 6.3 μM (data not shown). Therefore, all peptide hybrids reported here feature one or two prime position amino acids between the combretamin core and the Arg/Lys nonprime residues to allow complete hydrolysis.
The first objective was to verify that an activation of the assumed prodrugs is possible by the DENV protease in vitro. Therefore, compounds were incubated in a solution of the protease, and the degree of conversion was quantified by HPLC-UV. The metabolites were identified with HRMS-ESI. As shown in Table 1, compounds 6a, 6b, 6d, and 6f were hydrolyzed to a significant extent by the DENV protease in vitro. The cleavage efficiency was strongly influenced by the P1′ residue. Alanine and glycine were tolerated well in position P1′, but the nonproteinogenic β-alanine (6c) and the polar serine (6e, 6g) were not hydrolyzed efficiently. With respect to the serine residue, this observation was partially unexpected and in contrast to the prime-site preferences derived from the viral polyprotein cleavage sites (Figure 1).
Table 1. Biochemical and Antiviral Data of Combretastatin and Its Peptide Hybrids.

[DENV protease] = 1 μM, [compound] = 500 μM in buffer pH 9, incubated for 2 h at 37 °C. Each value was determined from triplicate wells.
[DENV protease] = 1 μM, [compound] from 100 μM up to1 mM in buffer pH 9, incubated for 10 min at rt. Each value was determined from triplicate wells.
Conditions: 4 mg/mL tubulin, 1 mM GTP, 10 μM compound, 80 mM PIPES, pH 6.9, 0.5 mM EGTA, 2 mM MgCl2, 1 h, 37 °C. The given number is the mean value of two or three independent measurements and the standard deviation. Calculated is the relative polymerization inhibition compared to an untreated (blank) measurement that counted as 0%. Negative numbers indicate increased polymerization.
Virus titer reduction was measured in a single point experiment. The virus containing supernatant was aliquoted into duplicates; after incubation, the average number of plaques was counted and the reduction was calculated in comparison to an untreated blank (see Table S3 in the Supporting Information).
The cytotoxic concentration (CC50) was determined after 24 h incubation in Huh-7 cells and after 72 h in HeLa and Huh-7 cells. Each value was determined from triplicate wells; the standard deviation was always below 10%. n.a. = not applicable (no hydrolysis site or no tubulin ligand).
Kinetic parameters (Km, vmax, and kcat) for the hydrolysis of the compounds were determined using the Lineweaver–Burk plot and compared to values obtained for other published substrates. Compound 6d had the highest affinity with a Km of 144 μM, and the Km values for 6a and 6f were around 200 μM. The natural cleavage sites of the protease are located between the various nonstructural proteins in the DENV polyprotein. Dodecapeptides that resemble the polyprotein cleavage sites were synthesized and assayed by Khumthong et al. and found to have affinities (Km) to DENV protease in the three-digit micromolar range as well (see Supporting Information).30 The data of our compounds were also compared to the FRET substrate that is commonly used for in vitro activity studies.29Km values were comparable to the Km of other substrates (Table 1). The turnover numbers (kcat) were up to 0.284 s–1 (for compound 6b) and thus comparable to the artificial FRET substrate with a kcat of 0.168 s–1. The resulting values were between one and two orders of magnitude higher than for the dodecapeptides. It is possible that the smaller size of the peptide hybrids and their metabolites lead to faster diffusion into and out of the active site of the enzyme and thus to a higher reaction rate.
Despite similar kinetic parameters, the FRET substrate was almost completely cleaved after two hours, whereas only a fraction of the peptide hybrids, about 40%, was hydrolyzed at this time point (Table 1). This effect can at least partially be explained by product inhibition. The hydrolysis product Bz-Arg-Arg-OH (from 6a, 6b, and 6d) shows 34.5% inhibition of the DENV protease activity at 250 μM, which is the assumed concentration of the product after half of the prodrug has been hydrolyzed. Product Bz-Lys-Arg-OH (from 6f) inhibits 12.2% of the protease activity at this concentration.
The second objective in creating compounds that could act as prodrugs was to decrease the cytotoxicity, relative to CA4, while maintaining antiviral activity. The cytotoxic concentration (CC50) was determined after 24 h and after 72 h of incubation in HeLa and Huh-7 cells. Like other inhibitors of tubulin polymerization, CA4 is a highly toxic molecule that inhibits mitosis, which eventually leads to apoptosis. After 24 h, however, mitosis has been initiated only in a fraction of cells, so the acute toxicity is moderate. The empirical doubling time is ca. 24 h for HeLa cells and ca. 48 h for Huh-7 cells. Consequently, the full toxic effect becomes evident only after extended incubation times. The readout for the antiviral effect (reduction of DENV and ZIKV titer in cells) was initiated after 24 h, and the antiviral activity was studied at compound concentrations lower than the CC50 (determined after 24 h).
The ability of CA4 and its derivatives to inhibit the polymerization of tubulin was determined with the isolated protein in vitro. The highest inhibitory effect was observed for CA4, which reduced the polymerization of tubulin by 60.3% at a concentration of 10 μM. In contrast, the peptide hybrids had almost no effect on tubulin polymerization (<10% inhibition). This indicates that, as intended, the bulky peptides attached to the combretastatin effectively prevent these compounds from binding into the tubulin subunit.
The reference compound CA4 had also the strongest cytotoxic effect, both after 24 and 72 h (Table 1). Its effective antiviral concentration against DENV was 870 nM in Huh-7 cells as reported earlier.11 Combretamin 3 showed similar antiviral effect against ZIKV, yet slightly lower anti-DENV activity and cytotoxicity. Despite the residual amino acids at position B-3, the primary metabolites 4a and 4b showed tubulin polymerization inhibition, cytotoxicity, and antiviral activity. It can be hypothesized that the residual amino acid is hydrolyzed intracellularly by host enzymes, such as exopeptidases from the cathepsin family.15 For example, cathepsin H, with preference for Arg-containing sequences, has been isolated from hepatocytes31 and is abundant in endosomes.32
The design goals were achieved in that all peptide hybrids had a clearly reduced toxicity compared to CA4, being about 20 to 2000 times less toxic in the 72 h measurement. All peptide hybrids (6a–h) were further tested in a virus titer reduction assay and showed an antiviral effect in the low micromolar range. It is noticeable that the compounds, which did not undergo hydrolysis by DENV protease in vitro, still have antiviral activity. Both findings can be explained by the fact that these compounds may also be inhibitors of the viral protease. The IC50 of compound 6e was exemplarily determined to be 25 μM (see SI). As already outlined above, other intracellular proteases may cleave this compound, delivering a CA4-derivative in the cell.
In order to confirm proteolytic cleavage and prodrug activation by the DENV protease, we employed HeLa cells stably transfected with DENV serotype 2 NS2B and NS3 proteins (DENV2proHeLa). Compounds that are hydrolyzed by the viral protease are expected to show increased cytotoxicity in these cells as compared to wildtype HeLa cells. In contrast, nonhydrolyzable compounds are expected to have similar effects in both cell lines. As shown in Figure 3, the reference compounds CA4 and 3 and the noncleavable compounds 6c, 6e, and 6g have practically identical toxicity in both cell lines. In contrast, two of the four hydrolyzable compounds (6a, 6b) show a remarkable increase of cytotoxicity in the DENV2proHeLa cells, which we regard as an indication of intracellular cleavage and proof-of-concept for the prodrug approach presented here. The relatively weak and probably insignificant increase of cytotoxicity for 6d and 6f can be caused by insufficient permeability, by variations of substrate recognition properties between the isolated NS2B-NS3 protein construct and the intracellular protease in the DENV2proHeLa cells, or by a combination thereof.
Figure 3.

Relative fold change in CC50 between wildtype HeLa and the DENV2proHeLa cells, which contain an intracellular DENV protease. Numerical cytotoxicity values are provided in Table S4 in the Supporting Information.
The lack of correlation between the biochemical effects (cleavage by isolated DENV protease and effect on tubulin polymerization) and the inhibition of viral replication can be due to three factors: (i) Cleavage of the prodrugs by nonviral intracellular proteases such as cathepsins or prohormone convertases. The extensive cleavage of compounds such as 6d and 6e by trypsin (cf. Table S2 in the Supporting Information) indicates that the prodrugs possess a certain degree of promiscuity toward nonflaviviral proteases. (ii) Pharmacokinetic effects, particularly a preferential or hindered uptake of prodrugs depending on the peptidic substituent. The mostly basic amino acids in the prodrug moiety are likely to favor, to a variable degree, binding to negatively charged cell membranes and subsequent cellular internalization by endocytosis. (iii) Involvement of as-yet undefined targets or mechanisms in the expression of the phenotypic effect of these compounds.
In conclusion, combretastatin peptide hybrids have been synthesized and found to be hydrolyzed by the DENV protease in vitro at a rate comparable to its natural substrates. Peptide hybrids and metabolites showed antiviral activity against DENV and ZIKV in hepatocyte-derived cells at subcytotoxic concentrations. However, not all assumed prodrugs were hydrolyzed, the SAR does not provide a clear relationship between prodrug activation and antiviral efficacy, and the therapeutic index is insufficient to consider further preclinical development. We therefore consider the present work as a conceptual study that sheds more light on the antiviral activity of tubulin inhibitors, especially from the combretastatin group, and prodrugs activated by viral factors.
Acknowledgments
We thankfully acknowledge the technical assistance of Natascha Stefan for the help in cell culture, Tobias Timmermann and Heiko Rudy for NMR and mass spectral data, Miriam Jaki and Irene Martinez for assistance in synthesis, and Veaceslav Boldescu for support with the tubulin assay. These persons are associated with Heidelberg University, IPMB Medicinal Chemistry. We also thank Stephanie Kallis (Department of Infectious Diseases, Heidelberg University) for support with the ZIKV antiviral assay.
Glossary
ABBREVIATIONS
- CA4
combretastatin A-4
- DENV
dengue virus
- DIPEA
diisopropylethylamine
- DMF
dimethylformamide
- EGTA
ethylene glycol-bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- NS 3
nonstructural protein 3
- PIPES
1,4-piperazinediethanesulfonic acid
- SAR
structure–activity relationship
- ZIKV
Zika virus
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00058.
Supplementary data (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Bhatt S.; Gething P. W.; Brady O. J.; Messina J. P.; Farlow A. W.; Moyes C. L.; Drake J. M.; Brownstein J. S.; Hoen A. G.; Sankoh O.; et al. The Global Distribution and Burden of Dengue. Nature 2013, 496 (7446), 504–507. 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard D. S.; Undurraga E. A.; Halasa Y. A.; Stanaway J. D. The Global Economic Burden of Dengue: A Systematic Analysis. Lancet Infect. Dis. 2016, 16, 935–941. 10.1016/S1473-3099(16)00146-8. [DOI] [PubMed] [Google Scholar]
- Stanaway J. D.; Shepard D. S.; Undurraga E. A.; Halasa Y. A.; Coffeng L. E.; Brady O. J.; Hay S. I.; Bedi N.; Bensenor I. M.; Castañeda-Orjuela C. A.; et al. The Global Burden of Dengue: An Analysis from the Global Burden of Disease Study 2013. Lancet Infect. Dis. 2016, 16 (6), 712–723. 10.1016/S1473-3099(16)00026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz L. M.; Halloran M. E.; Durbin A. P.; Longini I. M. The Dengue Vaccine Pipeline: Implications for the Future of Dengue Control. Vaccine 2015, 33 (29), 3293–3298. 10.1016/j.vaccine.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekly Epidemiological Report. World Heal. Organ. 2016, 30 (91), 349–364. [Google Scholar]
- Kaufmann S. H. E.; Dorhoi A.; Hotchkiss R. S.; Bartenschlager R. Host-Directed Therapies for Bacterial and Viral Infections. Nat. Rev. Drug Discovery 2018, 17 (1), 35–56. 10.1038/nrd.2017.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldescu V.; Behnam M. A. M.; Vasilakis N.; Klein C. D. Broad-Spectrum Agents for Flaviviral Infections: Dengue, Zika and Beyond. Nat. Rev. Drug Discovery 2017, 16 (8), 565–586. 10.1038/nrd.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo K. Y.; Chee H. Interaction between Flavivirus and Cytoskeleton during Virus Replication. BioMed Res. Int. 2015, 2015, 1–6. 10.1155/2015/427814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta E. G.; Castilla V.; Damonte E. B. Alternative Infectious Entry Pathways for Dengue Virus Serotypes into Mammalian Cells. Cell. Microbiol. 2009, 11 (July), 1533–1549. 10.1111/j.1462-5822.2009.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese M.; Goellner S.; Acosta E. G.; Neufeldt C. J.; Oleksiuk O.; Lampe M.; Haselmann U.; Funaya C.; Schieber N.; Ronchi P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18 (9), 2113–2123. 10.1016/j.celrep.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M.; Boldescu V.; Graf D.; Streicher F.; Dimoglo A.; Bartenschlager R.; Klein C. D. Synthesis, Biological Evaluation, and Molecular Docking of Combretastatin and Colchicine Derivatives and Their HCE1-Activated Prodrugs as Antiviral Agents. ChemMedChem 2019, 14 (4), 469–483. 10.1002/cmdc.201800641. [DOI] [PubMed] [Google Scholar]
- Mackow E.; Makino Y.; Zhao B. T.; Zhang Y. M.; Markoff L.; Buckler-White A.; Guiler M.; Chanock R.; Lai C. J. The Nucleotide Sequence of Dengue Type 4 Virus: Analysis of Genes Coding for Nonstructural Proteins. Virology 1987, 159 (2), 217–228. 10.1016/0042-6822(87)90458-2. [DOI] [PubMed] [Google Scholar]
- Yasobu N.; Kitajima M.; Kogure N.; Shishido Y.; Matsuzaki T.; Nagaoka M.; Takayama H. Design, Synthesis, and Antitumor Activity of 4-Halocolchicines and Their Pro-Drugs Activated by Cathepsin B. ACS Med. Chem. Lett. 2011, 2 (5), 348–352. 10.1021/ml100287y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidle U. H.; Tiefenthaler G.; Georges G. Proteases as Activators for Cytotoxic Prodrugs in Antitumor Therapy. Cancer Genomics and Proteomics 2014, 11, 67–80. [PubMed] [Google Scholar]
- Rawlings N. D.; Barrett A. J.; Thomas P. D.; Huang X.; Bateman A.; Finn R. D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46 (D1), D624–D632. 10.1093/nar/gkx1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J.-M.; Brenner S. E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14 (6), 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz M. O.; Prota A. E. Microtubule-Targeting Agents: Strategies To Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 1–17. 10.1016/j.tcb.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Ravelli R. B. G.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-like Domain. Nature 2004, 428 (6979), 198–202. 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- Gaspari R.; Prota A. E.; Bargsten K.; Cavalli A.; Steinmetz M. O. Structural Basis of Cis - and Trans -Combretastatin Binding to Tubulin. Chem. 2017, 2 (1), 102–113. 10.1016/j.chempr.2016.12.005. [DOI] [Google Scholar]
- Wittig G.; Schöllkopf U. Über Triphenyl-Phosphin-Methylene Als Olefinbildende Reagenzien (I. Mitteil. Chem. Ber. 1954, 87 (9), 1318–1330. 10.1002/cber.19540870919. [DOI] [Google Scholar]
- Medarde M.; Maya A. B. S.; Pérez-Melero C. Naphthalene Combretastatin Analogues: Synthesis, Cytotoxicity and Antitubulin Activity. J. Enzyme Inhib. Med. Chem. 2004, 19 (6), 521–540. 10.1080/14756360412331280473. [DOI] [PubMed] [Google Scholar]
- Mikstacka R.; Stefański T.; Rózański J. Tubulin-Interactive Stilbene Derivatives as Anticancer Agents. Cell. Mol. Biol. Lett. 2013, 18 (3), 368–397. 10.2478/s11658-013-0094-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. Medicinal Chemistry of Combretastatin A4: Present and Future Directions. J. Med. Chem. 2006, 49 (11), 3033–3044. 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Temple C.; Narayanan V. L.; Varma R.; Simpson M. J.; Boyd M. R.; Rener G. A.; Bansal N. Antineoplastic Agents 322. Synthesis of Combretastatin A-4 Prodrugs. Anti-Cancer Drug Des. 1995, 10 (4), 299–309. [PubMed] [Google Scholar]
- Nihei Y.; Suga Y.; Morinaga Y.; Ohishi K.; Okano A.; Ohsumi K.; Hatanaka T.; Nakagawa R.; Tsuji T.; Akiyama Y.; et al. A Novel Combretastatin A-4 Derivative, AC-7700, Shows Marked Antitumor Activity against Advanced Solid Tumors and Orthotopically Transplanted Tumors. Jpn. J. Cancer Res. 1999, 90 (9), 1016–1025. 10.1111/j.1349-7006.1999.tb00850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroch K.; Karolak M.; Górski P.; Jaroch A.; Krajewski A.; Ilnicka A.; Sloderbach A.; Stefański T.; Sobiak S. Combretastatins: In Vitro Structure-Activity Relationship, Mode of Action and Current Clinical Status. Pharmacol. Rep. 2016, 68 (6), 1266–1275. 10.1016/j.pharep.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Liu T.; Zhang D.; Song W.; Tang Z.; Zhu J.; Ma Z.; Wang X.; Chen X.; Tong T. A Poly(L-Glutamic Acid)-Combretastatin A4 Conjugate for Solid Tumor Therapy: Markedly Improved Therapeutic Efficiency through Its Low Tissue Penetration in Solid Tumor. Acta Biomater. 2017, 53, 179–189. 10.1016/j.actbio.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Ohsumi K.; Nakagawa R.; Fukuda Y.; Hatanaka T.; Morinaga Y.; Nihei Y.; Ohishi K.; Suga Y.; Akiyama Y.; Tsuji T. Novel Combretastatin Analogues Effective against Murine Solid Tumors: Design and Structure-Activity Relationships. J. Med. Chem. 1998, 41 (16), 3022–3032. 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- Nitsche C.; Klein C. D. Fluorimetric and HPLC-Based Dengue Virus Protease Assays Using a FRET Substrate. Methods Mol. Biol. 2013, 1030, 221–236. 10.1007/978-1-62703-484-5_18. [DOI] [PubMed] [Google Scholar]
- Khumthong R.; Niyomrattanakit P.; Chanprapaph S.; Angsuthanasombat C.; Panyim S.; Katzenmeier G. Steady-State Cleavage Kinetics for Dengue Virus Type 2 Ns2b-Ns3(pro) Serine Protease with Synthetic Peptides. Protein Pept. Lett. 2003, 10 (1), 19–26. 10.2174/0929866033408228. [DOI] [PubMed] [Google Scholar]
- Davidson E.; Poole B. Fractionation of the Rat Liver Enzymes That Hydrolyze Benzoyl-Arginine-2-Naphthylamide. BBA - Enzymol. 1975, 397 (2), 437–442. 10.1016/0005-2744(75)90133-3. [DOI] [PubMed] [Google Scholar]
- Claus V.; Jahraus A.; Tjelle T.; Berg T.; Kirschke H.; Faulstich H.; Griffiths G. Lysosomal Enzyme Trafficking between Phagosomes, Endosomes, and Lysosomes in J774 Macrophages. J. Biol. Chem. 1998, 273 (16), 9842–9851. 10.1074/jbc.273.16.9842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.