

Abstract
We present a practical and easy-to-run in silico workflow exploiting a structure-based strategy making use of docking simulations to derive highly predictive classification models of the androgenic potential of chemicals. Models were trained on a high-quality chemical collection comprising 1689 curated compounds made available within the CoMPARA consortium from the US Environmental Protection Agency and were integrated with a two-step applicability domain whose implementation had the effect of improving both the confidence in prediction and statistics by reducing the number of false negatives. Among the nine androgen receptor X-ray solved structures, the crystal 2PNU (entry code from the Protein Data Bank) was associated with the best performing structure-based classification model. Three validation sets comprising each 2590 compounds extracted by the DUD-E collection were used to challenge model performance and the effectiveness of Applicability Domain implementation. Next, the 2PNU model was applied to screen and prioritize two collections of chemicals. The first is a small pool of 12 representative androgenic compounds that were accurately classified based on outstanding rationale at the molecular level. The second is a large external blind set of 55450 chemicals with potential for human exposure. We show how the use of molecular docking provides highly interpretable models and can represent a real-life option as an alternative nontesting method for predictive toxicology.
Keywords: Molecular docking, classification model, applicability domain, androgen receptor, predictive toxicology
Graphical Abstract
1. Introduction
In the past several decades, the exposure of people and environmental species to xenobiotics has dramatically increased (Egeghy et al., 2012; Judson et al., 2009) due to the use and manufacture of tens of thousands of natural and synthetic chemicals, derived from agricultural and industrial processes as well as from medical applications. It is suspected that some of these are responsible for a broad spectrum of adverse health effects seen in humans and wildlife (Diamanti-Kandarakis et al., 2009; Mrema et al., 2013). In particular, risks are posed by exposure to chemicals interfering with the physiological functioning of the endocrine system whose dysregulation can cause adverse effects in an intact organism or its progeny (Kavlock et al., 1996; Testai et al., 2013; Zoeller et al., 2012). It is widely acknowledged that the mode of action of well-known endocrine disrupting chemicals (EDCs) consists of either mimicking the interaction of natural hormones at the receptor level or of altering synthesis, transport, and metabolism pathways (Schug et al., 2011). A well-known case is diethylstilbestrol (DES), a synthetic estrogenic drug (Bibbo et al., 1975) widely prescribed until the early 1970s to pregnant women to prevent spontaneous abortion and to stimulate fetal growth. DES was banned upon discovery of its causative role in affecting the development of the reproductive system and in inducing vaginal cancer after puberty in women exposed in utero.(Hoover et al., 2011)
The need to protect human health and the environment has led to the development of scientific and regulatory approaches to detecting potential EDCs. A key milestone in this area is REACH (EC, 2006), the European regulation whose Annexes explicitly mention endocrine disruption as an important toxicological end point. In parallel, the assessment of EDCs is a focus of the Endocrine Disruptor Screening Program (EDSP) of the US Environmental Protection Agency (US-EPA). Two recent projects carried out under the EDSP have combined (quantitative) structure activity relationship ((Q)SAR) models from multiple research groups to predict estrogen receptor (ER) and androgen receptor (AR) activity of tens of thousands of chemicals in the environment (ER, Collaborative Estrogen Receptor Activity Prediction Project (CERAPP) (Mansouri et al., 2016); and AR, Collaborative Modeling Project for Androgen Receptor Activity (CoMPARA) (Mansouri et al., 2017).
These projects are examples of collaborations between national governments, the chemical industry, and academic scientific researchers to develop and apply in silico methods, such as QSAR and read-across (Jacobs, 2004; Low et al., 2013; Nicolotti et al., 2009, 2014; Patlewicz et al., 2014; Raunio, 2011), as alternatives to animal experimentation, with the aim of limiting the in vivo testing of chemicals that is time-consuming, costly, and ethically questionable (Devillers, 2009). (Q)SAR and read-across can work well when there is a high similarity between a data-poor target chemical whose activity is to be predicted and a panel of structural analogues having known toxicological profiles (Devillers et al., 2006). (Q)SAR and read-across can often fail when the target chemical and its analogues are built from significantly different scaffolds (Jagiello et al., 2016).
To overcome this intrinsic limitation (i.e., lack of data rich analogues with close structural similarity to a given target chemical), a strategy that is often adopted in drug discovery programs with protein receptor or enzyme targets is to use 3-dimensional information on the interaction of the ligand and its protein binding site. This can be especially useful when the known active compounds (ones that bind to the protein target) span a range of structural classes or backbones. Following this approach, we have used molecular docking to help predict whether chemicals could be EDCs through interaction with AR. Molecular docking is a very effective method to predict the preferred posing of a given molecule approaching a specific target macromolecule to form an energetically stable complex. Docking employs the wealth of physicochemical information contained in experimentally solved structures of target proteins, in order to infer interactions and other mechanistic knowledge about structurally heterogeneous small molecules (Ferreira et al., 2015; Meng et al., 2011). Docking benefits from the growing availability of solved crystallographic protein structures (https://www.wwpdb.org/stats/deposition). These structures represent a great high quality resource for improving the state of the art of predictive toxicology relative to endocrine disruption and other modes of action irrespective of their chemical structural class (Nicolotti et al., 2008; Vedani and Smiesko, 2009). A docking model was included in the CERAPP collaboration (Mansouri et al., 2016; Trisciuzzi et al., 2015) which combined results of a large number of in silico models to predict the ability of chemicals to interact with the ER. The results of this collaboration are being used to prioritize further testing of potential EDCs.
Comparable to estrogenic hormones, androgens are involved in the development and maintenance of the sexual reproductive system and secondary physical changes associated with puberty (Gao et al., 2005; Wilson, 1999). Recent studies (Emmelot-Vonk et al., 2008; Matsumoto et al., 2013) demonstrated that chemicals interfering with AR or dysregulating androgen-dependent signaling pathways can be classified as EDCs, and are responsible for the onset of several pathological conditions (e.g., decreased sperm counts, increased infertility) (Luccio-Camelo and Prins, 2011) or diseases (e.g., testicular dysgenesis syndrome, prostate cancer) (Fisher, 2004; Kim et al., 2005).
The integrated approach of the CERAPP project for evaluating candidate estrogenic compounds (Browne et al., 2015; Mansouri et al., 2016) was subsequently adapted to identify potential AR active chemicals through the CoMPARA initiative, a large-scale modeling collaboration between 35 research groups in the Americas, Europe, and Asia.
Following the CERAPP approach of Mansouri et al. (2016) each participating group in CoMPARA developed one or more in silico models using high quality experimental data derived from multiple in vitro AR high-throughput screening (HTS) assays (Kleinstreuer et al., 2017). The ultimate goal of CoMPARA consisted of deriving consensus scores in order to prioritize compounds for further analysis regarding their potential to interact with AR (Mansouri et al., 2017)
Building on recently published work (Trisciuzzi et al., 2015), we herein demonstrate a practical and easy-to-run protocol employing molecular docking to derive classification models able to identify compounds that interact with AR. The models were developed within the CoMPARA program using a 3D collection of 1689 chemicals with high-quality AR data provided by the US-EPA (Kleinstreuer et al., 2017). Importantly, we illustrate how docking-based classification models should be carefully evaluated in the context of predictive toxicology and how to interpret related statistical parameters. Special attention is given to the definition and implementation of the applicability domain (AD). Interestingly, restricting predictions to chemicals within the AD allowed us to improve robustness and reliability of our classification model in distinguishing AR active from inactive chemicals. From a methodological point of view, this work thus represents the first attempt to extend the concept of AD, normally used in (Q)SAR and 3D-(Q)SAR strategies (Gissi et al., 2014; Sahigara et al., 2012), to predictive models based on docking simulations.
2. Material and Methods
2.1. Starting training set EPA-ARDB
The starting training data set provided by the US-EPA (subsequently referred to as EPA-ARDB) consisted of a curated collection of 1689 chemicals having high quality experimental data. For each chemical, the AR bioactivity was quantified based on the results of 11 AR-related in vitro assays, conducted in HTS measuring the androgen-related activity at multiple points along the AR signaling pathway (Kleinstreuer et al., 2017). This data set is taken from the Tox21 and ToxCast programs (Kavlock et al., 2012; Tice et al., 2013). For classification modeling, a binary (0/1) value was assigned to each compound as a function of biological response. The training set contained 205 compounds (i.e., class = 1) acting as binders (i.e., approximately 12%), and 1484 compounds (i.e., class = 0) acting as nonbinders (i.e., approximately 88%). It is worth noting that the EPA-ARDB includes a relatively small fraction of binders, thus resulting in a strongly unbalanced training set. The full set of 1689 chemicals was supplied as SMILES strings, provided in the Supporting Information.
The training data additionally classified chemicals as being agonist or antagonists, but in the current model, we treat all actives as a single class of binders. For the sake of clarity, we hereafter refer to androgenic/nonandrogenic compounds as binders/nonbinders, respectively.
2.2. Molecular docking
Docking simulations were conducted using nine AR crystal structures retrieved from the Protein Data Bank (PDB) (Berman et al., 2000) selected as follows: six crystal structures previously selected by Kolšek et al. (2014) based on their capability to provide highly predictive docking-based classification models, and two of the most recent AR X-ray solved crystals available in the PDB (PDB IDs: 5CJ6 (Saeed et al., 2016) and 4QL8 (Ullrich et al., 2014)). In addition, a crystal structure (PDB ID: 2HVC (Wang et al., 2006)) showing a noncanonical binding mode of the cognate ligand compared to the other selected X-ray complexes was selected. For further details, the chemical structures of the X-ray cognate ligands and their interactions patterns are reported in Table S1. The receptor structures considered as targets for docking studies are summarized in Table 1.
Table 1.
Detailed information of the nine selected X-ray crystal structures.
| PDB CODE |
Cognate ligand | Ligand effect |
Resolution(Å) | R- Value Free |
References |
|---|---|---|---|---|---|
| 2AM9 | testosterone | agonist | 1.64 | 0.230 | (Pereira de Jésus-Tran et al., 2006) |
| 2AX9 | (R)-3-bromo-2-hydroxy-2-methyl-n-[4-nitro-3(trifluoromethyl)phenyl]propanamide] | agonist | 1.65 | 0.249 | (Bohl et al., 2005) |
| 2PNU | (5S,8R,9S,10S,13R,14S,17S)-13-{2-[(3,5-difluorobenzyl)oxy]ethyl}-17-hydroxy-10-methylhexadecahydro-3h-cyclopenta[a]phenanthren-3-one | agonist | 1.65 | 0.205 | (Cantin et al., 2007) |
| 3B66 | 4-{[(1R,2S)-1,2-dihydroxy-2-methyl-3-(4-nitrophenoxy)propyl]amino}-2-(trifluoromethyl)benzonitrile | agonist | 1.65 | 0.242 | (Bohl et al., 2008) |
| 3L3X | 5-alpha-dihydrotestosterone | agonist | 1.55 | 0.206 | (Zhou et al., 2010) |
| 3RLJ | (2S)-3-(4-cyanophenoxy)-n-[4-cyano-3-(trifluoromethyl)phenyl]-2-hydroxy-2-methylpropanamide | antagonist | 1.90 | 0.265 | (Duke et al., 2011) |
| 5CJ6 | agonist 2-chloro-4-{[(1R,2R)-2-hydroxy-2-methylcyclopentyl]amino}-3-methylbenzonitrile | agonist | 2.07 | 0.222 | (Saeed et al., 2016) |
| 4QL8 | 2-chloro-4-[(3S,3aS,4S)-4-hydroxy-3-methoxy-3a,4,5,6-tetrahydro-3H-pyrrolo[1,2-b]pyrazol-2-yl]-3-methylbenzonitrile | agonist | 2.10 | 0.275 | (Ullrich et al., 2014) |
| 2HVC | 6-[bis(2,2,2,-trifluoroethyl)amino]-4-(trifluoromethyl)quinolin-2(1h)-one | agonist | 2.10 | 0.253 | (Wang et al., 2006) |
All protein structures were refined using Protein Preparation Wizard (Schrödinger Suite 2016–3) for correcting common problems such as missing hydrogen atoms, incomplete side chains and loops, ambiguous protonation states, and flipped residues. The 3D conformations of the EPA-ARDB were processed by LigPrep (Schrödinger Suite 2016–3) in order to properly generate all the possible tautomers and ionization states at a pH value of 7.0 ± 2.0. This procedure increased the database size to 2634 structures. Finally, all of the chemicals were docked into the nine pretreated AR protein structures using GOLD (Genetic Optimization for Ligand Docking) software (Jones et al., 1997). For all simulations, a spherical grid having a radius of 10 Å centered on the center of mass of the cognate ligands was used. All the default flexible ligand docking settings from Gold v.5.2 (Jones et al., 1997) and the fitness function ChemScore were used. Finally, all the compounds were ranked according to their docking scores.
In order to corroborate the robustness of our docking protocol, each original X-ray cognate ligand was redocked back into its corresponding protein binding site. Comparing the Cartesian coordinates of the corresponding heavy atoms of the obtained poses, the X-ray cognate ligands moved back to the original positions with a root mean square deviation (RMSD) < 2 Å (Wilantho et al., 2008) as reported in Table S2.
2.3. Applicability Domain (AD)
The AD represents the space of reliability of a model, where the predictions are the result of interpolations rather than extrapolations. Basically, the quality of the AD depends on the number of chemicals and, more importantly, on the set of molecular descriptors forming a model (Gadaleta et al., 2016; Sahigara et al., 2012). In our study, an initial pool of 237 2D physicochemical and topological molecular descriptors was generated for each chemical in the EPA-ARDB using CANVAS, a cheminformatics program available in the Schrödinger Suite (Canvas, Schrödinger Suite 2016–3). To compare the contribution of each descriptor to the model reliability, we calculated their statistical population variance. The molecular descriptors having population variance equal to zero were discarded, resulting in a final set of 162 descriptors. It is worth noting that the AD assessment is strongly influenced by the modeling approach and the characteristics of the starting training data set (Sahigara et al., 2012). In other words, the AD is determined case-by-case based on the pursued data modeling strategy (Nembri et al., 2016). In the present investigation, the AD was defined using a two-step approach. First, we applied a range-based method, known as the bounding-box (Gadaleta et al., 2016; Sahigara et al., 2012): a chemical is inside the AD if its set of molecular descriptors falls in the range bounded by the maximum and minimum values of the selected molecular descriptors calculated for the EPA-ARDB. In other words, a given predicted external compound showing even only one descriptor exceeding the range defined by the descriptors calculated over the training set chemicals is considered outside the AD (Jaworska et al., 2005). The maximum and minimum values for each molecular descriptor defining the bounding-box are listed in Table S3.
Chemicals within the initial AD were then further examined by using the convex-hull strategy (Sahigara et al., 2012). Following this geometric-based approach, an interpolation space based on the Cartesian coordinates of the top two principal components (PCs), obtained from the initial 162 descriptors, was defined.
The model AD is represented by the smallest convex area whose borders describe the perimeter of a polygon containing the training set compounds. In order to avoid the overestimation of underrepresented areas (Gissi et al., 2013), the polygon area was further restricted to an inner region containing the top 95% of EPA-ARDB chemicals on the basis of their closeness to the EPA-ARDB centroid in the PC space. In doing that, a given predicted external compound located outside the inner polygon is flagged as being outside of the AD.
2.4. Validation set
Three validation sets (VS) were obtained by extracting data from the DUD-E (Directory of Useful Decoys Enhanced) database (Mysinger et al., 2012). In particular, a total of 14619 chemicals relevant to AR interaction were available from the DUD-E web interface (the details of individual chemicals can be found at http://dude.docking.org/targets/andr). Three VS (hereafter referring to VS1, VS2, and VS3, respectively) comprised 2590 chemicals and were all equally sized. In particular, each benchmark database contained the same pool of 259 binders (10% of the set size), doped with three different groups of 2331 nonbinder compounds (90% of the set size) randomly chosen. Notably, duplicate compounds (same entry in both the VS and EPA-ARDB) were identified and removed from the validation set.
The 3D structures of all the compounds were processed by LigPrep (Schrödinger Suite 2016–3) in order to properly generate all the tautomers and ionization states at a pH value of 7.0 ± 2.0. This procedure increased the three VS sizes to 4323, 4332, and 4364 structures, respectively.
2.5. Performance evaluation
As elsewhere described (Trisciuzzi et al., 2015), a statistical confusion matrix was used to assess the goodness of fit of the molecular docking strategy to discern binders from nonbinders. As shown in Table S4, the confusion matrix contains the instances of experimental and predicted classes obtained from each considered classification model (Provost and Kohavi, 1998). More specifically, the confusion matrix reports the number of experimental positive and negative cases that were correctly predicted, i.e., true positives (TPs) and true negatives (TNs), as well as the number of experimental negative and positive cases that were incorrectly identified, otherwise called false positives (FPs) and false negatives (FNs), respectively. From a regulatory oriented conservative toxicological point of view, it is important to pay special attention to the FNs, whose number should be kept as low as possible to minimize the number of truly active compounds predicted to be inactive.
Sensitivity (SE) and specificity (SP) are used to measure the goodness of a classification model. They range from 0 to 1, represent the correctly classified proportion of binders and nonbinders, respectively, and are defined as parameters:
and
Furthermore, the performance of a classification model is assessed by the receiver operating curve (ROC) curve, which reports the variation of SE with respect to 1 – SP values, representing the TP and FP rates, respectively. In the present work, the area under the curve (AUC) and ROC-based enrichment factor (EF) were computed to evaluate and compare different docking-based classification models (Triballeau et al., 2005). The AUC curve provides an immediate idea of the goodness in the overall classification irrespective of any docking score used as cutoff. AUC values < 0.5 indicate that a given model is worse than a random classifier while AUC values > 0.5 indicate that a given model works better than a random classifier. The EF is defined as the percentage of binders found in a given early fraction of the ranked database, defined as follows:
where HSCR is the number of binders recovered at a specific percentage level of the binder/nonbinder ratio of the ranked database, HTOT is the total number of binders for a given target, DBSCR is the number of compounds screened at a specific percentage level of the database, and DBTOT is the total number of compounds in the database. Notably, we considered the percentage of binders found in the top 1% of the ranked EPA-ARDB (EF1%). Remarkably, EF1% depends on the binder to nonbinder ratio, and its value should be compared to the ideal EF (EFmax) obtained by dividing the total number of compounds of database by the total number of binders. The smaller the gap between EF1% and EFmax is, the higher the ranks of binders and thus the lower the number of FPs (Li et al., 2009). For the EPA-ARDB, the ideal EFmax value is equal to 8.2.
In addition to be useful for drawing ROC curves, SE can be employed to set user-definable thresholds (Kolšek et al., 2014). In particular, SE values equal to 0.25, 0.50 and 0.75 were chosen to define four classes of binding, for each AR crystal receptor, summarized as follows:
SE ≤ 0.25, the class with very high probability of binding (i.e., hazard molecules)
0.25 < SE ≤ 0.50, the class with high probability of binding (i.e., warning molecules)
0.50 < SE ≤ 0.75, the class with medium probability of binding (i.e., suspicious molecules)
SE > 0.75, the class with low probability of binding (i.e., safe molecules)
Based on the obtained rankings, the energetic docking scores better approximating the SE-based thresholds equal to 0.25, 0.50 and 0.75 were identified.
The goodness of the classification can be computed using the following parameters:
and
At a given threshold, PPV is related to the probability that a chemical predicted as a binder (over-threshold) is actually a binder, whereas NPV is related to the probability that a chemical predicted as a nonbinder (under-threshold) is actually a nonbinder. In the present work, NPV and PPV values were calculated for each SE threshold (SE = 0.25, SE = 0.50 and SE = 0.75). The prediction classes were generated automatically by using an in-house Python script implementing the computation of PPV and NPV from desired SE.
Given that the EPA-ARDB is strongly unbalanced (i.e., very low number of binders), two additional parameters were computed at each set SE threshold, which are the positive/negative likelihood ratio (+/− LR) and the Balanced Classification Rate (BCR).
The positive (+LR) and negative (-LR) likelihood ratio were computed for each SE threshold as follows:
and
A +LR value equal to 4 would indicate a four-fold increase of the probability of a compound being a binder with respect to the initial condition before the classification; similarly, a -LR = 0.4, shows that, for an under-threshold compounds, the probability to be a binder is equal to 4/10 with respect to that at the initial condition. The larger the +LR value (or the lower the -LR) is, at a given SE threshold, the better the performance of the classification model. Usually applied in the medicine to assess the accuracy of diagnostic tests, the likelihood ratios were herein adapted to evaluate the performance of classification models in the toxicological field.
The BCR is a modification of the well-established correct classification rate. BCR awards with higher scores those models showing an optimal balance between SE and SP (Sokolova and Lapalme, 2009). It is defined as follows:
For each given SE threshold (SE = 0.25, SE = 0.50 and SE = 0.75), the best model is selected as the one having the highest value of the BCR metric. (68).
3. Results & discussion
3.1. Models evaluation
The entire EPA-ARDB was docked into the binding sites of the nine selected X-ray crystal structures, and thereby nine docking-based classification models were derived. For the sake of clarity, each classification model is named using the same PDB code of the crystal structure used for docking simulations. Notice that the models were derived excluding a small fraction of compounds from the training set (i.e., percentage from 2.79% (2PNU) to 6.09% (2AM9) that are undocked or returning unrealistic (positive) values of docking score (see Table S5)). As reported in previous works (Kolšek et al., 2014; Trisciuzzi et al., 2015), the goodness of the docking protocol was preliminarily evaluated considering the AUC and ROC-based enrichment factor at 1% (EF1%) of the ranked database. ROC curves related to the nine classification models are reported in Figure 1 while the corresponding statistical parameters are summarized in Table 2.
Figure 1.
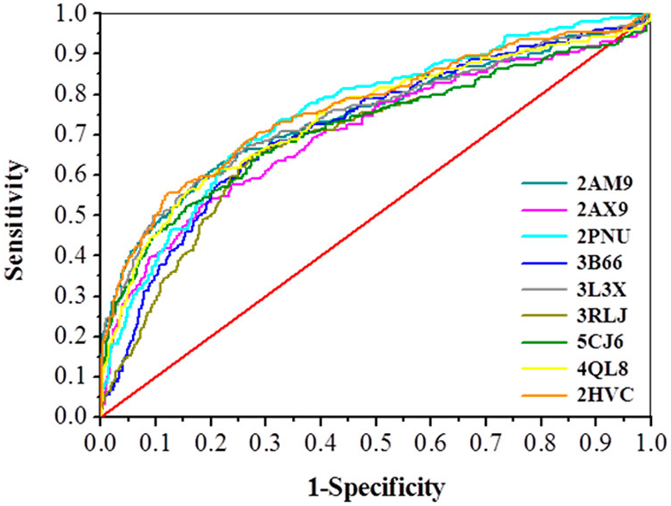
Receiver operating characteristic (ROC) curves derived from the nine selected AR structures (PDB IDs: 2AM9, 2AX9, 2PNU, 3B66, 3L3X, 3RLJ, 5CJ6, 4QL8 and 2HVC). Each classification model is named as the PDB code of the crystal structure employed to perform docking simulations. Red line: AUC = 0.50; black line: AUC = 1.0. All ROC-curves are colored as defined in the legend.
Table 2.
Area under the curve (AUC) of receiver operating characteristic (ROC) curves and enrichment factor at 1% (EF1%) relative to the Protein Data Bank entries (PDB IDs: 2AM9, 2AX9, 2PNU, 3B66, 3L3X, 3RLJ, 5CJ6, 4QL8 and 2HVC).
| PDB CODE |
AUC | EF1% |
|---|---|---|
| 2AM9 | 0.75 | 8.2 |
| 2AX9 | 0.70 | 3.2 |
| 2PNU | 0.76 | 6.7 |
| 3B66 | 0.72 | 4.7 |
| 3L3X | 0.75 | 7.6 |
| 3RLJ | 0.70 | 4.1 |
| 5CJ6 | 0.72 | 6.9 |
| 4QL8 | 0.74 | 6.4 |
| 2HVC | 0.76 | 8.0 |
Notably, the AUC values range from 0.70 to 0.76 demonstrating the high probability of all the models to catch binders (or nonbinders) with good accuracy with respect to a random choice. Likewise, the EF at 1% displays eligible values comparable to the ideal EFmax (i.e., 8.2).
At first glimpse, this preliminary analysis shows a good predictive power of the whole docking procedure for all the considered models. Indeed, albeit ligand based models showing even better performances are available in the literature (Li et al., 2010; Bohl et al., 2004; Jensen et al., 2011; Waller et al., 1996; Vinggaard et al., 2008) they cannot capture the wealth of information on the protein binding sites, thus, making structure-based models more easily interpretable according to regulatory purposes.
In particular, 2PNU and 2HVC returned the highest AUC values in agreement with the ROC curves depicted in Figure 1. For each AR crystal, the docking scores values corresponding to the considered SE thresholds are reported in Table 3.
Table 3.
Docking scores related to the applied SE-based thresholds (docking scores are expressed as kJ/mol).
| PDB CODE |
SE=0.25 | SE=0.50 | SE=0.75 |
|---|---|---|---|
| 2AM9 | −34.76 | −29.21 | −22.66 |
| 2AX9 | −32.90 | −28.13 | −22.63 |
| 2PNU | −37.72 | −31.34 | −25.67 |
| 3B66 | −35.22 | −30.47 | −24.18 |
| 3L3X | −33.94 | −29.47 | −23.01 |
| 3RLJ | −34.53 | −30.61 | −24.10 |
| 5CJ6 | −34.59 | −29.32 | −22.55 |
| 4QL8 | −34.99 | −31.05 | −24.68 |
| 2HVC | −36.22 | −31.51 | −24.69 |
For each AR crystal structure, the docking scores whose rank are closest to the SE thresholds will be designated as values for compound classification.
In this respect, compounds belonging to EPA-ARDB and ranked above the threshold SE = 0.25 were considered to have a high probability of binding; on the other hand, chemicals ranked below the threshold SE = 0.75 were predicted to have a low probability of binding. The remaining compounds were designated as substances difficult to classify and were placed into two diverse classes of prediction with two different degrees of potential toxicity, namely warning (0.25 < SE ≤ 0.50) and suspicious (0.50 < SE ≤ 0.75), according to their docking score values.
The PPVs and NPVs for each SE threshold were calculated (see Table 4). PPVs range from 29.1 to 64.4 for the first threshold (SE = 0.25) and from 16.8 to 22.3 for the third threshold (SE = 0.75). In drug discovery programs, PPVs play a key role to detect potential therapeutically active small molecules. Applied to computational toxicology, the focus shifts towards the latest fraction of the ranked database (that is below the threshold SE = 0.75). The ultimate goal is the prediction of potentially harmful chemicals while minimizing the number of FNs. In other words, we could consider acceptable a classification model even if it returns a high rate of FPs (low PPVs) but with a low number of FNs (high NPVs) at the SE threshold equals to 0.75. This is especially true in a screening and prioritization context where there will be follow-up studies to validate the initial positive calls (binders in this case). Keeping this in mind, we observe that the NPV values range from 94.0 in the case of 5CJ6 model to 95.1 in the case of 2PNU model, thus indicating a high general capability to minimize FNs. It can also be noted that PPVs displays a larger gap (computed considering the maximum and minimum value across all the models) with respect to NPVs. This apparent discrepancy is strictly connected to the already discussed unbalanced nature of the EPA-ARDB (only approximately 12% of the compounds are binders), which makes it difficult to estimate of the best performing classification model. To overcome this issue, we exploited more informative statistical parameters: the positive (+LR) and the negative (-LR) likelihood ratio and the Balanced Classification Rate (BCR). These parameters are indeed independent of the data distribution within the starting training set.
Table 4.
Negative and positive predictive values computed at three SE-thresholds (SE = 0.25, SE = 0.50 and SE = 0.75).
| PDB CODE |
SE=0.25 | SE=0.50 | SE=0.75 | |||
|---|---|---|---|---|---|---|
| PPV | NPV | PPV | NPV | PPV | NPV | |
| 2AM9 | 64.4 | 91.1 | 34.7 | 93.2 | 17.8 | 94.6 |
| 2AX9 | 48.0 | 90.8 | 27.7 | 92.7 | 16.8 | 94.1 |
| 2PNU | 42.1 | 90.3 | 29.5 | 92.5 | 22.3 | 95.1 |
| 3B66 | 33.1 | 90.3 | 27.1 | 92.5 | 18.5 | 94.4 |
| 3L3X | 58.8 | 91.0 | 36.5 | 93.2 | 19.3 | 94.9 |
| 3RLJ | 29.1 | 90.0 | 27.0 | 92.3 | 18.7 | 94.3 |
| 5CJ6 | 60.0 | 90.8 | 31.7 | 92.9 | 16.9 | 94.0 |
| 4QL8 | 47.5 | 90.7 | 34.3 | 93.0 | 19.8 | 94.8 |
| 2HVC | 60.0 | 90.9 | 39.3 | 93.3 | 20.2 | 95.0 |
As reported in Table 5, the 2AM9 crystal structure (+LR = 14.33 at SE = 0.25) is potentially able to catch binders with a good accuracy for further pharmacological investigations. However, as already mentioned, for our toxicological virtual screening application, the classification model should minimize the FNs. Consequently, model performance can be evaluated by computing –LR at SE = 0.75. Building on this evidence, one can see that 2PNU is the best performing model, returning the lowest –LR equal to 0.38 at SE = 0.75. The BCR values are similar for all the models at SE = 0.25 (ranging from 0.17 to 0.19) and SE = 0.50 (from 0.43 to 0.46). Conversely, BCR ranges from 0.47 (2AX9 and 5CJ6) to 0.62 (2PNU) at SE = 0.75, hence again confirming, among the different models, the best performance of 2PNU.
Table 5.
Positive (+LR) and the negative (−LR) likelihood ratio and the Balanced Classification Rate (BCR) computed at three SE-thresholds (SE = 0.25, SE = 0.50 and SE = 0.75).
| PDB CODE |
SE=0.25 | SE=0.50 | SE=0.75 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| +LR | −LR | BCR | +LR | −LR | BCR | +LR | −LR | BCR | |
| 2AM9 | 14.33 | 0.75 | 0.17 | 4.11 | 0.56 | 0.43 | 1.68 | 0.44 | 0.52 |
| 2AX9 | 6.97 | 0.77 | 0.18 | 2.92 | 0.60 | 0.45 | 1.53 | 0.48 | 0.47 |
| 2PNU | 5.38 | 0.77 | 0.18 | 3.06 | 0.59 | 0.45 | 2.10 | 0.38 | 0.62 |
| 3B66 | 3.69 | 0.79 | 0.19 | 2.76 | 0.61 | 0.45 | 1.69 | 0.44 | 0.52 |
| 3L3X | 11.09 | 0.76 | 0.17 | 4.39 | 0.56 | 0.43 | 1.83 | 0.42 | 0.56 |
| 3RLJ | 2.99 | 0.81 | 0.19 | 2.70 | 0.61 | 0.46 | 1.68 | 0.44 | 0.52 |
| 5CJ6 | 11.04 | 0.76 | 0.17 | 3.48 | 0.58 | 0.44 | 1.53 | 0.48 | 0.47 |
| 4QL8 | 6.86 | 0.77 | 0.17 | 3.94 | 0.56 | 0.43 | 1.86 | 0.41 | 0.56 |
| 2HVC | 11.55 | 0.76 | 0.17 | 4.93 | 0.55 | 0.43 | 1.92 | 0.40 | 0.58 |
From a methodological point of view, all the models could be ranked according to the sum of their ranking positions taking into account three performance parameters (NPV, BCR and -LR metrics at SE = 0.75) in order to clarify the best performing classification model).
As clearly shown in Figure 2, the classification model based on the 2PNU crystal structure ranks at the first position for all metrics. Consequently, we can conclude that 2PNU produced the best performing model.
Figure 2.

Summary of the crystals ranking obtained by the three statistical metrics (NPV, -LR and BCR) computed at SE = 0.75. NPV: Negative Predictive Value; -LR: Negative likelihood ratios; BCR: Balanced Classification Rate.
3.2. AD assessment using three validation sets
A key stage for the development of a predictive and robust classification model involves the validation step (Dearden et al., 2009; Mangiatordi et al., 2016). In this regard, we built three VS by extracting data from the DUD-E database. Following the computational details described in the Material and Methods section 2.2, all the compounds of each considered VS were first docked into the binding site of the selected 2PNU X-ray structure and then the AD two-step approach was implemented to assess its effect (see Material and Methods section 2.3).
Our attention was focused on the class of non-binding molecules set at SE>0.75, given our goal of minimizing the number of FNs. Within the non-binding class predicted by 2PNU model we detected 7.59% (VS1), 8.00% (VS2) and 8.56% (VS3) of binders thus indicating an almost random distribution (i.e., 10%). This disappointing result was reinvestigated by implementing the AD. Interestingly, the AD two-step approach had the effect of improving statistics significantly. As shown in Table 6, we observed a drop in the occurrence of the percentage of true binders (i.e. FNs) within the class of predicted non-binders (i.e., from 7.59% to 4.42%, from 8.00% to 4.59% and from 8.56% to 5.09% for VS1, VS2 and VS3 respectively) after applying both AD filters (VS – Bounding box/Convex hull). In other words, AD implementation simultaneously increased model reliability and model performance and reduced the FN rate.
Table 6.
Percentage of binders found at SE >0.75 in the class of predicted non-binders. Based on 2PNU model only (total VS), after the application of the first AD filter (VS – Bounding box) and after the application of both AD filters (VS – Bounding box/Convex hull) on the three VS.
| VS1 | VS2 | VS3 | |
|---|---|---|---|
| total VS | 7.59% | 8.00% | 8.56% |
| VS – Bounding box | 7.09% | 7.51% | 8.09% |
| VS – Bounding box/Convex hull | 4.42% | 4.59% | 5.09% |
The interested reader can look at Table S6 of the Supporting Information to inspect the amount of excluded compounds after the application of the first AD filter (VS – Bounding box) and after the application of both filters (VS – Bounding box/Convex hull) for all the three docked VS.
Figure 3 shows the projection of both EPA-ARDB and VS1 into the top two PCs obtained from the 162 descriptors previously computed for each compound of the EPA-ARDB. The perimeter of the polygon that defines the smallest convex area containing the top 95% of the EPA-ARDB compounds based on their closeness to the centroid is depicted in solid line. More specifically, a total number of 253 chemicals (9.76%) falls outside this polygon and thereby was excluded from the VS1. Additional details about VS2 and VS3 are reported in Figure S1.
Figure 3.
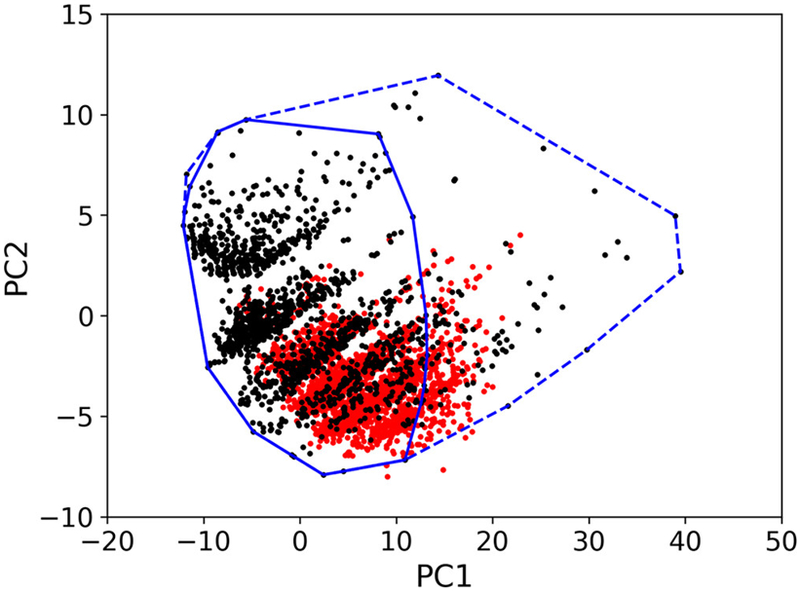
The projection of both EPA-ARDB and VS1 into the top two PCs obtained from the 162 descriptors computed for each compound of the EPA-ARDB. The outer polygon (dashed line) takes into accounts all the chemicals in the EPA-ARDB (black circles), while the inner polygon (solid line) retains the 95% of them. Chemicals of the VS1 (red circles) outside the inner 95% polygon are flagged as outside AD.
The performance of the previously selected classification model (2PNU) was assessed by evaluating how the percentage of binders decreases when moving from random selection (i.e., 10%) to the AD selection (see Table 6). In particular, the AD filtering effectiveness was assessed considering before (total VS) and after the application of the first filter (VS – Bounding box) and then adding also the Convex hull filter (VS – Bounding box/Convex hull). This allowed us to obtain the best performance in validation among different strategies used to define the AD (see Table S7 and the section “Supplementary methodological details” in the Supporting Information).
Taken together, these encouraging results make us confident about the high predictive power of the developed model. To the best of our knowledge, this work represents the first methodological attempt to integrate docking experiments and AD.
3.3. External predictions of twelve representative androgenic substances and of the CoMPARA blind set
To prove its real-life predictive power, our model was challenged with two additional tests. The first challenge consisted of the prediction of 12 reference androgenic substances. In this respect, two groups of chemicals were chosen: six drugs (R1881, nandrolone, ketoprofen, diclofenac, naproxen, and ibuprofen) and six compounds widely employed for industrial and commercial uses (bisphenol A, dichlorodiphenyldichloroethylene better known as p,p′-DDE, 2,2′,4,4′,5-pentabromodiphenyl ether better known as PBDE-99, methylparaben, propylparaben, and butylparaben).
R1881 and nandrolone are synthetic anabolic steroid analogues of testosterone. R1881, also known as methyltrienolone, binds the AR with a high affinity and therefore is used as a marker in prostatic tumors (Roediger et al., 2014); nandrolone and its ester form are employed for testosterone replacement therapy in hypogonadotropic hypogonadism and in AIDS-associated cachexia (de Souza and Hallak, 2011). Ketoprofen, diclofenac, naproxen, and ibuprofen are four nonsteroidal anti-inflammatory drugs (NSAIDs) mainly used in treatment of acute pain and chronic arthritis (Ezechiáš et al., 2016). Bisphenol A,(79–81) commonly abbreviated as BPA, is a key constituent of epoxy resins and one of the most common forms of polycarbonate plastic largely used in food containers and consumer products (i.e., water bottles for infants, kitchen utensils, and medical consumables) (Geens et al., 2012). p,p′-DDE, the main metabolite of DDT, is an organochlorine pesticide that bioaccumulates in the environment and directly interacts with the AR (Gray et al., 2006; Kelce et al., 1995). PBDE-99 is a flame retardant added to manufactured materials such as plastics, textiles, and surface finishes and coatings (Gray et al., 2006). The parabens are a class of parahydroxybenzoates or esters of parahydroxybenzoic acid with a broad spectrum of commercial applications such as personal care products, pharmaceuticals, and bactericidal and fungicidal preservatives (Rastogi et al., 1995).
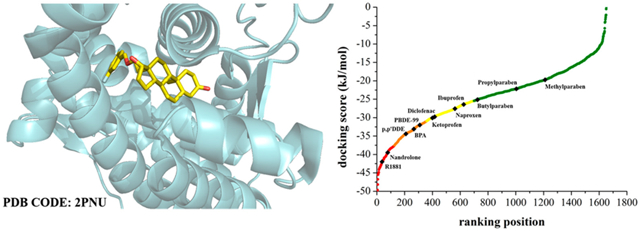
Interestingly, none of these 12 chemicals violated the AD two-step approach and are thus considered inside the AD of our model. We compared the docking score of each of these representative compounds with the values approximating the SE thresholds of the 2PNU classification model (see Table 3). Figure 4 reports the progression of the docking scores for each compound of EPA-ARDB docked on 2PNU as a function of ranking. Note that the graph is split according to the SE thresholds and translated into four color-coded regions corresponding to four binding classes of compounds with a different degree of binding probability: (i) high probability of binding (red class); (ii) warning molecules (orange class); (iii) suspicious molecules (yellow class); and (iv) nonbinding molecules (green class). Based on these criteria, we placed the 12 selected substances in the four binding classes as depicted in Figure 4.
Figure 4.

Progression of docking scores for EPA-ARDB compounds based on the 2PNU docking-based classification model. Twelve representative chemicals were assigned to a given binding class according to their docking scores. The curve is color-coded on the basis of binding class: the hazard, warning, suspicious and safe class is in red, orange, yellow and green, respectively.
For the sake of completeness, the values of docking scores obtained for each representative compound are reported in Table S8.
From a structural point of view, it is not surprising that R1881 and nandrolone are predicted to be binders as both are structurally similar to testosterone. Furthermore, it is also known that the wide abuse of the ester form of nandrolone (specifically nandrolone decanoate) by athletes in order to enhance their physical performance could be associated with infertility and testicular toxicity (Ahmed, 2015). Next, all four of the anti-inflammatory drugs were classified as suspicious molecules. In this regard, a recent study (Ezechiáš et al., 2016) warns of the intrinsic endocrine disrupting nature of these pharmaceuticals that contribute to explain a wide range of adverse health effects on human reproductive systems. For instance, ketoprofen is predicted with the best docking score value among the other NSAIDs in accordance with greatest ability to interact with the AR (Ezechiáš et al., 2016).
In the second group of industrial chemicals, three out of six are predicted to be warning compounds. The European Union and Canada have indeed banned the use of BPA in baby plastic bottles and infant formula packaging (Fischnaller et al., 2016; Kolšek et al., 2015). Additionally the use of p,p′-DDE and PBDE-99 are strictly restricted in several pieces of legislation, as they are persistent organic pollutants (POPs) that act as competitive inhibitors of AR binding (Gray et al., 2006; Kojima et al., 2013; Stoker et al., 2005). The parabens are chemicals with weak potential endocrine activity. It has been demonstrated that their endocrine activity increases with the size of the alkoxy ester group (Darbre and Harvey, 2008). It is worth noting that docking scores increase as a function of the length of the alkyl group (butyl > propyl > methyl). In particular, butylparaben has a docking score very close to the threshold of the class of suspicious molecules, whereas methylparaben and propylparaben are classified as nonbinding chemicals. Accordingly, a study (Kolšek et al., 2015) has recently documented that these two substances do not have androgenic activity.
Finally, encouraged by successful performance on the three VS and on 12 representative androgenic compounds, the 2PNU classification model was employed in the CoMPARA AR activity prediction challenge using a blind set consisting of 55450 chemicals provided by US-EPA.
First, the AD two-step approach was applied to check whether the external compounds were inside/outside AD. Specifically, 6912 chemicals (12.46%) were excluded by the application of both of the AD filters. Figure S2 depicts the projection of both EPA-ARDB and external data set compounds into the top two PCs obtained from the 162 descriptors.
Finally, the remaining 48538 chemicals were screened using the selected classification model. Considering the classification criteria, 3073 chemicals were classified as binders (SE ≤ 0.25) and 27730 substances as nonbinders (SE > 0.75). We also discerned the intermediate subclasses with different degree of binding affinity of 6725 and 10275 chemicals corresponding to warning (0.25 < SE ≤ 0.50) and suspicious (0.50 < SE ≤ 0.75) compounds, respectively.
4. Conclusion
In the present study, we proposed an innovative and practical in silico protocol which exploits the use of molecular docking to derive highly predictive classification models able to identify potential androgenic chemicals. The best performing model was derived from a starting training set of high-quality androgenic experimental data and was successfully challenged against a large blind external set. We also implemented the use of AD to define the areas of the chemical space where model predictions can be considered reliable. The AD implementation had the effect of significantly increasing the reliability of the docking model. To the best our knowledge, this is the first investigation aiming at integrating AD and docking-based toxicology studies.
The success of molecular docking in distinguishing binder from nonbinder chemicals with a high level of confidence can pave the road to its acceptance as a valuable nontesting method for regulatory purposes alongside QSAR and read-across approaches. More importantly, docking results can intrinsically provide an easier and more comprehensive mechanistic interpretation and thus add a valuable plus to the model beyond its mere statistical validity. This indeed matches the expectations of the OECD principles explicitly claiming “a mechanistic interpretation, if possible” for the validation of (Q)SAR (Gissi et al., in press; OECD, 2007).
Supplementary Material
Acknowledgements
This work was supported by FIRB [Futuro in Ricerca 2012, RBFR12SJA8_003] and the Programma IDEA 2011.
Footnotes
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Ahmed MAE, 2015. Amelioration of nandrolone decanoate-induced testicular and sperm toxicity in rats by taurine: effects on steroidogenesis, redox and inflammatory cascades, and intrinsic apoptotic pathway. Toxicol. Appl. Pharmacol 282, 285–296. doi: 10.1016/j.taap.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, 2000. The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibbo M, Al-Naqeeb M, Baccarini I, Gill W, Newton M, Sleeper KM, Sonek RN, Wied GL, 1975. Follow-up study of male and female offspring of DES-treated mothers a preliminary report. J. Reprod. Med 15, 29–32. [PubMed] [Google Scholar]
- Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT, 2005. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem 280, 37747–37754. doi: 10.1074/jbc.M507464200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohl CE, Wu Z, Chen J, Mohler ML, Yang J, Hwang DJ, Mustafa S, Miller DD, Bell CE, Dalton JT, 2008. Effect of B-ring substitution pattern on binding mode of propionamide selective androgen receptor modulators. Bioorg. Med. Chem. Lett 18, 5567–5570. doi: 10.1016/j.bmcl.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne P, Judson RS, Casey WM, Kleinstreuer NC, Thomas RS, 2015. Screening Chemicals for Estrogen Receptor Bioactivity Using a Computational Model. Environ. Sci. Technol 49, 8804–8814. doi: 10.1021/acs.est.5b02641 [DOI] [PubMed] [Google Scholar]
- Cantin L, Faucher F, Couture J-F, de Jésus-Tran KP, Legrand P, Ciobanu LC, Fréchette Y, Labrecque R, Singh SM, Labrie F, Breton R, 2007. Structural characterization of the human androgen receptor ligand-binding domain complexed with EM5744, a rationally designed steroidal ligand bearing a bulky chain directed toward helix 12. J. Biol. Chem 282, 30910–30919. doi: 10.1074/jbc.M705524200 [DOI] [PubMed] [Google Scholar]
- Commission. Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorization and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. J. Eur. Union Lett. 396, 1–849 (2006)]. [Google Scholar]
- Darbre PD, Harvey PW, 2008. Paraben esters: review of recent studies of endocrine toxicity, absorption, esterase and human exposure, and discussion of potential human health risks. J. Appl. Toxicol. JAT 28, 561–578. doi: 10.1002/jat.1358 [DOI] [PubMed] [Google Scholar]
- de Souza GL, Hallak J, 2011. Anabolic steroids and male infertility: a comprehensive review. BJU Int. 108, 1860–1865. doi: 10.1111/j.1464-410X.2011.10131.x [DOI] [PubMed] [Google Scholar]
- Dearden JC, Cronin MTD, Kaiser KLE, 2009. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR). SAR QSAR Environ. Res 20, 241–266. doi: 10.1080/10629360902949567 [DOI] [PubMed] [Google Scholar]
- Devillers J, 2009. Endocrine Disruption Modeling. CRC Press. [Google Scholar]
- Devillers J, Marchand-Geneste N, Carpy A, Porcher JM, 2006. SAR and QSAR modeling of endocrine disruptors. SAR QSAR Environ. Res 17, 393–412. doi: 10.1080/10629360600884397 [DOI] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Bourguignon J-P, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC, 2009. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev 30, 293–342. doi: 10.1210/er.2009-0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dud-e: a database of useful decoys: enhanced, http://dude.docking.org/targets/andr (last accessed date: 30/06/2017). [Google Scholar]
- Duke CB, Jones A, Bohl CE, Dalton JT, Miller DD, 2011. Unexpected binding orientation of bulky-B-ring anti-androgens and implications for future drug targets. J. Med. Chem 54, 3973–3976. doi: 10.1021/jm2000097 [DOI] [PubMed] [Google Scholar]
- Egeghy PP, Judson R, Gangwal S, Mosher S, Smith D, Vail J, Cohen Hubal EA, 2012. The exposure data landscape for manufactured chemicals. Sci. Total Environ 414, 159–166. doi: 10.1016/j.scitotenv.2011.10.046 [DOI] [PubMed] [Google Scholar]
- Emmelot-Vonk MH, Verhaar HJJ, Nakhai Pour HR, Aleman A, Lock TMTW, Bosch JLHR, Grobbee DE, van der Schouw YT, 2008. Effect of testosterone supplementation on functional mobility, cognition, and other parameters in older men: a randomized controlled trial. JAMA 299, 39–52. doi: 10.1001/jama.2007.51 [DOI] [PubMed] [Google Scholar]
- Ezechiáš M, Janochová J, Filipová A, Křesinová Z, Cajthaml T, 2016. Widely used pharmaceuticals present in the environment revealed as in vitro antagonists for human estrogen and androgen receptors. Chemosphere 152, 284–291. doi: 10.1016/j.chemosphere.2016.02.067 [DOI] [PubMed] [Google Scholar]
- Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H, Xie Q, Perkins R, Owens W, Sheehan DM, 2003. Study of 202 Natural, Synthetic, and Environmental Chemicals for Binding to the Androgen Receptor. Chem. Res. Toxicol 16, 1338–1358. doi: 10.1021/tx030011g [DOI] [PubMed] [Google Scholar]
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD, 2015. Molecular docking and structure-based drug design strategies. Mol. Basel Switz. 20, 13384–13421. doi: 10.3390/molecules200713384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischnaller M, Bakry R, Bonn GK, 2016. A simple method for the enrichment of bisphenols using boron nitride. Food Chem. 194, 149–155. doi: 10.1016/j.foodchem.2015.07.117 [DOI] [PubMed] [Google Scholar]
- Fisher JS, 2004. Environmental anti-androgens and male reproductive health: focus on phthalates and testicular dysgenesis syndrome. Reprod. Camb. Engl 127, 305–315. doi: 10.1530/rep.1.00025 [DOI] [PubMed] [Google Scholar]
- Gadaleta D, Mangiatordi GF, Catto M, Carotti A, Nicolotti O, 2016. Applicability Domain for QSAR Models: Where Theory Meets Reality. Int. J. Quant. Struct.-Prop. Relatsh. IJQSPR 1, 45–63. doi: 10.4018/IJQSPR.2016010102 [DOI] [Google Scholar]
- Gao W, Bohl CE, Dalton JT, 2005. Chemistry and Structural Biology of Androgen Receptor. Chem. Rev 105, 3352–3370. doi: 10.1021/cr020456u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geens T, Aerts D, Berthot C, Bourguignon J-P, Goeyens L, Lecomte P, Maghuin-Rogister G, Pironnet A-M, Pussemier L, Scippo M-L, Van Loco J, Covaci A, 2012. A review of dietary and non-dietary exposure to bisphenol-A. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc 50, 3725–3740. doi: 10.1016/j.fct.2012.07.059 [DOI] [PubMed] [Google Scholar]
- Gissi A, Gadaleta D, Floris M, Olla S, Carotti A, Novellino E, Benfenati E, Nicolotti O, 2014. An alternative QSAR-based approach for predicting the bioconcentration factor for regulatory purposes. ALTEX 31, 23–36. doi: 10.14573/altex.1305221 [DOI] [PubMed] [Google Scholar]
- Gissi A, Mangiatordi GF, Sobański T, Netzeva T, Nicolotti O Non-test methods for REACH legislation, 3rd ed, Comprehensive Medicinal Chemistry. Chackalamannil Samuel Rotella David Ward Simon. [Google Scholar]
- Gissi A, Nicolotti O, Carotti A, Gadaleta D, Lombardo A, Benfenati E, 2013. Integration of QSAR models for bioconcentration suitable for REACH. Sci. Total Environ 456–457, 325–332. doi: 10.1016/j.scitotenv.2013.03.104 [DOI] [PubMed] [Google Scholar]
- Gray LE, Wilson VS, Stoker T, Lambright C, Furr J, Noriega N, Howdeshell K, Ankley GT, Guillette L, 2006. Adverse effects of environmental antiandrogens and androgens on reproductive development in mammals. Int. J. Androl 29, 96–104; discussion 105–108. doi: 10.1111/j.1365-2605.2005.00636.x [DOI] [PubMed] [Google Scholar]
- Jacobs MN, 2004. In silico tools to aid risk assessment of endocrine disrupting chemicals. Toxicology 205, 43–53. doi: 10.1016/j.tox.2004.06.036 [DOI] [PubMed] [Google Scholar]
- Jagiello K, Grzonkowska M, Swirog M, Ahmed L, Rasulev B, Avramopoulos A, Papadopoulos MG, Leszczynski J, Puzyn T, 2016. Advantages and limitations of classic and 3D QSAR approaches in nano-QSAR studies based on biological activity of fullerene derivatives. J. Nanoparticle Res 18, 256. doi: 10.1007/s11051-016-3564-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworska J, Nikolova-Jeliazkova N, Aldenberg T, 2005. QSAR applicabilty domain estimation by projection of the training set descriptor space: a review. Altern. Lab. Anim. ATLA 33, 445–459. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R, 1997. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 267, 727–748. doi: 10.1006/jmbi.1996.0897 [DOI] [PubMed] [Google Scholar]
- Judson R, Richard A, Dix DJ, Houck K, Martin M, Kavlock R, Dellarco V, Henry T, Holderman T, Sayre P, Tan S, Carpenter T, Smith E, 2009. The toxicity data landscape for environmental chemicals. Environ. Health Perspect 117, 685–695. doi: 10.1289/ehp.0800168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavlock R, Chandler K, Houck K, Hunter S, Judson R, Kleinstreuer N, Knudsen T, Martin M, Padilla S, Reif D, Richard A, Rotroff D, Sipes N, Dix D, 2012. Update on EPA’s ToxCast program: providing high throughput decision support tools for chemical risk management. Chem. Res. Toxicol 25, 1287–1302. doi: 10.1021/tx3000939 [DOI] [PubMed] [Google Scholar]
- Kavlock RJ, Daston GP, DeRosa C, Fenner-Crisp P, Gray LE, Kaattari S, Lucier G, Luster M, Mac MJ, Maczka C, Miller R, Moore J, Rolland R, Scott G, Sheehan DM, Sinks T, Tilson HA, 1996. Research needs for the risk assessment of health and environmental effects of endocrine disruptors: a report of the U.S. EPA-sponsored workshop. Environ. Health Perspect 104 Suppl 4, 715–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelce WR, Stone CR, Laws SC, Gray LE, Kemppainen JA, Wilson EM, 1995. Persistent DDT metabolite p,p’-DDE is a potent androgen receptor antagonist. Nature 375, 581–585. doi: 10.1038/375581a0 [DOI] [PubMed] [Google Scholar]
- Kim H-J, Park YI, Dong M-S, 2005. Effects of 2,4-D and DCP on the DHT-Induced Androgenic Action in Human Prostate Cancer Cells. Toxicol. Sci 88, 52–59. doi: 10.1093/toxsci/kfi287 [DOI] [PubMed] [Google Scholar]
- Kleinstreuer NC, Ceger P, Watt ED, Martin M, Houck K, Browne P, Thomas RS, Casey WM, Dix DJ, Allen D, Sakamuru S, Xia M, Huang R, Judson R, 2017. Development and Validation of a Computational Model for Androgen Receptor Activity. Chem. Res. Toxicol 30, 946–964. doi: 10.1021/acs.chemrestox.6b00347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Takeuchi S, Itoh T, Iida M, Kobayashi S, Yoshida T, 2013. In vitro endocrine disruption potential of organophosphate flame retardants via human nuclear receptors. Toxicology 314, 76–83. doi: 10.1016/j.tox.2013.09.004 [DOI] [PubMed] [Google Scholar]
- Kolšek K, Gobec M, Mlinarič Raščan I, Sollner Dolenc M, 2015. Screening of bisphenol A, triclosan and paraben analogues as modulators of the glucocorticoid and androgen receptor activities. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 29, 8–15. doi: 10.1016/j.tiv.2014.08.009 [DOI] [PubMed] [Google Scholar]
- Kolšek K, Mavri J, Sollner Dolenc M, Gobec S, Turk S, 2014. Endocrine disruptome--an open source prediction tool for assessing endocrine disruption potential through nuclear receptor binding. J. Chem. Inf. Model 54, 1254–1267. doi: 10.1021/ci400649p [DOI] [PubMed] [Google Scholar]
- Li H, Zhang H, Zheng M, Luo J, Kang L, Liu X, Wang X, Jiang H, 2009. An effective docking strategy for virtual screening based on multi-objective optimization algorithm. BMC Bioinformatics 10, 58. doi: 10.1186/1471-2105-10-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low Y, Sedykh A, Fourches D, Golbraikh A, Whelan M, Rusyn I, Tropsha A, 2013. Integrative Chemical-Biological Read-Across Approach for Chemical Hazard Classification. Chem. Res. Toxicol 26. doi: 10.1021/tx400110f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luccio-Camelo DC, Prins GS, 2011. Disruption of androgen receptor signaling in males by environmental chemicals. J. Steroid Biochem. Mol. Biol 127, 74–82. doi: 10.1016/j.jsbmb.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiatordi GF, Alberga D, Altomare CD, Carotti A, Catto M, Cellamare S, Gadaleta D, Lattanzi G, Leonetti F, Pisani L, Stefanachi A, Trisciuzzi D, Nicolotti O, 2016. Mind the Gap! A Journey towards Computational Toxicology. Mol. Inform 35, 294–308. doi: 10.1002/minf.201501017 [DOI] [PubMed] [Google Scholar]
- Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, Zakharov A, Worth A, Richard AM, Grulke CM, Trisciuzzi D, Fourches D, Horvath D, Benfenati E, Muratov E, Wedebye EB, Grisoni F, Mangiatordi GF, Incisivo GM, Hong H, Ng HW, Tetko IV, Balabin I, Kancherla J, Shen J, Burton J, Nicklaus M, Cassotti M, Nikolov NG, Nicolotti O, Andersson PL, Zang Q, Politi R, Beger RD, Todeschini R, Huang R, Farag S, Rosenberg SA, Slavov S, Hu X, Judson RS, 2016. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect 124, 1023–1033. doi: 10.1289/ehp.1510267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Sakari M, Okada M, Yokoyama A, Takahashi S, Kouzmenko A, Kato S, 2013. The androgen receptor in health and disease. Annu. Rev. Physiol 75, 201–224. doi: 10.1146/annurev-physiol-030212-183656 [DOI] [PubMed] [Google Scholar]
- Meng X-Y, Zhang H-X, Mezei M, Cui M, 2011. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des 7, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrema EJ, Rubino FM, Brambilla G, Moretto A, Tsatsakis AM, Colosio C, 2013. Persistent organochlorinated pesticides and mechanisms of their toxicity. Toxicology 307, 74–88. doi: 10.1016/j.tox.2012.11.015 [DOI] [PubMed] [Google Scholar]
- Mysinger MM, Carchia M, Irwin JJ, Shoichet BK, 2012. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem 55, 6582–6594. doi: 10.1021/jm300687e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nembri S, Grisoni F, Consonni V, Todeschini R, 2016. In Silico Prediction of Cytochrome P450-Drug Interaction: QSARs for CYP3A4 and CYP2C9. Int. J. Mol. Sci 17. doi: 10.3390/ijms17060914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolotti O, Benfenati E, Carotti A, Gadaleta D, Gissi A, Mangiatordi GF, Novellino E, 2014. REACH and in silico methods: an attractive opportunity for medicinal chemists. Drug Discov. Today 19, 1757–1768. doi: 10.1016/j.drudis.2014.06.027 [DOI] [PubMed] [Google Scholar]
- Nicolotti O, Giangreco I, Miscioscia TF, Carotti A, 2009. Improving quantitative structure-activity relationships through multiobjective optimization. J. Chem. Inf. Model 49, 2290–2302. doi: 10.1021/ci9002409 [DOI] [PubMed] [Google Scholar]
- Nicolotti O, Miscioscia TF, Carotti Andrea, Leonetti F, Carotti Angelo, 2008. An integrated approach to ligand- and structure-based drug design: development and application to a series of serine protease inhibitors. J. Chem. Inf. Model 48, 1211–1226. doi: 10.1021/ci800015s [DOI] [PubMed] [Google Scholar]
- OECD, 2007, n.d. OECD, 2007. Guidance 69. GUIDANCE DOCUMENT ON THE VALIDATION OF (QUANTITATIVE) STRUCTURE-ACTIVITY RELATIONSHIP [(Q)SAR] MODELS. Paris Organisation for Economic Cooperation and Development. [Google Scholar]
- Patlewicz G, Ball N, Becker RA, Booth ED, Cronin MTD, Kroese D, Steup D, van Ravenzwaay B, Hartung T, 2014. Read-across approaches--misconceptions, promises and challenges ahead. ALTEX 31, 387–396. [DOI] [PubMed] [Google Scholar]
- Pereira de Jésus-Tran K, Côté P-L, Cantin L, Blanchet J, Labrie F, Breton R, 2006. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. Publ. Protein Soc 15, 987–999. doi: 10.1110/ps.051905906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost F, Kohavi R, 1998. Guest Editors’ Introduction: On Applied Research in Machine Learning. Mach. Learn. 30, 127–132. doi: 10.1023/A:1007442505281 [DOI] [Google Scholar]
- Rastogi SC, Schouten A, de Kruijf N, Weijland JW, 1995. Contents of methyl-, ethyl-, propyl-, butyl- and benzylparaben in cosmetic products. Contact Dermatitis 32, 28–30. [DOI] [PubMed] [Google Scholar]
- Raunio H, 2011. In Silico Toxicology – Non-Testing Methods. Front. Pharmacol 2. doi: 10.3389/fphar.2011.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roediger J, Hessenkemper W, Bartsch S, Manvelyan M, Huettner SS, Liehr T, Esmaeili M, Foller S, Petersen I, Grimm M-O, Baniahmad A, 2014. Supraphysiological androgen levels induce cellular senescence in human prostate cancer cells through the Src-Akt pathway. Mol. Cancer 13. doi: 10.1186/1476-4598-13-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BS, 2011. Bisphenol A: an endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol 127, 27–34. doi: 10.1016/j.jsbmb.2011.05.002 [DOI] [PubMed] [Google Scholar]
- Saeed A, Vaught GM, Gavardinas K, Matthews D, Green JE, Losada PG, Bullock HA, Calvert NA, Patel NJ, Sweetana SA, Krishnan V, Henck JW, Luz JG, Wang Y, Jadhav P, 2016. 2-Chloro-4-[[(1R,2R)-2-hydroxy-2-methyl-cyclopentyl]amino]-3-methyl-benzonitrile: A Transdermal Selective Androgen Receptor Modulator (SARM) for Muscle Atrophy. J. Med. Chem 59, 750–755. doi: 10.1021/acs.jmedchem.5b01168 [DOI] [PubMed] [Google Scholar]
- Sahigara F, Mansouri K, Ballabio D, Mauri A, Consonni V, Todeschini R, 2012. Comparison of different approaches to define the applicability domain of QSAR models. Mol. Basel Switz. 17, 4791–4810. doi: 10.3390/molecules17054791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Rodríguez A, Pérez-Castillo Y, Schürer SC, Nicolotti O, Mangiatordi GF, Borges F, Cordeiro MNDS, Tejera E, Medina-Franco JL, Cruz-Monteagudo M, 2017. From flamingo dance to (desirable) drug discovery: a nature-inspired approach. Drug Discov. Today. doi: 10.1016/j.drudis.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2016–3: Canvas, Schrödinger, LLC, New York, NY, 2016. [Google Scholar]
- Schrödinger Release 2016–3: LigPrep, Schrödinger, LLC, New York, NY, 2016. [Google Scholar]
- Schrödinger Suite 2016–3 Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2016; Impact, Schrödinger, LLC, New York, NY, 2016; Prime, Schrödinger, LLC, New York, NY, 2016. [Google Scholar]
- Schug TT, Janesick A, Blumberg B, Heindel JJ, 2011. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol 127, 204–215. doi: 10.1016/j.jsbmb.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova M, Lapalme G, 2009. A Systematic Analysis of Performance Measures for Classification Tasks. Inf Process Manage 45, 427–437. doi: 10.1016/j.ipm.2009.03.002 [DOI] [Google Scholar]
- Stoker TE, Cooper RL, Lambright CS, Wilson VS, Furr J, Gray LE, 2005. In vivo and in vitro anti-androgenic effects of DE-71, a commercial polybrominated diphenyl ether (PBDE) mixture. Toxicol. Appl. Pharmacol 207, 78–88. doi: 10.1016/j.taap.2005.05.010 [DOI] [PubMed] [Google Scholar]
- Teng C, Goodwin B, Shockley K, Xia M, Huang R, Norris J, Merrick BA, Jetten AM, Austin CP, Tice RR, 2013. Bisphenol A affects androgen receptor function via multiple mechanisms. Chem. Biol. Interact 203, 556–564. doi: 10.1016/j.cbi.2013.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testai E, Galli CL, Dekant W, Marinovich M, Piersma AH, Sharpe RM, 2013. A plea for risk assessment of endocrine disrupting chemicals. Toxicology 314, 51–59. doi: 10.1016/j.tox.2013.07.018 [DOI] [PubMed] [Google Scholar]
- Tice RR, Austin CP, Kavlock RJ, Bucher JR, 2013. Improving the human hazard characterization of chemicals: a Tox21 update. Environ. Health Perspect 121, 756–765. doi: 10.1289/ehp.1205784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triballeau N, Acher F, Brabet I, Pin J-P, Bertrand H-O, 2005. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem 48, 2534–2547. doi: 10.1021/jm049092j [DOI] [PubMed] [Google Scholar]
- Trisciuzzi D, Alberga D, Mansouri K, Judson R, Cellamare S, Catto M, Carotti A, Benfenati E, Novellino E, Mangiatordi GF, Nicolotti O, 2015. Docking-based classification models for exploratory toxicology studies on high-quality estrogenic experimental data. Future Med. Chem 7, 1921–1936. doi: 10.4155/fmc.15.103 [DOI] [PubMed] [Google Scholar]
- Ullrich T, Sasmal S, Boorgu V, Pasagadi S, Cheera S, Rajagopalan S, Bhumireddy A, Shashikumar D, Chelur S, Belliappa C, Pandit C, Krishnamurthy N, Mukherjee S, Ramanathan A, Ghadiyaram C, Ramachandra M, Santos PG, Lagu B, Bock MG, Perrone MH, Weiler S, Keller H, 2014. 3-alkoxy-pyrrolo[1,2-b]pyrazolines as selective androgen receptor modulators with ideal physicochemical properties for transdermal administration. J. Med. Chem 57, 7396–7411. doi: 10.1021/jm5009049 [DOI] [PubMed] [Google Scholar]
- Vedani A, Smiesko M, 2009. In silico toxicology in drug discovery - concepts based on three-dimensional models. Altern. Lab. Anim. ATLA 37, 477–496. [DOI] [PubMed] [Google Scholar]
- Wang F, Liu X, Li H, Liang K, Miner JN, Hong M, Kallel EA, van Oeveren A, Zhi L, Jiang T, 2006. Structure of the ligand-binding domain (LBD) of human androgen receptor in complex with a selective modulator LGD2226. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun 62, 1067–1071. doi: 10.1107/S1744309106039340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilantho A, Tongsima S, Jenwitheesuk E, 2008. Pre-docking filter for protein and ligand 3D structures. Bioinformation 3, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JD, 1999. The Role of Androgens in Male Gender Role Behavior. Endocr. Rev 20, 726–737. doi: 10.1210/edrv.20.5.0377 [DOI] [PubMed] [Google Scholar]
- wwPDB: Worldwide Protein Data Bank. Statistics For PDB Structures That Are Deposited And Processed By Year And Site. https://www.wwpdb.org/stats/deposition (last accessed date: 30/06/2017).
- Zhou XE, Suino-Powell KM, Li J, He Y, Mackeigan JP, Melcher K, Yong E-L, Xu HE, 2010. Identification of SRC3/AIB1 as a preferred coactivator for hormone-activated androgen receptor. J. Biol. Chem 285, 9161–9171. doi: 10.1074/jbc.M109.085779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoeller RT, Brown TR, Doan LL, Gore AC, Skakkebaek NE, Soto AM, Woodruff TJ, Vom Saal FS, 2012. Endocrine-disrupting chemicals and public health protection: a statement of principles from The Endocrine Society. Endocrinology 153, 4097–4110. doi: 10.1210/en.2012-1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.