

Abstract

Antibody–drug conjugates (ADCs) that incorporate potent indolinobenzodiazepine DNA alkylators as the payload component are currently undergoing clinical evaluation. In one ADC design, the payload molecules are linked to the antibody through a peptidase-labile l-Ala-l-Ala linker. In order to determine the role of amino acid stereochemistry on antitumor activity and tolerability, we incorporated l- and d-alanyl groups in the dipeptide, synthesized all four diastereomers, and prepared and tested the corresponding ADCs. Results of our preclinical evaluation showed that the l-Ala-l-Ala configuration provided the ADC with the highest therapeutic index (antitumor activity vs toxicity).
Keywords: Antibody−drug conjugates, ADCs, indolinobenzodiazepines
Antibody–drug conjugates (ADCs) seek to improve the therapeutic index of highly cytotoxic drugs by linking them to tumor-selective monoclonal antibodies.1−4 For several years, the majority of ADCs undergoing clinical evaluation used the microtubule interacting agents maytansinoids and auristatins as the cytotoxic component.5,6 In recent years, the emphasis has shifted to drugs with alternative mechanisms of action, such as Topoisomerase 1 inhibition (camptothecins) or DNA cross-linking (pyrrolobenzodiazepines, PBDs).7,8 Emerging clinical data for ADCs of the PBD class indicate that doses achieved in patients are quite low (0.04–0.3 mg/kg), with systemic toxicity often limiting dosing beyond two cycles.9,10 We previously reported on a new class of DNA interacting compounds, the indolinobenzodiazepines (termed IGNs).11,12 While the parent compounds in this class were potent DNA cross-linkers, we found that simple controlled reduction of one of the two imine moieties could change the mechanism of action to DNA alkylation without the ability to cross-link. ADCs of these alkylating IGNs (e.g., anti-FRα DGN549 shown in Figure 1) were found to be only slightly less potent in vitro and in vivo than those of the corresponding DNA cross-linkers, while being less toxic in vivo. Importantly ADCs of alkylating IGNs were devoid of the phenomenon of delayed toxicity noted for the cross-linking version in mice.12
Figure 1.

Structures of the anti-FRα DGN549 ADC and its main catabolite 13.
Based on these findings, three ADCs of DNA alkylating IGNs have been advanced to the clinic. The first one, IMGN779, uses a disulfide linker, while the other two, IMGN632 and TAK164, incorporate a dipeptide (l-Ala-l-Ala) linker.11,12 Previous reports on ADCs of auristatins and maytansinoids with peptide linkages indicate that the stereochemistry of the amino acids played a significant role in determining in vivo tolerability. In the case of auristatin ADCs, a number of different dipeptide linkages was evaluated and, in general, conjugates that bore one d-amino acid in the linker were better tolerated in mice. In a head-to-head comparison, an auristatin F ADC with an l-Asn-d-Lys linker had similar in vitro potency to that with an l-Asn-l-Lys, but the former was 2-fold better tolerated in mice.13 Also, the ADC with the l-Asn-d-Lys linkage displayed the highest antitumor activity in vivo, among a panel of eight different linkers tested. In the case of maytansinoids, ADCs with the l-Ala-l-Ala or the d-Ala-l-Ala linkage showed similar in vitro potency and in vivo efficacy. However, the ADC with the d-Ala isomer in the linkage was significantly less toxic in mice.14
Based on these reports, incorporation of one d-amino acid in the dipeptide linker provides a tolerability advantage without loss of antitumor activity. The current study was designed to determine if these findings could be extended to ADCs containing IGNs that only alkylate DNA and are incapable of cross-linking. We have previously shown that the anti-FRα DGN549 ADC is catabolized upon internalization into antigen-expressing cells, resulting in scission of the amide bond between the anilino nitrogen and the l-alanyl moiety to give the potent anilino metabolite 13.12 We have also shown that this catabolite, when generated in antigen-positive cells, can diffuse into, and kill, proximal antigen negative cells, through a phenomenon called “bystander killing”, which results in enhanced antitumor activity in vivo. Since bystander killing relies on the release of a hydrophobic catabolite, such as 13, we used it as a surrogate to assess whether the amide bond between the anilino nitrogen and the amino acid is being cleaved in cells.12 The goal of this study was to gauge the effect of the amino acid stereochemistry in the dipeptide linker on bystander killing, in vivo efficacy, and tolerability of the ADCs. We synthesized IGNs bearing all four diastereomers of the Ala-Ala linker (ll, dl, ld, dd) and conjugated them to a monoclonal antibody. Here, we report the results of our in vitro and in vivo evaluation of the ADCs.
An approach to the synthesis of ADCs (11a–d) bearing the four diastereomeric dipeptides is shown in Scheme 1, exemplified by the preparation of the l-Ala-l-Ala linker bearing compound IGN 10, which contains a N-hydroxysuccinimide (NHS) ester for conjugation to an antibody. Coupling of (S)-tert-butyl 2-aminopropanoate (1) with (S)-2-(((benzyloxy)carbonyl)amino)propanoic acid (2) using HOBt followed by hydrogenolysis with palladium on carbon gave the desired enantiomerically pure protected dipeptide 3. Further coupling of 3 with monomethyladipate under similar HOBt conditions and deprotection of the t-butyl ester with TFA gave the free carboxylic acid 4, which was in turn treated with (5-amino-1,3-phenylene)dimethanol in the presence of EEDQ to give diol 5. Conversion to the activated bis-mesylate 6 was accomplished through the treatment of diol 5 with methanesulfonic acid in the presence of triethylamine. With the synthesis of the enantiomerically pure dipeptide spacer in hand, the diimine containing IGN dimer 8 was prepared in modest yield by treating dimesylate 6 with two equivalents of the imine containing IGN monomer 7 in the presence of potassium iodide and potassium carbonate in dimethylacetamide.
Scheme 1. Synthesis of ADCs 11a–d.

Reagents and conditions: (a) EDC, HOBt, DIPEA, DMF, 85%; (b) H2, Pd/C, CH3OH, 97%; (c) monomethyladipate, EDC, HOBt, DIPEA, DMF, 100%; (d) TFA, water, 100%; (e) (5-amino-1,3-phenylene)dimethanol, EEDQ, CH3OH, DCM, 36%; (f) methanesulfonic anhydride, Et3N, DCM/DMF, 85%; (g) K2CO3, KI, DMA, 52%; (h) NaBH(OAc)3, DCE, ∼25%; (i) LiOH, THF/water; (j) N-hydroxysuccinimide, EDC, DCM, 26% over two steps.
We found that the best results to prepare the desired monoimine containing dimer 9 were achieved using 0.95 equiv of sodium triacetoxyborohydride in dichloroethane followed by C18 purification to separate it from unreacted starting material and the over-reduced diamine product. Finally, hydrolysis of the methyl ester with lithium hydroxide and treatment with N-hydroxysuccinimide gave the desired NHS ester 10 after lyophilization. Through a similar set of chemical transformations using the different enantiomers of amino acids 1 and 2, we were able to prepare the other three diastereomeric NHS esters (dl, ld, dd) of 10. With the successful preparation of the required NHS esters, each diastereomer was conjugated separately with an anti-FRα antibody via lysine residues as described previously, giving the resulting ADCs 11a–d in high yield.12 All conjugates were found to have an average of ∼2.7 IGNs per antibody, with >96% monomeric purity by SEC analysis and contained less than 0.5% unconjugated IGN (free drug).
All four anti-FRα ADCs were evaluated in vitro (Table 1) against three cell lines expressing various levels of the targeted antigen. In the higher expressing cell lines, KB and T47D, all four ADCs demonstrated similarly high potency with IC50 values ranging from 5 to 40 pM. This cytotoxic effect was shown to be antigen-specific since the addition of excess unconjugated antibody (1 μM) abolished this activity. Interestingly, in the lower antigen expressing cell line, NCI-H2110, there was an apparent preference for anti-FRα ADC 11a (l-Ala-l-Ala) as it was found be ∼3-fold more potent than ADCs 11b (d,l) and 11c (l,d), and almost 90-fold more potent than the completely unnatural d-Ala-d-Ala containing dipeptide ADC 11d. These data seem to indicate that peptide cleavage in the H2110 cell line to release the fully active metabolite (13) occurs at different rates, favoring the natural dipeptide configuration, though other factors are possibly involved as well.
Table 1. In Vitro Potency of Anti-FRα ADCs 11a–db.
| anti-FRα
ADC IC50, pM |
|||||
|---|---|---|---|---|---|
| cell line | ABCa | 11a | 11b | 11c | 11d |
| KB | >2000k | 7 | 8 | 5 | 9 |
| T47D | 100k | 30 | 20 | 20 | 40 |
| NCI-H2110 | 50k | 80 | 300 | 250 | 7000 |
Number of antibody molecules bound per cell.
Cancer cell lines were incubated with ADCs for 5 days at 37 °C. IC50 values were determined using a WST-based cell viability assay.
We next evaluated the impact of the dipeptide linker stereochemistry on bystander activity, using a mixture of FRα-positive and FRα-negative 300.19 cells as previously described.12 As shown in Figure 2, treatment with each of the four anti-FRα IGN ADCs 11a–d at 0.2 nM resulted in 100% killing of the antigen positive 300.19 cells (black bars), whereas the antigen negative cells (blue bars) were unaffected. However, when FRα-antigen positive and negative cells were cocultured together (a mixture containing 1000 antigen positive and 2000 antigen negative cells) and then treated with the anti-FRα IGN ADCs 11a–d, only ADC 11a (l-Ala-l-Ala) demonstrated strong bystander activity resulting in complete killing of the proximal antigen negative cells (green bars). Of all the anti-FRα IGN ADCs with an unnatural amino acid (11b–d), only ADC 11c, possessing the natural l-alanyl peptide adjacent to the aniline nitrogen, which may be slightly susceptible to peptidase cleavage, exhibited a low degree of bystander activity where approximately 30% of the antigen negative cells were killed, whereas 11b and 11d were entirely devoid of any bystander activity.
Figure 2.
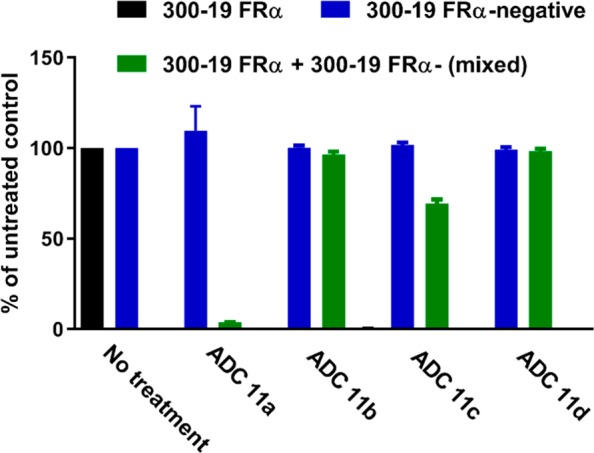
Impact of stereochemistry on bystander activity.
Encouraged by the similar in vitro potency of anti-FRα IGN ADCs 11a and 11b toward target cells, and their significant difference in bystander activity, we were interested in exploring the impact the dipeptide linker had on in vivo efficacy and tolerability. Our hope was that 11b would provide an ADC with activity similar to that of 11a but with improved tolerability. Thus, the antitumor activity of these two ADCs was evaluated in SCID mice using the NCI-H2110 subcutaneous nonsmall cell lung xenograft model. As shown in Figure 3, treatment with a single i.v. dose of either 0.3 or 1.5 mg/kg ADC (equivalent to 5 and 25 μg/kg linked IGN, respectively) resulted in drastically different antitumor activity. The anti-FRα IGN ADC 11a was highly active at both doses exhibiting 5/6 CRs at the lower dose, and all animals were tumor-free at study end for the high dose. In contrast, anti-FRα IGN ADC 11b, while highly active at the higher dose of 1.5 mg/kg, showed only 1/6 CRs and was found to be inactive at the lower dose of 0.3 mg/kg. The inactivity of 11b can presumably be attributed to its lack of bystander activity as the NCI-H2110 model expresses antigen in a heterogeneous manner (H-Score of 110) and thus requires bystander killing to kill nonexpressing cells.12 Interestingly, despite these antitumor activity differences, both ADCs 11a and 11b were found to demonstrate a similar maximum tolerated dose in mice of ∼6 mg/kg (equivalent to ∼100 μg/kg linked IGN).
Figure 3.
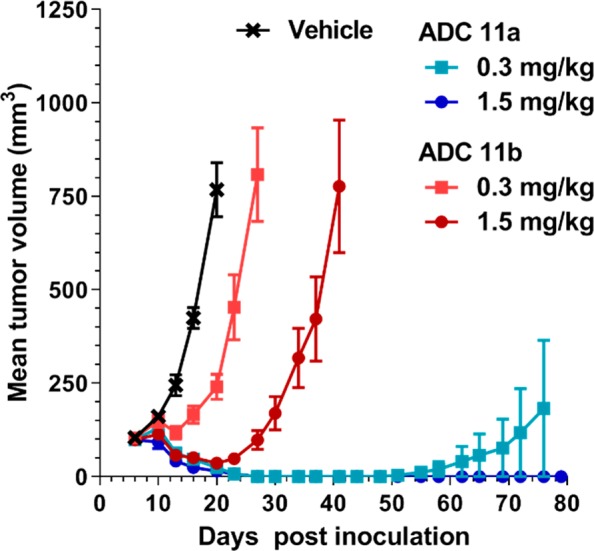
In vivo antitumor activity of ADCs 11a and 11b in nonsmall cell lung cancer NCI-H2110 xenografts in SCID mice.
In an effort to understand the cellular metabolism of these anti-FRα IGN ADCs, FRα positive cells were treated with either ADC 11a or 11b at 37 °C for 24 h, and the catabolites were affinity captured, extracted with acetone, and concentrated. Analysis of the catabolites isolated by UPLC/MS revealed that the only identifiable catabolite for ADC 11b (d-Ala-l-Ala) was the IGN with the lysine-linked intact dipeptide 12 (Figure 4). As this carboxylic acid containing catabolite would not be expected to be cell permeable, this could explain the decreased bystander activity and antitumor activity of anti-FRα IGN ADC 11b. In contrast, analysis of the catabolites for ADC 11a showed only a small amount of the lysine-linked catabolite, with the majority of the catabolite found to be the fully dipeptide cleaved aniline 13, which is known to be exquisitely potent (4–20 pM) in many cell lines.12 Furthermore, aniline 13 was identified as the only catabolite found when SCID mice bearing FRα tumors were treated with anti-FRα IGN ADC 11a when those tumors were excised and homogenized, and the catabolites were extracted. Taken together, the presence of this cell permeable and highly potent catabolite both in vitro and in vivo leads to the observed high bystander and exceptional antitumor activity described above.
Figure 4.

Structure of catabolite 12.
In conclusion, we prepared ADCs of IGNs possessing all four diastereomers (11a–d) of an Ala-Ala dipeptide linker attached at a central aniline of the potent catabolite 13. All ADCs, regardless of stereochemistry, were found to be potent in vitro in high antigen expressing cell lines; however, in lower expressing cell lines and in bystander studies, there was a strong preference for 11a containing the natural l-Ala-l-Ala dipeptide. Based on previous studies for both the maytansinoids and auristatins, where improved tolerability with similar antitumor activity was observed for dipeptides possessing an unnatural d-amino acid, we explored this comparison for anti-FRa ADCs 11a (l,l) and 11b (d,l). In our studies we found that the antitumor activity was quite different for these two ADCs in a NCI-H2110 xenograft where 11a was highly active at both doses tested and 11b was only active at the higher dose of 1.5 mg/kg. Interestingly, the decreased antitumor activity for 11b did not lead to improved tolerability as 11a and 11b were found to have identical MTDs in vivo. It is well-known that only a small percent of the ADC localizes at the tumor, and bystander killing is an important contributor to efficacy, especially in a heterogeneous model. Thus, 11a is more efficacious than 11bin vivo. A majority of the administered ADC is taken up through pinocytosis by a number of normal tissues, notably liver, where release of active catabolite results in toxicity that turns out to be similar for 11a and 11b.
Glossary
ABBREVIATIONS
- ADC
antibody–drug conjugate
- PBD
pyrrolobenzodiazepine
- IGN
indolinobenzodiazepine
- NHS
N-hydroxysuccinimide ester
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DCM
dichloromethane
- DMA
dimethylacetmide
- DCE
dichloroethane
- Et3N
trimethylamine
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOBt
hydroxybenzotriazole
- TFA
trifluoroacetic acid
- EEDQ
N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline
- SEC
size-exclusion chromatography
- CR
complete regression
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00240.
Detailed experimental procedures for all compounds, ADCs, and catabolite studies (PDF)
Author Present Address
† Pharmaron, Inc., Waltham, MA 02451.
Author Present Address
‡ H3 Biomedicine, Cambridge, MA 02139.
Author Present Address
§ Foley Hoag, LLP, Boston, MA 02210-2600.
Author Present Address
⊥ Takeda Pharmaceuticals, Cambridge, MA 02139.
Author Contributions
The manuscript was written through contributions of all authors. All authors of submitting affiliation have given approval to the final version of the manuscript.
This study was supported by ImmunoGen, Inc.
The authors declare no competing financial interest.
Supplementary Material
References
- Chari R. V.; Miller M. L.; Widdison W. C. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem., Int. Ed. 2014, 53, 3796–827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discovery 2017, 16, 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Polakis P. Antibody Drug Conjugates for Cancer Therapy. Pharmacol. Rev. 2016, 68, 3–19. 10.1124/pr.114.009373. [DOI] [PubMed] [Google Scholar]
- Coats S.; Williams M.; Kebble B.; Dixit R.; Tseng L.; Yao N.; Tice D. A.; Soria J.-C. Antibody Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 10.1158/1078-0432.CCR-19-0272. [DOI] [PubMed] [Google Scholar]
- Chari R. V. Expanding the Reach of Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2016, 7, 974–976. 10.1021/acsmedchemlett.6b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers E. L.; Senter P. D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- Ogitani Y.; Aida T.; Hagihara K.; Yamaguchi J.; Ishii C.; Harada N.; Soma M.; Okamoto H.; Oitate M.; Arakawa S.; Hirai T.; Atsumi R.; Nakada T.; Hayakawa I.; Abe Y.; Agatsuma T. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. 10.1158/1078-0432.CCR-15-2822. [DOI] [PubMed] [Google Scholar]
- Mantaj J.; Jackson P. J.; Rahman K. M.; Thurston D. E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew. Chem., Int. Ed. 2017, 56, 462–488. 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E. M.; Walter R. B.; Erba H. P.; Fathi A. T.; Advani A. S.; Lancet J. E.; Ravandi F.; Kovacsovics T.; DeAngelo D. J.; Bixby D.; Faderl S.; Jillella A. P.; Ho P. A.; O’Meara M. M.; Zhao B.; Biddle-Snead C.; Stein A. S. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood 2018, 131, 387–396. 10.1182/blood-2017-06-789800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin C. M.; Pietanza M. C.; Bauer T. M.; Ready N.; Morgensztern D.; Glisson B. S.; Byers L. A.; Johnson M. L.; Burris H. A. 3rd; Robert F.; Han T. H.; Bheddah S.; Theiss N.; Watson S.; Mathur D.; Vennapusa B.; Zayed H.; Lally S.; Strickland D. K.; Govindan R.; Dylla S. J.; Peng S. L.; Spigel D. R. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. 10.1016/S1470-2045(16)30565-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. L.; Fishkin N. E.; Li W.; Whiteman K. R.; Kovtun Y.; Reid E. E.; Archer K. E.; Maloney E. K.; Audette C. A.; Mayo M. F.; Wilhelm A.; Modafferi H. A.; Singh R.; Pinkas J.; Goldmacher V.; Lambert J. M.; Chari R. V. A New Class of Antibody-Drug Conjugates with Potent DNA Alkylating Activity. Mol. Cancer Ther. 2016, 15, 1870–8. 10.1158/1535-7163.MCT-16-0184. [DOI] [PubMed] [Google Scholar]
- Miller M. L.; Shizuka M.; Wilhelm A.; Salomon P.; Reid E. E.; Lanieri L.; Sikka S.; Maloney E. K.; Harvey L.; Qiu Q.; Archer K. E.; Bai C.; Vitharana D.; Harris L.; Singh R.; Ponte J. F.; Yoder N. C.; Kovtun Y.; Lai K. C.; Ab O.; Pinkas J.; Keating T. A.; Chari R. V. J. A DNA-Interacting Payload Designed to Eliminate Cross-Linking Improves the Therapeutic Index of Antibody-Drug Conjugates (ADCs). Mol. Cancer Ther. 2018, 17, 650–660. 10.1158/1535-7163.MCT-17-0940. [DOI] [PubMed] [Google Scholar]
- Doronina S. O.; Bovee T. D.; Meyer D. W.; Miyamoto J. B.; Anderson M. E.; Morris-Tilden C. A.; Senter P. D. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjugate Chem. 2008, 19, 1960–3. 10.1021/bc800289a. [DOI] [PubMed] [Google Scholar]
- Widdison W. C.; Ponte J. F.; Coccia J. A.; Lanieri L.; Setiady Y.; Dong L.; Skaletskaya A.; Hong E. E.; Wu R.; Qiu Q.; Singh R.; Salomon P.; Fishkin N.; Harris L.; Maloney E. K.; Kovtun Y.; Veale K.; Wilhelm S. D.; Audette C. A.; Costoplus J. A.; Chari R. V. Development of Anilino-Maytansinoid ADCs that Efficiently Release Cytotoxic Metabolites in Cancer Cells and Induce High Levels of Bystander Killing. Bioconjugate Chem. 2015, 26, 2261–78. 10.1021/acs.bioconjchem.5b00430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.