

Abstract
A series of pyrrolidine amino nitrile DPP1 inhibitors have been developed and characterized. The S2 pocket structure–activity relationship for these compounds shows significant gains in potency for DPP1 from interacting further with target residues and a network of water molecules in the binding pocket. Herein we describe the X-ray crystal structures of several of these compounds alongside an analysis of factors influencing the inhibitory potency toward DPP1 of which stabilization of the water network, demonstrated using Grand Canonical Monte Carlo simulations and free energy calculations, is attributed as a main factor.
Keywords: Cathepsin C, dipeptidyl peptidase I, Cathepsin C inhibitor, DPP1 inhibitor, structural studies, water mediated interaction, Grand Canonical Monte Carlo
Dipeptidyl peptidase I (DPP1, Cathepsin C, EC:3.4.14.1) is a lysosomal cysteine protease that plays a key role in the activation of the pro-inflammatory neutrophil serine proteases (NSPs) neutrophil elastase, cathepsin G, and proteinase-3.1,2 DPP1 is an exopeptidase that removes dipeptide moieties from the N-terminus of the pro-form of the target proteins, leading to their activation. This subsequently triggers degradation of the extracellular matrix, which may lead to tissue damage and inflammation. Knockout studies in mice and pharmacological inhibition using DPP1 inhibitors have confirmed a central role for DPP1 in the activation of the NSPs and strongly support the therapeutic strategy of targeting DPP1 inhibition in diseases that carry a high neutrophilic burden, such as chronic obstructive pulmonary disease.3−5
A published X-ray structure reveals that human DPP1 is a tetrameric enzyme where each of the identical subunits comprise three independent protein chains: the heavy chain, the light chain, and the exclusion domain.6 The active site that harbors the reactive cysteine residue (Cys 234) is located at the interface of the three chains. DPP1 has an open S1 pocket positioned at the entrance of the active site, while the S2 pocket is large but enclosed within the protein and mainly hydrophobic in nature. At the base of the S2 pocket is a chloride ion that is required for activity.7 The exclusion domain blocks access to substrates beyond the S2 site. The side chain of Asp 1 in the exclusion domain governs substrate recognition by interacting with the terminal α-amino group of the substrate, as illustrated by the crystal structure of a substrate inhibitor complex.8,9 The architecture of the substrate site allows binding of a range of different groups and is thus consistent with the broad DPP1 substrate specificity.10
The potential therapeutic utility and medicinal chemistry of DPP1 has been reviewed, as have attempts to develop inhibitors of this enzyme.1,11 Most DPP1 inhibitors reported to date are believed to interact through covalent bond formation with the active site cysteine. Despite the relatively high interest in DPP1, there are relatively few reports describing characterization of protein–inhibitor complexes by X-ray crystallography.8,9,12−15
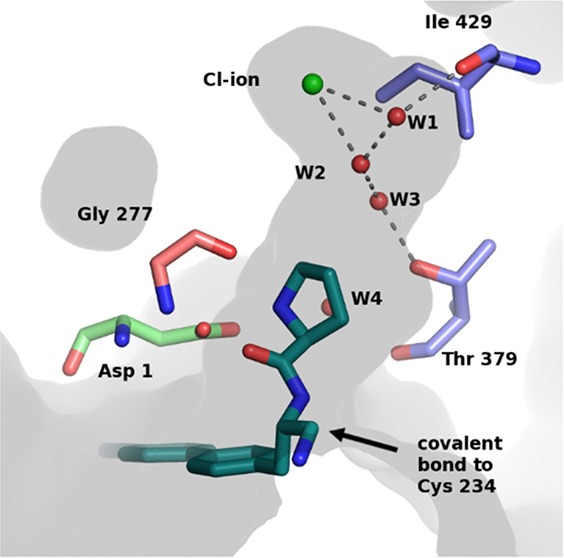
As described,12,13 AstraZeneca has developed potent selective and metabolically stable amino nitrile based DPP1 inhibitors, exemplified in Chart 1. Furthermore, a piperidinyl substituted series has been reported, Figure 1.12 Exploration in this series identified a potential hydrogen bond to the hydroxyl group of Thr 379. Targeting this interaction with a 4S-hydroxyl moiety on the piperidine ring offered a substantial potency gain for compound 1 compared with the unsubstituted compound 2. A crystal structure shows that the added hydroxyl group is within hydrogen bond distance of Thr 379,12Figure 1.
Chart 1. Examples of Previously Reported Amino Nitrile Based DDP1 Inhibitors.

Figure 1.
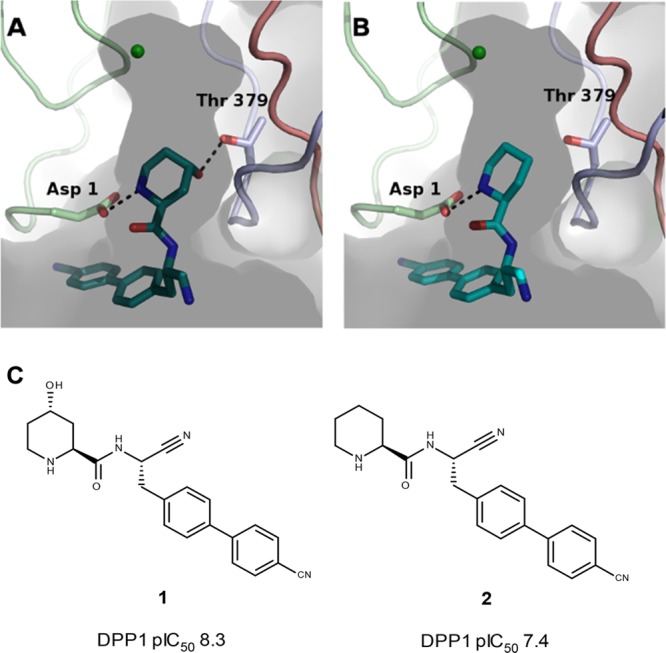
(A) X-ray structure of compound 1 binding to the active site of DPP1, PDB: 4CDF. (B) X-ray structure of compound 2 binding to the active site of DPP1, PDB 4CDD. Distinct colors denote individual chains of DPP1. (C) Chemical structure of compounds 1 and 2.
A second related series of pyrrolidine amino nitrile compounds, exemplified by compound 3, has subsequently been explored, Table 1. As with previous DPP1 inhibitors,12 changing the group occupying the S1 pocket from 4-biphenyl to 4′-cyano-biphenyl led to an increase in potency, comparing compound 3 to compound 4. Unlike in the piperidine scaffold, the addition of the equivalent (3R) hydroxy moiety had no effect on DPP1 potency, as is observed between compounds 4 and 5. However, the 3S-hydroxy isomer, compound 6, showed increased potency when compared to these two. Based on our perceived knowledge of the DDP1 S2 pocket, the 3S-hydroxy functionality points away from Thr 379 and into the S2 pocket, suggesting a marked difference in structure–activity relationship compared to the piperidine compounds. This observation motivated us to pursue a detailed analysis of the interactions within the S2 pocket of DPP1.
Table 1. Structure–Activity Relationship for Substituted Pyrrolidine Aminonitriles.

| compound | R1 | R2 | R3 | DPP1 enzyme, pIC50a | calc. Log D | LLEc | pKacalcd |
|---|---|---|---|---|---|---|---|
| 3 | H | OH | H | 6.8b | 1.8 | 5.0 | 7.6 |
| 4 | H | OH | CN | 7.3b | 1.3 | 6.0 | 7.6 |
| 5 | H | H | CN | 7.3 | 1.8 | 5.5 | 8.4 |
| 6 | OH | H | CN | 8.2 | 1.3 | 6.9 | 7.6 |
| 7 | H | F | CN | 6.2 | 2.3 | 3.9 | 6.6 |
| 8 | F | H | CN | 8.8 | 2.3 | 6.5 | 6.6 |
| 9 | F | F | CN | 7.1 | 3.1 | 4.0 | 4.8 |
| 10 | MeS | H | CN | 9.4 | 2.8 | 6.6 | 7.4 |
pIC50 determined by cleavage of fluorescence substrate H-Gly-Arg-AFC, unless stated.
H-Gly-Arg-AMC substrate was used. The good correlation, R2 = 0.85, between assays run with different substrates allows direct comparison (data not shown).
LLE = pIC50 – calculated LogD.17
pKa was calculated using ACD/Laboratories software version 2015 (see www.acdlabs.com) applying an internal correction library.
In this study, compounds 3–10 were synthesized. The preparation of compound 3 has already been described.16 Compounds 4–10 were prepared as outlined in Scheme 1.
Scheme 1. Synthetic Route of Compounds 4 to 10.

Reagents and conditions: (a) 11b,c,g, HOPO, EDCI, DMF; (b) 11d–f, T3P, NEt3, DMF; (c) HCO2H, 50 °C; (d) 11a, TBTU, NEt3, DMF; (e) Burgess’ reagent, DCM. For compound 4, coupling of (2S,4R)-4-tert-butoxy-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (11a with (S)-2-amino-3-(4′-cyano-[1,1′-biphenyl]-4-yl)propanamide 14(16) gave intermediate amide 15. Dehydration of the amide 15 using Burgess’ reagent, gave the nitrile 13a, and subsequent global deprotection using formic acid gave the target compound 4. For compounds 5–10, starting from the appropriate pyrrolidine analogue 11b–g, coupling with 4-[4-[(2S)-2-amino-2-cyano-ethyl] phenyl] benzonitrile 12(13) using either conditions (a) or (b) gave the intermediate Boc-protected nitriles 13b–g. Deprotection using formic acid gave the target compounds 5–10.
The compounds were characterized in an enzymatic assay, previously described, based on cleavage of a synthetic substrate, see Table 1.12
Crystal structures were generated for compounds 3, 5, 6, 8, and 10 in complex with DPP1 (Figure 2) and show that the binding mode for the pyrrolidine-derived amino nitrile compound series is similar to that of the piperidine amino nitriles. As expected, the key hydrogen bond interactions between the amine nitrogen and the backbone atoms of Gly 277, Asn 380, and to the side chain of Asp 1 are conserved with respect to piperidinyl compounds, 1 and 2, as is the covalent bond to Cys 234 (Figure S1). Interestingly, an S2 pocket water network was observed to vary significantly across the structures in this series.
Figure 2.
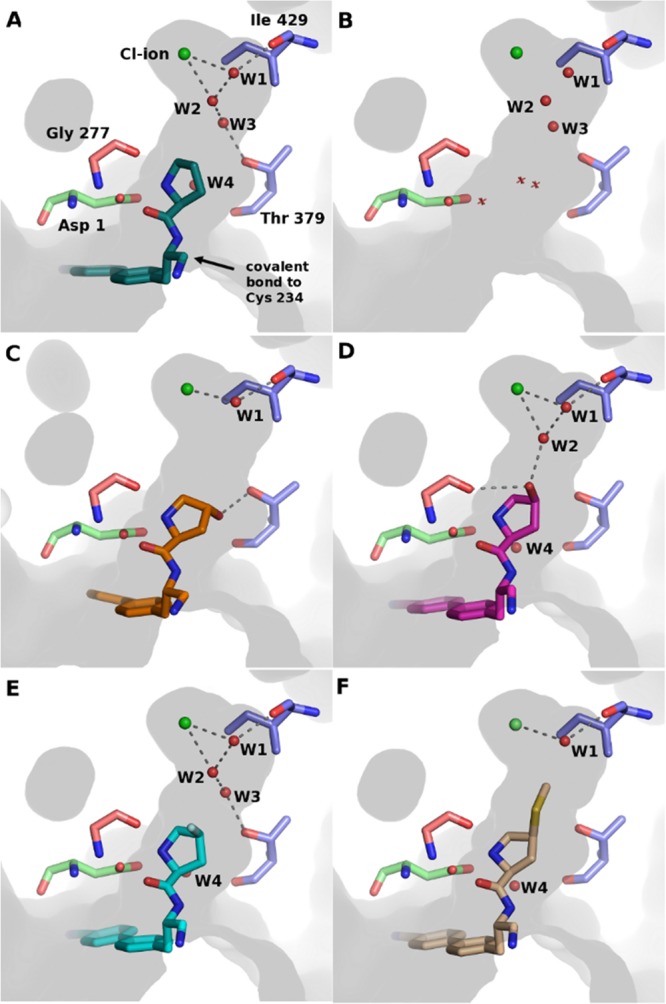
Crystal structures of DPP1 in complex with ligands and the position of the identified waters in the deep part of the S2 pocket. The amino acids are colored according to the DPP1 chain: green = exclusion domain, blue = heavy chain, salmon = light chain. (A) Compound 5 in complex with DPP1. (B) Active site of DPP1 in absence of inhibitor (PDB: 1K3B), water positions discussed herein are depicted as spheres, while additional waters are displayed as crosses. (C) DPP1 in complex with compound 3, (D) compound 6, (E) compound 8, and (F) compound 10.
The crystal structure of the unsubstituted pyrrolidine compound 5 in complex with DPP1 revealed a network of three water molecules (W1–W3) at the top of the S2 pocket (Figure 2a). Water W1 bridges from Ile 429 to the Cl–, while water W2 is positioned to form hydrogen bonds with the Cl– and water W3, which in turn is within hydrogen bonding distance of Thr 379. Waters have previously been identified in these positions for the apo structure of human DDP1 (PDB: 1K3B) Figure 2b.6 Similarly, Mølgaard et al. concluded that a network of water molecules was conserved in the deep S2 pocket.8 The X-ray structure of compound 4 has not been solved; however, the structure of the close analogue 3 shows that the 4R-hydroxyl substitution of compound 3 is well positioned for a hydrogen bond to Thr 379 (Figure 2c) as was observed for piperidine compound 1. While a similar substitution on the piperidine compound 1 results in a 10-fold potency gain, suggesting that a productive interaction with Thr 379 side-chain has been formed, the 4R-hydroxyl substitution on compound 4 does not increase the potency, see Table 1. We hypothesized that the energetics of this network of water molecules might be contributing to the observed difference in SAR between the two series. We utilized Grand Canonical Monte Carlo (GCMC) simulations to explore the role of these waters. GCMC can locate waters within protein binding sites and estimate their binding affinities and is especially suited to study networks of waters.18−21
The simulations correctly identify the location of these three waters, while giving insight into their cooperativity and energetics as a function of the bound ligand. The free energy for sequentially adding waters W1, W2, and W3 to DPP1 in the presence of ligand is shown in Table 2. The simulations demonstrate that the binding free energy of W1 is independent of the ligand, whereas the free energy of adding W2 to the system varies significantly in the presence of the different ligands. Cumulative addition of W3 to the system is unfavorable in all cases; however, the net free energy of the water network is still favorable. Furthermore, the simulations indicate that the water network is most stabilized in the presence of ligand 6.
Table 2. Accumulative Free Energies for Sequential Addition of W1-3 to DPP1, Calculated Using GCMCa.
| compoundb | ΔGW1 | ΔGW2 | ΔGW3 |
|---|---|---|---|
| 4 | –7.56 | –10.56 | –5.47 ± 0.25 |
| 5 | –7.75 | –11.21 | –7.60 ± 0.24 |
| 6 | –7.63 | –13.48 | –10.74 ± 0.10 |
| 7 | –6.80 | –6.46 | –2.82 ± 0.30 |
| 8 | –6.99 | –6.71 | –4.39 ± 0.10 |
| 9 | –6.90 | –6.53 | –3.68 ± 0.21 |
| 10 | –7.74 ± 0.28 | ||
Energies are in kcal/mol. Errors represent the uncertainty in the free-energy for adding W1–3 (for ligands 4–9) or W1 (ligand 10) to DPP1, calculated as the standard deviation from 1000 bootstrap samples.
Note that the protonation state of the pyrrolidine N is charged for compounds 4–6 and 10, and neutral for compounds 7, 8, and 9.
Having demonstrated that the energetics of the water network do differ significantly across ligands, we used alchemical free energy calculations19,20 in conjunction with the GCMC simulations to study the effect of the water network on the ligand binding free energies. These were used to construct a free energy cycle, shown in Figure 3.
Figure 3.

Free energy cycles for the conversion of 5 to 4 and 6. Green arrows highlight the relative difference in binding affinity calculated using the Replica Exchange Thermodynamic Integration method (RETI),23 while orange arrows highlight the legs calculated using GCMC. The blue dot on the top leg indicates that the perturbation was performed in the presence of waters W1 and W2. The free-energy cycle closure error is shown in the black box, calculated using the Bennett Acceptance Ratio (BAR) method.
The free energy transformation from 5 to 4, in the presence of DPP1, was calculated in the absence of water and in the presence of waters W1 and W2. A separate free energy transformation from 5 to 4 was also performed in bulk solvent, which allows the calculation of the relative binding affinity between the two ligands as the difference between the protein and solvent legs. As shown in the left panel of Figure 3, the relative binding affinity is predicted to be slightly favorable in both cases suggesting that the formation of an H-bond to Thr 379 brings only a small affinity gain for these compounds.
When viewed alongside the GCMC simulations, it becomes clear that any minor benefit from making an H-bond to Thr 379 is counteracted by a slight destabilization in the water network consistent with 4 and 5 being equipotent. The presence of the hydroxyl functionality in the opposite configuration, i.e., the 4S-hydroxypyrrolidine, compound 6, results in a 10-fold increase in DPP1 potency. The 4S-hydroxyl substituent of 6 is positioned to donate a hydrogen bond to the main-chain carbonyl of Gly 277 and accept a hydrogen bond from water W2 (Figure 2d). The GCMC simulations show that the binding affinity of W2 is significantly more favorable for 6 compared to 4 and 5. The analogous free energy simulations performed for the transformation from 5 to 6 were found to be significantly more favorable in the presence than in the absence of the water network, highlighted in the right panel of Figure 3. This provides evidence for the increased potency of 6 compared to 5, arising primarily from the stabilization of the network of waters. Pleasingly, for both the 5 to 4 and 6 to 5 transformations, the difference between the alchemical perturbation legs and the GCMC simulations was less than 0.6 kcal/mol, demonstrating good convergence and lending confidence to the calculations.
The influence of the bound water molecules on the potency of the 4R- and 4S-fluoro substituted pyrrolidine compounds, compounds 7 and 8, respectively, was subsequently investigated. This pair showed a similar but more pronounced effect compared to the hydroxyl-substituted compounds, with compound 8 being 32-fold more potent than compound 5 in the DPP1 enzyme assay, while the 4R-fluoro substituted analogue 7 lost nearly 10-fold in potency, compared to 5. The 4,4-difluoro analogue, compound 9, was prepared and found to have a similar potency to the unsubstituted pyrrolidine, compound 5, Table 1.
The crystal structure of the 4S-fluoro-analogue 8 confirmed that it bound in a conformation very similar to that of 6 (Figure 2e). The fluoro substituent of 8 cannot hydrogen-bond to Gly 277 as it lacks a donor, and it is slightly too far away to form a hydrogen-bond to water W2. Thus, water W2 is free to hydrogen-bond with water W3, maintaining the network seen in the structure of compound 5.
Comparing compounds 4–6 to 7–9 from a computational perspective is challenging. Based upon the pKa calculations shown in Table 1 compounds 7–9 are expected to be neutral in a buffered protein environment, while 4–6 are expected to be positively charged. The change in pyrrolidine protonation state means that alchemical transformations between the species cannot be easily performed, while there is no justification for simulating 5 as neutral or 7/8 as charged based upon pKa. Applying an electrostatic correction post simulation offers one strategy to modeling ligands with different charge states, although such corrections are challenging to perform.22 Instead, we sought to understand the SAR within the 7–9 subseries.
GCMC simulations reveal the energies for adding W1 and W2 to the complexes of 7, 8, and 9 are all comparable, while some changes are seen in the stability of W3. A large destabilization effect (+3.64 kcal/mol) is predicted in the presence of 7, which has a fluorine in the 4R-position, while 8, with fluorine in the opposite configuration, shows a much-reduced destabilization effect (+2.32 kcal/mol).
The stabilization effect for 9 was +2.85 kcal/mol, broadly the average destabilization effect of 7/8. While these simulations go somewhat to explaining the SAR, we also considered the conformations adopted by the fluorinated pyrrolidines.
Low-Mode MD was performed upon compounds 7, 8, and 9 to identify the low-energy conformations and compare them to the crystallographic bioactive conformation. Distinct differences were seen between the three; 91% of the low-energy conformations of 8 was found to be within 0.2 Å RMSD of the bioactive conformation, while only 41% of the conformations for 7 was within 0.2 Å. Compound 9 showed 75% of the conformations within 0.2 Å. The lowest-energy conformation of 7 places the F axially (Figure 4), which is incompatible with the bioactive conformation where it is equatorial. An increased entropic cost is therefore required to adopt the bioactive conformations for 7 and 9 compared to 8, offering a further explanation for their decrease in potency alongside the GCMC evidence.
Figure 4.
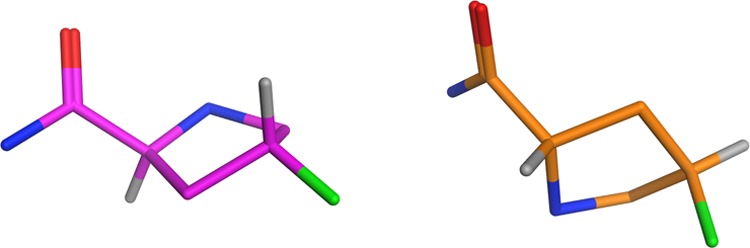
Lowest energy conformation for the pyrrolidine ring of compound 7 (right) compared to the bioactive conformation (left).
We noted in the literature that the presence of a 4S-methylsulfanylpyrrolidine in the S2 pocket was reported to increase potency by approximately 150-fold compared to the unsubstituted analogue and decided to make the corresponding amino nitrile compound 10 for comparison.24 Gratifyingly we observed a substantial (126-fold) increase in potency compared to unsubstituted compound 5. The crystal structure with 10 shows that the methylsulfanyl group is buried deep in the hydrophobic S2 pocket where it displaces waters W2 and W3 and makes additional hydrophobic contacts (Figure 2f). The methyl is positioned 3.7 Å from water W1, too far to form an additional hydrogen bond. As seen in Table 2, only W1 was observed during the GCMC simulations, consistent with the crystallographic evidence and the displacement of W2 and W3. The improvement in potency is likely due to the entropy gain from releasing two waters to bulk solvent and replacing them with a large, lipophilic group. The polarizable sulfur and delta positive methyl hydrogens may contribute to the charge stabilization of the ligand amine and chloride ion in the absence of water W2.
We have studied a set of pyrrolidine amino nitrile DPP1 inhibitors for which small modifications have a relatively large and at a first glance, unexpected effect on the potency. The modifications to the molecules are localized to the part which binds in a deep, buried, hydrophobic pocket of DPP1 harboring a chloride ion. Interrogation of the 3D crystal structures and the application of GCMC suggests that the presence of the water network in the S2 pocket, in particular water W2 linking the charged groups of the ligand and chloride ions, plays a significant role in the SAR of this series of inhibitors. GCMC is likely to be of high use in similar case studies where networks of waters are in close proximity to ligands.
Acknowledgments
We thank Hongwei Guo for establishing the crystallization protocol for DPP1. We also thank Martin Packer and Kurt Pike for useful discussions and reviewing the manuscript.
Glossary
ABBREVIATIONS
- DPP1
dipeptidyl peptidase I
- NSPs
neutrophil serine proteases
- LLE
lipophilic ligand efficiencies
- SAR
structure–activity relationship
- GCMC
Grand Canonical Monte Carlo
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00261.
Experimental details for chemistry, macromolecular X-ray crystallography, and computational analyses (PDF)
Author Present Address
⊥ Medicinal Chemistry, Oncology, R&D, AstraZeneca, Cambridge, UK.
Author Present Address
# Astex Pharmaceuticals, Cambridge, UK.
The authors declare no competing financial interest.
Supplementary Material
References
- Guay D.; Beaulieu C.; Percival M. D. Therapeutic utility and medicinal chemistry of cathepsin C inhibitors. Curr. Top. Med. Chem. 2010, 10, 708–716. 10.2174/156802610791113469. [DOI] [PubMed] [Google Scholar]
- Korkmaz B.; Caughey G. H.; Chapple I.; Gauthier F.; Hirschfeld J.; Jenne D. E.; Kettritz R.; Lalmanach G.; Lamort A.-S.; Lauritzen C.; Łȩgowska M.; Lesner A.; Marchand-Adam S.; McKaig S. J.; Moss C.; Pedersen J.; Roberts H.; Schreiber A.; Seren S.; Thakker N. S. Therapeutic targeting of cathepsin C: from pathophysiology to treatment. Pharmacol. Ther. 2018, 190, 202–236. 10.1016/j.pharmthera.2018.05.011. [DOI] [PubMed] [Google Scholar]
- Methot N.; Rubin J.; Guay D.; Beaulieu C.; Ethier D.; Reddy T. J.; Riendeau D.; Percival M. D. Inhibition of the activation of multiple serine proteases with a cathepsin C inhibitor requires sustained exposure to prevent pro-enzyme processing. J. Biol. Chem. 2007, 282, 20836–20846. 10.1074/jbc.M702615200. [DOI] [PubMed] [Google Scholar]
- Gardiner P.; Wikell C.; Clifton S.; Shearer J.; Benjamin A.; Peters S. A. Neutrophil maturation rate determines the impact of dipeptidyl peptidase 1 inhibition on neutrophil serine protease activity. Br. J. Pharmacol. 2016, 173, 2390–2401. 10.1111/bph.13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino C.; Hamon Y.; Croix C.; Lamort A. S.; Dallet-Choisy S.; Marchand-Adam S.; Lesner A.; Baranek T.; Viaud-Massuard M. C.; Lauritzen C.; Pedersen J.; Heuze-Vourc’h N.; Si-Tahar M.; Firatli E.; Jenne D. E.; Gauthier F.; Horwitz M. S.; Borregaard N.; Korkmaz B. Prolonged pharmacological inhibition of cathepsin C results in elimination of neutrophil serine proteases. Biochem. Pharmacol. 2017, 131, 52–67. 10.1016/j.bcp.2017.02.009. [DOI] [PubMed] [Google Scholar]
- Turk D.; Janjic V.; Stern I.; Podobnik M.; Lamba D.; Dahl S. W.; Lauritzen C.; Pedersen J.; Turk V.; Turk B. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001, 20, 6570–6582. 10.1093/emboj/20.23.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigic B.; Pain R. H. Location of the binding site for chloride ion activation of cathepsin C. Eur. J. Biochem. 1999, 264, 944–51. 10.1046/j.1432-1327.1999.00697.x. [DOI] [PubMed] [Google Scholar]
- Molgaard A.; Arnau J.; Lauritzen C.; Larsen S.; Petersen G.; Pedersen J. The crystal structure of human dipeptidyl peptidase I (cathepsin C) in complex with the inhibitor Gly-Phe-CHN2. Biochem. J. 2007, 401, 645–650. 10.1042/BJ20061389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubach J. K.; Cui G.; Schneck J. L.; Taylor A. N.; Zhao B.; Smallwood A.; Nevins N.; Wisnoski D.; Thrall S. H.; Meek T. D. The Amino-Acid Substituents of Dipeptide Substrates of Cathepsin C Can Determine the Rate-Limiting Steps of Catalysis. Biochemistry 2012, 51, 7551–7568. 10.1021/bi300719b. [DOI] [PubMed] [Google Scholar]
- Tran T. V.; Ellis K. A.; Kam C. M.; Hudig D.; Powers J. C. Dipeptidyl peptidase I: importance of progranzyme activation sequences, other dipeptide sequences, and the N-terminal amino group of synthetic substrates for enzyme activity. Arch. Biochem. Biophys. 2002, 403, 160–170. 10.1016/S0003-9861(02)00217-5. [DOI] [PubMed] [Google Scholar]
- Laine D. I.; Busch-Petersen J. Inhibitors of cathepsin C (dipeptidyl peptidase I). Expert Opin. Ther. Pat. 2010, 20, 497–506. 10.1517/13543771003657172. [DOI] [PubMed] [Google Scholar]
- Furber M.; Tiden A.-K.; Gardiner P.; Mete A.; Ford R.; Millichip I.; Stein L.; Mather A.; Kinchin E.; Luckhurst C.; Barber S.; Cage P.; Sanganee H.; Austin R.; Chohan K.; Beri R.; Thong B.; Wallace A.; Oreffo V.; Hutchinson R.; Harper S.; Debreczeni J.; Breed J.; Wissler L.; Edman K. Cathepsin C inhibitors: Property optimization and identification of a clinical candidate. J. Med. Chem. 2014, 57, 2357–2367. 10.1021/jm401705g. [DOI] [PubMed] [Google Scholar]
- Doyle K.; Lönn H.; Käck H.; Van de Poël A.; Swallow S.; Gardiner P.; Connolly S.; Root J.; Wikell C.; Dahl G.; Stenvall K.; Johannesson P. Discovery of Second Generation Reversible Covalent DPP1 Inhibitors Leading to an Oxazepane Amidoacetonitrile Based Clinical Candidate (AZD7986). J. Med. Chem. 2016, 59, 9457–9472. 10.1021/acs.jmedchem.6b01127. [DOI] [PubMed] [Google Scholar]
- Laine D.; Palovich M.; McCleland B.; Petitjean E.; Delhom I.; Xie H.; Deng J.; Lin G.; Davis R.; Jolit A.; Nevins N.; Zhao B.; Villa J.; Schneck J.; McDevitt P.; Midgett R.; Kmett C.; Umbrecht S.; Peck B.; Davis A. B.; Bettoun D. Discovery of novel cyanamide-based inhibitors of cathepsin C. ACS Med. Chem. Lett. 2011, 2, 142–147. 10.1021/ml100212k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz B.; Lesner A.; Wysocka M.; Gieldon A.; Håkansson M.; Gauthier F.; Logan D. T.; Jenne D. E.; Lauritzen C.; Pedersen J. Structure-based design and in vivo anti-arthritic activity evaluation of a potent dipeptidyl cyclopropyl nitrile inhibitor of cathepsin C. Biochem. Pharmacol. 2019, 164, 349–367. 10.1016/j.bcp.2019.04.006. [DOI] [PubMed] [Google Scholar]
- WO2009/074829 (AstraZeneca).
- Bruneau P.; McElroy N. R. logD7.4 Modeling Using Bayesian Regularized Neural Networks. Assessment and Correction of the Errors of Prediction. J. Chem. Inf. Model. 2006, 46, 1379–1387. 10.1021/ci0504014. [DOI] [PubMed] [Google Scholar]
- Bodnarchuk M. S. Water, water, everywhere··· It’s time to stop and think. Drug Discovery Today 2016, 21, 1139–1146. 10.1016/j.drudis.2016.05.009. [DOI] [PubMed] [Google Scholar]
- Ross G. A.; Bodnarchuk M. S.; Essex J. W. Water sites, networks, and free energies with grand canonical monte carlo. J. Am. Chem. Soc. 2015, 137, 14930–14943. 10.1021/jacs.5b07940. [DOI] [PubMed] [Google Scholar]
- Bodnarchuk M. S.; Viner R.; Michel J.; Essex J. W. Strategies to Calculate Water Binding Free Energies in Protein–Ligand Complexes. J. Chem. Inf. Model. 2014, 54, 1623–1633. 10.1021/ci400674k. [DOI] [PubMed] [Google Scholar]
- Ross G. A.; Bruce Macdonald H. E.; Cave-Ayland C.; Cabedo Martinez A. I.; Essex J. W. Replica-Exchange and Standard State Binding Free Energies with Grand Canonical Monte Carlo. J. Chem. Theory Comput. 2017, 13, 6373–6381. 10.1021/acs.jctc.7b00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif M.; Oostenbrink C. Net charge changes in the calculation of relative ligand-binding free energies via classical atomistic molecular dynamics simulation. J. Comput. Chem. 2014, 35, 227–243. 10.1002/jcc.23490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C. J.; Essex J. W.; King M. A. The development of replica-exchange-based free-energy methods. J. Phys. Chem. B 2003, 107, 13703–13710. 10.1021/jp0356620. [DOI] [Google Scholar]
- Guay D.; Beaulieu C.; Jagadeeswar Reddy T.; Zamboni R.; Methot N.; Rubin J.; Ethier D.; David Percival M. Design and synthesis of dipeptidyl nitriles as potent, selective, and reversible inhibitors of cathepsin C. Bioorg. Med. Chem. Lett. 2009, 19, 5392–5396. 10.1016/j.bmcl.2009.07.114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.