

Abstract

GSK2798745, a clinical candidate, was identified as an inhibitor of the transient receptor potential vanilloid 4 (TRPV4) ion channel for the treatment of pulmonary edema associated with congestive heart failure. We discuss the lead optimization of this novel spirocarbamate series and specifically focus on our strategies and solutions for achieving desirable potency, rat pharmacokinetics, and physicochemical properties. We highlight the use of conformational bias to deliver potency and optimization of volume of distribution and unbound clearance to enable desirable in vivo mean residence times.
Keywords: GSK2798745, TRPV4, congestive heart failure, conformational bias, volume of distribution
Transient receptor potential vanilloid 4 (TRPV4) is a cation channel that mediates the influx of Ca2+ across plasma membranes.1 The channel can be activated by heat, hypotonicity, physical stress, and small molecules.2−5 TRPV4 activation caused by heightened pulmonary vascular pressure increases the permeability of the alveolar septal barrier that leads to pulmonary edema.6,7
In preclinical congestive heart failure models, increased pressure in the pulmonary vascular system causes the development of pulmonary edema, which is reduced with TRPV4 channel inhibition. Therefore, a TRPV4 inhibitor is hypothesized to prevent channel activation and reduce pulmonary edema in heart failure patients.8,9
GlaxoSmithKline (GSK) previously reported TRPV4 antagonists series, quinolines and benzimidazoles, failed to deliver clinical drug candidates necessitating the need to identify an alternative series.10,19
Herein we report the discovery of GSK2798745 (30), a clinical candidate for the inhibition of TRPV4. GSK2798745 (30) arose from a lead optimization campaign aimed at identifying a small molecule oral drug candidate capable of inhibiting the TRPV4 ion channel at or above unbound IC50 for 24 h per day from a low once a day oral dose.
To achieve this target profile, we optimized the molecule for both potency and pharmacokinetics (PK). TRPV4 inhibition was measured via a FLIPR HEK cellular assay with a targeted IC50 ≤ 10 nM required for successful candidates. PK optimization was performed in Sprague–Dawley rats.10 We focused our attention on identification of compounds with robust oral bioavailability (>30%) and mean residence time (MRT). An extended MRT is highly desirable as it minimizes the peak to trough concentrations during a dose, which improves therapeutic index and enables a lower overall dose. A minimum MRT of 2 h in rat was projected to scale to enable once a day dosing in human.11,12
In addition to the potency and PK targets, we sought to identify a compound with desirable physicochemical properties to mitigate selectivity and attrition liabilities.13,14 Thus, we targeted compounds exhibiting a cLogP of <3.5 and measured chromatographic log D at pH 7.4 (ChromLogD) of <4.0.13
The spirocarbamate lead 1 (Figure 1) was derived from an HTS screening hit molecule from the GSK screening collection.15 Compound 1 demonstrated robust potency against TRPV4 (IC50 = 16 nM) and an attractive physicochemical properties starting point, with a ChromLogD = 4.5 and LLE = 3.4 (LLE = pIC50 – ChromLogD), especially compared to earlier reported TRPV4 inhibitors.10,16−19
Figure 1.

Spirocarbamate lead. (a) TRPV4 IC50 values are averages of at least two measurements. (b) DMPK properties are averages of measurements taken in at least two Sprague–Dawley rats. (c) Rat PPB fu values are averages of at least two measurements.
The initial focus of our lead optimization effort was to improve the rat PK of 1, which demonstrated a short MRT (0.7 h). MRT is the quotient of volume of distribution (Vdss) and clearance (CL).12,20 It can thus be extended by either increased Vdss or reduced clearance. Our initial strategy was to address the high in vivo clearance (64 mL/min/kg) of 1 by removing metabolic liabilities.
Oxidation adjacent to nitrogen is a well-documented pathway of metabolism and was observed in an in vitro metabolism ID study.21 To address potential sites of metabolism in 1, we targeted four analogs (2–5) specifically aimed at sterically blocking oxidative metabolism (Table 1).22
Table 1. Initial Analogs To Address Clearance.

TRPV4 IC50 is an average of at least two measurements.
DMPK properties are averages of measurements taken in at least two Sprague–Dawley rats.
Rat PPB fu is an average of at least two measurements.
Installing a gemdimethyl group adjacent to the benzimidazole nitrogen (2) had no benefit on MRT (0.3 h) and reduced TRPV4 activity significantly (IC50 > 25 000 nM). An alternative approach to sterically block metabolism at the methylene center through methyl introduction onto the cyclohexane ring (3) similarly had no benefit on MRT (0.5 h). Interestingly, we noted a profound 32-fold TRPV4 potency increase resulting from this change (IC50 = 0.5 nM) over compound 1. The enhanced potency offered by the methyl group (∼2 kcal/mol) is beyond that of lipophilic binding energy, as indicated by LLE increase (1 = 3.4, 3 = 4.7), and is suggestive of a conformational effect, perhaps reducing the conformational flexibility of the cyclohexane or benzimdazole.23,24
Compound 4 was designed to address potential oxidation near the carbamate nitrogen through substitution of the neopentyl group with tert-butyl but had no benefit on MRT (0.5 h) and demonstrated weak TRPV4 potency (IC50 = 5200 nM). In contrast, N-aryl substitution (5) provided a significant increase in both MRT (3.1 h) and oral bioavailability (100%).
The increased MRT of 5 is a result of both reduced clearance and more importantly increased Vdss. The three-fold clearance reduction can be fully attributed to a five-fold increase in plasma protein binding for 5 (fu = 6.5%) compared to 1 (fu = 32%). The unbound clearance (CLu = CL/PPB fu,1 = 200, 5 = 350) indicates that there is minimal change in the free drug clearance between the N-neopentyl (1) and N-aryl (6). In contrast, the volume of distribution increase is very substantial.25 The five-fold increase in PPB fu between the N-neopentyl (1) and N-aryl (5) would have been expected to elicit a proportional reduction in the Vdss.24 Instead, we observed a 1.5-fold increase in Vdss. This indicates that the unbound drug volume of distribution (Vdss/fu, 1 = 8.1, 5 = 61) has increased by eight times and is the underlying driver for the increased MRT. The Vdss increase cannot be attributed to any change in lipophilicity with ChromLogD remaining unchanged at 4.5. Unfortunately, the aryl substitution producing 5 led to a substantial loss in TRPV4 potency compared to 1 (IC50 = 1900 nM vs 16 nM).
Initial attempts to address the potency loss of 5 via N-aryl substitutions led to multiple analogs that improved potency by ∼10-fold (7–10) but did not meet the 10 nM target.
This led us to incorporate the potency-enhancing methyl observed in 3 into the cyclohexane ring of 5 to give 14. This modification led to the expected improvement in potency (IC50 = 96 nM), and the excellent rat PK was maintained (Figure 2). On the basis of its improved potency, we were optimistic that 14 could be improved to reach the target potency at TRPV4 (IC50 < 10 nM) by leveraging the established N-aryl substituent SAR (Table 2).
Figure 2.

Early methyl-cyclohexane N-aryl analog. (a) TRPV4 IC50 values are averages of at least two measurements. (b) DMPK properties are an average of measurements taken in at least two Sprague–Dawley rats.
Table 2. N-Aryl Potency SAR on the 5 Desmethylcyclohexane Core.
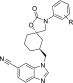
| cmpd | R | hTRPV4 IC50 ± SEM (nM)a | ChromLogD |
|---|---|---|---|
| 5 | p-F (±) | 1,900 ± 610 | 4.5 |
| 6 | H | 820 ± 200 | 4.6 |
| 7 | p-OEt | 140 ± 7.9 | 4.8 |
| 8 | m-OEt | 74 ± 11 | 5.0 |
| 9 | m-Cl | 240 ± 51 | 5.3 |
| 10 | o-Cl | 63 ± 2.9 | 4.3 |
| 11 | o-F | 540 ± 150 | 4.2 |
| 12 | o-Me | 310 ± 29 | 4.6 |
| 13 | o-OMe | 340 ± 85 | 4.2 |
TRPV4 IC50 is an average of at least two measurements.
The combination of N-aryl substituents with the methylcyclohexane core led to many highly potent (<10 nM) compounds (Table 3) but also highlighted that highly divergent SAR existed between the desmethylcyclohexane and methylcyclohexane cores. Para- and meta-ethoxy substitutions (16 and 17) demonstrated excellent potency (IC50 = 5.2 and 1.6 nM), which can be attributed to the potency of corresponding desmethylcyclohexane analogs 7 and 8 (IC50 = 140 and 74 nM) supplemented by the expected ∼30-fold potency increase from the methylcyclohexane core. Meta-chloro substitution (18) demonstrated 50 nM potency, and the minimal and unexpected five-fold potency increase over desmethylcyclohexane 6 revealed that divergent SAR existed between the two structurally similar cores. The disparate SAR was most apparent in compounds bearing ortho-aryl substituents (11–14). Ortho-substituted compounds on the desmethylcyclohexane core (10–13) typically had minimal effects on potency. However, installing the methylcyclohexane core on these ortho-substituted analogs produced activity increases of up to 280-times to give compounds such as 22 with TRPV4 IC50 = 1.2 nM. We noted that the efficiency for the methylcyclohexane core introduction with ortho-substituents also correlated with increased electron donating capacity of the substituent. (σpara Cl = +0.23, F = +0.06, Me = −0.17, OMe = −0.27). We hypothesized that electron donating ortho-substituents elicit a strong conformational effect on the N-aryl rotation, which was highly complementary to the conformational effects elicited by the methylcyclohexane core.21,23,24 Of interest, the compound containing a meta-Cl (18) substituent exhibited a much higher measured lipophilicity (ChromLogD = 5.7) compared to the ortho-Cl (19) analog (ChromLogd = 4.8), which may arise from the proposed N-aryl conformational effects caused by the ortho-substituents.
Table 3. N-Aryl Methylcyclohexane TRPV4 Potency SAR.
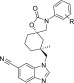
| cmpd | R | hTRPV4 IC50 ± SEM (nM)a | ChromLogDb | LLE | Me effectc |
|---|---|---|---|---|---|
| 14 | p-F | 96 ± 17 | 4.9 | 2.1 | 20× |
| 15 | H | 49 ± 5.9 | 5.0 | 2.3 | 17× |
| 16 | p-OEt | 5.2 ± 0.3 | 5.4 | 2.9 | 26× |
| 17 | m-OEt | 1.6 ± 0.2 | 5.3 | 3.5 | 45× |
| 18 | m-Cl | 48 ± 7.5 | 5.7 | 1.6 | 5× |
| 19 | o-Cl | 2.5 ± 0.3 | 4.8 | 3.8 | 25× |
| 20 | o-F | 5.2 ± 1.0 | 4.9 | 3.4 | 100× |
| 21 | o-Me | 1.7 ± 0.2 | 4.8 | 4.0 | 200× |
| 22 | o-OMe | 1.2 ± 0.2 | 4.6 | 4.3 | 280× |
Unfortunately, the introduction of the potency enhancing alkoxy groups led to reduced MRT values by differing mechanisms as exemplified by 16 and 22 (Table 4). The reduced MRT of 22 (1.0 h) is the result of reduced volume of distribution (1.4 L/kg) with clearance (26 mL/min/kg) remaining similar to para-fluoro 14 (39 mL/min/kg). Apparently, the potency enhancing conformational effect of the ortho-methoxy substituent (22) led to reduced tissue affinity versus 14, which would be difficult to correct while maintaining potency. In contrast, the reduced MRT of 16 (0.9 h) is a function of increased clearance (140 mL/min/kg) with volume of distribution remaining unchanged (7.2 L/kg) relative to para-fluoro 14 (6.5 L/kg). The para-ethoxy group presumably introduced a metabolic liability, but without changing the underlying volume of distribution advantage observed with the early N-aryls 5 and 14.
Table 4. Potent N-Aryl Compounds with Reduced MRTs.

TRPV4 IC50 is an average of at least two measurements.
DMPK properties are an average of measurements taken in two Sprague–Dawley rats.
Rat PPB fu is an average of at least two measurements.
Consistent with this conclusion, we could reduce clearance and restore the MRT of 16 via the introduction of heterocycles (23–25), which are thought to attenuate oxidative metabolism of the aryl ring (Table 5).26
Table 5. Rat PK SAR for Pyridyl and Diazine Analogs.
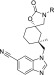

TRPV4 IC50 is an average of at least two measurements.
DMPK properties are an average of measurements taken in two Sprague–Dawley rats.
Rat PPB fu is an average of at least two measurements.
The 2-ethoxy pyridyl (23) improved potency (IC50 = 3.1 nM), lowered clearance (24 mL/min/kg), and enhanced MRT (1.6 h) versus 16, which prompted efforts to incorporate an additional nitrogen to produce alkoxy diazines. Ethoxy pyridazine (24) led to a further reduction in clearance (14 mL/min/kg) and increase in MRT (1.9 h) but also reduced the TRPV4 potency (IC50 = 60 nM). In contrast, the ethoxy pyrazine (25) demonstrated robust TRPV4 potency (IC50 = 4.2 nM) and a long MRT (5.2 h) but with similar clearance (17 mL/min/kg) to 23 and 24. Overall, our conclusion is that para-ethoxy substitution (16) enhanced or introduced arene oxidation and that this can be attenuated or eliminated by pyridyl nitrogen insertion. We hypothesized that the nitrogen containing heterocycles may be reducing metabolism either through reduced cytochrome P450 binding affinity or by reducing aryl oxidative capacity.26
Having established our target TRPV4 potency and rat PK in compounds such as 25 (Figure 3), we also noted that the replacement of phenyl (16) with pyrazine (25) dramatically lowered cLogP from 4.5 to 3.1. Therefore, we progressed 25 to candidate selection, but it was subsequently surpassed by a superior compound, which resulted from lowering measured lipophilicity.
Figure 3.

Potent TRPV4 inhibitor with desirable rat PK. (a) TRPV4 IC50 is an average of at least two measurements. (b) DMPK properties are an average of measurements taken in two Sprague–Dawley rats. (c) Rat PPB fu is an average of at least two measurements.
Interestingly, the pyrazine had no effect on measured lipophilicity as ChromLogD was nearly identical for 16 (5.4) and 25 (5.5). We sought to reduce the measured lipophilicity recognizing that this value was more relevant for positively impacting off-target activity and solubility.14 Specifically for this series we had noted that the unbound clearance of 25 was inferior to 1 and 5, both of which demonstrated lower ChromLogD. We hypothesized that by decreasing lipophilicity we could reduce unbound clearance, presumably through attenuating binding affinity to metabolizing enzymes. An improvement in unbound clearance while maintaining potency and MRT is highly desirable as it leads to increased free drug concentration that lowers the overall clinical dose.27
We modulated lipophilicity by varying the pyrazine substituent of 25 (Table 6). Contraction from ethoxy (25) to methoxy (26) reduced ChromLogD (5.5 to 4.8) and CLu (460 to 400) but at the expense of potency (IC50 = 14 nM). Exchange of ethoxy for a methyl substituent (27) reduced ChromLogD to 4.2 but again led to modest TRPV4 potency (IC50 = 60 nM). Target potency could be increased through larger alkyl groups such as cyclo-propyl (28) and tert-butyl (29) to 1.5 nM and 1.2 nM, respectively, with the former demonstrating slightly improved CLu relative to 25. The alkyl substituent data (27–29) illustrated the importance of lipophilicity for obtaining excellent TRPV4 potency but also demonstrated that a potency/lipophilicity plateau had been reached since 29 gave no activity increase over 28. The latter observation led us to replace one of tert-butyl methyls in 29 with a hydroxy group to give isopropyl alcohol (30), which demonstrated excellent TRPV4 potency (IC50 = 1.8 nM) along with significantly reduced ChromLogD (4.0) (Figure 4). The reduced lipophilicity led to a six-fold improvement in CLu compared to 25. We hypothesize that the hydroxy may be internally hydrogen-bonded to the adjacent pyrazine nitrogen when bound to TRPV4, thereby masking some its polarity and leading to the excellent potency. However, the polarity of the hydroxy group is apparently dynamically exposed in aqueous solvent, leading to its reduced ChromLogD and unbound clearance.28
Table 6. Aryl Substituent Lipophilicity SAR.
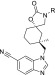

TRPV4 IC50 is an average of at least two measurements.
DMPK properties are an average of measurements taken in two Sprague–Dawley rats.
Rat PPB fu is an average of at least two measurements. CLu = CL × PPB fu.
Figure 4.

GSK2798745 TRPV4 oral candidate. (a) TRPV4 IC50 is an average of at least two measurements. (b) DMPK properties are an average of measurements taken in two Sprague–Dawley rats. (c) Rat PPB fu is an average of at least two measurements.
Overall, compound 30 demonstrated excellent TRPV4 potency, owing largely to the methyl-cyclohexane core, and showed exceptional rat PK. The long MRT (4.9 h) and low unbound clearance (81) could be attributed to the metabolic stability of the N-aryl pyrazine and low overall lipophilicity of the compound. Compound 30 (designated GSK2798745) demonstrated efficacy in an in vivo rat PK/PD model of pulmonary edema, excellent TRP selectivity (M5, A1, C3, and C6 > 25 μM), and an adequate preclinical safety profile that enabled its candidate selection and subsequent progression to clinical trials.9,29
The spirocarbamate TRPV4 inhibitors were accessed via the synthetic method described in Supporting Information Schemes 1 and 2.30
In summary, a lead optimization effort beginning with spirocarbamate 1 identified GSK2798745 (30), a clinical candidate for the inhibition of TRPV4. The lead optimization began by successfully addressing the poor rat PK of the lead but resulted in a significant reduction in TRPV4 potency. The TRPV4 potency loss was addressed primarily through the introduction of a highly efficient methyl group. This was then followed by subtle refinement of TRPV4 potency, rat pharmacokinetics, and physiochemical properties to achieve a desirable oral candidate profile. GSK2798745 is a clinical candidate that is being used to assess the role of TRPV4 in human diseases.
Acknowledgments
We thank all members of the TRPV4 Program Team at GlaxoSmithKline for their contribution. We thank Remy Gebleux and Lina Ding for synthetic contributions; J. P. Jarworski and Michael Klein for TRPV4 FLIPR data; and Katrina Rivera, Stephen Eisennagel, and Kevin Baptiste for rat PK data.
Glossary
Abbreviations
- ChromLogD
chromatographic logD at pH 7.4
- cmpd
compound
- CL
clearance
- CLu
clearance of unbound drug
- FLIPR
fluorometric imaging plate reader
- IV
intravenous
- Fpo
oral bioavailability
- MRT
mean residence time
- PK
pharmacokinetic
- PPB Fu
plasma protein binding fraction unbound
- SAR
structure–activity relationship
- TRPV
transient receptor potential vanilloid
- Vdss
volume of distribution at steady state
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00274.
Compound synthesis and spectroscopic characterization of 1, 16, 25, and 30 (PDF)
All studies involving the use of animals were conducted after review by the GlaxoSmithKline (GSK) Institutional Animal Care and Use Committee and in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals.
The authors declare the following competing financial interest(s): All authors are current or past employees of GlaxoSmithKline and/or stockholders of GlaxoSmithKline.
Supplementary Material
References
- Voets T.; Prenen J.; Vriens J.; Watanabe H.; Janssens A.; Wissenbach U.; Bödding M.; Droogmans G.; Nilius B. Molecular determinants of permeation through the cation channel TRPV4. J. Biol. Chem. 2002, 277, 33704–33710. 10.1074/jbc.M204828200. [DOI] [PubMed] [Google Scholar]
- Güler A. D.; Lee H.; Lida T.; Shimizu I.; Tominaga M.; Caterina M. Heat-evoked activation of the ion channel, TRPV4. J. Neurosci. 2002, 22, 6408–6414. 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strotmann R.; Harteneck C.; Nunnenmacher K.; Schultz G.; Plant T. D. OTRPC, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2000, 2, 695–702. 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- Delany N. S.; Hurle M.; Facer P.; Alnadaf T.; Plumpton C.; Kinghorn I.; See C. G.; Costigan M.; Anand P.; Woolf C. J.; Crowther D.; Sanseau P.; Tate S. N. Identification and characterization of a novel human vanilloid receptor-like protein, VRL-2. Physiol. Genomics 2001, 4, 165–174. 10.1152/physiolgenomics.2001.4.3.165. [DOI] [PubMed] [Google Scholar]
- Thorneloe K. S.; Sulpizio A. C.; Lin Z.; Figueroa D. J.; Clouse A. K.; McCafferty G. P.; Chendrimada T. P.; Lashinger E. S.; Gordon E.; Evans L.; Misajet B. A.; Demarini D. J.; Nation J. H.; Casillas L. N.; Marquis R. W.; Votta B. J.; Sheardown S. A.; Xu X.; Brooks D. P.; Laping N. J.; Westfall T. D. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)- 1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J. Pharmacol. Exp. Ther. 2008, 326, 432–442. 10.1124/jpet.108.139295. [DOI] [PubMed] [Google Scholar]
- Jian M. Y.; King J. A.; Al-Mehdi A. B.; Liedtke W.; Townsley M. I. High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am. J. Respir. Cell Mol. Biol. 2008, 38, 386–392. 10.1165/rcmb.2007-0192OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez D. F.; King J. A.; Weber D.; Addison E.; Liedtke W.; Townsley M. I. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ. Res. 2006, 99, 988–995. 10.1161/01.RES.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willette R. N.; Bao W.; Nerurkar S.; Yue T. L.; Doe C. P.; Stankus G.; Turner G. H.; Ju H.; Thomas H.; Fishman C. E.; Sulpizio A.; Behm D. J.; Hoffman S.; Lin Z.; Lozinskaya I.; Casillas L. N.; Lin M.; Trout R. E.; Votta B. J.; Thorneloe K.; Lashinger E. S.; Figueroa D. J.; Marquis R.; Xu X. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J. Pharmacol. Exp. Ther. 2008, 326, 443–452. 10.1124/jpet.107.134551. [DOI] [PubMed] [Google Scholar]
- Thorneloe K. S.; Cheung M.; Bao W.; Alsaid H.; Lenhard S.; Jian M.-Y.; Costell M.; Maniscalco-Hawk K.; Krawiec J. A.; Olzinski A.; Gordon E.; Lozinskaya I.; Elefante L.; Qin P.; Matasic D. S.; James C.; Tunstead J.; Donovan B.; Kallal L.; Waszkiewicz A.; Vaidya K.; Davenport E. A.; Larkin J.; Burgert M.; Casillas L. N.; Marquis R. W.; Ye G.; Eidam H. S.; Goodman K. B.; Toomey J. R.; Roethke T. J.; Jucker B. M.; Schnackenberg C. G.; Townsley M. I.; Lepore J. J.; Willette R. N. An Orally Active TRPV4 Channel Blocker Prevents and Resolves Pulmonary Edema Induced by Heart Failure. Sci. Transl. Med. 2012, 4, 159ra148. 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- Hilfiker M. A.; Hoang T. H.; Cornil J.; Eidam H.; Matasic D.; Roethke T. J.; Klein M.; Thorneloe K. S.; Cheung M. Optimization of a Novel Series of TRPV4 Antagonists with In vivo Activity in a Model of Pulmonary Edema. ACS Med. Chem. Lett. 2013, 4, 293–296. 10.1021/ml300449k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward K. W.; Smith B. R. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetics parameters from rat, dog, and monkey to humans. I. Clearance. Drug metabolism and disposition 2004, 32 (6), 603–611. 10.1124/dmd.32.6.603. [DOI] [PubMed] [Google Scholar]
- Ward K. W.; Smith B. R. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetics parameters from rat, dog, and monkey to humans. II. Volume of distribution and mean residence time. Drug metabolism and disposition 2004, 32 (6), 612–619. 10.1124/dmd.32.6.612. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 2011, 16, 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Eidam H.; Cheung M.; Hammond M. Unpublished work.
- Skerratt S. E.; Mills J. E.; Mistry J. Identification of false positives in “HTS hits to lead”: the application of Bayesian models in HTS triage to rapidly deliver a series of selective TRPV4 antagonists. MedChemComm 2013, 4, 244–251. 10.1039/C2MD20259J. [DOI] [Google Scholar]
- Duncton M. A. J.Small Molecule Agonists and Antagonists of TRPV4. TRP Channels as Therapeutic Targets; Szallasi A., Ed., 2015; pp 205–219. [Google Scholar]
- Fabian V.; Duncton M. A. J. TRPV4 Agonists and Antagonists. Curr. Top. Med. Chem. 2011, 11, 2216–2226. 10.2174/156802611796904861. [DOI] [PubMed] [Google Scholar]
- Cheung M.; Bao W.; Behm D. J.; Brooks C. A.; Bury M. J.; Dowdell S. E.; Eidam H. S.; Fox R. M.; Goodman K. B.; Holt D. A.; Lee D.; Roethke T. J.; Willette R. N.; Xu X.; Ye G.; Thorneloe K. S. Discovery of GSK2193874: an Orally Active, Potent and Selective Blocker of Transient Receptor Potential Vanilloid 4. ACS Med. Chem. Lett. 2017, 8, 549–554. 10.1021/acsmedchemlett.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Jusko W. Application of mean residence-time concepts to pharmacokinetics systems with noninstantaneous input and nonlinear elimination. Pharm. Res. 1989, 6 (1), 4–12. 10.1023/A:1015883131875. [DOI] [PubMed] [Google Scholar]
- Davis A.; Ward S. E.. The Handbook of Medicinal Chemistry Principles and Practice; Royal Society of Chemistry, 2015. [Google Scholar]
- Compounds 2, 4, and 5 were prepared as racemic trans mixtures for ease of synthesis and profiling. The eutomer of compound 5 demonstrated hTRPV4 IC50 = 1600 nM.
- Cernak T.; Schonherr H. Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem., Int. Ed. 2013, 52, 12256–12267. 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- Leung C. S.; Leung S. S. F.; Tirado-Rives J.; Jorgensen W. L. Methyl effects on protein-ligand binding. J. Med. Chem. 2012, 55, 4489–4500. 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58, 5691–5698. 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- Smith D. A.Metabolism, Pharmacokinetics and Toxicity of Functional Groups, Impact of Chemical Building Blocks on ADMET; RSC, 2010. [Google Scholar]
- Gunaydin H.; Altman M. D.; Ellis J. M.; Fuller P.; Johnson S. A.; Lahue B.; Lapointe B. Strategy for Extending Half-life in Drug Design and Its Significance. ACS Med. Chem. Lett. 2018, 9, 528–533. 10.1021/acsmedchemlett.8b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi Sebastiano M.; Doak B. C.; Backlund M.; Poongavanam V.; Over B.; Ermondi G.; Caron G.; Matsson P.; Kihlberg J. Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. 10.1021/acs.jmedchem.8b00347. [DOI] [PubMed] [Google Scholar]
- Goyal N.; Skrdla P.; Schroyer R.; Kumar S.; Fernando D.; Oughton A.; Norton N.; Sprecher D. L.; Cheriyan J. Clinical pharmacokinetics, safety, and tolerability of a novel, first-in-class TRPV4 ion channel inhibitor, GSK2798745, in healthy and heart failure subjects. Am. J. Cardiovasc. Drugs 2019, 19, 335–342. 10.1007/s40256-018-00320-6. [DOI] [PubMed] [Google Scholar]
- For general synthesis of spirocarbamate compounds see:Brooks C. A.; Cheung M.; Goodman K. B.; Hammond M.. TRPV4 antagonists, Patent WO2012174340 A1; GlaxoSmithKline, 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.