

Abstract
Protein arginine deiminases (PADs) are calcium-dependent enzymes that mediate the post-translational conversion of arginine into citrulline. Dysregulated PAD activity is associated with numerous autoimmune disorders and cancers. In breast cancer, PAD2 citrullinates histone H3R26 and activates the transcription of estrogen receptor (ER) target genes. However, PAD2 lacks a canonical Nuclear Localization Sequence (NLS), and it is unclear how this enzyme is transported into the nucleus. Here, we show for the first time that PAD2 translocates into the nucleus in response to calcium signaling. Using BioID2, a proximity-dependent biotinylation method for identifying interacting proteins, we found that PAD2 preferentially associates with ANXA5 in the cytoplasm. Calcium binding to PAD2 weakens this cytoplasmic interaction, which generating a pool of calcium bound PAD2 that can interact with Ran. We hypothesize that this latter interaction promotes the translocation of PAD2 into nucleus. These findings highlight a critical role for ANXA5 in regulating PAD2 and identify an unusual mechanism whereby proteins translocate between the cytosol and nucleus.
Keywords: PAD2, ANXA5, Ran, BioID2, translocation, calcium dependency
INTRODUCTION
Protein arginine deiminases (PADs) are a family of enzymes that catalyze the post-translational conversion of a positively charged arginine residue into a neutral citrulline. This modification disrupts ionic and hydrogen bonding interactions within substrate proteins and can result in alterations to their secondary and tertiary structures as well as potential effects upon protein function and protein-protein interactions.1–5 There are five mammalian PADs, which are denoted as PAD1, 2, 3, 4 and 6.6 Amongst them, PAD2 is thought to be the ancestral homologue; the remaining PADs were derived from PAD2 via a series of gene duplication events.2
PAD2 is widely expressed in brain, spleen, skeletal muscle, and leukocytes.7, 8 Dysregulated PAD2 activity is associated with neurodegenerative and inflammatory diseases as well as breast cancers.9, 10 Like the other PAD isozymes, PAD2 is a calcium-dependent enzyme and is usually inactive under physiological conditions where the calcium concentration ranges from 10−8 – 10−6 M.11 Notably, citrullination only occurs in vitro in the presence of significantly higher calcium concentrations (i.e., 10−4 – 10−3 M), as the concentration of calcium required for half-maximum activity, i.e., the K0.5, is 1.8 × 10−4 M.12 These concentrations can only be reached after membrane rupture or in cells treated with calcium ionophores, which suggests that citrullination can occur only under ‘extreme’ conditions, such as during apoptosis or terminal differentiation of the epidermis. Recently, however, PAD2 was also found to function in gene regulation under homeostatic calcium levels,13, 14 suggesting that there must be a mechanism to modulate the calcium dependence of PAD2 in live cells.
PAD4 is the only PAD to contain a canonical nuclear localization signal (NLS) and was long believed to be the only PAD that can localize to the nucleus.15, 16 Recent work from the Coonrod group has shown, however, that PAD2 localizes to the nucleus where it citrullinates arginine 26 in histone H3 upon estrogen stimulation.13 This modification is strongly correlated with the increased transcription of ER (estrogen receptor) target genes. Herein, we show for the first time that PAD2 translocates from the cytoplasm into the nucleus upon stimulation with calcium. Using BioID2, a proximity-dependent biotinylation method for identifying interacting proteins, we found that ANXA5 preferentially associates with PAD2 in the cytoplasm. Calcium binding to PAD2 weakens this interaction and promotes PAD2 binding to Ran, which, we hypothesize, shuttles PAD2 into nucleus. The FF motif, a beta-turn that extends upon calcium binding, is important for mediating the preferential localization of PAD2 to the nucleus. Overall, our findings strongly implicate a critical role for ANXA5 and Ran in the activation of nuclear PAD2 activity and highlight an unusual mechanism whereby proteins translocate between the cytosol and nucleus.
MATERIALS AND METHODS
Reagents and Cell Lines.
HEK 293 and 293T cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS), and 1% penicillin streptomycin and maintained in a humidified atmosphere with 5% CO2 at 37 °C. A complete list of reagents and suppliers is provided in Table S1. A list of proteins is provide in Table 1.
Table 1:
Proteins used in these studies.
| Protein | UniProt ID | Link |
|---|---|---|
| Biotin [acetyl-CoA-carboxylase] ligase (birA) | O66837 | https://www.uniprot.org/uniprot/O66837 |
| Protein-arginine deiminase type-2 (PADI2) | Q9Y2J8 | https://www.uniprot.org/uniprot/Q9Y2J8 |
| Annexin A5 (ANXA5) | P08758 | https://www.uniprot.org/uniprot/P08758 |
| GTP-binding nuclear protein Ran (RAN) | P62826 | https://www.uniprot.org/uniprot/P62826 |
Protein Purification.
PAD2 was purified as previously described.12 Briefly, Escherichia coli BL21(DE3)pLysS cells transformed with our PAD2 expression construct17 were grown in LB with ampicillin (100 μg/mL) and chloramphenicol (50 μg/mL) at 37 °C until the cultures reached an OD600 of 0.8. PAD2 expression was induced by the addition of IPTG (isopropyl-β-D-thiogalactopyranoside) (0.4 mM final) and allowed to proceed overnight at 16 °C. Cells were harvested by centrifugation at 4800g for 10 min at 4 °C. The pellet was resuspended in lysis buffer (20 mM Tris-HCl pH 7.6, 500 μM TCEP, 5 mM imidazole, and 500 mM NaCl) and lysed by six cycles of sonication. The lysate was clarified by centrifugation (30 min at 35,000 rpm) and the supernatant was incubated with Ni-NTA−agarose resin pre-equilibrated with lysis buffer for 45 min at 4 °C with gentle stirring. Wash buffer (50 mL of 20 mM Tris-HCl pH 8.0, 10% glycerol, 500 μM TCEP, 50 mM imidazole, and 500 mM NaCl) and elution buffer (20 mL of 20 mM Tris-HCl pH 8.0, 10% glycerol, 500 μM TCEP, 250 mM imidazole, and 500 mM NaCl) were then applied. Fractions from the Ni-NTA column were analyzed by 4–12% SDS-PAGE and the fraction containing pure PAD2 was concentrated and then dialyzed (20 mM Tris, pH 7.6, 500 mM NaCl, 500 μM TCEP, and 10% glycerol), flash frozen in liquid nitrogen, and stored at −80 °C.
E. coli BL21(DE3) cells, transformed with ANXA5 or RAN expression constructs, were grown in a shaker incubator (225 rpm) at 37 °C to an OD600 of 0.4 – 0.6 with ampicillin (100 μg/mL). To induce protein expression, IPTG was added to the culture to a final concentration of 500 μM. The cells were incubated for 3 h at 37 °C under continuous shaking at 225 rpm. The bacterial culture was then harvested at 4,000g for 15 min, flash frozen in liquid nitrogen, and resuspended in lysis buffer (20 mM Tris pH 7.6, 0.5 M NaCl, 0.5 mM TCEP, protease inhibitor tablets) and lysed by sonication using a Sonic Dismembrator (Fisher Scientific). Lysates were cleared by centrifugation at 30,000g for 30 min at 4 °C followed by affinity purification at 4 °C using Ni-NTA resin and washing using a stepwise gradient of 20 mM Tris pH 7.6, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP, and 0.02−0.1 M imidazole followed by elution from the resin with 20 mM Tris pH 7.6, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP, and 0.25 M imidazole. Samples containing the target protein were pooled and dialyzed into a long-term storage buffer (LTSB: 20 mM Tris pH 7.6, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP), assayed for purity, and stored at −80°C.
PAD2 Calcium Dependence Assay.
The calcium dependence of PAD2 in the presence of various proteins was characterized using a discontinuous activity assay.12 Briefly, Reaction Buffer, containing 100 mM Tris pH 7.6, 50 mM NaCl, 2 mM DTT, 10 mM benzoylarginine ethylester (BAEE), and varying concentrations of CaCl2 (0–10 mM), was preincubated for 10 min at 37 °C, followed by the addition of PAD2 and RAN or PAD2 and ANXA5, which had been preincubated at 37 °C for 1 h (0.2 μM final) to initiate the reaction. After 15 min, the reactions were quenched and citrulline production was quantified as previously described.18, 19 The data obtained were fit to Equation 1,
| (Equation 1), |
using GraphPad Prism version 7.0c, where v is the initial rate, Vmax is the maximum rate, K0.5 is the concentration of calcium that yields half maximal activity, and n is the hill coefficient. The experiment was carried out in duplicate.
Transfection of BioID2 and BioID2-PAD2 into HEK293 Cells.
The BioID2 and BioID2-PAD2 constructs were transfected into HEK293 cells using Lipofectamine 2000 according to the manufacturer’s protocol. Stable transfection was established using neomycin as a selection marker. Briefly, HEK293 cells were seeded onto a tissue culture flask 1 day prior to transfection. Upon reaching 70 – 80% confluence, cells were transfected with 20 μg of myc-BioID2-MCS (with stop codon) or myc-BioID2-PAD2-MCS with Lipofectamine 2000. Cells were cultured for 48 h post-transfection and then selected with 1 mg/mL neomycin.
RIBFA labeling.
Cells were grown to ∼80% confluence in a Nunc™ cell culture treated EasyFlask T175 flask and harvested by centrifugation at 1000g for 3 min. The cell pellets were then washed 4× with 1×PBS and cell pellets were then resuspended in 1× PBS with 1% Triton X-100 and incubated for 1 h on ice. Lysates were cleared by centrifugation at 21,000g for 15 min. The supernatant was isolated and quantified by the Detergent Compatible (DC) assay. Lysates (2 μg/μL, 50 μL) were then treated with 2 mM CaCl2 and 2 μM RIBFA20 and incubated at 37 °C for 1 h. The reaction was quenched with 6× SDS loading buffer and separated by SDS−PAGE (4−15% gradient gel). The bands were visualized by scanning the gel in an Amersham Imager 600 (excitation/emission maxima of 546/579, respectively).
Cellular Fractionation Experiments.
Biochemical fractionation was adapted from the REAP protocol.21 Briefly, 1×107 cells per cell line were collected, washed in PBS, and lysed by incubation in Buffer A (1 × PBS, 0.1% NP 40, Pierce protease inhibitor tablets) for 10 min on ice. Cytoplasmic proteins were separated from nuclei by centrifugation at 1,300g for 5 min at 4 °C. Pelleted nuclei were washed in Buffer A three times, and then resuspended in Buffer A before sonication for 10 s using a Sonic Dismembrator. Protein concentration in the lysates was measured by DC Assay (Bio-Rad) to ensure equal protein loading. 6X-SDS protein buffer was added into the lysates to yield a final concentration of 1X buffer and lysates were then boiled at 95 °C for 5 min. The lysates were subjected to SDS-PAGE and subsequently transferred to PVDF membranes (BioRad). Membranes were blocked in 5% BSA in PBS buffer for 1 h at room temperature. Primary antibodies were incubated overnight at 4 °C and then membranes were washed in PBS-T, followed by a 1 h incubation at room temperature with 1:5,000 Licor IRDye secondary antibodies. All blots were washed for 30 min (3 × 10 min) with PBS-T after secondary antibody incubation and then visualized using the Licor Odyssey imaging system. Cleanliness of fractionation was determined with antibodies for Histone H3 (nuclear) and superoxide dismutase 4 (SOD4) (cytoplasmic). The blot was imaged and band intensities were quantified using the Licor Imager Software (700 nm and 800 nm).
Sample preparation for proteomics.
HEK293 cells expressing BioID2 or BioID2-PAD2 were grown in DMEM media minus L-lysine and L-arginine supplemented with 10% dialyzed FBS for SILAC and either 84 μg/mL [13C/15N]-L-arginine (R10) and 146 μg/mL [13C/15N]-L-Lysine (K8) or 84 μg/mL L-arginine (R0) and 146 μg/mL L-lysine (K0) at 37 °C and 5% CO2 for a minimum of 6 passages.
Lysed samples were diluted in PBS buffer to a final concentration of 2 mg/mL, and then 500 μL of heavy lysate (2 mg/mL), and 500 μL of light lysate (2 mg/mL) were combined. All the samples were prepared in duplicate. The samples were then incubated with 20% trichloroacetic acid (100 μL of 100% TCA), and proteins were precipitated by incubation at −80 °C for 1 h followed by centrifugation (15,000 rpm, 10 min) at 4 °C. The supernatants were discarded, and the protein pellet was washed twice with ice-cold acetone and dried.
Protein pellets were then resuspended in 1.2% SDS in PBS and solubilized by heating (65 °C for 5 min) and sonication (bath sonicator for 10 min). Once dissolved, samples were diluted in PBS buffer to 0.2% final SDS concentration and incubated with streptavidin agarose slurry (170 μL) overnight at 4 °C and for a further 3 h at room temperature. Streptavidin beads were then washed with PBS containing 0.2% SDS (1X), with PBS alone (3X), and water (3X). After washing, the beads were incubated at 65 °C for 20 min in a PBS buffer containing 6 M urea and 10 mM DTT. The beads were further treated with iodoacetamide (20 mM) for 30 min at 37 °C. On-bead trypsin digestion was performed in a PBS solution containing 2 M urea, 1 mM CaCl2 and 2 μg trypsin overnight at 37 °C. The beads were spun down and the supernatant, containing the peptide digests was collected. Formic acid was added to the supernatant and samples were stored at −20 °C until MS analysis.
Validation of Isotope Incorporation Efficiency in SILAC.
Samples were diluted in PBS buffer to a final concentration of 1 mg/mL, and then incubated with 10% trichloroacetic acid, and proteins were precipitated by incubation at −80 °C for 1 h followed by centrifugation (15,000 rpm, 10 min) at 4 °C. The supernatants were discarded, and the protein pellet was washed twice with ice-cold acetone and dried.
Protein pellets were then resuspended in 8 M urea in PBS and solubilized by heating (65 °C for 5 min) and sonication (bath sonicator for 10 min). Once dissolved, 10 mM DTT was added and the samples were incubated at 65 °C for 15 min. Next the samples were treated further with iodoacetamide (20 mM) for 30 min at 37 °C. After diluting the urea concentration to 2 M with PBS, 1 mM CaCl2 and 2 μg trypsin were then added to the sample and incubated overnight at 37 °C. Formic acid was then added to quench trypsin and samples were centrifuged at 15,000 rpm for 20 min to pellet precipitated proteins. The supernatants were stored at −20 °C until MS analysis. The heavy/light intensity ratio (RH/L) is set to 20 if a mass corresponding to a peptide with light lysine or arginine was not found. Our observation that most peptides had a RH/L of 20 confirmed the complete incorporation of heavy arginine and lysine.
LC/LC-MS/MS and Data Processing.
Mass spectrometry was performed on an LTQ Orbitrap Discovery (Thermo Fisher) coupled to an Agilent 1200 series HPLC. Protein digests were pressure loaded onto a 250 μm fused silica desalting column packed with 4 cm Aqua C18 reverse phase resin (Phenomenex). Peptides were eluted onto a 100 μm fused silica biphasic column packed with 4 cm of strong cation exchange resin (Watman) and 10 cm of C18 resin using a five-step multidimensional LC/LC-MS/MS protocol (MudPIT) 22. Each of the five runs begins with a salt push (0%, 25%, 50%, 80%, and 100% buffer C), followed by a gradient 0–100% buffer B in buffer A (buffer A: 95% water, 5% acetonitrile, 0.1% formic acid; buffer B: 20% water, 80% acetonitrile, 0.1% formic acid; buffer C 95% water, 5% acetonitrile, 500 mM ammonium acetate). The flow rate was ~0.25 μL/min and the spray voltage was set to 2.75 kV. One full MS1 scan (400–1800 m/z) was followed by 8 data dependent scans of the nth most intense ion.
Peptides were assigned to MS2s using the SEQUEST23 algorithm, searching against a human reverse-concatenated non-redundant protein database. Data sets were searched independently for peptides containing isotopically light and heavy amino acids. For the light search the default masses of lysine and arginine were used, for the heavy search a static modification was specified for lysine (+8.0142 m/z) and arginine (+10.00826 m/z). A static modification was specified for cysteine residues (+57.0215 m/z, iodoacetamide alkylation) in both heavy and light samples. MS2 matches were assembled by protein and filtered to achieve a false discovery rate of 5% using DTASelect 2.0 24.
Light/heavy intensity ratios (RL/H) quantification.
Light/heavy intensity ratios (RL/H) were quantified using the CIMAGE quantification package as described previously.22 CIMAGE reports peptide RL/H ratio for co-eluting light and heavy peptides. The peptides are grouped by protein and a representative RL/H for each protein is calculated from the median RL/H for each peptide identified for that protein.
Detection of PAD2 interacting proteins by Western blot and Immunoprecipitation.
Cells were lysed with a microprobe sonicator in PBS buffer containing Pierce protease inhibitor tablets (one tablet in 10 mL of buffer). Protein concentration in the lysates was measured by DC Assay (Bio-Rad) to ensure equal protein loading. 6X-SDS protein buffer was added into the lysates to yield a final concentration of 1X-buffer and lysates were then boiled at 95 °C for 5 min. The lysates were subjected to SDS-PAGE and subsequently transferred to PVDF membranes (BioRad). Membranes were blocked in 5% BSA in PBS buffer for 1 h at room temperature. Primary antibodies were incubated overnight at 4 °C and then membranes were washed in PBS-T, followed by a 1 h incubation at room temperature with 1:5,000 Licor IRDye secondary antibody. All blots were washed for 30 min (3 X 10 min) with PBS-T after secondary antibody incubation and then visualized using the Licor Odyssey imaging system. For immunoprecipitation, lysates were incubated with the indicated antibodies overnight at 4 °C and then incubated with Protein A/G PLUS–Agarose for 2 h at room temperature. The bound proteins were eluted by boiling for 10 min at 1X-SDS loading buffer and subjected to Western blotting.
Detection of PAD2 Interaction with ANXA5 by Size Exclusion Chromatography (SEC).
An AKTA Pure series liquid chromatography system (GE Healthcare Life Science) equipped with Superose 6 10/300 GL (GE Healthcare Life Science) was used for chromatographic measurements. We used a flow rate of 1 mL/min for all measurements. The eluent was the same as the long-term storage buffer.
Treatment of HEKPAD2 with Importazole.
HEK293T PAD2 cells were grown to ~80% confluence in Nunc Cell Culture Treated EasYFlask™ T25 flasks. Complete media was removed from flasks and carefully replaced with serum free medium. Cells were treated with either DMSO or importazole (40 μM final concentration) and incubated at 37 °C with 5% CO2 for 1 h. Cells were then treated with ionomycin (5 μM final concentration) and incubated at 37 °C with 5% CO2 for another 1 h.
Quantification And Statistical Analysis.
Data presented herein was replicated in separate experiments. Unless otherwise stated, data points shown in figures are the mean of independent data, and the error represents standard deviation (SD). All data fitting and statistical analysis was performed using GraphPad Prism software (version 7.0c).
RESULTS
Calcium binding induces the nuclear localization of PAD2.
Prior structural work from our lab12 demonstrated that calcium binding to PAD2 triggers a series of conformational changes where the pseudosubstrate R347 flips out of the active site and W348 moves in to form one wall of the substrate binding pocket. C647 also moves into the active site and adopts a conformation that is competent for catalysis. In addition to these changes, calcium binding to the 3, 4 and 5 sites cause another dramatic conformational change that results in two hydrophobic residues, F221 and F222, extending into solution (Figure 1A, compare left and middle panels). This FF-motif adopts a nearly perfect β-turn in the PAD2–10 mM Ca2+ structure and undergoes a cis/trans prolyl isomerization upon the binding of calcium at the Ca2 site, which generates the fully active holoenzyme (Figure 1A, right panel). Notably the FF motif is unique to PAD2 (Figure 1B), suggesting that it may play a novel role in regulating PAD2 activity. To assess that possibility, we first evaluated the effect of mutating or deleting this loop on both the activity and calcium dependence of PAD2. Interestingly, the F221A/F222A (FFA) double mutant neither alters PAD2 catalytic efficiency nor calcium dependence (Figure 1C). Two and four residue deletions of the loop also had no effect on these parameters (Figure 1C).
Figure 1. The FF-Motif Extends Upon Calcium Binding and Does not Alter PAD2 Activity.
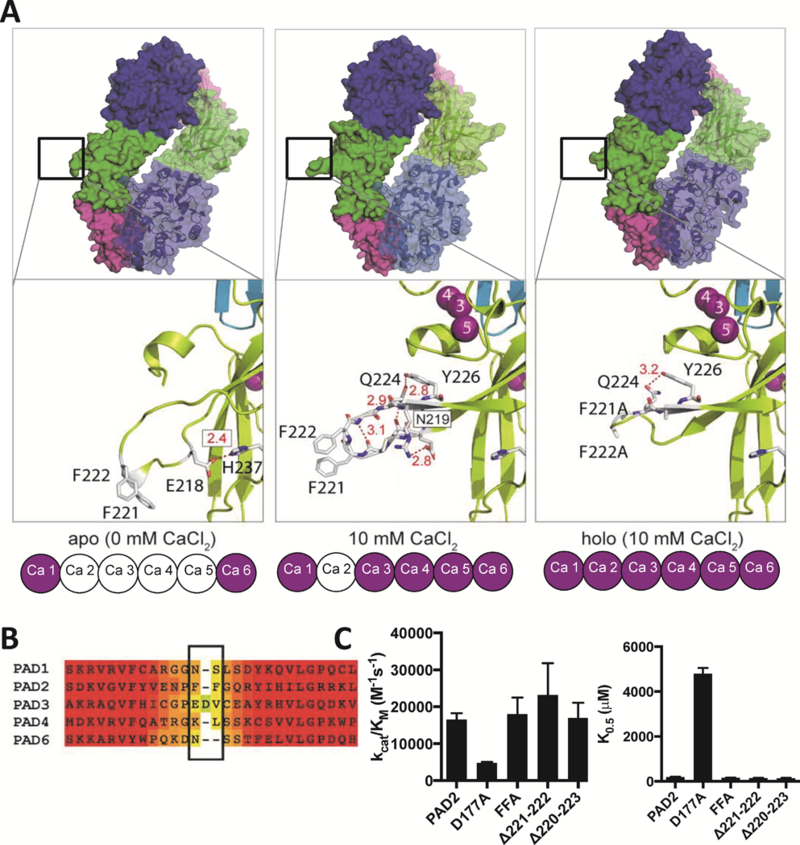
(A) Surface (upper) and stick-cartoon (below) representations of apoPAD2 (PDB: 4N2O), 10 mM Ca2+-PAD2 (PDB: 4N2B) and holoFFA (PDB: 4N2C).12 (B) Heat map shows highly conserved (red) and the hyper-variable region (yellow) in PADs that encompasses the FF motif (boxed) in PAD2. (C) Catalytic efficiency and calcium dependence of PAD2, D177A, FFA, Δ221–222 and Δ220–223. Data are represented as mean ± SEM.
To further investigate the role of this motif, we next compared the subcellular localization of wild type PAD2 to the FFA mutant. We generated nuclear and cytoplasmic fractions and compared the subcellular localization of wild type and mutant PAD2 (Figure 2A–D). Accuracy of cell fractionation was confirmed by probing the same PVDF membrane with antibodies for Superoxide Dismutase 4 (SOD4), a cytoplasmic protein, and histone H3, a nuclear protein. Nuclear PAD2 was clearly visible 1 h after ionomycin stimulation. With increasing stimulation time, the fraction of cytoplasmic wild type PAD2 entering the nucleus increased, whereas the amount of cytoplasmic FFA remained relatively constant. We also evaluated the subcellular localization of the D177A mutant (Figure 2E and F). Since D177 coordinates to both Ca3 and 5 and its mutation leads to a 26-fold increase in the K0.5 (Figure 1C) 12, we hypothesized that the mutant would also be retained in the cytoplasm since calcium binding triggers the extension of the FF motif. Consistent with our hypothesis, the D177A mutant was retained in the cytoplasm even after a 3 h incubation with ionomycin.
Figure 2. Calcium promotes the nuclear localization of PAD2.
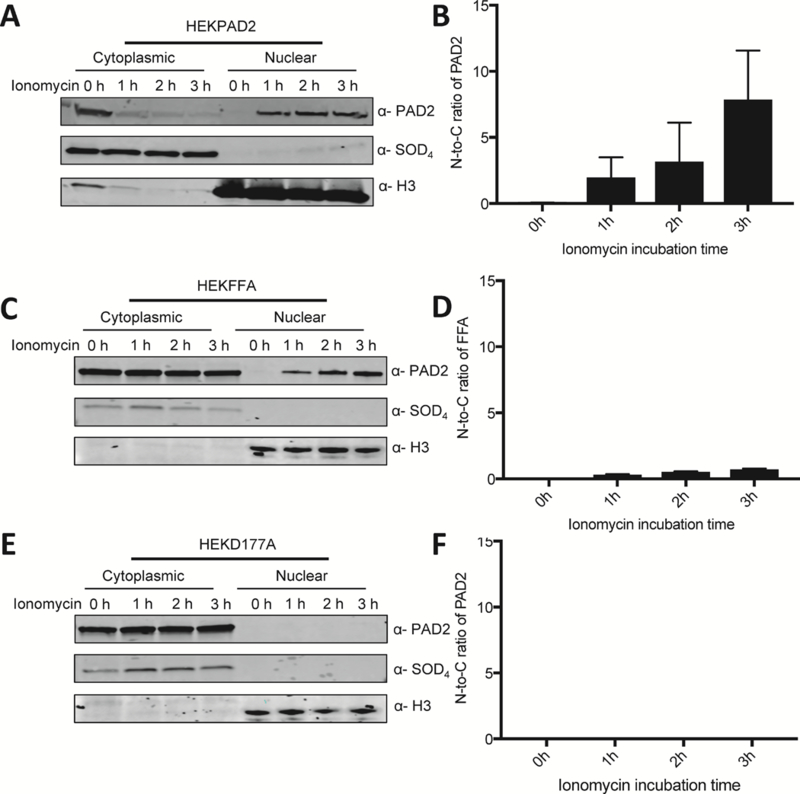
(A) HEK293T PAD2, (C) HEK293T FFA and (E) HEKD177A cellular proteins were fractionated into cytoplasmic and nuclear lysates and the presence of PAD2 was examined by western blotting. Cleanliness of fractionation was determined using antibodies for Histone H3 (nuclear) and SOD4 (cytoplasmic) protein. The ratio of normalized density of (B) PAD2, (D) PAD2FFA and (F) PAD2D177A levels in nucleus over those in the cytoplasm is shown on the left to the corresponding western blots (n = 3).
Calcium binding to the PAD2 fusion protein also triggers its nuclear localization.
Given the above data, we hypothesized that calcium binding triggers a conformational change that extends the FF-motif out into solution where it interacts with an unknown protein(s) (or disrupts interactions) that promotes nuclear uptake. To evaluate this hypothesis, we used the BioID2 approach to identify PAD2 interacting proteins. BioID2 is an improved version of the BioID platform,25 where a smaller promiscuous biotin ligase is fused in frame to the protein of interest to facilitate the biotinylation of proximal proteins in live cells. BioID2 uses the Aquifex aeolicu biotin ligase,26 in which a conserved residue, Arg40 in the catalytic domain, is replaced with Gly; this mutation increases the promiscuity of biotinylation. Compared to traditional genetic and biochemical methods, the BioID2 technique is particularly useful for studying insoluble candidates and weak or transient interactions in the native cellular environment.
For these studies, wild type PAD2 was cloned in frame to the C-terminus of the BioID2 biotin ligase and the constructs (i.e., BioID2PAD2) as well as the control BioID2 construct, were stably transfected into HEK293 cells. To ensure that these fusion constructs retained native-like behavior, we investigated their functional properties. First, we compared the catalytic activity of wild type PAD2 and the fusion protein using a PAD-targeted activity-based probe, i.e. RIBFA, that specifically modifies PAD2 20 (Figure S1A). Briefly, cell lysates were incubated with RIFBA for 1 h at 37 °C in the presence of calcium chloride. Next, proteins were resolved by SDS-PAGE and fluorescently tagged proteins imaged. Compared to HEKPAD2, the activity of BioID2PAD2 remained broadly the same. Equal protein loading was demonstrated by blotting the same gel to a PVDF membrane and PAD2 levels detected with anti-PAD2 antibody. Next, we confirmed that BioID2PAD2 possessed similar subcellular localization properties. Cellular fractionation experiments (Figure S1B and C) confirmed that like the full-length protein, ionomycin stimulation triggers the translocation of BioIDPAD2 from the cytoplasm to the nucleus. In total, these data demonstrate that the fusion protein retains native-like activity and subcellular distribution.
To initially demonstrate the feasibility of identifying PAD2 interacting proteins, we compared the biotinylation patterns present in cells expressing BioID2 alone to those present in the BioID2PAD2 expressing cells (Figure S2). As previously reported,26 the levels of biotinylated proteins increase with the time of exposure to biotin. The dense bands at 25 and 100 kDa represent BioID2 and BioID2PAD2, respectively. The identities of these two bands were confirmed using anti-MYC and anti-PAD2 antibodies (Figure S2, lower panels). A comparison of the western blots of proteins from cells expressing biotin ligase alone to those expressing the fusion protein, show distinct patterns, demonstrating the differential proximity-dependent biotinylation of BioID2 and BioID2PAD2 associated proteins. Moreover, ionomycin, which activates PAD2, further modulated the biotinylation patterns, consistent with a change in subcellular localization.
Combined BioID2 and SILAC approaches identify PAD2 interacting proteins.
To identify proximal and direct PAD2-interacting proteins in resting cells and ionomycin-stimulated cells, we performed a stable isotope labeling in cell culture (SILAC)27 experiment to quantitatively compare biotinylated proteins in BioID2PAD2 and control BioID2 expressing cells (Figure 3A). For these experiments, BioID2 expressing HEK293 cells were SILAC-labeled with heavy (13C and 15N substituted) lysine and arginine and compared to light BioID2PAD2 cells. This approach was taken to maximize the amount of information from a single set of experiments and to minimize the need to SILAC label multiple cell lines. Cells were incubated with excess biotin in the culture media to induce biotinylation of interacting proteins. The cells were then lysed with SDS, light and heavy cell lysates mixed together, and biotinylated proteins bound to streptavidin-agarose. An on-bead trypsin digestion was performed, and the digest was analyzed by LC/LC-MS/MS (Figure 3A, Table S2).
Figure 3. Identification of PAD2 Interacting Proteins.
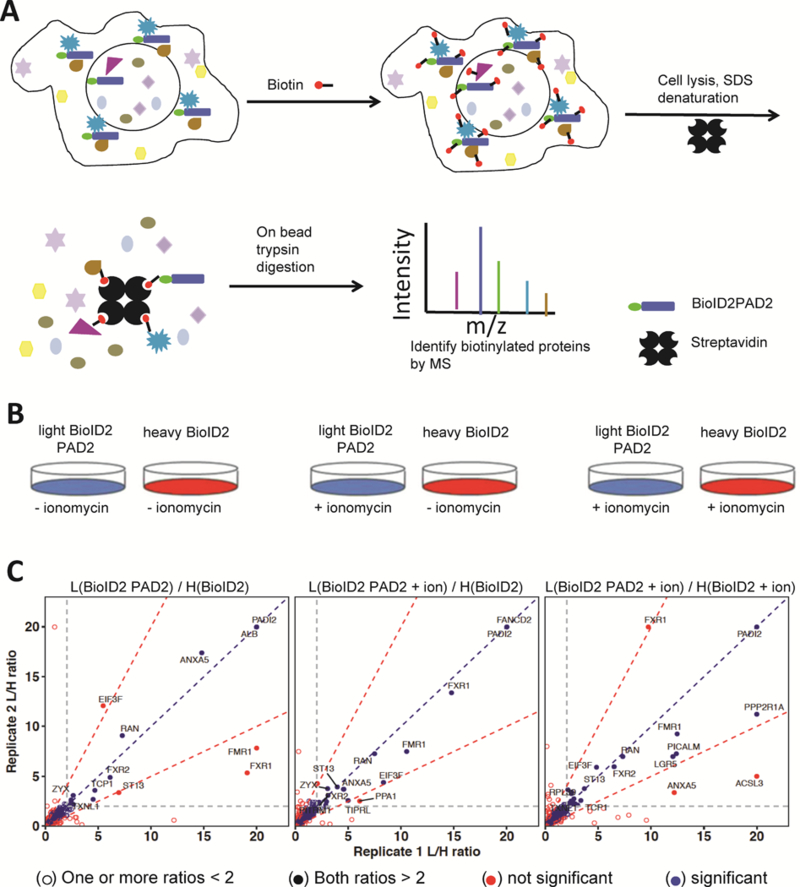
(A) Schematic describing the proximity-dependent biotinylation method and identification of proteins associated with BioID2-PAD2. (B) Three sets of samples were used in proteomic experiments. (C) Mass spectrometry analysis of proximity biotinylated proteins associated with PAD2 with or without ionomycin.
Three combinations of samples were analyzed as shown in Figure 3B. The direct comparison of BioID2 and BioID2PAD2 lysates will identify PAD2-interacting proteins that occur in the absence of calcium. By contrast, the BioID2 and BioID2PAD2 plus ionomycin comparison identifies proteins that interact with calcium-bound PAD2; ionomycin was added at the same time of biotin addition to increase intracellular calcium concentration. BioID2 plus ionomycin and BioID2PAD2 plus ionomycin serves as a control for global calcium-mediated proteomic changes as well as a secondary verification of proteins interacting with calcium bound PAD2. For each condition, the data were obtained from two biological replicates. The SILAC light/heavy (L/H) ratios for proteins in replicate 1 were plotted on the x-axis and the ratio for replicate 2 were plotted on the y-axis (Figure 3C). Those proteins having ratios higher than 2 in both replicates were considered significant. Proteins which fell outside of the region between the red dotted lines with slopes of 2 and 0.5 were not considered to be significant because their ratios were not consistent across the replicates.
The small GTPase Ran, which plays an important role in nucleocytoplasmic shuttling,28, 29 showed a high L/H ratio in all three data sets. By contrast, the cytoplasmic protein ANXA530-32 had a high L/H ratio only in the BioID2 and BioID2PAD2 comparison (Figure 3C left panel). The L/H ratio of FXR1, which localizes in the nucleus,33–35 increased in the presence of calcium, consistent with our finding that calcium triggers the entry of PAD2 into the nucleus. To evaluate our data more broadly, we performed a gene ontology analysis to analyze the subcellular localization of each protein identified in the plus/minus ionomycin datasets (Figure 4A). Consistent with calcium triggering the nuclear localization of PAD2, the number of nuclear proteins identified in the BioID2PAD2 samples increased in the presence of ionomycin. To obtain a list of candidate proteins that preferentially interact with BioID2PAD2 in the presence of calcium, the ratio of BioID2PAD2 plus ionomycin over BioID2PAD2 was obtained by dividing the BioID2PAD2/BioID2 L/H ratio by the BioID2-PAD2 plus ionomycin/BioID2 L/H ratio (Figure 4B). The proteins located in the upper left corner preferentially interact with calcium-bound PAD2 whereas those proteins located in right corner preferentially interact with the metal-free form. Notably, ANXA5 strongly associates with apo PAD2.
Figure 4. PAD2 interacting proteins are modulated by calcium and PAD2 physically interacted with ANXA5.
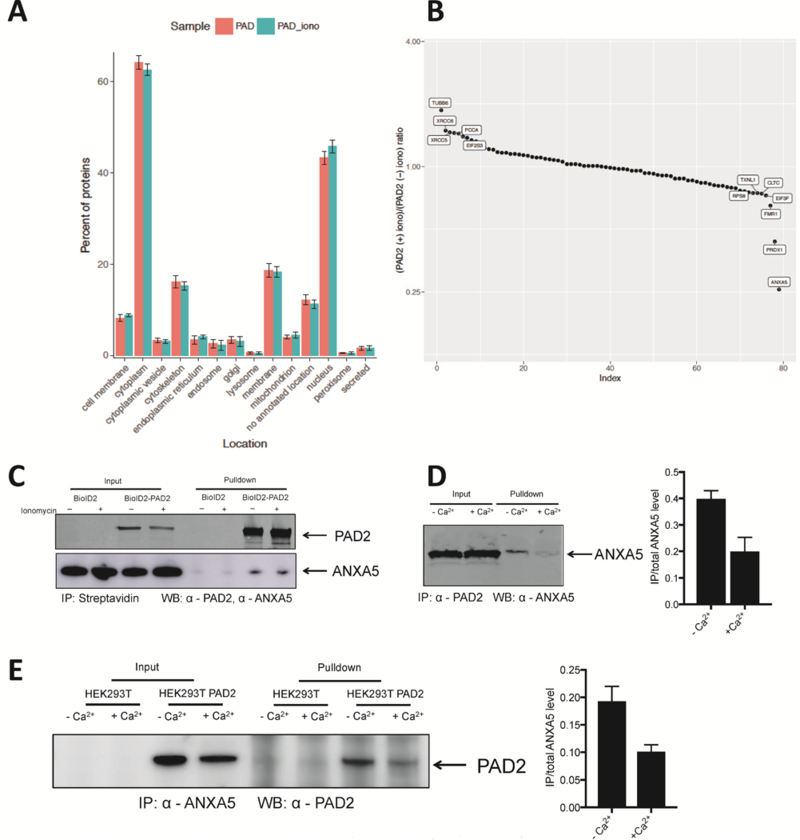
(A) Subcellular localization of biotinylated proteins associated with PAD2 with or without ionomycin stimulation. (B) Ratio of ratio generated from Figure 3C left two graphs. (C) BioID2 and BioID2-PAD2 transfected HEK-293T cells were treated with biotin with or without ionomycin. Biotinylated proteins were isolated using streptavidin-agarose and detected by anti-PAD2 and anti-ANXA5. (D) Recombinant PAD2 and ANXA5 were incubated at 37 °C for 2 h. anti-PAD2 was used to pull down ANXA5. Normalized density of precipitated ANXA5 is on the left (n = 3). (E) Immunoprecipitation of PAD2 transfected HEK-293T cells treated with or without ionomycin by anti-ANXA5 and detected with an anti-PAD2 antibody. Normalized density of precipitated ANXA5 is on the left (n = 2).
ANXA5 preferential binds to apo PAD2.
Given that ANXA5 preferentially interacts with calcium-free PAD2, we initially focused on this protein. ANXA5 is traditionally thought of as a calcium-dependent phospholipid binding protein.36, 37 ANXA5 was also proposed to function as a calcium channel after elucidation of the first ANXA5 crystal structure.38 There is a central pore present at the core of the protein structure, which is lined by residues E95 and E112. Mutational studies showed that these residues are determinants of calcium-ion selectivity and voltage sensitivity.39 Planar lipid bilayer experiments also showed that the ion-channel like properties of ANXA5 are similar to those of other calcium channels.40 Notably, ANXA5 needs calcium to initiate a conformational change and bind to the target membrane. Once attached, it is able to penetrate and form a channel that conducts Ca2+ ions.40
To begin to validate the interaction between ANXA5 and PAD2, BioID2PAD2 expressing HEK-293T cells were incubated with 50 μM of biotin for 20 h. Cell lysates were then prepared, and biotinylated proteins isolated by streptavidin agarose. Biotinylated ANXA5 and PAD2 were detected by western blotting. Compared to BioID2 expressing HEK-293T cells, we were only able to isolate ANXA5 and PAD2 from BioID2-PAD2 expressing cells (Figure 4C). We did not see a difference in the biotinylation pattern between the plus and minus ionomycin samples. This result was expected because biotinylation occurs prior to ionomycin signaling and is a stable PTM. Next, we determined whether the interaction between ANXA5 and PAD2 is modulated by calcium. For these experiments, we determined whether purified recombinant PAD2 physically interacts with ANXA5 by coimmunoprecipitation using an anti-PAD2 antibody (Figure 4D). The direct binding between these two proteins was confirmed using an anti-ANXA5 antibody. Notably, this interaction was inhibited by the addition of calcium. These data confirm our proteomic data and demonstrate that ANXA5 preferentially interacts with calcium-free PAD2.
We also evaluated the interaction between PAD2 and ANXA5 in PAD2 expressing HEK-293T cells (Figure 4E). Anti-ANXA5 was able to immunoprecipitate a ~75-kDa protein from PAD2-overexpressing HEK-293T cells, but not from control HEK-293T cells. Consistent with our in vitro data, treatment with a calcium ionophore attenuated this interaction. Size exclusion chromatography (SEC) was next used to monitor how ANXA5 modulates the quaternary structure of PAD2. PAD2 was incubated with ANXA5 for 1 h with or without 2 mM calcium at 37 °C before injecting onto a SEC. Compared to the mixture with calcium, a new peak appeared in the SEC spectrum of the mixture without calcium (Figure S3A). Western blotting showed that this new peak was comprised of PAD2 (Figure S3B). This peak has a smaller retention time than the PAD2 peak in the control sample, indicating that ANXA5 promotes the formation of a higher order PAD2 oligomer. Notably, this new peak was not present when PAD2 was incubated with ANXA5 in the presence of 2 mM calcium (Figure S3). Notably, we do not observe ANXA5 coeluting with this oligomer. One possible explanation is that substoichiometric amounts of ANXA5 are required to promote oligomer formation and these levels are below the limit of detection.
PAD2 interacts with Ran.
The nuclear pore complex (NPC) controls the exchange of macromolecules between the cytoplasm and nucleoplasm.41, 42 The NPC is composed of approximately thirty proteins, termed nucleoporins (nups), which are highly enriched in Phenylalanine-Glycine (FG) repeats.43, 44 FG-repeats adopt an unfolded conformation that fills the pore, thereby making it impermeable to proteins larger than ∼40 kDa.45, 46 PAD2 has a molecular weight of ∼75 kDa,47 and, as such, it cannot diffuse through the pore. Therefore, a carrier-mediated mechanism is required to transport PAD2 across the NPC. The small GTPase Ran is necessary for the transport of most proteins across the NPC.48–50 From our proteomics dataset (Figure 3C), Ran interacts with both apo PAD2 and calcium-bound PAD2.
Streptavidin pulldown assays confirmed the biotinylation of Ran in BioID2PAD2 cells (Figure 5A). The physical interaction between Ran and PAD2 was confirmed in PAD2 expressing cells (Figure 5B). For these studies, mouse anti-PAD2 antibody was used to immunoprecipitate Ran from the PAD2 expressing cells. The interaction between Ran and PAD2 was detected with a rabbit anti-Ran antibody. Ran Q69L, a GTPase-deficient Ran mutant locked in the GTP-bound state51 and Ran T24N, which exists primarily in the GDP-bound state52, 53 were purified. Their interactions with PAD2 were also detected (Figure 5C). PAD2 bound similarly to both forms of Ran indicating that the interaction is independent of which nucleotide is bound to Ran. Notably, Ran preferentially interacts with calcium bound PAD2 both in vitro (Figure 5C) and in cells (Figure 5B).
Figure 5. PAD2 physically interacted with Ran.
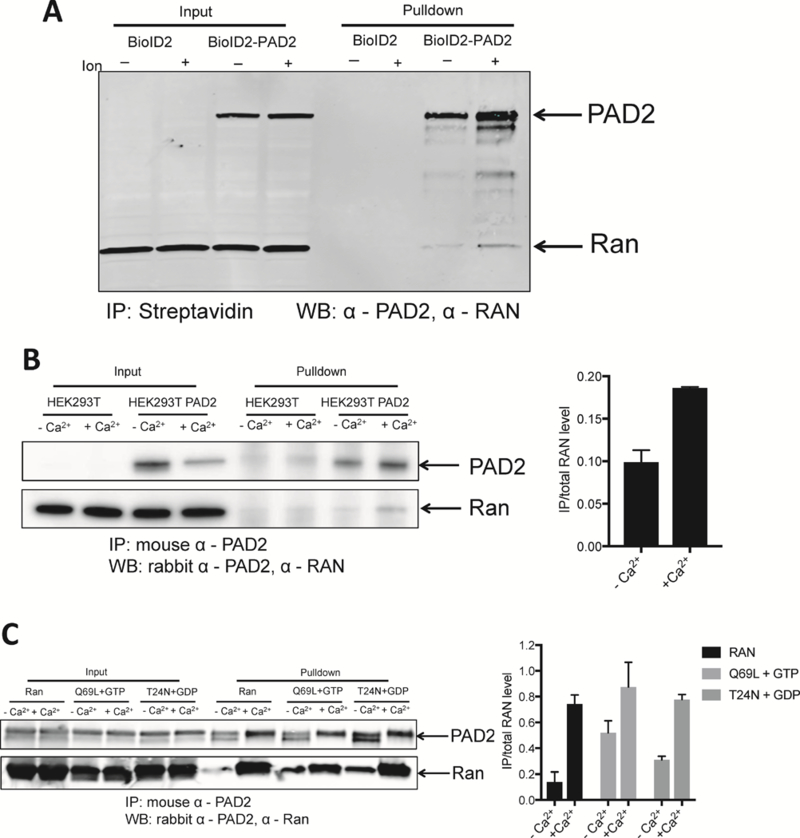
(A) BioID2 and BioID2-PAD2 transfected HEK-293 cells were treated with biotin with or without ionomycin. Biotinylated proteins were isolated on streptavidin agarose and proteins detected by western blotting using anti-PAD2 and anti-Ran antibodies. (B) IP was performed in PAD2 transfected HEK-293 cells treated with or without ionomycin by mouse anti-PAD2 and detected by rabbit anti-Ran and anti-PAD2. Normalized density of precipitated RAN is on the left (n = 2). (C) Recombinant PAD2 and Ran, RanQ69L, RanT24N were incubated at 37 °C for 2 h. anti-PAD2 was used to pull down Ran. Normalized density of precipitated RAN is on the left (n = 2).
Interactions between wild type and mutant PAD2 with Ran and ANXA5.
Given our data showing that ANXA5 preferentially binds apo-PAD2 and Ran preferentially interacts with calcium-bound PAD2, we next evaluated the effect of the D177A and FFA mutants on this interaction using in vitro immunoprecipitation experiments with purified proteins. Notably, neither mutation altered the interaction with Ran consistent with our proteomic data showing that Ran is constitutively associated with PAD2 (Figure 6A–B). By contrast, the D177A mutation prevented the loss of interaction with ANXA5 that occurs upon calcium binding, consistent with our data showing that ANXA5 preferentially interacts with apoPAD2 (Figure 6C–D). Given that the supraphysiological levels of calcium are required to activate PAD2 activity, we considered the possibility that ANXA5 or Ran might alter the activity or calcium dependence of PAD2. However, neither protein altered the activity or calcium dependence of PAD2 (Table S3).
Figure 6. Interactions between wild type and mutant PAD2 with Ran and ANXA5.
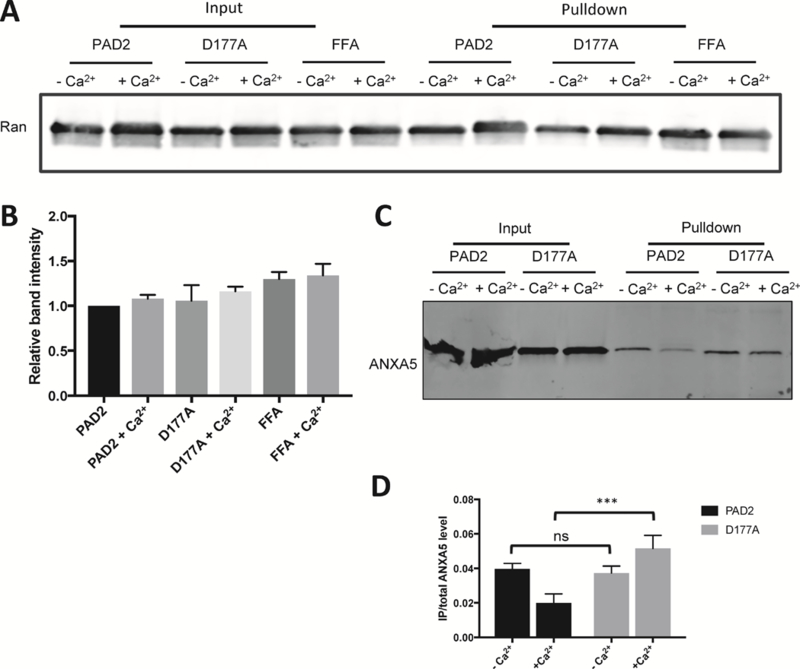
(A) anti-PAD2 antibody was used to pull down Ran incubated with wild type PAD2 and the D177A and FFA mutants in the absence or presence of Ca2+. (B) Normalized density of Ran proteins in pull down assays. (C) In vitro anti-PAD2 pull down ANXA5 bound to PAD2 and the D177A mutant. (D) Normalized density of precipitated ANXA5.
DISCUSSION
Calcium has long been known to regulate the catalytic activity of the PADs by triggering a conformational change that boosts enzyme activity by greater than 10,000-fold.16 However, we show here for the first time that calcium also regulates the subcellular distribution of PAD2 and promotes its translocation into the nucleus. This result is significant because PAD4 has long been considered to be the only nuclear PAD because it possesses a nuclear localization signal and in localization studies using GFP-tagged PADs 1, 2, 3, and 4, only PAD4 localized to the nucleus.15 However, these experiments never evaluated the calcium dependence of nuclear uptake. These results are also significant because they add an additional layer of evidence to support a role for PAD2 in regulating gene expression as demonstrated by us and others.10, 13, 54
In addition to demonstrating that calcium regulates the nuclear localization of PAD2, we used the BioID2 platform to identify PAD2 interacting proteins, with one goal being the identification of the factors that control the calcium dependent nuclear uptake of PAD2. The results of these studies were revealing and, consistent with our cellular fractionation experiments, calcium signaling shifts the PAD2 interactome from one that is dominated by cytoplasmic proteins to one that is more nuclear oriented. A comparison of those proteins that preferentially interact with apo PAD2 and calcium-bound PAD2 highlighted ANXA5 and Ran as key players regulating the cellular distribution of PAD2.
ANXA5 is a calcium dependent, phospholipid-binding protein. Although, there is only one example where ANXA5 functions as a Ca2+ channel in vivo55 (i.e., in the case of bone formation by matrix vesicles), the voltage-sensitive Ca2+ channel activity of ANXA5 was verified with patch clamp and planar lipid bilayer experiments,56–58 as well as in a calcium influx assays that showed that ANXA5 mediates the movement of Ca2+ into large unilamellar vesicles.59 In addition, the crystal structure of ANXA539 proved its typical ion channel essential properties.
Our studies show that ANXA5 preferentially interacts with apo PAD2 (Figure 3C left panel and Figure 4B). After treatment with 1 μM ionomycin, the abundance of ANXA5 became negligible in BioID2PAD2 expressing cell (Figure 3C middle and right panels). Immunoprecipitation of ANXA5 and PAD2 in vitro (Figure 4D) and in HEK293PAD2 cells (Figure 4E) confirmed that ANXA preferentially interacts with apo PAD2. We also examined the interaction between ANXA5 and the D177A mutant (Figure 6C–D). The data indicate that ANXA5 remains associated with the D177A mutant in the presence of calcium. Given that the D177A mutant is not fully saturated with calcium under our experimental conditions (i.e., 2 mM Ca2+ versus a K0.5 of 4.8 ± 0.2 mM for the D177A mutant), it is not surprising that calcium could not perturb the interaction between ANXA5 and this calcium binding mutant. The interaction between ANXA5 and PAD2 was also confirmed by size exclusion chromatography (Figure S3). In the absence of calcium, a new species with a shorter retention time appeared in the mixture of ANXA5 and PAD2. Western blotting confirmed that this signal belonged to PAD2 (Figure S3B). These data imply that ANXA5 induces a conformational change in PAD2 that triggers the formation of a higher order oligomer. This oligomer may prevent the import of PAD2 into the nucleus.
Another critical protein identified from our dataset is Ran. Ran is a small GTPase,28, 60 that directs protein translocation across the NPC. GTP-bound Ran is high in the nucleus whereas Ran-GDP levels are high in the cytoplasm.42 There are two mechanisms for protein transport across the NPC: passive diffusion and karyopherin-mediated transport.61–65 Small proteins with molecular weights smaller than ~40 kDa can passively diffuse through the pore,42, 61 whereas proteins >40 kDa rely on carrier-mediated entry. Since the PAD2 monomer which is ~75 kDa, it can only be transported through the pore with assistance of a carrier.
Most of the proteins that carry cargos through the NPC are members of the karyopherin family.66 Karyopherins consist mainly of HEAT or Armadillo repeats stacked on top of one another.67–70 This common structural feature facilitates their ability to bind to the FG domains of nucleoporins.71–73 We did identify one karyopherin protein in our dataset, i.e., KPNB1. However, immunoprecipitation experiments did not support an interaction between KPNB1 and PAD2 in vitro or in cells (data not shown). Moreover, importazole, a KPNB1 inhibitor 74, did not block the nuclear accumulation of PAD2 (Figure S4). These data are consistent with a KPNB1-independent mechanism of nuclear import. This finding is also consistent with the fact that PAD2 lacks a canonical NLS that is required to interact with a karyopherin 75–77.
Given the abundance NLS-containing proteins, many assume that this pathway is the only mechanism for protein transport into the nucleus; however, it is known that other pathways account for a large amount of nuclear traffic. For example, bioinformatic analysis revealed that amongst the 1515 proteins whose localization is restricted to the nucleus in Saccharomyces cerevisiae, ~ 43% use an alternative mechanism for nuclear entry.77 Indeed, certain amphiphilic proteins (e.g., actinin-4, βI-spectrin, and β-catenin) undergo a conformational change that increases their hydrophobic surface area to promote spontaneous passage through the hydrophobic NPC.78 Experiments also showed that chemically modified BSA with a higher surface hydrophobicity is sufficient to overcome the selectivity barrier of the NPC.79
Given the fact that Ran is highly abundant in our experimental data (Figure 3C) and is involved in nuclear import, we considered that it might play a novel role in the transport of PAD2 into the nucleus. Biotinylation of Ran in HEK293 BioID2PAD2 samples but not in HEK293 BioID2 samples (Figure 5A) confirmed the proximity of Ran to PAD2. Surprisingly, pull down experiments performed with HEK293T-PAD2 cell lysate and with recombinant proteins showed a significant direct interaction between PAD2 and Ran without any adaptor (Figure 5B and C). This result suggests that Ran directly binds and imports PAD2 into the nucleus. The calcium induced conformational change in PAD2 enhanced their interaction. We also evaluated the interaction of PAD2 with GTPase-deficient RanQ69L mutant and GDP bound mutant RanT24N (Figure 5C). Compared with wild type Ran, there is no significant difference between the interactions with calcium free PAD2 and calcium-bound PAD2. Therefore, PAD2 shows no differential binding to nuclear Ran (GTP bound) or cytoplasmic Ran (GDP bound). Then how does nuclear PAD2 accumulate in the presence of ionomycin? Comparing the structures of holoPAD2 with apoPAD2 (Figure 1A),12 one significant change that occurs is in the position of the FF motif. Upon calcium binding, the FF motif extends into solvent. And this change neither alters PAD2 catalytic efficiency nor calcium dependence (Figure 1C). When these two residues were replaced with alanine, the ratio of nuclear PAD2 to cytoplasmic PAD2 decreased dramatically (Figure 2C and D). Since this change has no influence on the interaction between PAD2 and Ran (Figure 6A), we suggest that extension of the FF motif disrupts an interaction that sequesters PAD2 in the cytoplasm. In turn, the binding of PAD2 to Ran facilitates nuclear uptake.
In summary, we show for the first time that calcium triggers the nuclear accumulation of PAD2. Based on our data, we suggest that ANXA5 restricts the location of PAD2 to the cytoplasm. Upon calcium influx, calcium-bound PAD2 is released from ANXA5 and interacts directly with Ran, which shuttles continuously between nucleus and cytoplasm, and promotes the nuclear entry of PAD2 into nucleus. This interaction promotes the Ran-dependent transport of PAD2 into the nucleus where it can citrullinate histones and modulate gene expression programs (Figure 7). Future experiments will focus on uncovering how Ran interacts with PAD2 and evaluating nuclear transport in physiologically relevant cell lines.
Figure 7. PAD2 transport between the cytoplasm and nucleus.
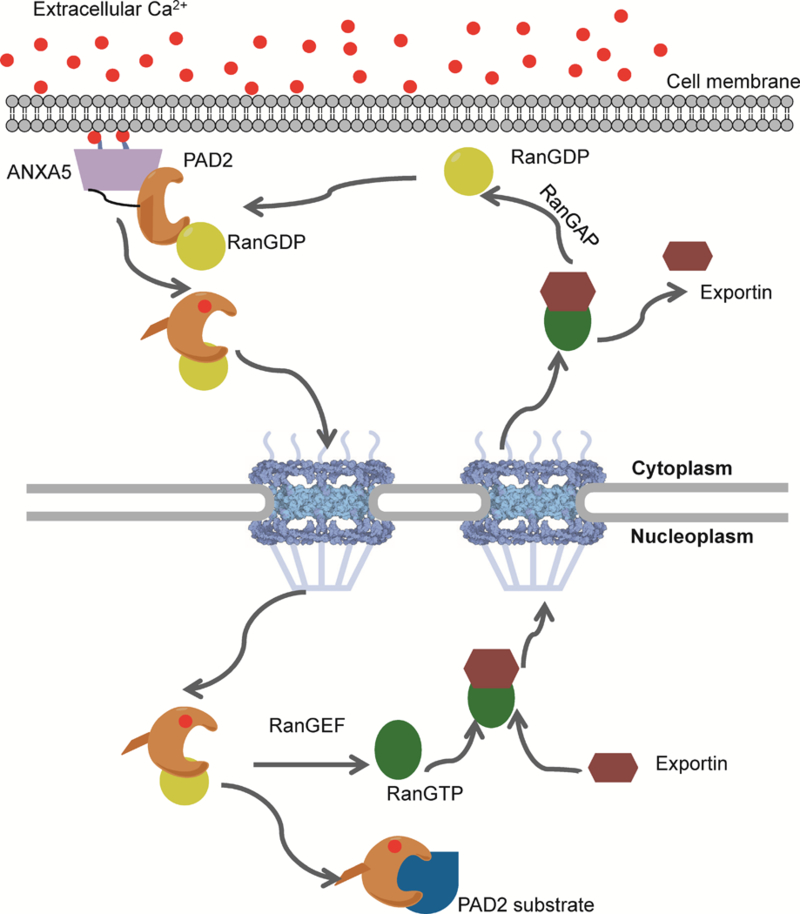
ANXA5 is activated and binds to the membrane to facilitate calcium influx. PAD2 binds calcium, which triggers a conformational change that promotes its release from ANXA5 and subsequent transport into the nucleus via its interaction with Ran. Once in the nucleus, PAD2 citrullinates histones and other nuclear proteins to modulate gene transcription.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported in part by NIH grant R35 GM118112 (P.R.T.).
Footnotes
ASSOCIATED CONTENT
Supporting information. The Supporting Information is available free of charge on the ACS Publications website
The authors declare the following competing financial interest(s): P.R.T. founded Padlock Therapeutics and is entitled to payments from Bristol Myers Squibb if certain milestones are met. P.R.T. is a consultant for Celgene and Disarm Therapeutics. E.W. is a consult for Celgene.
REFERENCES
- (1).Slade DJ, Subramanian V, and Thompson PR (2014) Pluripotency: citrullination unravels stem cells, Nat Chem Biol 10, 327–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Vossenaar ER, Zendman AJ, van Venrooij WJ, and Pruijn GJ (2003) PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease, Bioessays 25, 1106–1118. [DOI] [PubMed] [Google Scholar]
- (3).Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA, Bertone P, Silva JC, Zernicka-Goetz M, Nielsen ML, Gurdon JB, and Kouzarides T (2014) Citrullination regulates pluripotency and histone H1 binding to chromatin, Nature 507, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tilvawala R, Nguyen SH, Maurais AJ, Nemmara VV, Nagar M, Salinger AJ, Nagpal S, Weerapana E, and Thompson PR (2018) The Rheumatoid Arthritis-Associated Citrullinome, Cell Chem Biol. 25, 691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nemmara VV, Tilvawala R, Salinger AJ, Miller L, Nguyen SH, Weerapana E, and Thompson PR (2018) Citrullination Inactivates Nicotinamide-N-methyltransferase, ACS Chem Biol 13, 2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chavanas S, Mechin MC, Takahara H, Kawada A, Nachat R, Serre G, and Simon M (2004) Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6, Gene 330, 19–27. [DOI] [PubMed] [Google Scholar]
- (7).Lamensa JW, and Moscarello MA (1993) Deimination of human myelin basic protein by a peptidylarginine deiminase from bovine brain, J Neurochem 61, 987–996. [DOI] [PubMed] [Google Scholar]
- (8).Watanabe K, Akiyama K, Hikichi K, Ohtsuka R, Okuyama A, and Senshu T (1988) Combined biochemical and immunochemical comparison of peptidylarginine deiminases present in various tissues, Biochim Biophys Acta 966, 375–383. [DOI] [PubMed] [Google Scholar]
- (9).Musse AA, Li Z, Ackerley CA, Bienzle D, Lei H, Poma R, Harauz G, Moscarello MA, and Mastronardi FG (2008) Peptidylarginine deiminase 2 (PAD2) overexpression in transgenic mice leads to myelin loss in the central nervous system, Dis Model Mech 1, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cherrington BD, Zhang X, McElwee JL, Morency E, Anguish LJ, and Coonrod SA (2012) Potential role for PAD2 in gene regulation in breast cancer cells, PLoS One 7, e41242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Takahara H, Okamoto H, and Sugawara K (1986) Calcium-dependent Properties of Peptidylarginine Deiminase from Rabbit Skeletal Muscle, Agricultural and Biological Chemistry 50, 2899–2904. [Google Scholar]
- (12).Slade DJ, Fang P, Dreyton CJ, Zhang Y, Fuhrmann J, Rempel D, Bax BD, Coonrod SA, Lewis HD, Guo M, Gross ML, and Thompson PR (2015) Protein arginine deiminase 2 binds calcium in an ordered fashion: implications for inhibitor design, ACS Chem Biol 10, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang X, Bolt M, Guertin MJ, Chen W, Zhang S, Cherrington BD, Slade DJ, Dreyton CJ, Subramanian V, Bicker KL, Thompson PR, Mancini MA, Lis JT, and Coonrod SA (2012) Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor alpha target gene activation, Proc Natl Acad Sci U S A 109, 13331–13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gyorgy B, Toth E, Tarcsa E, Falus A, and Buzas EI (2006) Citrullination: a posttranslational modification in health and disease, Int J Biochem Cell Biol 38, 1662–1677. [DOI] [PubMed] [Google Scholar]
- (15).Nakashima K, Hagiwara T, and Yamada M (2002) Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes, J Biol Chem 277, 49562–49568. [DOI] [PubMed] [Google Scholar]
- (16).Fuhrmann J, Clancy KW, and Thompson PR (2015) Chemical biology of protein arginine modifications in epigenetic regulation, Chem Rev 115, 5413–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dreyton CJ, Knuckley B, Jones JE, Lewallen DM, and Thompson PR (2014) Mechanistic studies of protein arginine deiminase 2: evidence for a substrate-assisted mechanism, Biochemistry 53, 4426–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kearney PL, Bhatia M, Jones NG, Yuan L, Glascock MC, Catchings KL, Yamada M, and Thompson PR (2005) Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis, Biochemistry 44, 10570–10582. [DOI] [PubMed] [Google Scholar]
- (19).Knipp M, and Vasak M (2000) A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline, Anal Biochem 286, 257–264. [DOI] [PubMed] [Google Scholar]
- (20).Muth A, Subramanian V, Beaumont E, Nagar M, Kerry P, McEwan P, Srinath H, Clancy K, Parelkar S, and Thompson PR (2017) Development of a Selective Inhibitor of Protein Arginine Deiminase 2, J Med Chem 60, 3198–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, and Riabowol K (2010) REAP: A two minute cell fractionation method, BMC Res Notes 3, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes, Nature 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Eng JK, McCormack AL, and Yates JR (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database, J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- (24).Tabb DL, McDonald WH, and Yates JR 3rd. (2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics, J Proteome Res 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Roux KJ, Kim DI, and Burke B (2013) BioID: a screen for protein-protein interactions, Curr Protoc Protein Sci 74, Unit 19.23. [DOI] [PubMed] [Google Scholar]
- (26).Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, and Roux KJ (2016) An improved smaller biotin ligase for BioID proximity labeling, Mol Biol Cell 27, 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics, Mol Cell Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (28).Moore MS, and Blobel G (1993) The GTP-binding protein Ran/TC4 is required for protein import into the nucleus, Nature 365, 661–663. [DOI] [PubMed] [Google Scholar]
- (29).Okada N, and Sato M (2015) Spatiotemporal Regulation of Nuclear Transport Machinery and Microtubule Organization, Cells 4, 406–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Degrelle SA, Gerbaud P, Leconte L, Ferreira F, and Pidoux G (2017) Annexin-A5 organized in 2D-network at the plasmalemma eases human trophoblast fusion, Scientific Reports 7, 42173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rand JH, Wu XX, Lin EY, Griffel A, Gialanella P, and McKitrick JC (2012) Annexin A5 binds to lipopolysaccharide and reduces its endotoxin activity, MBio 3, e00292–00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, and Henson PM (1997) Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase, J Biol Chem 272, 26159–26165. [DOI] [PubMed] [Google Scholar]
- (33).Eberhart DE, Malter HE, Feng Y, and Warren ST (1996) The Fragile X Mental Retardation Protein is a Ribonucleoprotein Containing Both Nuclear Localization and Nuclear Export Signals, Human Molecular Genetics 5, 1083–1091. [DOI] [PubMed] [Google Scholar]
- (34).Adams-Cioaba MA, Guo Y, Bian C, Amaya MF, Lam R, Wasney GA, Vedadi M, Xu C, and Min J (2010) Structural studies of the tandem Tudor domains of fragile X mental retardation related proteins FXR1 and FXR2, PLoS One 5, e13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zarnescu DC, and Gregorio CC (2013) Fragile hearts: New insights into translational control in cardiac muscle, Trends Cardiovasc Med 23, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Moss SE (1992) The Annexins, Portland Press, London; Chapel Hill, NC. [Google Scholar]
- (37).Gerke V, Creutz CE, and Moss SE (2005) Annexins: linking Ca2+ signalling to membrane dynamics, Nat Rev Mol Cell Biol 6, 449–461. [DOI] [PubMed] [Google Scholar]
- (38).Berendes R, Voges D, Demange P, Huber R, and Burger A (1993) Structure-function analysis of the ion channel selectivity filter in human annexin V, Science 262, 427–430. [DOI] [PubMed] [Google Scholar]
- (39).Demange P, Voges D, Benz J, Liemann S, Gottig P, Berendes R, Burger A, and Huber R (1994) Annexin V: the key to understanding ion selectivity and voltage regulation?, Trends Biochem Sci 19, 272–276. [DOI] [PubMed] [Google Scholar]
- (40).Raynal P, and Pollard HB (1994) Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins, Biochim Biophys Acta 1197, 63–93. [DOI] [PubMed] [Google Scholar]
- (41).Feldherr CM, Kallenbach E, and Schultz N (1984) Movement of a karyophilic protein through the nuclear pores of oocytes, J Cell Biol 99, 2216–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gorlich D, and Mattaj IW (1996) Nucleocytoplasmic transport, Science 271, 1513–1518. [DOI] [PubMed] [Google Scholar]
- (43).Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, and Chait BT (2000) The yeast nuclear pore complex: composition, architecture, and transport mechanism, J Cell Biol 148, 635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lott K, and Cingolani G (2011) The importin beta binding domain as a master regulator of nucleocytoplasmic transport, Biochim Biophys Acta 1813, 1578–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Denning DP, Patel SS, Uversky V, Fink AL, and Rexach M (2003) Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded, Proc Natl Acad Sci U S A 100, 2450–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lim RY, Huang NP, Koser J, Deng J, Lau KH, Schwarz-Herion K, Fahrenkrog B, and Aebi U (2006) Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport, Proc Natl Acad Sci U S A 103, 9512–9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Watanabe K, and Senshu T (1989) Isolation and characterization of cDNA clones encoding rat skeletal muscle peptidylarginine deiminase, J Biol Chem 264, 15255–15260. [PubMed] [Google Scholar]
- (48).Ullman KS, Powers MA, and Forbes DJ (1997) Nuclear export receptors: from importin to exportin, Cell 90, 967–970. [DOI] [PubMed] [Google Scholar]
- (49).Izaurralde E, and Adam S (1998) Transport of macromolecules between the nucleus and the cytoplasm, RNA 4, 351–364. [PMC free article] [PubMed] [Google Scholar]
- (50).Mattaj IW, and Englmeier L (1998) Nucleocytoplasmic transport: the soluble phase, Annu Rev Biochem 67, 265–306. [DOI] [PubMed] [Google Scholar]
- (51).Coutavas E, Ren M, Oppenheim JD, D’Eustachio P, and Rush MG (1993) Characterization of proteins that interact with the cell-cycle regulatory protein Ran/TC4, Nature 366, 585–587. [DOI] [PubMed] [Google Scholar]
- (52).Kornbluth S, Dasso M, and Newport J (1994) Evidence for a dual role for TC4 protein in regulating nuclear structure and cell cycle progression, J Cell Biol 125, 705–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Dasso M, Seki T, Azuma Y, Ohba T, and Nishimoto T (1994) A mutant form of the Ran/TC4 protein disrupts nuclear function in Xenopus laevis egg extracts by inhibiting the RCC1 protein, a regulator of chromosome condensation, EMBO J 13, 5732–5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Clancy KW, Russell AM, Subramanian V, Nguyen H, Qian Y, Campbell RM, and Thompson PR (2017) Citrullination/Methylation Crosstalk on Histone H3 Regulates ER-Target Gene Transcription, ACS Chem Biol 12, 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Rojas E, Arispe N, Haigler HT, Burns AL, and Pollard HB (1992) Identification of annexins as calcium channels in biological membranes, Bone Miner 17, 214–218. [DOI] [PubMed] [Google Scholar]
- (56).Rojas E, Pollard HB, Haigler HT, Parra C, and Burns AL (1990) Calcium-activated endonexin II forms calcium channels across acidic phospholipid bilayer membranes, J Biol Chem 265, 21207–21215. [PubMed] [Google Scholar]
- (57).Pollard HB, Guy HR, Arispe N, de la Fuente M, Lee G, Rojas EM, Pollard JR, Srivastava M, Zhang-Keck ZY, and Merezhinskaya N (1992) Calcium channel and membrane fusion activity of synexin and other members of the Annexin gene family, Biophys J 62, 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Arispe N, Rojas E, Genge BR, Wu LN, and Wuthier RE (1996) Similarity in calcium channel activity of annexin V and matrix vesicles in planar lipid bilayers, Biophys J 71, 1764–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Berendes R, Burger A, Voges D, Demange P, and Huber R (1993) Calcium influx through annexin V ion channels into large unilamellar vesicles measured with fura-2, FEBS Lett 317, 131–134. [DOI] [PubMed] [Google Scholar]
- (60).Melchior F, Paschal B, Evans J, and Gerace L (1993) Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor, J Cell Biol 123, 1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Marfori M, Mynott A, Ellis JJ, Mehdi AM, Saunders NF, Curmi PM, Forwood JK, Boden M, and Kobe B (2011) Molecular basis for specificity of nuclear import and prediction of nuclear localization, Biochim Biophys Acta 1813, 1562–1577. [DOI] [PubMed] [Google Scholar]
- (62).Adam EJ, and Adam SA (1994) Identification of cytosolic factors required for nuclear location sequence-mediated binding to the nuclear envelope, J Cell Biol 125, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Gorlich D, Prehn S, Laskey RA, and Hartmann E (1994) Isolation of a protein that is essential for the first step of nuclear protein import, Cell 79, 767–778. [DOI] [PubMed] [Google Scholar]
- (64).Gorlich D, Vogel F, Mills AD, Hartmann E, and Laskey RA (1995) Distinct functions for the two importin subunits in nuclear protein import, Nature 377, 246–248. [DOI] [PubMed] [Google Scholar]
- (65).McLane LM, Pulliam KF, Devine SE, and Corbett AH (2008) The Ty1 integrase protein can exploit the classical nuclear protein import machinery for entry into the nucleus, Nucleic Acids Res 36, 4317–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Macara IG (2001) Transport into and out of the nucleus, Microbiol Mol Biol Rev 65, 570–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Bayliss R, Littlewood T, and Stewart M (2000) Structural basis for the interaction between FxFG nucleoporin repeats and importin-beta in nuclear trafficking, Cell 102, 99–108. [DOI] [PubMed] [Google Scholar]
- (68).Bayliss R, Littlewood T, Strawn LA, Wente SR, and Stewart M (2002) GLFG and FxFG nucleoporins bind to overlapping sites on importin-beta, J Biol Chem 277, 50597–50606. [DOI] [PubMed] [Google Scholar]
- (69).Liu SM, and Stewart M (2005) Structural basis for the high-affinity binding of nucleoporin Nup1p to the Saccharomyces cerevisiae importin-beta homologue, Kap95p, J Mol Biol 349, 515–525. [DOI] [PubMed] [Google Scholar]
- (70).Conti E, and Izaurralde E (2001) Nucleocytoplasmic transport enters the atomic age, Curr Opin Cell Biol 13, 310–319. [DOI] [PubMed] [Google Scholar]
- (71).Iovine MK, Watkins JL, and Wente SR (1995) The GLFG repetitive region of the nucleoporin Nup116p interacts with Kap95p, an essential yeast nuclear import factor, J Cell Biol 131, 1699–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Bailer SM, Siniossoglou S, Podtelejnikov A, Hellwig A, Mann M, and Hurt E (1998) Nup116p and nup100p are interchangeable through a conserved motif which constitutes a docking site for the mRNA transport factor gle2p, EMBO J 17, 1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Marelli M, Aitchison JD, and Wozniak RW (1998) Specific binding of the karyopherin Kap121p to a subunit of the nuclear pore complex containing Nup53p, Nup59p, and Nup170p, J Cell Biol 143, 1813–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Soderholm JF, Bird SL, Kalab P, Sampathkumar Y, Hasegawa K, Uehara-Bingen M, Weis K, and Heald R (2011) Importazole, a small molecule inhibitor of the transport receptor importin-beta, ACS Chem Biol 6, 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Goldfarb DS, Gariepy J, Schoolnik G, and Kornberg RD (1986) Synthetic peptides as nuclear localization signals, Nature 322, 641–644. [DOI] [PubMed] [Google Scholar]
- (76).Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, and Chook YM (2006) Rules for nuclear localization sequence recognition by karyopherin beta 2, Cell 126, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, and Corbett AH (2007) Classical nuclear localization signals: definition, function, and interaction with importin alpha, J Biol Chem 282, 5101–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Kumeta M, Yamaguchi H, Yoshimura SH, and Takeyasu K (2012) Karyopherin-independent spontaneous transport of amphiphilic proteins through the nuclear pore, J Cell Sci 125, 4979–4984. [DOI] [PubMed] [Google Scholar]
- (79).Naim B, Zbaida D, Dagan S, Kapon R, and Reich Z (2009) Cargo surface hydrophobicity is sufficient to overcome the nuclear pore complex selectivity barrier, EMBO J 28, 2697–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.