

Abstract
In the designed compounds, either a biarylamide or biarylurea moiety or an N-substituted piperazine motif was linked to position 1 of the phthalazine core. The anti-proliferative activity of the synthesised compounds revealed that eight compounds (6b, 6e, 7b, 13a, 13c, 16a, 16d and 17a) exhibited excellent broad spectrum cytotoxic activity in NCI 5-log dose assays against the full 60 cell panel with GI50 values ranging from 0.15 to 8.41 µM. Moreover, the enzymatic assessment of the synthesised compounds against VEGFR-2 tyrosine kinase showed the significant inhibitory activities of the biarylureas (12b, 12c and 13c) with IC50s of 4.4, 2.7 and 2.5 μM, respectively, and with 79.83, 72.58 and 71.6% inhibition of HUVEC at 10 μM, respectively. Additionally, compounds (7b, 13c and 16a) were found to induce cell cycle arrest at S phase boundary. Compound 7b triggered a concurrent increase in cleaved caspase-3 expression level, indicating the apoptotic-induced cell death.
Keywords: Substituted phthalazines, VEGFR-2 kinase inhibitors, anti-proliferative, apoptosis
1. Introduction
Cancer is a leading cause of death worldwide 1 . Epidemiological studies revealed that cancer accounts for one of every five deaths. Moreover, it is estimated that the annual number of deaths due to cancers will increase from 7.6 million in 2008 to 13 million in 2030 2 .
Apoptosis, or programed cell death, plays a crucial role in maintaining the normal body health. However, malignant cells are generally less sensitive to these stresses and tend to evade apoptosis. Genetic regulation, initiation and effector mechanisms can be regarded as stages of apoptosis in its simplest model. Gamma and ultraviolet irradiation, anticancer drugs, deprivation of survival factors such as interleukin-1 (IL-1), or other cytokines that activate “death receptors” are considered as initiators of apoptosis 3 . Hence, the identification of apoptosis inducers has evolved as an attractive approach for the development of potential anticancer agents.
From another point of view, angiogenesis, the process of sprouting of new blood vessel formations from pre-existing ones, is also regarded as an important hallmark of cancer development. Vascular endothelial growth factor family (VEGFs) are known to be one of the key regulators of angiogenesis. Their biologic effects is exerted by binding to extracellular domain.
Multiple reports have detailed several small-molecule inhibitors of VEGFR-2 acting by binding to the ATP-binding site in the intracellular kinase domain resulting in diminished VEGF signal transduction, these inhibitors may be broadly categorised into two main types: type I; inhibitors represent ATP competitors that generally bind in or around the catalytic site of the kinase in its active conformation, in the region originally occupied by the adenine moiety of ATP 4 . On the other hand, type-II inhibitors stabilise the inactive conformation of the enzyme derived upon the movement of the DFG motif (i.e. Asp-Phe-Gly), hence, they exploit new interactions with the lipophilic pocket revealed in this new rearrangement 5 .
In the past few years, a number of VEGFR-2 inhibitors have proven success as targeted anticancer therapeutics. Among these is the 1,4-disubstituted phthalazine inhibitor, vatalanib. (PTK-787) (I) (IC50 VEGFR-2 = 43 nM) (Figure 1) 6 .
Figure 1.

Structures of some known VEGFR-2 kinase inhibitors.
The biarylurea-based inhibitor, sorafenib (II) (IC50-VEGFR-2 = 90 nM) (Figure 1), has been approved for the treatment of advanced renal cell carcinoma 7 . Sorafenib was proved to suppress tumour proliferation, metastasis and angiogenesis; in addition, it was also found to be capable of inducing apoptosis, both in vitro and in vivo, in a host of cancer cells. However, the underlying mechanisms seemed to vary depending on cancer cell types 8–10 . Interest in developing novel biarylurea-based VEGFR-2 inhibitors has been increasingly highlighted with the clinical success of sorafenib and its analogs, such as regorafenib (III) 11 .
Moreover, the biarylamide, (AAL993) (IV), a hybrid-design type-II inhibitor derived from the type-I inhibitor vatalanib, was reported as a potent inhibitor of VEGFR family with VEGFR-2 IC50 of 23 nM 12 .
Also, telatinib (V), the furopyridazine derivative, emerged as a potent and orally available inhibitor of VEGFR-2, VEGFR-3 with IC50s 6 and 4 nM, respectively. It is currently in clinical trials for gastric and colorectal cancer 13 .
From another point of view, the piperazinyl moiety, the small rigid heterocyclic motif, has been successfully incorporated in several potent kinase inhibitors 14–17 . The aryl piperazine-based derivative (VI) (Figure 1) was reported to exhibit potent single-digit nanomolar inhibitory activity on specific kinases 18 , 19 .
2. Rationale and design
Based on the above findings, three series of phthalazine-based derivatives linked to a biarylamide (series A) or biarylurea tail (series B, C) at position 1 of the phthalazine core via an amino or ether linkage were designed and synthesised as potent anti-proliferative derivatives as well as apoptosis inducers in tumour cells. Different substitution patterns were introduced to the terminal aromatic ring aiming to better occupy the hydrophobic pocket revealed by the kinase in its DFG-out conformation (Figure 2). The design of these series was based on attaining new hybrid structures from sorafenib and vatalanib while maintaining the structure activity relationships as well as common pharmacophoric features shared by several VEGFR inhibitors which involve the following three features; (1) a flat heteroaromatic ring system to better occupy the ATP-binding region of the kinase, (2) a hydrogen bond donor–acceptor pair represented by the urea or amide moieties that usually form hydrogen bonds with Glu 885 and Asp 1046 residues, (3) a substituted terminal aryl moiety to occupy the hydrophobic pocket revealed by the movement of the DFG motif of the kinase in its inactive conformation 20 .
Figure 2.

Strategy for the design of the target compounds.
Furthermore, two other series of vatalanib analogs (series D, E) were designed having a 4-chloroaniline moiety at position 1 of the phthalazine and an anilino or phenoxy motif linked to the position 4 via a methylene spacer.
In addition, a sixth series of substituted piperazine-based derivatives (series F) was prepared as well, via linking the two major fragments: the phthalazine core recognised as a kinase-privileged fragment, to the piperazine moiety aiming to offer more rigidity and to facilitate deriving spatial–activity relationships 15 .
3. Chemistry
The pathways adopted for the synthesis of the new substituted phthalazine derivatives are depicted in Schemes (1–3). The key intermediate 1-aryl-3–(4-hydroxyphenyl)urea (1a–c), were prepared following the literature methods as illustrated in (Scheme 1(a)) 21 . On the other hand, cyclisation of the respective o-substituted benzoic acid derivative with hydrazine hydrate afforded the corresponding phthalazinones (2a,b). Chlorination of 2a,b with phosphorous oxychloride afforded the corresponding 1-chlorophthalazines (3a,b), which were used to prepare the target series of 1-substituted phthalazines via reaction with various nucleophiles. Thus, the reaction of 3a,b with p-phenylenediamine in butanol via the displacement of the chloro atom of phthalazine with the suitable nitrogen nucleophile such as p-phenylenediamine afforded the N 1-(phthalazin-1-yl)benzene-1,4-diamines (4a,b). The latter compounds (4a,b) were further reacted with the respective benzoyl chloride in acetonitrile, in the presence of triethylamine to afford the target phthalazines bearing the biarylamide tail (5a–d) (series A). Moreover, the reaction of (4a,b) with different arylisocyanates in DMF yielded the biarylureas (6a–f) (series B). On the other hand, 1-aryl-3–(4-(4-substituted phthalazin-1-yloxy)phenyl)ureas (7a–f) (series C) were obtained via reacting the respective 1-chlorophthalazines (3a,b) with the appropriate intermediates (1a–c) in refluxing acetonitrile. This latter reaction proceeds via ipso addition by the nucleophile that gives an anion with a highly delocalised charge then followed by leaving of the chlorine atom of the respective 1-chlorophthalazines (3a,b) to afford the alkoxyphthalazines (7a–f). Refluxing the 1-chlorophthalazines (3a,b) with the appropriate piperazine derivative in ethanol yielded the target phthalazines bearing the substituted piperazinyl tail (8a–j) (series F) in good yields (Scheme 1(b)).
Scheme 1.

(a) Reagents and conditions: (i) dioxane, rt, 1 h. (b) Reagents and conditions: (i) NH2NH2 99%, propanol, 2 h (ii) POCl3, reflux 70 °C, 2 h (iii) p-phenylenediamine, butanol, 110 °C, 1 h, (iv) benzoyl chlorides, acetonitile, triethylamine, 6 h (v) arylisocyanates, DMF, reflux, 8 h, (vi) 1a–c, Cs2CO3, acetonitrile, reflux, 6 h, (vii) piperazines, K2CO3, KI, ethanol, reflux, 3 h.
Scheme 2.

Reagents and conditions: (i) NH2NH2 acetic, reflux, 3 h (ii) POCl3, reflux 110 °C, 1 h (iii) p-phenylenediamine, butanol, 110 °C, 1 h, (iv) phenylisocyanates, DMF, reflux, 8–10 h, (v) 1a–c, Cs2CO3, acetonitrile, reflux, 6–8 h.
Scheme 3.

Reagents and conditions: (i) aniline derivatives, butanol, K2CO3, KI, reflux, 3 h (ii) NBS, dibenzoyl chloride, reflux, 24 h (iii) aniline derivatives, acetone, K2CO3, KI, reflux, 6 h (iv) phenol derivatives, NaH, acetone, reflux, 8 h.
As for the synthesis of the biarylureas (12a–c and 13a–c) having a chloro substituent at 4 position of the phthalazine (Scheme 2), it was started by refluxing phthalic anhydride with hydrazine hydrate in acetic acid, followed by reflux with POCl3 to give the 1,4-dichlorophthalazine intermediate (10). Thereafter, the target compounds (12a–c and 13a–c) were obtained in a manner similar to that adopted in Scheme 1(b).
Compounds (16a–d, and 17a,b) (series D, E) were synthesised via initial bromination of the key intermediate (14) with NBS in presence of catalytic amount of dibenzoyl peroxide. The reaction of the bromo derivative (15) with the respective aniline or phenol in acetone then afforded the titled compounds (Scheme 3).
4. Results and discussion
4.1. Biological evaluation
4.1.1. In vitro anticancer screening at full NCI 60 cell panel
Aiming to evaluate the general antitumour activity of the synthesised phthalazines, the structures of the final compounds were submitted to National Cancer Institute “NCI” (www.dtp.nci.nih.gov), Bethesda, Maryland, USA. In vitro NCI anticancer screening involves two-stage process, the first stage started with the evaluation of the selected candidates against the full NCI 60 cell lines panel representing leukaemia, non-small cell lung cancer, melanoma, colon, CNS, breast, ovarian, renal and prostate cancers at a single dose of 10 µM. NCI selection is based on degree of structure variation and application of computer modelling techniques to prioritise compounds based on their ability to add diversity to the NCI small molecule compound collection.
Eighteen of the submitted phthalazines (5b, 5d, 6b, 6e, 7b, 7e, 8a, 8d, 8f, 8g, 8h, 8i, 13a, 13b, 13c, 16a, 16d, and 17a) were selected by NCI under drug discovery programme for screening against the full NCI 60 cell panel at single dose 10 µM. Among the investigated 18 compounds, eight compounds (6b, 6e, 7b, 13a, 13c, 16a, 16d and 17a) were selected by NCI for further screening at 5-log dose molar range due to their prominent cell growth inhibition against various cell lines.
4.1.1.1. Primary in vitro antineoplastic single-dose assay
Primary in vitro single-dose anticancer assay was evaluated against full NCI 60 cell panel. Results for each compound were presented as a mean graph of the growth per cent of the treated cells compared to the untreated control cells. Eight of the investigated phthalazine-based derivatives (6b, 6e, 7b, 13a, 13c, 16a, 16d and 17a) showed a distinctive pattern of sensitivity against various NCI cell lines (Tables S1 and S2 in Supplementary material ). Thus, the biarylurea-based derivatives having a terminal 4-chloro substituent (6b, and 6e) exhibited remarkable broad spectrum cell growth inhibition above 90% against various cell lines including leukaemia, non-small cell lung cancer, melanoma, prostate cancer and breast cancer cell lines. Compound 6e even exhibited lethal effect (above 100% inhibition) against several leukaemia, melanoma, renal cancer cell lines. Interestingly, it showed highly potent inhibitory or lethal effects against most of the tested leukaemia and melanoma cell lines.
Furthermore, their analogue (7b) linked to the phthalazine nucleus via an ether linkage also showed excellent broad spectrum inhibitory or lethal activity against most of the tested NCI cell lines representing all the nine tumour subpanels specially those of leukaemia, colon, melanoma and breast cancer cell lines, with mean growth inhibition of 106.99%. However, its analogue (7e) having a methyl substituent at 4-position of phthalazine core only showed moderate cell growth inhibition against leukaemia and breast cell lines (Table S3 in Supplementary material).
Thus, the unsubstituted biarylurea derivative (13a) displayed broad spectrum cell growth inhibition above 90% against various cell lines including non-small cell lung cancer, melanoma, CNS cancer, melanoma and breast cancer cell lines. Interestingly, its disubstituted analogue (13c) exhibited an excellent broad spectrum lethal activity against most of the tested NCI cell lines representing all the nine tumour subpanels specially those of non-small lung cancer, colon, CNS and melanoma cell lines, with mean growth inhibition of 145.09%. Surprisingly, the 4-chloro substituted derivative (13b) did not show any significant cell line growth inhibition (Table S3 in Supplementary material).
For the vatalanib analogues having a 4-anilino (16a, and 16d) or 4-phenoxy substituent (17a) linked to the phthalazine via methylene spacer; It was found that all the three tested derivatives displayed remarkable broad spectrum cell growth inhibition above 90% against various cell lines including leukaemia, melanoma and breast cancer cell lines, with the meta methoxy analogue (16d) showing the most prominent inhibition against most of the tested cell lines with mean growth inhibition of 116.00%.
Neither of the biarylamides (5b, and 5d) nor the piperazines (8a, 8d, 8f, 8g, 8h and 8i) showed any significant cell line growth inhibition under the same test conditions (Table S3 in Supplementary material).
4.1.1.2. In vitro 5 log dose full NCI 60 cell panel assay
Eight compounds (6b, 6e, 7b, 13a, 13c, 16a, 16d and 17a) were selected by NCI for further 5 log dose screening against full NCI 60 cell panel. All the 60 cell lines, representing nine tumour subpanels (leukaemia, non-small cell lung cancer, colon cancer, CNS cancer, prostate cancer, melanoma, ovarian cancer, renal cancer and breast cancer) which were incubated at five different concentrations (0.01, 0.1, 1, 10 and 100 µM) of each of the test compounds. The outcomes were used to create log concentration vs % growth inhibition curves, then three response parameters (GI50, TGI and LC50) were calculated for each cell line, (Tables S4 and S5 in Supplementary material).
All investigated biarylureas (6b, 6e, 7b, 13a, and 13c) and vatalanib analogues (16a, 16d, and 17a) revealed potent antiproliferative activity against most of the tested cell lines representing the nine different subpanels with GI50 values between 0.15–8.41 µM, except for 16a which was insensitive to prostate and MCF-7 breast cancer cell lines.
With regard to the sensitivity against the tested cell lines, compound (7b and 13c) exhibited the highest broad spectrum submicromolar inhibitory activity against most of the tested cell lines specially those of leukaemia, colon, melanoma, breast cancer and renal (13c only) cell panels with GI50 values of 0.15–2.81 µM and 0.2–2.66 µM for 7b and 13c, respectively. It is worth noting that 13c showed submicromolar GI50s against all tested leukaemia and melanoma cell lines.
As for the selectivity of the test compounds towards some specific tumour subpanels, which is calculated based on the ratio obtained by dividing the full panel MID (the average sensitivity of all cell lines towards the test agent) by their individual subpanel MID (the average sensitivity of all cell lines of a particular subpanel towards the test agent) 24 , 25 . As per this criterion, the test compounds were regarded to be more selective against leukaemia, renal, melanoma, colon and breast cancer subpanels.
4.1.2. In vitro cytotoxic activity against MCF-7, HCT-116 and HepG-2 cancer cell lines
The growth inhibition of the rest of synthesised compounds (not selected by NCI) (5a, 5c, 6a, 6c, 6d, 6f, 7c, 7d, 7f, 8b, 8c, 8e and 8j) was also evaluated against two specific cell lines, namely MCF-7 and HCT-116 using doxorubicin as a reference drug. In addition, compounds (12a, 12b, 12c, 16b, 16c and 17b) were evaluated against a third cell line (HepG-2) These first two cell lines were selected based on the sensitivity of the previously NCI-tested derivatives towards them amongst the 60 NCI panel. The growth inhibition is expressed as the median growth inhibitory concentration (IC50) which corresponds to the concentration required for 50% inhibition of cell viability and are provided in (Table S6 in supplementary material).
Investigation of the results of the growth inhibition of the tested molecules revealed that the biarylurea-based phthalazines (series B and C) generally exhibited moderate to significant cytotoxicity on both cell lines. This was relatively in accordance with the NCI findings for the same series of compounds.
Further analysis of the inhibitory results within the biarylurea-based derivatives linked to phthalazine core via an NH-linker (series B) revealed that, the 4-chloro-3-trifluoromethyl derivatives (6c and 6f), bearing an electron withdrawing substitution pattern similar to that of sorafenib, displayed enhanced growth inhibition on both cell lines compared to their unsubstituted analogues (6a and 6d) (IC50s = 6.2, 6.0 and 3.2, 3.1 µM compared to 67.6, 91.2 and 67.6 and 57.5 µM respectively).
As for the biarylureas bearing an ether linker (series 7), the 4-chloro-3-trifluoromethyl derivative (7c) similarly displayed significant inhibiton on both cell lines, (IC50s = 2.5 and 2.6 µM). Surprisingly, its methyl analogue (7f) demonstrated weaker activity (IC50s = 31 and 49 µM) compared to its unsubstituted analogue (7d, IC50s = 7.8 and 7.2 µM). However, the amide-based derivatives (series A) and the piperazine-based derivatives have not shown any interesting cell line inhibition which was also consistent our previous results.
Unfortunately, neither the 4-chlorophthalazines (12a–c) nor the vatalanib analogues (series D and E) showed any significant cell line inhibition (Table S6 in Supplementary Material).
4.1.3. In vitro VEGFR-2 tyrosine kinase activity
4.1.3.1. VEGFR-2 kinase activity at single-dose 10 μM concentration
The VEGFR-2 tyrosine kinase assay was performed in an attempt to investigate the mechanism of potent antiproliferative activity exerted by the synthesised compounds. The VEGFR tyrosine kinase assays were performed at BPS Bioscience (www.bpsbioscience.com). All the synthesised compounds representing the six series were evaluated for their ability to inhibit VEGFR-2 tyrosine kinase at single dose of 10 µM.
Investigation of the results of VEGFR-2 inhibitory activities among the synthesised phthalazines revealed that: Within the biaryl ureas (series B, C): derivatives having a 4-chloro-3-trifluoromethyl substitution pattern on the terminal phenyl ring similar to that of sorafenib (6c, 6f, 7c, 12c and 13c) tended to exhibit enhanced VEGFR-2 inhibitory activity as excepted from cell line results compared to their monosubstituted or unsubstituted analogues.
In accordance with cell lines results, among these biaryl ureas, the introduction of a 4-chloro group to the phthalazine core replacing the 4-CH3 or unsubstituted derivatives, resulted in favourable increase in VEGFR-2 inhibitory activity (12a–c, 13a–c compared to 6a–f, 7a–f), which might be attributed to enhanced lipophilicity of the chloro group. Thus, compounds 6c, 12b, 12c and 13c showed 70–100% inhibition at 10 µM concentration (Table 1).
Table 1.
The VEGFR-2 inhibition per cent of the synthesised phthalazines (series A, B, C) at 10 μM concentration.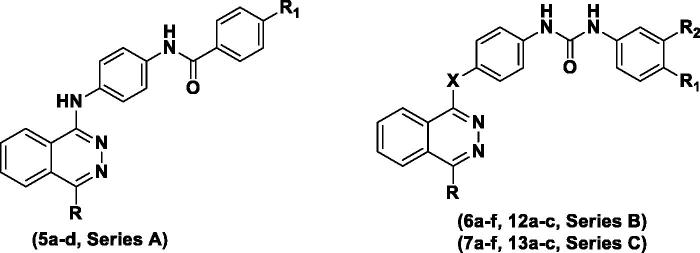
| Cpd No | X | R | R1 | R2 | % inhibition |
|---|---|---|---|---|---|
| 5a | – | H | H | – | 4 |
| 5b | – | H | Cl | – | 8 |
| 5c | – | CH3 | H | – | 12 |
| 5d | – | CH3 | Cl | – | 15 |
| 6a | NH | H | H | H | 13 |
| 6b | NH | H | Cl | H | 20 |
| 6c | NH | H | Cl | CF3 | 70 |
| 6d | NH | CH3 | H | H | 15 |
| 6e | NH | CH3 | Cl | H | 19 |
| 6f | NH | CH3 | Cl | CF3 | 32 |
| 12a | NH | Cl | H | H | 26 |
| 12b | NH | Cl | Cl | H | 70 |
| 12c | NH | Cl | Cl | CF3 | 78 |
| 7a | O | H | H | H | 5 |
| 7b | O | H | Cl | H | 13 |
| 7c | O | H | Cl | CF3 | 47 |
| 7d | O | CH3 | H | H | 10 |
| 7e | O | CH3 | Cl | H | 14 |
| 7f | O | CH3 | Cl | CF3 | 21 |
| 13a | O | Cl | H | H | 24 |
| 13b | O | Cl | Cl | H | 38 |
| 13c | O | Cl | Cl | CF3 | 100 |
| Staurosporin | 100 |
The bold values are the most active compounds.
However, the vatalanib analogues (16a–d and 17a,b) (series D, E), they generally exhibited weak to moderate activity, with the derivatives having unsubstituted anilino or phenoxy moiety (16a, 17a) showing relatively better activity than their meta substituted analogues (Table 2).
Table 2.
The VEGFR-2 inhibition per cent of the synthesised phthalazines (series D, E, F) at 10 μM concentration. 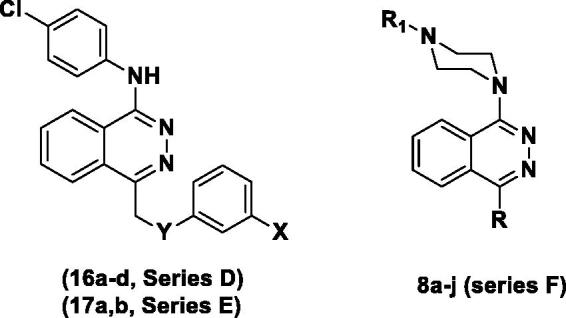
| Cpd No | Y | X | R | R1 | % inhibition |
|---|---|---|---|---|---|
| 16a | NH | H | – | – | 47 |
| 16b | NH | Cl | – | – | 21 |
| 16c | NH | CH3 | – | – | 19 |
| 16d | NH | OCH3 | – | – | 24 |
| 17a | O | H | – | – | 31 |
| 17b | O | CH3 | – | – | 15 |
| 8a | – | – | H | ethoxycarbonyl | 13 |
| 8b | – | – | H | phenyl | 8 |
| 8c | – | – | H | 2-fluorophenyl | 9 |
| 8d | – | – | H | pyridyl | 5 |
| 8e | – | – | H | 2-furoyl | 9 |
| 8f | – | – | CH3 | ethoxycarbonyl | 14 |
| 8g | – | – | CH3 | phenyl | 13 |
| 8h | – | – | CH3 | 2-fluorophenyl | 9 |
| 8i | – | – | CH3 | pyridyl | 6 |
| 8j | – | – | CH3 | 2-furoyl | 10 |
| Staurosporin | – | – | – | – | 100 |
The bold values are the most active compounds.
As expected from the cell lines results, neither of the amide-based derivatives (5a–d) (series A) nor the piperazines (8a–j)(series F) showed any significant VEGFR-2 inhibition in accordance with cell lines results.
4.1.3.2. Measurement of potential VEGFR-2 inhibitory activity (IC50)
Further investigation for the promising candidates, (6c, 12b, 12c, 13c), which exhibited VEGFR-2 inhibition percent above 70% at 10 µM concentration, were evaluated at five different concentrations (0.01, 0.1, 1, 10 and 100 µM) to calculate their IC50 values (Table 3 and Figure 3).
Table 3.
The IC50 values for compounds (6c, 12 b, 12c, 13c).
| Compound ID | VEGFR-2 IC50 |
|---|---|
| 6c | 13.4 μM |
| 12b | 4.4 μM |
| 12c | 2.7 μM |
| 13c | 2.5 μM |
Figure 3.

IC50 curves for compounds (6c, 12b, 12c and 13c) against the VEGFR-2 kinase.
Results confirmed the significant inhibition of 4-chlorophthalazines bearing a biarylurea tail with a terminal 4-chloro substitution (12b) or a terminal 4-chloro-3-CF3 substitution (12c, 13c) with IC50s of 4.4, 2.7 and 2.5 µM respectively.
4.1.4. In vitro HUVEC anti-proliferative assay
In order to further assess the antiangiogenic activity of the promising candidates, three compounds (12b, 12c and 13c) showing best VEGFR-2 IC50s were selected to be tested for their ability to in vitro inhibit VEGF-induced HUVEC cell line proliferation at single dose of 10 µM concentration.
The HUVEC anti-proliferative assay was also carried out in BPS Bioscience Corporation, San Diego, CA, USA (www.bpsbioscience.com).
Angiogenesis process comprises endothelial cell (EC) outgrowth from the parent vessel, followed by proliferation, migration, tube formation, alignment, and anastomosis to other vessels. Several in vitro models have attempted to recreate this complex progression of events 22 . The development of atherosclerotic plaques and angiogenesis, the role of the endothelium in the response of the blood vessel wall to shear forces and stretch in addition to creating a model system for the study of the regulation of endothelial cell function had emphasised the importance of using human umbilical vein endothelial cells (HUVECs). As most endothelial cell assays utilise human umbilical vein endothelial cells (HUVECs) or bovine aortic endothelial cells (BAECs) as being relatively easy to harvest from large blood vessels and also as representatives of vascular endothelial cells in vivo 23 .
The test compounds (12b, 12c and 13c) manifested significant inhibition of HUVEC cell lines of 79.83%, 72.58% and 71.60%, respectively, which may support their potential antiangiogenic activity (Table 4 and Figure 4).
Table 4.
The effect of Compounds (12 b, 12c, 13c) on HUVEC proliferation.
| Compound ID | % Cell growth | % Cell inhibition |
|---|---|---|
| 12b | 20.17 | 79.83 |
| 12c | 27.42 | 72.58 |
| 13c | 28.40 | 71.60 |
Figure 4.
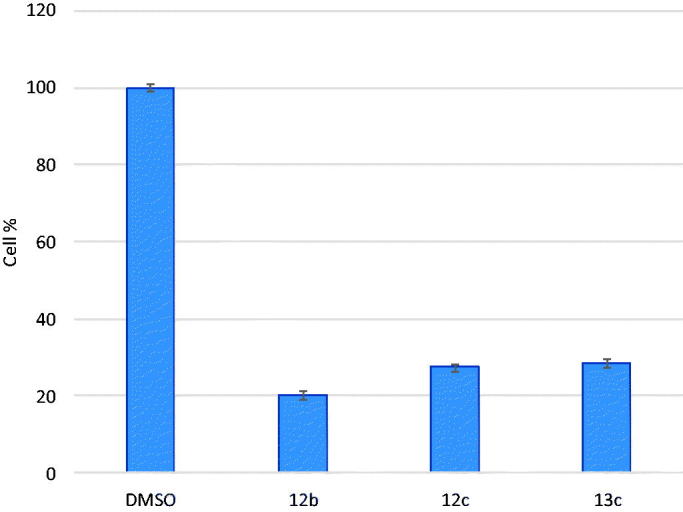
The bar graphs showing the HUVECs growth percentage after treatment with the target compounds.
4.1.5. Cell-cycle analysis
In an attempt to elucidate the potential mechanism of action of the synthesised phthalazines that exhibited potent anti-proliferative activity against the NCI cell panel but weak or moderate VEGFR-2 inhibitory activity, compounds (7b and 16a) were selected to investigate their effect on cell-cycle progression, and induction of apoptosis using breast cancer adenocarcinoma cell line (MCF-7) and colon cancer cell line (HCT-116), respectively.
From another point of view, since the inhibition of VEGF pathway has a greater effect on the induction of endothelial cell apoptosis and the decrease of vascular tubulogenesis, as the repair of endothelial lining defects caused by endothelial apoptosis is coordinated by growth factors, mainly by vascular endothelial growth factor (VEGF), which conveys survival signals to endothelial cells and acts as a potent antiapoptotic, proliferation stimulating factor 26 ; thus, the biarylurea derivative (13c) (VEGFR-2 IC50 = 2.5 µM) was selected to test its effect on cell cycle analysis and induction of apoptosis using breast cancer adenocarcinoma cell line (MCF-7).
Cell cycle is a chain of events that takes place in a cell leading to its division and replication. G1 phase, S phase (synthesis), G2 phase (collectively known as interphase) and M phase (mitosis) are considered as the four main distinct phases of the cell cycle. The G1-phase, also called post-mitotic pre-synthesis phase, directly follows cell division. The S- or synthesis phase is characterised by the process of DNA replication. The G2-, premitotic or post-synthetic phase is the time when the cell prepares to split off in two cells, the actual division. Finally, in the M- or mitosis-phase division, the doubled DNA organised in chromosomes is separated 27 .
The effect of compound (7b and 13c) on the normal cell-cycle progression was characterised using flow cytometric analysis of the DNA ploidy in MCF-7 cells using BD FACSDIVA TM software. Exposure of MCF-7 cells to either 7b or 13c at their GI50 concentration (0.32 µM, 0.57 µM, respectively) for 24 h and 48 h was shown to induce a remarkable disruption in cell cycle profile and cell-cycle arrest at S-phase boundary with concurrent time-dependent increase in pre-G cell population (Figures 5 and 6). 13c showed excellent disruption and even more prominent increase in sub-G1 cell population than 7b. The observed increase in sub-G1 cell population may imply DNA fragmentation and apoptosis as a potential mechanism for 7b/13c-induced cancer cell death.
Figure 5.

Effect of compound 7b on DNA-ploidy flow cytometric analysis of MCF7 cells. MCF-7cells were treated with DMSO 0.01% or compound 7b at its GI50 (0.32 µM), for 24 h and 48 h, and the harvested cells were subjected to Cell-cycle analysis using a FACS Calibur flow cytometer (A,B and C). Bar chart shows percentage of MCF7 cells at each phase of cell cycle in DMSO and compound 7b treated cells.
Figure 6.

Effect of compound 13c on DNA-ploidy flow cytometric analysis of MCF7 cells. MCF-7cells were treated with DMSO 0.01% or compound 13c at its GI50 (0.57 µM), for 24 h and 48 h, and the harvested cells were subjected to cell-cycle analysis using a FACS Calibur flow cytometer (A, B and C). Bar chart shows percentage of MCF7 cells at each phase of cell cycle in DMSO and compound 13c treated cells.
Furthermore, exposure of HCT-116 cells to 16a at GI50 concentration (1.20 µM) for 24 h and 48 h also seemed to induce a significant action at both S phase and sub-G1 cell population (Figure 7).
Figure 7.

Effect of compound 16a on DNA-ploidy flow cytometric analysis of HCT-116 cells. HCT-116 cells were treated with DMSO 0.01% or compound 16a at its GI50 (1.20 µM), for 24 h and 48 h, and the harvested cells were subjected to Cell-cycle analysis using a FACS Calibur flow cytometer (A,B and C). Bar chart shows percentage of HCT-116 cells at each phase of cell cycle in DMSO and compound 16a treated cells.
4.1.6. Apoptosis determination
To further investigate the ability of compounds (7b, 13c and 16a) to induce apoptosis, an Annexin V (conjugated to FITC) apoptosis detection kit was employed. This assay detects phosphatidylserine (PS) expressed on the surface of apoptotic cells and fluoresces green after interacting with the labelled Annexin V. During early apoptosis, membrane asymmetry is lost, and PS translocates from the cytoplasmic side of the membrane to the external leaflet. Propidium iodide (PI), the counter stain used in this assay, has the ability to cross only compromised membranes to intercalate into the DNA. Therefore, PI is used to detect the late stages of apoptosis by the presence of red fluorescence. Exposure of MCF-7 cells to 7b at its GI50 (0.32 µM) for 24 h and 48 h increased the percentage of Annexin-V positive cells indicating an early (lower right quadrant) or late (upper right quadrant) apoptosis in a time-dependent manner compared to DMSO treated cells (Figure 8). However, 13c and 16a increased the percentage of Annexin-V positive cells in lower right quadrant indicating early apoptosis (Figures 9 and 10).
Figure 8.

Effect of compound 7b treatment on induction of apoptosis. MCF-7 cells were treated with DMSO 0.01% or compound 7b at its GI50 (0.32 µM) for 24 or 48h, the harvested cells were stained with Annexin V-FITC apoptosis detection kit and Analyzed on a Flow cytometer.
Figure 9.

Effect of compound 13c treatment on induction of apoptosis. MCF-7 cells were treated with DMSO 0.01% or compound 13c at its GI50(0.57 µM) for 24 or 48h, the harvested cells were stained with Annexin V-FITC apoptosis detection kit and Analyzed on a Flow cytometer.
Figure 10.

Effect of compound 16a treatment on induction of apoptosis. HCT-116 cells were treated with DMSO 0.01% or compound 16a at its GI50(1.20 µM) for 24 or 48h, the harvested cells were stained with Annexin V-FITC apoptosis detection kit and Analyzed on a Flow cytometer.
4.1.7. Effects of 7b on the cellular and nuclear morphology
To further examine the apoptosis inducing effect of 7b, the cellular as well as nuclear morphological changes for MCF-7 cells treated with 7b (0.32 µM) were studied following Dapi staining. The effect of 7b on cell morphology was initially analysed with light microscopy. While control cells treated with DMSO exhibited normal morphological features, cells treated with 7b for 24 and 48 h showed deteriorated morphological changes including cell rounding, detachment and cellular fragmentation (Figure 11(A)). Similarly, fluorescence staining further indicated that, while DMSO-treated control cells exhibited uniformly dispersed chromatin, 7b-treated cells showed typical apoptotic characteristics; including condensation of chromatin (brightly stained cells), blubbing with the appearance of nuclear fragmentation (arrow heads indicate apoptotic nucleus) (Figure 11(B)). These results collectively confirms the ability of compound (7b) to induce cellular apoptosis in MCF-7 cells as a mechanism of cancer cell death.
Figure 11.

Effect of compound 7b on the cellular and nuclear morphology. MCF-7 cells were seeded on cover slips in a 6- well tissue culture plate, the cells then were treated with DMSO 0.01% or compound 7b (0.32 µM) for 24 or 48h, the cells were imaged with the light microscope to examine the cellular morphology (A) or stained with the nuclear stain Dapi to study the nuclear morphology using fluorescence microscope (B).
4.1.8. Effect of compound 7b treatment on the expression level of cleaved caspase-3
Caspase-3 is a member of the cysteine-aspartic acid protease (caspase) family. Sequential activation of caspases is considered as one of the important keys in cellular apoptosis process. Actually, caspases exists as inactive proenzymes that changed to the active form by undergoing proteolytic processing. Cleaved caspase-3 further activates a cascade of caspases that induce cell death. Thus, the ability of compound 7b to increase the expression of cleaved caspase-3 was studied as a driving force in 7b-induced apoptotic cell death. Exposure of MCF-7 cells to compound (7b) (0.32 µM) for 24 h and 48 h resulted in time-dependent increase in cleaved caspase-3 protein expression, indicating a pivotal involvement of caspases in the induced cancer cell death (Figure 12).
Figure 12.

Effect of compound 7 b on cleaved caspase-3 protein expression. MCF-7 cells were seeded on cover slips in a 6-well tissue culture plate, the cells then were treated with DMSO 0.01% or compound 7b at its GI50(0.32 µM) for 48h, the cells were then permiabilised, fixed, blocked and stained with the primary and secondary antibodies, the nuclear stain Dapi was used as a counter stain. The image were taken using a fluorescence microscope.
4.2. Molecular modelling studies
Molecular docking study was performed using AutoDock Vina 28 through docking of the synthesised compounds in the VEGFR-2 kinase active site. Docking study aimed to interpret the VEGFR-2 inhibitory activity of the investigated molecules and gain further insight into their relative binding affinities and binding interactions with the kinase active site. The coordinates of the VEGFR-2 structure were obtained from the crystal structure of VEGFR complexed with sorafenib as its inhibitor (PDB code 4ASD), which revealed the hydrogen bond interactions between the NH and CO motifs of urea moiety with the backbone of Asp1046 and the carboxylic acid moiety of Glu885, respectively, as well as a H-bond with Cys919 residue in the hinge region of the kinase active site 20 .
Owing to the fact that the chloro group of the co-crystallised ligand (sorafenib) is in the far proximity to any halogen-bond 29–31 acceptor in the binding site (Figure S7 in Supplementary material), we excluded investigating halogen-bond contacts in our docking-based assessment.
The best-scored pose of the biarylurea derivative (6c) reproduced the key interactions of the co-crystallised ligand (sorafenib) which includes H-bonding interactions of Glu885 and Asp1046 with the urea group of 6c. In addition, other hydrophobic interactions can be observed for the hydrophobic side chains of Ile892, Leu1019 and Val898 with the halogen groups (chloro and trifluoromethyl) of the phenyl moiety of 6c. Nevertheless, losing the H-bonding interaction of the Cys 919 with the terminal amide group of sorafenib can explain the lower activity of 6c compared to sorafenib (Figure 13(A)).
Figure 13.

Overlay of the best-scored docking poses of some relevant compounds on the co-crystallised ligand Sorafenib (cyan sticks) in the binding site of VEGFR2 (PDB: 4ASD):. (A) The docking pose of 6c as magenta sticks, (B) the docking poses of 12b and 12c as yellow and purple sticks, respectively, and (C) the docking pose of 13c as gold sticks. (D) The docking poses of 12c and 13c at a different perspective of the binding site showing the extended hydrophobic contact of the chloro and trifluoromethyl phenyl moiety with the side chains of PHE-918 and LEU-840. The yellow dashed-lines represent the polar contacts (H-bonding interactions). Non-polar hydrogen atoms were omitted for clarity. All presented docking poses are the best-scored by AutoDock Vina. 28
The relative higher inhibitory activity of 6c compared to similar analogues of 6a and 6b is most probably attributable to the fact that 6c possessing additional halogen group (trifluoromethyl group) on the phenyl moiety which contributes to an additional hydrophobic interaction, this also agrees with the predicted score of the best poses by AutoDock Vina 28 (Table 5). More information about rationalising the docking poses of the 6d, 6e and 6f can be found in (Figure S8 in Supplementary material).
Table 5.
The score of best-scored poses of some relevant compounds. The score is expressed as a binding affinity (kcal/mol) as an output of AutoDock Vina. a
| Compound Number | Score (SD) | Average Number of Poses Per Run |
|---|---|---|
| 6a | −11.4 (0) | 4.6 |
| 6b | −11.5 (0) | 4.6 |
| 6c | −12.4 (0.06) | 5.3 |
| 12a | −11.0 (0) | 4.3 |
| 12b | −11.1 (0.06) | 5 |
| 12c | −11.5 (0) | 8.3 |
| 13a | −10.7 (0) | 9 |
| 13b | −10.8 (0) | 9 |
| 13c | −11.0 (0) | 9 |
aThe shown score is the mean of three consecutive runs. The docking method was validated by successful pose-retrieval docking experiment of Sorafenib (score: −12.3(0)). SD is the standard deviation.
Unlike 6c, the predicted best scored poses of both biarylurea derivatives 12b and 12c showed a flip of 180 degrees compared to sorafenib and biarylurea compound (6c) (Figure 13(B)). This implies that the chlorophthalazine moiety of both derivatives 12b and 12c is directed towards the 4-chloro-3-trifluoromethylphenyl moiety of sorafenib. Avoiding a possible clash of the chloro group with the backbone of Cys 919 can explain this flipping behaviour (Figure S9 in Supplementary material). The flipped pose showed interaction pattern in the binding site that maintained the H-bonding interaction of Glu885, however, in this case with the amino group of 12b and 12c. Also, chlorophthalazine ring was packed in the hydrophobic region of Ile892, Val898 and Leu1019. Similarly, the chlorophenyl moiety of 12b and the 4-chloro-3-trifluoromethylphenyl moiety of 12c showed hydrophobic contacts with the side chains of Phe918 and Leu840 (Figure 13(B,D)). Owing to the existence of additional hydrophobic contacts of 12c (extra halogens) compared to 12a and 12b, the predicted scores showed relative improvement (Table 5). This also agrees with the marginal improvement of the observed inhibitory activity of 12c compared to similar analogs. Additional Figures of the docked poses of 12a, 12b and 12c can be found in the (Figure S10 in Supplementary material).
Like 12c, the predicted pose of 13c showed the same flip and relatively the same interaction pattern. Only exception is that the isosteric ether link in 13c showed H-bonding interaction with Asp1046 instead of Glu885 with 12c as shown in (Figure 13(C)). This agrees with the biological activity observed for both 12c and 13c.
5. Conclusion
Four series of phthalazine-based derivatives bearing biarylamides (5a–d), biarylurea (6a–f, 7a–f, 12a–c and 13a–c) or a substituted piperazine moiety at position 1 of the phathalazine nucleus (8a–j), in addition to two other series of vatalanib analogs (16a–d, and 17a,b) were designed, synthesised as anticancer agents exerting potent anti-proliferative activity as well as apoptosis inducers in tumour cells. Eight of the phthalazines derivatives; namely the biarylureas (6b, 6e, 7b, 13a and 13c) and the vatalanib analogues (16a, 16d and 17a) exhibited excellent broad spectrum anti-proliferative activity in NCI 5-log dose screening against 60 cell panel with GI50 values ranging from 0.15 to 8.41 µM, especially on leukaemia, renal, melanoma and breast cancer cell lines. Accordingly, the three compounds (7b, 13c and 16a) were selected to further investigate the potential underlying mechanisms that induced cancer cell death. The latter three compounds were found to induce cell-cycle arrest with concurrent increase in pre-G cell population and increase the apoptotic cell population in time-dependent manner. Moreover, compound (7b) was found to disrupt the normal cellular and nuclear morphology and increase the expression of cleaved caspase-3, the pro-apoptotic protein, in MCF-7 cell lines which collectively indicate the involvement of these compounds in apoptotic-induced cell death. Moreover, all the synthesised phthalazines were evaluated for their VEGFR-2 inhibitory activity, which revealed the significant inhibition of the 4-chlorophthalazines having a biarylurea tail with either a terminal 4-chloro (12b) or a terminal 4-chloro-3-trifhuoromethyl substituents (12c and 13c) with IC50 of 4.4, 2.7 and 2.5 µM, respectively. 4-chloropthalazines were found to exhibit better VEGFR-2 activity than their corresponding 4-methyl or 4-unsubstituted analogues. 12b, 12c and 13c also displayed significant inhibition of VEGF-stimulated proliferation of human umbilical vein endothelial cells (HUVEC) with 79.83, 72.58 and 71.6% inhibition, respectively, at 10 µM concentration. Finally, molecular docking study of the active biarylurea compounds (6c, 12b, 12c and 13c) in VEGFR-2 kinase active site revealed their ability to form the essential H bond interactions with Glu 885 or Asp1046 key residues in the VEGFR-2 active site.
6. Experimental
6.1. Chemistry
Starting materials and reagents were purchased from Sigma – Aldrich or Acros Organics. Melting points were recorded on Gallen Kamp apparatus and were uncorrected. FT-IR spectra were recorded on a Shimadzu IR 435 spectrophotometer. 1HNMR spectra were recorded in δ scale given in ppm on a Varian 400 MHz spectrophotometer or a Varian 300 MHz spectrophotometer. Coupling patterns are described as follows: s, singlet; d, doublet, dd, doubled doublet; t, triplet; m, multiplet. J describes a coupling constant. The coupling constants were rounded off to one decimal place. MS spectra mass were recorded on Hewlett Packard 5988 spectrometer (70 eV). Elemental analyses were performed at the Microanalytical Center, Al-Azhar University. Compounds 1a–c 21 , 2a,b 32 and 3a,b 33 , 9 and 10 34 were prepared following reported procedures.
6.1.1. Synthesis of N1-(4-substituted-phthalazin-1-yl)benzene-1,4-diamine (4a,b)
General procedure:
p-Phenylenediamine (0.33 g, 3.12 mmol, 3equiv) and the respective chlorophthalazine (2.08 mmol, 2equiv) were treated with 2-BuOH (7.5 ml) in a tube and heated to 110 °C. The reaction quickly became a solid, yellow mass. After four hours, the reaction was cooled and diluted with water. The resultant slurry was then partitioned between equal volumes of DCM and 1 N NaOH (15 ml). The aqueous layer was extracted into DCM (2x15 ml). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The resulting orange solid was crystallised from (EtOAc/pet ether 1:1) to afford the title compounds (4a,b) as an orange crystals.
6.1.1.1. N1-(Phthalazin-1-yl)benzene-1,4-diamine (4a)
Yield (0.21 g, 91%); mp 284–286 °C; 1HNMR (300 MHz, DMSO-d6): δ 9.15 (1H, s, –NH D2O exchangeable), 9.04 (2H, s, –NH2 D2O exchangeable), δ 8.95–8.92 (2H, d, J = 9 Hz, phthalazine), δ 8.18–8.15 (2H,d, J = 9 Hz, phthalazine), δ 7.97 (1H, s, phthalazine), δ 7.65–7.62 (2H,d, J = 9 Hz, ArH), δ 6.97–6.83 (2H,d, J = 9 Hz, ArH); FT-IR (ύ max, cm−1): 3400 (NH2), 3329 (NH); MS (Mwt.: 236.11): m/z 236.00 (M+, 43.00%), 212.00 (100.00%), 118.00 (54.00%), 56.00 (70.00%); Anal. Calcd for C14H12N4: C, 71.17; H, 5.12; N, 23.71; Found: C, 71.34; H, 5.17; N, 23.96
6.1.1.2. N1-(4-Methylphthalazin-1-yl)benzene-1,4-diamine (4b)
Yield (0.22 g, 88%); mp 240–242 °C; 1HNMR (300 MHz, DMSO-d6): δ 8.88 (1H, s, –NH D2O exchangeable), 8.85 (2H, s, –NH2 D2O exchangeable), δ 8.13–8.08 (2H, d, J = 7 Hz, phthalazine), δ 7.95–7.92 (2H, d, J = 7 Hz, phthalazine), δ 7.46–7.43 (2H, d, J = 9 Hz, ArH), δ 6.83–6.76 (2H, d, J = 9 Hz, ArH), δ 2.78 (3H, s, CH3); 13CNMR (100 MHz, DMSO) : δ 162.4, 150.5, 135.2, 134.5, 131.2, 130.7, 128.0, 127.3, 125.0, 122.8, 120.7, 120.2, 115.6, 115.3, 18.2; FT-IR (ύ max, cm−1): 3385 (NH2), 3321 (NH); MS (Mwt.: 250.12): m/z 250.00 (M+, 50.00%), 249.00 (100.00%), 108.00 (60.92%), 65.00 (77.59%); Anal. Calcd for C15H14N4: C, 71.98; H, 5.64; N, 22.38; Found: C, 72.09; H, 5.78; N, 22.52.
6.1.2. Synthesis of N-(4–(4-Substitutedphthalazin-1-ylamino)phenyl)-4-substituted-benzamides (5a-d)
General procedure:
To a stirred solution of the respective N1-(4-arylphthalazin-1-yl)benzene-1,4-diamine (4a,b) (10.0 mmol, 1equiv) and triethylamine (1.1 ml, 10.0 mmol) in acetonitrile (20 ml), the respective benzoyl chloride (viz.; benzoyl chloride, 4-chlorobenzoyl chloride) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 6 h, until the disappearance of starting material as judged by TLC (CHCl3/CH3OH 9.5:0.5). The reaction mixture was filtered and then the filtrate was concentrated in vacuo, to afford the crude product which was purified by flash column chromatography (EtOAc/pet ether 9:1) to afford 5a–d as yellow crystals.
6.1.2.1. N-(4-(Phthalazin-1-ylamino)phenyl)benzamide (5a)
Yield (0.17 g; 50%), mp 142–144 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.66 (1H, s, –NH D2O exchangeable), δ 9.12 (1H, s, –NH D2O exchangeable), δ 8.62 (1H, s, J = 8 Hz, phthalazine), δ 8.11–8.08 (2H, d, J = 7.6 Hz, Ar-H), δ 7.98–7.96 (2H, d, J = 8 Hz, phthalazine), δ 7.93–7.88 (2H, m, phthalazine), δ 7.64–7.62 (1H, m, Ar-H), δ 7.62–7.60 (2H, d, J = 7.6 Hz, Ar-H), δ 7.59–7.57 (2H, d, J = 6.8 Hz, Ar-H), δ 7.50–7.48 (2H, m, Ar-H); FT-IR (ύ max, cm−1): 3550, 3510 (2NH), 1649 (C=O); MS (Mwt.: 340.38): m/z 340.15 [M+, 1.58%), 138.05 (2.70%), 86.10 (100%); Anal. Calcd for C21H16N4O: C, 74.10; H, 4.74; N, 16.46; Found: C, 74.34; H, 4.80; N, 16.58.
6.1.2.2. 4-Chloro-N-(4-(phthalazin-1-ylamino)phenyl)benzamide (5b)
Yield (0.20 g; 54%), mp 156–158 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.50 (1H, s, –NH D2O exchangeable), δ 9.15 (1H, s, –NH D2O exchangeable), δ 8.99 (1H, s, phthalazine), δ 8.11–8.08 (2H, d, J = 9 Hz, Ar-H), δ 7.94–7.92 (2H,d, J = 7.6 Hz, phthalazine), δ 7.91–7.90 (2H, d, J = 7.6 Hz phthalazine), δ 7.56–7.54 (2H,d, J = 9 Hz, Ar-H), δ 7.53–7.51 (2H,d, J = 7.5 Hz, Ar-H), δ 7.31–7.28 (2H, m, Ar-H); FT-IR (ύ max, cm−1): 3355, 3310 (2NH), 1640 (C=O); MS (Mwt.: 374.82): m/z 375.00 (M+, 1.57%), 245.00 (2.41%), 139.05 (46.44%), 111.10 (25.30), 64.00 (100.00%); Anal. Calcd for C21H15ClN4O: C, 67.29; H, 4.03; N, 14.95; Found: C, 67.52; H, 4.08; N, 15.22.
6.1.2.3. N-(4–(4-Methylphthalazin-1-ylamino)phenyl)benzamide(5c)
Yield (0.24 g; 68%), mp 283–285 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.50 (1H, s, –NH D2O exchangeable), δ 10.40 (1H, s, –NH D2O exchangeable), δ 8.27–8.24 (2H, d, J = 7.6 Hz, phthalazine), δ 8.02–8.00 (2H, m, Ar-H), δ 7.99–7.97 (2H,d, J = 7.6 Hz, phthalazine), δ 7.77–7.73 (1H, d, J = 9.2 Hz, Ar-H), δ 7.63–7.61 (2H,d, J = 7.2 Hz, Ar-H), δ 7.59–7.56 (2H,d, J = 7.6 Hz, Ar-H), δ 7.54–7.51 (2H, m, Ar-H), δ 2.91 (3H,s, CH3); FT-IR (ύ max, cm−1): 3331, 3257 (2NH), 1649 (C=O); MS (Mwt.: 354.15): m/z 354.95 (M+, 1.18%), 315.95 (15.90%), 105.00 (100.00%); Anal. Calcd for C22H18N4O: C, 74.56; H, 5.12; N, 15.81; Found: C, 74.78; H, 5.19; N, 16.04
6.1.2.4. 4-Chloro-N-(4–(4-methylphthalazin-1-ylamino)phenyl)benzamide (5d)
Yield (0.20 g; 51%), mp 147–149 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.65 (1H, s, –NH D2O exchangeable), δ 10.45 (1H, s, –NH D2O exchangeable), δ 8.05–8.03 (2H, d, J = 8 Hz, phthalazine), δ 7.97–7.95 (2H,d, J = 7.6 Hz, Ar-H), δ 7.88–7.86 (2H, d, J = 8 Hz, phthalazine), δ 7.63–7.61 (2H, d, J = 7.6 Hz, Ar-H), δ 7.59–7.57 (2H,d, J = 7.6 Hz, Ar-H), δ 7.31–7.28 (2H, m, Ar-H), δ 2.91 (3H, s,CH3); 13CNMR (100 MHz, DMSO) : δ 166.9, 165.0, 151.5, 138.45, 138.2, 137.5, 137.0, 136.9, 134.6, 133.9, 131.6, 130.9, 130.2, 130.2, 130.1, 129.2, 128.9, 128.1, 125.3, 121.5, 121.2, 18.9; FT-IR (ύ max, cm-1): 3300, 3192 (2NH), 1678 (C=O); MS (Mwt.: 388.85): m/z 388.90 (M+, 7.10%), 279.90 (7.84%), 248.95 (25.65%), 139.00 (100%); Anal. Calcd for C22H17ClN4O: C, 67.95; H, 4.41; N, 14.41; Found: C, 68.24; H, 4.48; N, 14.49.
6.1.3. Synthesis of 1-Aryl-3–(4-(4-substitutedphthalazin-1-ylamino)phenyl)ureas (6a-f)
General procedure:
To a stirred solution of the respective N1-(4-arylphthalazin-1-yl)benzene-1,4-diamine (4a,b) (10.0 mmol, 1equiv) in DMF (20 ml), the respective phenylisocyanate (viz.; phenyl- isocyanate, 4-chlorophenylisocyanate, 3-trifluoromethyl-4-chlorophenylisocyanate) (10.0 mmol, 1 equiv) was added and the mixture was heated under reflux for 6 h, after which TLC (CHCl3/CH3OH 9:1) showed no starting material. The reaction mixture was poured over ice water, the formed precipitate was allowed to settle, then filtered off and dried to afford the crude products(6a-f) which was further crystallised from EtOAc.
6.1.3.1. 1-Phenyl-3–(4-(phthalazin-1-ylamino)phenyl)urea (6a)
Yield (0.10 g; 30%), mp 145– 147 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.22 (1H, s,–NH D2O exchangeable), δ 9.16 (1H,s, –NH D2O exchangeable), δ 9.09 (1H, s, –NH D2O exchangeable), δ 8.74 (1H, s, phthalazine), δ 8.68–8.66 (2H, d, J = 8 Hz, phthalazine), δ 8.02–8.00 (2H, d, J = 8 Hz, phthalazine), δ 7.58–7.56 (2H, m, Ar-H), δ 7.48–7.46 (2H, d, J = 7.6 Hz, Ar-H), δ 7.31–7.29 (2H,d, J = 7.2 Hz, Ar-H), δ 7.19–7.17 (1H, m, Ar-H), δ 6.98–6.96 (2H, d, J = 7.2 Hz, Ar-H) FT-IR (ύ max, cm−1): 3300, 3047 (3NH), 1700 (C=O); MS (Mwt.: 355.14): m/z 355.10 (M+, 8.88%), 289.10 (9.51%), 123.10 (15.43%), 69.00(100.00%); Anal. Calcd for C21H17N5O: C, 70.97; H, 4.82; N, 19.71; Found: C, 71.21; H, 4.89; N, 19.88
6.1.3.2. 1–(4-Chlorophenyl)-3–(4-(phthalazin-1-ylamino)phenyl)urea (6b)
Yield (0.20 g; 53%), mp 168–171 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.33 (1H, s, –NH D2O exchangeable), δ 10.07 (1H, s, –NH D2O exchangeable), δ 9.16 (1H, s, –NH D2O exchangeable), δ 8.96 (1H, s, phthalazine), δ 8.09–8.07 (2H, d, J = 9.2 Hz, phthalazine), δ 7.63–7.61 (2H, d, J = 9.2 Hz, phthalazine), δ 7.49–7.47 (2H, d, J = 8.8 Hz, Ar-H), δ 7.44–7.42 (2H, d, J = 8 Hz, Ar-H), δ 7.33–7.31 (d, J = 8.8 Hz, 2H, Ar-H), δ 6.56–6.54 (2H,d, J = 8 Hz, Ar-H); 13CNMR (100 MHz, DMSO) : δ 160.2, 153.0, 145.4, 139.4, 137.6, 135.8, 134.8, 133.2, 132.6, 129.7, 129.2, 128.9, 127.7, 122.6, 122.4, 121.8, 121.5, 120.3, 119.9, 118.6, 115.6; FT-IR (ύ max, cm−1): 3396, 3341 (3NH), 1672 (C=O); MS (Mwt.: 389.10): m/z 389.00 (M+, 1.51%), 281.95 (19.41%), 253.95(100.00%), 152.95 (13.00%); Anal. Calcd for C21H16ClN5O: C, 64.70; H, 4.14; N, 17.96; Found: C, 64.89; H, 4.21; N, 18.17.
6.1.3.3. 1–(4-Chloro-3-(trifluoromethyl)phenyl)-3–(4-(phthalazin-1-ylamino)phenyl)urea (6c)
Yield (0.22 g; 49%), mp 288–291 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.08 (1H, s, –NH D2O exchangeable), δ 9.36 (1H, s, –NH D2O exchangeable), δ 9.11 (1H, s, –NH D2O exchangeable), δ 8.22 (1H, s, Ar-H), 8.09–8.07 (2H, d, J = 8.8 Hz, phthalazine), δ 7.97–7.95 (2H, d, J = 8.8 Hz, phthalazine), δ 7.79 (1H, s, Ar-H), δ 7.69–7.67 (2H, d, J = 9.6 Hz, Ar-H), δ 7.52–7.50 (2H, d, J = 8.4 Hz, Ar-H), δ 7.39–7.37 (2H, d, J = 8.4 Hz, Ar-H); FT-IR (ύ max, cm−1): 3321, 3263, 3126 (3NH), 1649 (C=O); MS (Mwt.: 457.09): m/z 457.90 (M+, 1.06%), 415.90 (7.68%), 194.95(100.00%); Anal. Calcd for C22H15ClF3N5O: C, 57.71; H, 3.30; N, 15.30; Found: C, 57.93; H, 3.28; N, 15.48
6.1.3.4. 1–(4-(4-Metyhlphthalazin-1-ylamino)phenyl)3-phenylurea(6d)
Yield (0.06 g; 20%), mp 284–286 °C; 1HNMR (300 MHz, DMSO-d6): δ 9.20(1H, s, –NH D2O exchangeable), δ 9.14 (1H, s,–NH D2O exchangeable), δ 9.06 (1H, s,–NH D2O exchangeable), δ 8.93–8.91 (1H, d, J = 7 Hz, phthalazine), δ 8.77–8.75 (1H, d, J = 7 Hz, phthalazine), δ 8.22–8.20 (2H, d, J = 7 Hz, phthalazine), δ 7.85–7.83 (2H, d, J = 8.4 Hz, Ar-H), δ 7.52–7.49 (2H, d, J = 8.4 Hz, Ar-H), δ 7.43–7.40 (2H, d, J = 7.2 Hz, Ar-H), δ 7.25–7.23 (1H, m, Ar-H), δ 6.95–6.93 (2H, d, J = 7.2 Hz, Ar-H), δ 2.81 (3H, s, CH3);) ; FT-IR (ύ max, cm−1): 3292, 3194, 3132 (3NH), 1651 (C=O); MS (Mwt.: 369.16): m/z 369.00 (M+, 2.99%), 212.00 (6.90%), 93.00 (100.00%);Anal. Calcd for C22H19N5O: C, 71.53; H, 5.18; N, 18.96; Found: C, 71.69; H, 5.27; N, 19.21
6.1.3.5. 1–(4-Chlorophenyl)-3–(4-(4-methylphthalazin-1-ylamino)phenyl)urea (6e)
Yield (0.20 g; 50%), mp 178–180 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.20(1H, s, –NH D2O exchangeable), δ 9.17 (1H, s, –NH D2O exchangeable), δ 9.03 (1H, s, –NH D2O exchangeable), δ 8.18–8.16 (2H, d, J = 7.6 Hz, phthalazine), δ 8.09–8.05 (2H, d, J = 7.6 Hz, phthalazine), δ 7.86–7.84 (2H, d, J = 8.8 Hz, Ar-H), δ 7.73–7.71 (2H, d, J = 8.8 Hz, Ar-H), δ 7.49–7.47 (2H, d, J = 8.8 Hz, Ar-H), δ 7.33–7.30 (2H, d, J = 8.8 Hz, Ar-H), δ 2.80 (3H, s,CH3); FT-IR (ύ max, cm−1): 3294, 3188, 3168 (3NH), 1681 (C=O); MS (Mwt.: 403.86): m/z 403.00 (M+, 1.51%), 276.95 (17.01%), 127.00 (100.00%); Anal. Calcd for C22H18ClN5O: C, 65.43; H, 4.49; N, 17.34; Found: C, 65.62; H, 4.52; N, 17.52.
6.1.3.6. 1–(4-Chloro-3-(trifluoromethyl)phenyl)-3–(4-(4-methylphthalazin-1-ylamino)phenyl)-urea (6f)
Yield (0.30 g; 63%), mp 258–260 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.43(1H, s, –NH D2O exchangeable), δ 9.14 (1H, s, –NH D2O exchangeable), δ 9.02 (1H, s, –NH D2O exchangeable), δ 8.88–8.86 (2H, d, J = 8 Hz, phthalazine), δ 8.24–8.22 (2H, d, J = 8 Hz, phthalazine), δ 8.13 (1H, s,Ar-H), δ 7.97–7.95 (1H, d, J = 9.2 Hz, Ar-H), δ 7.88–7.86 (1H, d, J = 9.2 Hz, Ar-H), δ 7.63–7.61 (2H, d, J = 10 Hz, Ar-H), δ 6.78–6.76 (2H, d, J = 10 Hz, Ar-H), δ 2.80 (3H, s, CH3); 13CNMR (100 MHz, DMSO) : δ 160.8, 153.0, 140.7, 139.4, 134.0, 132.7, 131.9, 127.7, 127.3, 127.1, 125.6, 124.8, 123.9, 122.1, 121.8, 121.4, 119.8, 119.5, 118.8, 118.6, 117.6, 115.4, 19.6; FT-IR (ύ max, cm−1): 3363, 3255, 3153 (3NH), 1681(C=O); MS (Mwt.: 471.86): m/z 472.10(M+, 0.30%), 223.00 (43.56%), 195.00 (70.63%), 52.05 (100%); Anal. Calcd for C23H17ClF3N5O: C, 58.54; H, 3.63; N, 14.84; Found: C, 58.61; H, 3.61; N, 15.03
6.1.4. 1-Aryl-3–(4-(4-substituted-phthalazin-1-yloxy)phenyl)ureas (7a-f)
General procedure:
To a stirred solution of the respective chlorophthalazine derivative (3a,b) (10.0 mmol, 1equiv), and cesium carbonate (6.5 g, 20.0 mmol, 2equiv) in acetonitrile (20 ml), the appropriate 1-aryl-3–(4-hydroxyphenyl)urea (1a-c) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 6 h, after which TLC (CHCl3/CH3OH 9:1) showed no starting material. The filtrate was evaporated in-vacuo to afford the crude product (7a-f) which was further purified by column chromatography (using gradient elution starting from EtOAc then 1% MeOH/EtOAc).
6.1.4.1. 1-Phenyl-3–(4-(phthalazin-1-yloxy)phenyl)urea (7a)
Yield (0.22 g; 64%), mp 180–182 °C; 1HNMR(300 MHz, DMSO-d6): δ 9.57 (1H, s, –NH D2O exchangeable), δ 9.37 (1H, s, –NH D2O exchangeable), δ 8.36 (1H, s, phthalazine), δ 8.10–8.00 (2H, m, phthalazine), δ 7.93–7.91 (2H, d, J = 7 Hz, phthalazine), δ 7.91–7.90 (2H, m, Ar-H), δ 7.49–7.47 (2H, d, J = 7.8 Hz, Ar-H), δ 7.19–7.25 (m, 3H, Ar-H), δ 6.89–6.86 (1H, d, J = 7.5 Hz, Ar-H), δ 6.67–6.65 (1H, d, J = 7.8 Hz, Ar-H); FT-IR (ύ max, cm−1): 3304, 3235 (2NH), 1641 (C=O); MS (Mwt.: 356.38): m/z 355.00 (M-1+, 0.11%), 146.05 (23.71%), 118.05 (8.47%), 79.95 (100.00%); Anal. Calcd for C21H16N4O2: C, 70.77; H, 4.53; N, 15.72; Found: C,71.02; H, 4.61; N, 15.89
6.1.4.2. 1–(4-Chlorophenyl)-3–(4-(phthalazin-1-yloxy)phenyl)urea (7b)
Yield (0.19 g; 50%), mp 166–168 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.72 (1H, s, –NH D2O exchangeable), δ 9.44 (1H, s, –NH D2O exchangeable), δ 8.37 (1H, s, phthalazine), δ 8.28–8.26 (2H, d, J = 8 Hz, phthalazine), δ 8.18–8.16 (2H, d, J = 8 Hz, phthalazine), δ 7.58–7.56 (2H, d, J = 8.8 Hz, Ar-H), δ 7.56–7.53 (2H, d, J = 8.8 Hz, Ar-H), δ 7.36–7.34 (2H, d, J = 7.2 Hz, Ar-H), δ 7.14–7.12 (2H, d, J = 7.2 Hz, Ar-H); FT-IR (ύ max, cm−1): 3278, 3161 (2NH), 1701 (C=O); MS (Mwt.: 390.82): m/z 390.10 (4.47%), 365.10 (24.02%), 75.00 (100.00%);Anal. Calcd for C21H15ClN4O2: C, 64.54; H, 3.87; N, 14.34; Found: C, 64.69; H, 4.01; N, 14.62
6.1.4.3. 1–(4-Chloro-3-(trifluoromethyl)phenyl)-3–(4-(phthalazin-1-yloxy)phenyl)urea (7c)
Yield (0.22 g; 49%), mp 158–160 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.91 (1H, s, –NH D2O exchangeable), δ 9.08 (s, 1H, –NH D2O exchangeable), δ 8.39 (s, 1H, phthalazine), δ 8.24–8.21 (d, J = 8 Hz, 2H, phthalazine), δ 8.17–8.15 (d, J = 8 Hz, 2H, phthalazine), δ 7.99 (s, 1H, Ar-H), δ 7.85–7.83 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.76–7.74 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.28–7.26 (d, J = 8.4 Hz, 2H, Ar-H), δ 6.76–6.74 (d, J = 8.4 Hz, 1H, Ar-H); 13CNMR (100 MHz, DMSO) : δ171.0, 153.7, 153.2, 141.1, 138.7, 133.8, 132.7, 132.0, 131.7, 129.5, 129.3, 129.1, 128.5, 127.0, 125.8, 124.7, 121.4, 121.2, 121.0, 119.6, 118.7, 115.6; FT-IR (ύ max, cm−1): 3300, 3153 (2NH), 1679 (C=O); MS (Mwt.: 458.82): m/z 458.10 (2.63%), 271.05 (7.73%), 195.00 (100.00%); Anal. Calcd for C22H14ClF3N4O2: C, 57.59; H, 3.08; N, 12.21; Found: C, 57.82; H, 3.06; N, 12.57
6.1.4.4. 1–(4-(4-Methylphthalazin-1-yloxy)-3-phenylurea (7d)
Yield (0.20 g; 54%), mp 174–176 °C; 1HNMR (400 MHz, DMSO-d6): δ 8.76 (1H, s, –NH D2O exchangeable), δ 8.69 (1H, s, –NH D2O exchangeable), δ 8.39–8.37 (1H, d, J = 8 Hz, phthalazine), δ 8.23–8.21 (1H, d, J = 8 Hz, phthalazine), δ 8.11–8.09 (1H, d, J = 8 Hz, phthalazine), δ 8.08–8.06 (1H, d, J = 8 Hz, phthalazine), δ 7.55–7.52 (2H, d, J = 8.8 Hz, Ar-H), δ 7.46–7.43 (2H, d, J = 8.8 Hz, Ar-H), δ 7.33–7.30 (2H, d, J = 7.6 Hz, Ar-H), δ 7.22–7.20 (2H, d, J = 8.8 Hz, Ar-H), δ 6.69–6.67 (1H, d, J = 7.6 Hz, Ar-H), δ 2.96 (3H, s,CH3); FT-IR (ύ max, cm−1): 3302, 3141 (2NH), 1643 (C=O); MS (Mwt.: 370.40): m/z 370.00 [M+, 2.28%), 277.95 (19.33%), 251.00 (100.00%); Anal. Calcd for C22H18N4O2: C, 71.34; H, 4.90; N, 15.13; Found: C, 71.49; H, 4.97; N, 15.29.
6.1.4.5. 1–(4-Chlorophenyl)-3–(4-(4-methylphthalazin-1-yloxy)phenyl)urea (7e)
Yield (0.17 g; 42%), mp 169–171 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.06 (1H, s, –NH D2O exchangeable), δ 10.05 (1H, s, –NH D2O exchangeable), δ 8.38–8.36 (1H, d, J = 7.6 Hz, phthalazine), δ 8.20–8.18 (1H, d, J = 7.6 Hz, phthalazine), δ 8.07–8.04 (2H, d, J = 7.6 Hz, phthalazine), δ 7.58–7.56 (2H, d, J = 8.4 Hz, Ar-H), δ 7.29–7.22 (2H, d, J = 8.4 Hz, Ar-H), δ 7.20–7.18 (2H, d, J = 9.2 Hz, Ar-H), δ 6.66–6.63 (2H, d, J = 9.2 Hz, Ar-H), δ 2.81 (3H, s, CH3); FT-IR (ύ max, cm−1): 3300, 3153 (2NH), 1643 (C=O); MS (Mwt.: 404.85): m/z 404.90(M+, 2.04%), 385.85 (1.29%), 267.90 (4.75), 250.95 (28.16), 127.00 (100.00%); Anal. Calcd for C22H17ClN4O2: C, 65.27; H, 4.23; N, 13.84; Found: C, 65.52; H, 4.28; N, 13.97.
6.1.4.6. 1–(4-Chloro-3-(trifluoromethyl)phenyl)-3–(4-(4-metyhlphthalazin-1-yloxy)phenyl)urea (7f)
Yield (0.20 g; 42%), mp 150–152 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.31 (1H, s, –NH D2O exchangeable), δ 10.30 (1H, s, –NH D2O exchangeable), δ 8.37 (1H, s, Ar-H), δ 8.19–8.16 (2H, d, J = 8 Hz, phthalazine), δ 8.07–8.05 (1H, d, J = 7.6 Hz, Ar-H), δ 7.78–8.75 (1H,d, J = 8 Hz, phthalazine), δ 7.61–7.59 (2H, d, J = 7.6 Hz, Ar-H), δ 7.56–7.54 (1H, d, J = 8.8 Hz, Ar-H), δ 7.24–7.21(2H, d, J = 8.8 Hz, Ar-H), δ 2.80 (3H, s, CH3); FT-IR (ύ max, cm−1): 3300, 3153 (2NH), 1679 (C=O); MS (Mwt.: 472.85): m/z 472.80 (M+, 2.07%), 391.95 (10.86), 335.85 (25.53), 250.95 (86.76%), 194.90 (100%); Anal. Calcd for C23H16ClF3N4O2: C, 58.42; H, 3.41; N, 11.85; Found: C, 58.49; H, 3.39; N, 12.01
6.1.5. Synthesis of ethyl 4–(4-substitutedphthalazine-1-yl)piperazine-1-carboxylate and 1-Substituted-4-(arylpiperazine-1-yl)phthalazine (8a–j)
General procedure:
To a stirred mixture of the respective 1-chlorophthalazine (3a,b) (10.0 mmol, 1equiv), potassium carbonate (0.27 g, 20.0 mmol, 2equiv), potassium iodide (0.01 g, 0.01 mmol, 0.1equiv) in absolute ethanol (20 ml), the respective piperazine (viz.; ethyl piperazinecarboxylate, phenylpiperazine, 2-flourophenylpiperazine, 2-pyridylpiperazine, 2-furoylpiperazine) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 2 h, after which TLC (CHCl3/CH3OH 9:1) showed no starting material. The mixture was then concentrated in vacuo, the residue was washed with water, extracted with EtOAc, dried over anhydrous Na2SO4. The crude product was recrystallised from EtOAc.
6.1.5.1. Ethyl 4-(phthalazin-1-yl)piperazine-1-carboxylate (8a)
Yield (0.19 g; 68%), mp 271–273 °C; 1HNMR (300 MHz, DMSO): δ 8.10 (1H, s, phthalazine), δ 7.98–7.96 (2H, d, J = 7 Hz, phthalazine), δ 7.93–7.91 (2H, d, J = 7 Hz, phthalazine), δ 4.07 (2H, q,CH2-CH3), δ 3.63–3.62 (2H, m, piperazine ring), δ 2.86–2.85 (2H, m, piperazine ring), δ 2.55–2.54 (2H, m, piperazine ring), δ 2.27–2.26 (2H, m, piperazine ring), δ 1.14 (3H, t, CH2-CH3); 13CNMR (100 MHz, DMSO) : δ 179.9, 154.9, 149.2, 134.1, 132.3, 129.6, 127.2, 123.1, 122.7, 61.6, 43.1, 41.2, 30.1, 30.0, 14.6; FT-IR (ύ max, cm−1): 1672 (C=O); MS (Mwt.: 286.33): m/z 286.05 (M+, 0.95%), 270.00 (3.77%), 56.00 (100.00%); Anal. Calcd for C15H18N4O2: C, 62.92; H, 6.34; N, 19.57; Found: C, 63.23; H, 6.38; N, 19.71
6.1.5.2. 1–(4-Phenylpiperazin-1-yl)phthalazine (8b)
Yield (0.20 g; 69%), mp 242–244 °C; 1HNMR (300 MHz, DMSO): δ 9.72–7.96 (1H, s, phthalazine), δ 8.17–8.15 (1H, d, J = 8 Hz, phthalazine), δ 8.12–8.10 (1H, d, J = 8 Hz, phthalazine), δ 8.00–7.95 (2H, d, J = 8 Hz, phthalazine), δ 7.28–7.24 (2H, d, J = 7.6 Hz, Ar-H), δ 7.05–7.03 (2H, d, J = 7.6 Hz, Ar-H), δ 6.84–6.81 (1H, d, J = 7.6 Hz, Ar-H), δ 3.58–3.56 (4H, m, piperazine ring), δ 3.44–3.42 (4H, m, piperazine ring); 13CNMR (100 MHz, DMSO) : δ151.5, 148.2, 129.5, 128.6, 127.3, 124.3, 119.7, 116.0, 51.2, 48.7; FT-IR (ύ max, cm−1): 3428 (CH aromatic), 2991, 2919 (CH aliphatic); MS (Mwt.: 290.36): m/z 290.10 (M+, 0.41%), 162.10 (8.10%), 120.10 (100.00%), Anal. Calcd for C18H18N4: C, 74.46; H, 6.25; N, 19.30; Found: C, 74.62; H, 6.37; N, 19.54.
6.1.5.3. 1–(4-(2-Fluorophenyl)piperazin-1-yl)phthalazine (8c)
Yield (0.21 g; 70%), mp 88–90 °C; 1HNMR (300 MHz, DMSO): δ 8.61 (1H, s, phthalazine), δ 8.27–8.24 (2H, d, J = 9 Hz, phthalazine), δ 8.14–8.11 (2H, d, J = 9 Hz, phthalazine), δ 7.19–7.17 (1H, d, J = 7.6 Hz, Ar-H), δ 7.15–7.13 (1H, d, J = 7.6 Hz, Ar-H), δ 7.08–7.06 (1H, d, J = 7.6 Hz, Ar-H), δ 6.99–6.97 (1H, d, J = 7.6 Hz, Ar-H), δ 3.70–3.69 (2H, m, piperazine ring), δ 3.31–3.30 (2H, m, piperazine ring), δ 3.23–3.21 (4H, m, piperazine ring); 13CNMR (100 MHz, DMSO) : δ 175.9, 159.2,145.3, 138.9, 138.3, 134.8, 134.0, 130.0, 127.6, 125.6, 123.3, 122.8, 122.7, 116.0, 50.8, 49.9, 47.0, 46.9; FT-IR (ύ max, cm−1): 3039 (CH aromatic), 2924 (CH aliphatic); MS (Mwt.: 308.35): m/ z 308.05 (M+, 1.81%), 180.05 (19.88%), 138.10 (100.00%); Anal. Calcd for C18H17FN4: C, 70.11; H, 5.56; N, 18.17; Found: C, 70.29; H, 5.60; N, 18.28.
6.1.5.4. 1–(4-(Pyridin-2-yl)piperazin-1-yl)phthalazine (8d)
Yield (0.20 g; 69%), mp 286–288 °C; 1HNMR (300 MHz, DMSO): δ 8.14–8.12 (1H, d, J = 7 Hz, pyridine ring), δ 7.93–7.91 (3H, m, phthalazine), δ 7.59–7.56 (2H, d, J = 9 Hz, phthalazine), δ 7.56–7.54 (1H, d, J = 7 Hz, pyridine ring), δ 6.69–6.67 (2H, d, J = 7 Hz, pyridine ring), δ 3.67–3.65 (4H, m, piperazine ring), δ 3.06–3.04 (4H, m, piperazine ring); FT-IR (ύ max, cm−1): 3030 (CH aromatic), 2926 (CH aliphatic); MS (Mwt.: 291.35): m/z 291.10 (M+, 0.19%), 279.05 (0.28%), 160.10 (27.24%), 95.05 (100.00); Anal. Calcd for C17H17N5: C, 70.08; H, 5.88; N, 24.04; Found: C, 70.32; H, 5.64; N, 24.31
6.1.5.5. Furan-2-yl-(4-(phthalazin-1-yl)piperazin-1-yl)methanone (8e)
Yield (0.23 g; 77%), mp 90–92 °C; 1HNMR (300 MHz, DMSO): δ 8.36 (1H, s, phthalazine), δ 8.12–8.10 (1H, d, J = 7 Hz, furoyl ring), δ 8.03–8.01 (2H, d, J = 7.8 Hz, phthalazine), δ 7.98–7.96 (2H, d, J = 7.8 Hz, phthalazine), δ 6.99–6.98 (2H, d, J = 7 Hz, furoylring), δ 3.69–3.67 (4H, m,piperazine ring), δ 2.91–2.90 (4H, m, piperazine ring); 13CNMR (100 MHz, DMSO) : δ 175.9, 159.8, 154.6, 154.2, 142.3, 135.4, 133.9, 128.5, 127.8, 125.7, 123.2, 122.2, 117.2, 111.7, 50.2, 42.8, 40.2, FT-IR (ύ max, cm−1): 1679 (C=O); MS (Mwt.: 308.33): m/z 308.05 (M+, 4.64%), 274.00 (6.66%), 158.05 (57.70%), 95.00 (100.00%) Anal. Calcd for C17H16N4O2: C, 66.22; H, 5.23; N, 18.17; Found: C, 66.41; H, 5.32; N, 18.40
6.1.5.6. Ethyl 4–(4-methylphthalazin-1-yl)piperazine-1-carboxylate (8f)
Yield (0.22 g; 73%), mp 95–97 °C; 1HNMR (300 MHz, CDCl3): δ 8.43–8.41 (2H, d, J = 8.1 Hz, phthalazine), δ 8.36–8.34 (2H, d, J = 8.1 Hz, phthalazine), δ 4.19 (2H, q, CH2-CH3), δ 3.75–3.74 (4H, m, piperazine ring), δ 3.65–3.64 (4H, m, piperazine ring), δ 3.37 (3H, s, CH3), δ 1.33 (3H, t,CH3-CH2); FT-IR (ύ max, cm−1): 1693 (C=O); MS (Mwt.: 300.36): m/z 300.05(M+, 5.71%), 244.05 (3.15%), 184.55 (13.85%), 172.30 (48.47%), 55.65 (100%); Anal. Calcd for C16H20N4O2: C, 63.98; H, 6.71; N, 18.65; Found: C, 64.17; H, 6.83; N, 19.01
6.1.5.7. 1-Methyl-4–(4-phenylpiperazin-1-yl)phthalazine (8 g)
Yield (0.15 g; 56%), mp 229–231 °C; 1HNMR (300 MHz, DMSO): δ 7.28–7.26(2H, d, J = 7.2 Hz, phthalazine), δ 7.25–7.23 (2H, d, J = 7.2 Hz, phthalazine), δ 6.99–6.97 (2H, d, J = 7.8 Hz, Ar-H), δ 6.88–6.86 (1H, m, Ar-H), δ 6.85–6.83 (2H, d, J = 7.8 Hz, Ar-H), δ 3.34–3.32 (4H, m, piperazine ring), δ 3.22–3.20 (4H, m, piperazine ring), δ 2.48 (3H, s, CH3); FT-IR (ύ max, cm−1): 3070 (CH aromatic), 2954 (CH aliphatic); MS (Mwt.: 304.39): m/z 304.00(M+, 1.73%), 284.60 (2.19%), 192.35 (5.24%), 105.20 (34.33%), 55.65 (100.00%); Anal. Calcd for C19H20N4: C, 74.97; H, 6.62; N, 18.41; Found: C, 80.32; H, 6.70; N, 18.48.
6.1.5.8. 1–(4-(2-Fluorophenyl)piperazin-1-yl)-4-methylphthalazine (8 h)
Yield (0.12 g; 71%), mp 149–151 °C; 1HNMR (300 MHz, CDCl3): δ 8.15–8.13 (1H, d, J = 9.6 Hz, phthalazine), δ 8.05–8.01 (1H, d, J = 9.6 Hz, phthalazine), δ 7.87–7.83 (2H, d, J = 9.6 Hz, phthalazine), δ 7.07–7.04 (2H, d, J = 8.7 Hz, Ar-H), δ 7.01–7.00 (2H, d, J = 8.7 Hz, Ar-H), δ 3.69–3.67 (4H, m, piperazine ring), δ 3.39–3.36 (4H, m, piperazine ring), δ 2.92 (3H, s, CH3); 13CNMR (100 MHz, DMSO) : δ 173.5, 158. 9, 153.1, 137.4, 131.8, 131.5, 127.2, 125.2, 123.2, 123.1, 122.3, 120.4, 119.3, 116.1, 115.8, 50.9, 50.0, 47.0, 19.0, FT-IR (ύ max, cm−1): 3035 (CH aromatic), 2951 (CH aliphatic); MS (Mwt.: 322.38): m/z 323.50 [M + 1H]+ (9.74%), 172.15 (48.77%,), 122.05 (100.00%); Anal. Calcd for C19H19FN4: C, 70.79; H, 5.94; N, 17.38; Found: C, 71.03; H, 5.99; N, 18.48.
6.1.5.9. 1-Methyl-4–(4-(pyridin-2-yl)piperazin-1-yl)phthalazine (8i)
Yield (0.10 g; 59%), mp 226–228 °C; 1HNMR (300 MHz, DMSO): δ 8.16–8.13(1H, d, J = 8.7 Hz, pyridyl ring), δ 7.62–7.60 (2H, d, J = 8.4 Hz, phthalazine), δ 7.58–7.56 (2H, d, J = 8.4 Hz, phthalazine), δ 6.91–6.89 (1H, d, J = 8.7 Hz, pyridyl ring), δ 6.74–6.72 (2H, d, J = 8.7 Hz, pyridyl ring), δ 3.75–3.73 (4H, m, piperazine ring), δ 3.12–3.10 (4H, m, piperazine ring), δ 2.08 (3H, s, CH3); FT-IR (ύ max, cm−1) 3132 (CH aromatic), 2954 (CH aliphatic); MS (Mwt.: 305.38): m/z 305.15 (M+, 1.16%), 185.65 (16.50%), 172.35 (41.23%,), 55.55 (100.00%); Anal. Calcd for C18H19N5: C, 70.80; H, 6.27; N, 22.93; Found: C, 71.04; H, 6.38; N, 23.18.
6.1.5.10. Furan-2-yl-(4–(4-methylphthalazin-1-yl)piperazin-1-yl)methanone (8j)
Yield (0.11 g; 60%), mp 90–92 °C; 1HNMR (300 MHz, DMSO): δ 8.32–8.30 (2H, d, J = 7 Hz, phthalazine), δ 8.29–8.27 (2H, d, J = 7 Hz, phthalazine), δ 7.82–7.81 (1H, d, J = 7.2 Hz, furoyl ring), δ 6.97–6.96(1H, d, J = 7.2 Hz, furoyl ring), δ 6.62–6.60 (1H, d, J = 7.2 Hz, furoyl ring), δ 3.64–3.62 (4H, m, piperazine ring), δ 3.41–3.39 (4H, m, piperazine ring), δ 2.92 (3H, s, CH3); FT-IR (ύ max, cm-1): 1610 (C=O); MS (Mwt.: 322.36): m/z 322.00 (M+, 6.60%), 184.00 (31.70%), 172.00 (100.00%); Anal. Calcd for C18H18N4O2: C, 67.07; H, 5.63; N, 17.38; Found: C, 67.21; H, 5.69; N, 17.52.
6.1.6. Synthesis of N1-(4-chlorophthalazin-1-yl)benzene-1,4-diamine (11)
General procedure:p-Phenylenediamine (0.33 g, 3.12 mmol, 3equiv) and the dichlorophthalazine (2.08 mmol, 2equiv) were treated with 2-BuOH (7.5 ml) in a tube and heated to 110 °C. The reaction quickly became a solid, yellow mass. After four hours, the reaction was cooled and diluted with water. The resultant slurry was then partitioned between equal volumes of DCM and 1 N NaOH (15 ml). The aqueous layer was extracted into DCM (2 × 15 ml). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The resulting orange solid was crystallised from (EtOAc/pet ether 1:1) to afford the title compound (11) as an orange crystals.
Yield 0.22 g, (88%); mp 170–172 °C; 1HNMR (300 MHz, DMSO-d6): δ 13.21 (s, 1H, –NH D2O exchangeable), 9.27 (s, 2H, 2H, –NH2 D2O exchangeable), δ 8.30–8.28 (d, J = 7 Hz, 2H, phthalazine), δ 8.07–8.04 (d, J = 7 Hz, 2H, phthalazine), δ 7.46–7.43 (d, J = 9 Hz, 2H, ArH), δ 6.83–6.76 (d, J = 9 Hz, 2H, ArH), δ 2.48 (s, 3H, CH3); FT-IR (υ max, cm−1): 3385 (NH2), 3321(NH); MS (Mwt.: 270.72): m/z 250.00 (M+, 50.00%), 249.00 (100.00%), 108.00 (60.92%), 65.00 (77.59%); Anal. Calcd for C14H11ClN4: C, 62.11; H, 4.10; N, 20.70; Found: C, 62.37; H, 4.18; N, 20.94.
6.1.7. Synthesis of 1-aryl-3–(4-(4-substituted phthalazin-1-ylamino)phenyl)urea (12a-c)
General procedure:
To a stirred solution of the N1-(4-chlorophthalazin-1-yl)benzene-1,4-diamine (11) (10.0 mmol, 1equiv) in DMF (20 ml), the respective phenyl isocyanate (viz.; phenyl isocyanate, 4-chloro phenyl isocyanate, 3-trifluromethyl-4-chlorophenyl isocyanate) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 8–10 h, after which TLC (CH2Cl2/CH3OH 9:1) showed no starting material. The reaction mixture was poured over ice-water; the formed precipitate was allowed to settle, then filtered off and dried to afford the crude product (12a-c) which was further crystallised from EtOAc.
6.1.7.1. 1–(4-(4Chlorophthalazin-1-ylamino)phenyl)-3-phenylurea (12a)
Yield 0.12 g; (68%), mp 225–227 °C; 1HNMR (400 MHz, DMSO-d6): δ 12.85 (s, 3H, –NH D2O exchangeable), δ 8.63 (d, J = 10 Hz 1H, phthalazine), δ 8.30–8.27 (d, J = 10 Hz, 2H, phthalazine), δ 8.08–8.05 (d, J = 10 Hz, 1H, phthalazine), δ 8.03–7.02 (d, J = 8 Hz, 2H, Ar-H), δ 7.99–7.97 (d, J = 8 Hz, 2H, Ar-H), δ 7.95–7.94 (d, J = 8 Hz, 1H, Ar-H), δ 7.46–7.43 (d, J = 9.2 Hz, 2H, Ar-H), δ 7.29–7.24 (d, J = 9.2 Hz, 1H, Ar-H), δ 6.98–6.94 (d, J = 9.2 Hz, 1H, Ar-H); 13CNMR (100 MHz, DMSO) : δ 159.0, 152.5, 146.1, 139.6, 137.0, 134.4, 132.9, 128.7, 128.3, 126.4, 125,4, 121.7, 120.5, 119.5, 118.1; FT-IR (υ max, cm−1): 3296, 3286, 3155 (3NH), 1668 (C=O); MS (Mwt.: 389.84): m/z 389.28 (M+, 2.84%), 367.27 (6.23%), 322.10 (11.84%), 128.10(100.00%); Anal. Calcd for C21H16ClN5O: C, 64.70; H, 4.14; N, 14.96; Found: C, 64.71; H, 3.89; N, 14.52.
6.1.7.2. 1–(4-Chlorophenyl)-3–(4-(4-chlorophthalazin-1-ylamino)phenyl)urea (12b)
Yield 0.20 g; (53%), mp 236–238 °C; 1HNMR (400 MHz, DMSO-d6): δ 12.84 (s, 2H, –NH D2O exchangeable), δ 8.81 (s, 1H, –NH D2O exchangeable), δ 8.29–8.27 (d, J = 8.8 Hz, 1H, phthalazine), δ 8.07–8.05 (d, J = 8.8 Hz, 1H, phthalazine), δ 8.02–8.00 (d, J = 8.8 Hz, 1H, phthalazine), δ 7.98–7.96 (d, J = 8.8 Hz, 1H, phthalazine), δ 7.95–7.94 (d, J = 6.4 Hz, 2H, Ar-H), δ 7.57–7.56 (d, J = 6.4 Hz, 2H, Ar-H), δ 7.48–7.47 (d, J = 6.4 Hz, 2H, Ar-H), δ 7.31–7.30 (m, 2H, Ar-H); FT-IR (υ max, cm−1): 3446, 3300, 3157 (3NH), 1670 (C=O); MS (Mwt.: 423.07): m/z 424.88 [M + 1H]+ (3.74%), 423.50 (M+, 9.84%), 328.27 (52.58%), 252.97 (20.15%), 119.04(100.00%); Anal. Calcd for C21H15Cl2N5O: C, 59.45; H, 3.56; N, 16.51; Found: C, 59.49; H, 3.37; N, 16.29.
6.1.7.3. 1–(4-Chloro-3-(trifluoromethyl)phenyl)-3–(4-(4-chlorophthalazin-1-ylamino)phenyl)-urea (12c)
Yield 0.22 g; (49%), mp 187–189 °C; 1HNMR (400 MHz, DMSO-d6): δ 12.84 (s, 2H, –NH D2O exchangeable), δ 9.36 (s, 1H, –NH D2O exchangeable), δ 8.29–8.27 (d, J = 8.8 Hz, 2H, phthalazine), δ 8.27–8.25 (d, J = 8.8 Hz, 2H, phthalazine), δ 8.09 (s, 1H, Ar-H), 8.05–8.03 (d, J = 9.6 Hz, 1H, Ar-H), δ 8.01–7.97 (d, J = 9.6 Hz, 1H, Ar-H), δ 7.59–7.50 (d, J = 8.4 Hz, 2H, Ar-H), δ 7.39–7.37 (d, J = 8.4 Hz, 2H, Ar-H); FT-IR (υ max, cm−1): 3387, 3344, 3290 (3NH), 1664(C=O); MS (Mwt.: 492.28): m/z 493.06 [M + 1H]+ (2.63%), 395.08 (32.95%), 372.90 (35.24%), 137.07 (100.00%); Anal. Calcd for C22H14Cl2F3N5O: C, 53.68; H, 2.87; N, 14.23; Found: C, 53.72; H, 2.68; N, 14.44.
6.1.8. Synthesis of 1-aryl-3–(4-(4-chlorophthalazin-1-yloxy)phenyl)urea (13a-c)
General procedure:
To a stirred solution of 1,4 dichlorophthalazine derivative (10) (10.0 mmol, 1equiv), and cesium carbonate (6.5 g, 20.0 mmol, 2equiv) in acetonitrile (20 ml), the appropriate 1-aryl-3–(4-hydroxyphenyl)urea (1a-c) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 6 h, after which TLC (CHCl3/CH3OH 9:1) showed no starting material. The solvent was evaporated under vacuum and the resultant solid was stirred with cold 1 M NaOH solution (20 ml), filtered off, dried and recrytallised from acetonitrile to afford compounds (13a-c).
6.1.8.1. 1–(4-(4-Chlorophthalazin-1-yloxy)phenyl)-3-phenylurea (13a)
Yield 0.12 g; (46%), mp 191–193 °C; 1HNMR (300 MHz, DMSO-d6): δ 10.48 (s, 1H, –NH D2O exchangeable), δ 10.31 (s, 1H, –NH D2O exchangeable), δ 8.46–8.44 (d, J = 8 Hz, 2H, phthalazine), δ 8.29–8.27 (d, J = 8 Hz, 2H, phthalazine), δ 7.90–7.88 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.69–7.66 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.40–7.37 (d, J = 8 Hz, 1H, Ar-H), δ 7.27–7.25 (d, J = 8 Hz, 2H, Ar-H), δ 6.94–6.92 (d, J = 8 Hz, 2H, Ar-H); 13CNMR (100 MHz, DMSO) : δ 164.7, 158.8, 153. 8, 152.4, 141.3, 136.6, 135.3, 132.3, 128.4, 128.0, 127.3, 125. 7, 123.8, 121.6, 120.6, 120.2, 118.1, 115.1; FT-IR (υ max, cm−1): 3325, 3295 (2NH), 1662 (C=O); MS (Mwt.: 390.82): m/z 390.97.00 (M+, 2.24%), 346.01 (5.06%), 268.11 (18.11%), 179.11 (100.00%); Anal. Calcd for C21H15ClN4O2: C, 64.54; H, 3.87; N, 14.43; Found: C, 64.97; H, 4.21; N, 14.12.
6.1.8.2. 1–(4-Chlorophenyl)-3–(4-(4-chlorophthalazin-1-yloxy)phenyl)urea (13b)
Yield 0.19 g; (50%), mp 172–174 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.06 (s, 1H, –NH D2O exchangeable), δ 8.54 (s, 1H,–NH D2O exchangeable), δ 8.34–8.32 (d, J = 8.4 Hz, 2H, phthalazine), δ 8.18–8.16 (d, J = 8.4 Hz, 2H, phthalazine), δ 7.44–7.42 (d, J = 8.8 Hz, 2H, Ar-H), δ 7.35–7.33 (d, J = 8.8 Hz, 2H, Ar-H), δ 7.27–7.25 (d, J = 7.2 Hz, 2H, Ar-H), δ 6.78–6.76 (d, J = 7.2 Hz, 2H, Ar-H); FT-IR (υ max, cm−1): 3278, 3155 (2NH), 1666 (C=O); MS (Mwt.: 425.27): m/z 425.28 (M+, 5.29%), 305.13 (7.74%), 201.08 (11.73%), 63.03 (100.00%); Anal. Calcd for C21H14Cl2N4O2: C, 59.31; H, 3.32; N, 13.17; Found: C, 59.62; H, 3.60; N, 13.65.
6.1.8.3. 1–(4-Chloro-3-(trifluoromethyl)phenyl)-3–(4-(phthalazin-1-yloxy)phenyl)urea (13c)
Yield 0.22 g; (49%), mp 210–212 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.20 (s, 1H, –NH D2O exchangeable), δ 10.13 (s, 1H, –NH D2O exchangeable), δ 8.28–8.26 (d, J = 9 Hz, 2H, phthalazine), δ 8.25–8.23 (d, J = 9 Hz, 2H, phthalazine), δ 8.09 (s, 1H, Ar-H), δ 7.93–7.92 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.76–7.74 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.54–7.51 (d, J = 8.4 Hz, 2H, Ar-H), δ 6.69–6.66 (d, J = 8.4 Hz, 2H, Ar-H); FT-IR (υ max, cm−1): 3294, 3155(2NH), 1662 (C=O); MS (Mwt.: 493.27): 493.46 (M+, 1.24%), 281.00 (2.26%), 194.97(100.00%); Anal. Calcd for C22H13Cl2F3N4O2: C, 53.57; H, 2.66; N, 11.36; Found: C, 53.81; H, 2.90; N, 11.3.
6.1.9. Synthesis of N-(4-chlorophenyl)-4-methylphthalazin-1-amine (14)
General procedure:
To a stirred mixture of the 1-chloro-4-methyl-phthalazine (3b) (1.78, 10.0 mmol, 1 equiv) in butanol (50 ml), triethylamine (1 ml, 10.0 mmol, 1 equiv) the respective amine (viz.; aniline, p-Cl aniline) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 2 h, after which TLC (CHCl3/CH3OH 9:1) showed no starting material. The mixture was then concentrated in vacuo, the crude product was recrystallised from methanol.
Yield 1.56 g; (88%), mp 213–215 °C; 1HNMR (400 MHz, DMSO-d6): δ 10.47 (s, 1H, –NH D2O exchangeable), δ 9.10–9.07 (d, J = 9 Hz, 1H, phthalazine), δ 8.45 –8.44 (d, J = 9 Hz, 1H, phthalazine), δ 8.31–8.29 (d, J = 9 Hz, 1H, phthalazine), δ 8.23–8.20 (d, J = 9 Hz, 1H, phthalazine), δ 7.85–7.82 (d, J = 8 Hz, 1H, Ar-H) , δ 7.51–7.49 (d, J = 8 Hz, 1H, Ar-H), δ 7.10–7.07 (d, J = 8 Hz, 1H, Ar-H), δ 6.70–6.68 (d, J = 8 Hz, 1H, Ar-H) , δ 2.97 (s, 3H, CH3); FT-IR (υ max, cm−1) 3360 (NH), 3050 (CH aromatic); MS (Mwt.: 269.73): m/z 269.28 (M+, 8.26%), 239.18 (15.43%), 125.22 (24.70%), 90.97(100.00%); Anal. Calcd for C15H12ClN3: C, 66.79; H, 4.48; N, 15.58; Found: C, 67.03; H, 4.57; N, 15.75.
6.1.10. Synthesis of 4-(bromomethyl)-N-(4-chlorophenyl)phthalazin-1-amine (15)
General procedure:
A mixture of the N-aryl-4-methylphthalazin-1-amine (14b) (0.26 mmol, 1 equiv), N-bromo-succinimide (4.6 g, 0.26 mmol, 1 equiv) and dibenzoyl peroxide (0.44 g, 0.018 mmol, 0.07 equiv) in carbon tetrachloride (150 ml) was refluxed for 24 h. The reaction mixture was cooled to 40 °C, and then filtered. The filtrate was concentrated under vacuum to afford the crude product, which was triturated with ether to give a yellowish solid product.
Yield 0.10 g; (42%), mp 172.-174 °C; 1HNMR (400 MHz, DMSO-d6): δ 11.06 (s, 1H, –NH D2O exchangeable), δ 8.07–8.05 (d, J = 7.2 Hz, 2H, phthalazine), δ 7.99 –7.97 (d, J = 7.2 Hz, 2H, phthalazine), δ 7.62–7.60 (d, J = 7.6 Hz, 2H, Ar-H), δ 7.45–7.44 (d, J = 7.6 Hz, 2H, Ar-H), δ 4.02 (s, 2H, CH2); FT-IR (υ max, cm−1) 3450 (NH), 3053 (CH aromatic); MS (Mwt.: 348.62): m/z 350.00 (M+2, 2.86%), 348.00 (M+, 9.89%), 268.04 (65.94), 102.03 (100.00%); Anal. Calcd for C15H11BrClN3: C, 51.68; H, 3.18; N, 12.05; Found: C, 51.90; H, 3.16; N, 12.18.
6.1.11. Synthesis of N-(4-chlorophenyl)-4–(3-substitutedphenylamino)methyl)phthalazin-1-amines (16a-d)
General procedure:
To a stirred solution of 4-(bromomethyl)-N-(4-chlorophenyl)phthalazin-1-amine (15) (10.0 mmol, 1 equiv), potassium carbonate (0.27 g, 20.0 mmol, 2equiv), potassium iodide (0.01 g, 0.01 mmol, 0.1equiv) in acetone (20 ml), the respective amine (viz.; aniline, m-Chloroaniline, m-touilidine, m-anisidine) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 4–6 h, after which TLC (CH2Cl2/CH3OH 99:1) showed no starting material. The mixture was then filtered and the filtrate concentrated in vacuo, the residue was crystallised from acetone.
6.1.11.1. N-(4-Chlorophenyl)-4-((phenylamino)methyl)phthalazin-1-amine (16a)
Yield 0.10 g; (42%), mp 120–122 °C; 1HNMR (400 MHz, DMSO-d6): δ 11.03 (s, 1H, –NH D2O exchangeable), δ 9.65 (s, 1H, –NH D2O exchangeable), δ 8.75–8.72 (d, J = 9.2 Hz, 2H, phthalazine), δ 8.28 –8.25 (d, J = 9.2 Hz, 2H, phthalazine), δ 7.90–7.88 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.85–7.87 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.46–7.43 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.05–7.03 (d, J = 7.6 Hz, 2H, Ar-H), δ 6.83–6.81 (d, J = 7.6 Hz, 2H, Ar-H), δ 3.89 (s, 2H, CH2); FT-IR (υ max, cm−1) 3446, 3383 (2NH), 3068 (CH aromatic), 2976 (CH aliphatic); MS (Mwt.: 360.84): m/z 360.02 (M+, 2.87%), 345.88 (13.64%), 268.02 (49.93%), 75.01 (100%); Anal. Calcd for C21H17 ClN4: C, 69.90; H, 4.75; N, 15.53; Found: C, 70.12; H, 4.82; N, 15.69.
6.1.11.2. N-(4-Chlorophenyl)-4-((3-chlorophenylamino)methyl)phthalazin-1-amine (16b)
Yield 0.10 g; (42%), mp 164–166 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.90 (s, 2H, –NH D2O exchangeable), δ 8.86–8.84 (d, J = 9.2 Hz, 2H, phthalazine), δ 8.34 –8.32 (d, J = 9.2 Hz, 2H, phthalazine), δ 8.20–8.18 (d, J = 8.6 Hz, 2H, Ar-H), δ 8.12–8.09 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.88–7.86 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.53–7.51 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.44–7.42 (d, J = 7.6 Hz, 2H, Ar-H), δ 3.79 (s, 2H, CH2); FT-IR (υ max, cm−1) 3379 (2NH), 3080 (CH aromatic), 2954 (CH aliphatic); MS (Mwt.: 395.28) m/z 395.99 (M+, 2.17%), 271.01 (16.80%), 268.01 (100%); Anal. Calcd for C21H16Cl2N4: C, 63.81; H, 4.08; N, 14.17; Found: C, 63.94; H, 4.12; N, 14.28.
6.1.11.3. N-(4-Chlorophenyl)-4-((m-tolylamino)methyl)phthalazin-1-amine (16c)
Yield 0.10 g; (42%), mp 130–132 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.70 (s, 2H, –NH D2O exchangeable), δ 8.86–8.84 (d, J = 9.2 Hz, 2H, phthalazine), δ 8.34 –8.32 (d, J = 9.2 Hz, 2H, phthalazine), δ 8.20–8.18 (d, J = 8.6 Hz, 2H, Ar-H), δ 8.12–8.09 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.88–7.86 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.53–7.51 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.44–7.42 (d, J = 7.6 Hz, 2H, Ar-H), δ 3.79 (s, 2H, CH2), δ 2.69 (s, 3H, CH3);): 13CNMR (100 MHz, DMSO): δ 162.2, 152.9, 151.1, 138.9, 135.0, 133.8, 131.7, 127.3, 127.1, 126.8, 126.4, 126.3, 124.7, 124.3, 124.1, 120.5, 120.2, 117.4, 113.2, 110.5, 45.5, 16.7, FT-IR (υ max, cm−1) 3446, 32992 (2NH), 3050 (CH aromatic), 2922 (CH aliphatic); MS (Mwt.: 374.13): m/z 374.75 (M+, 2.65%), 268.06 (21.45%), 189.95 (18.36%), 77.06 (100%); Anal. Calcd for C22H19ClN4: C,70.49; H, 5.11; N, 14.95; Found: C, 70.73; H, 5.19; N, 15.08.
6.1.11.4. N-(4-Chlorophenyl)-4-((3-methoxyphenylamino)methyl)phthalazin-1-amine (16d)
Yield 0.10 g; (42%), mp 175–177 °C; 1HNMR (400 MHz, DMSO-d6): δ 11.34 (s, 1H, –NH D2O exchangeable), δ 9.78 (s, 1H, –NH D2O exchangeable), δ 8.73–8.70 (d, J = 9.6 Hz, 2H, phthalazine), δ 8.48 –8.46 (d, J = 9.6 Hz, 2H, phthalazine), δ 8.06–8.04 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.87–84 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.68–7.66 (d, J = 7.5 Hz, 1H, Ar-H), δ 7.46–7.44 (d, J = 7.5 Hz, 2H, Ar-H), δ 7.14–7.12 (d, J = 7.5 Hz, 1H, Ar-H), δ 4.29 (s, 2H, CH2), δ 3.63 (s, 3H, OCH3); 13CNMR (100 MHz, DMSO) : δ 164.2, 163.9, 148.7, 148.8, 139.2, 132.3, 131.3, 129.6, 128.5, 127.9, 123.19, 122.1, 120.4, 120.4, 115.1, 110.3, 109.2, 106.0, 105.7, 105.0, 51.2, 45.7;FT-IR (υ max, cm−1) 3412, 3385 (2NH), 3041(CH aromatic), 2976 (CH aliphatic); MS (Mwt.: 390.87): m/z 391.95 (M+, 7.73%), 350.00 (29.42%), 348.01 (100.00%), 313.04 (10.74%); Anal. Calcd for C22H19 ClN4O: C, 67.60; H, 4.90; N, 14.33; Found: C, 67.91; H, 4.95; N, 14.50.
6.1.12. Synthesis of N-(4-chlorophenyl)-4-(aryloxymethyl)phthalazin-1-amine (17a,b)
General procedure:
To a stirred solution of 4-(bromomethyl)-N-(4-chlorophenyl)phthalazin-1-amine (15) (10.0 mmol, 1 equiv), sodium hydride (0.27 g, 20.0 mmol, 2equiv) in acetone (20 ml), the respective phenol (viz.; phenol, m-cresol) (10.0 mmol, 1equiv) was added and the mixture was heated under reflux for 6–8 h, after which TLC (CH2Cl2/CH3OH 99:1) showed no starting material. The mixture was then filtered and the filtrate concentrated in vacuo, the residue was crystallised from acetone.
6.1.12.1. N-(4-Chlorophenyl)-4-(phenoxymethyl)phthalazin-1-amine (17a)
Yield 0.10 g; (42%), mp 242–244 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.33 (s, 1H,–NH D2O exchangeable), δ 8.74–8.75 (d, J = 9.2 Hz, 2H, phthalazine), δ 8.30 –8.28 (d, J = 9.2 Hz, 2H, phthalazine), δ 8.11–8.09 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.89–7.87 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.49–7.47 (d, J = 7.6 Hz, 2H, Ar-H), δ 7.17–7.15 (d, J = 7.6 Hz, 1H, Ar-H), δ 6.76–6.74 (d, J = 7.6 Hz, 2H, Ar-H), δ 3.89 (s, 2H, CH2); FT-IR (υ max, cm−1) 3379 (NH), 3066 (CH aromatic), 2978 (CH aliphatic); MS (Mwt.: 361.82): m/z 361.00 (M+, 5.29%), 349.01 (17.15%), 310.24 (12.19%), 268.11 (64.63%), 133.44 (100.00%); Anal. Calcd for C21H16ClN3O: C, 69.71; H, 4.46; N, 11.61; Found: C, 69.88; H, 4.39; N, 11.69.
6.1.12.2. N-(4-Chlorophenyl)-4-(m-tolyloxymethyl)phthalazin-1-amine (17b)
Yield 0.10 g; (42%), mp 193–195 °C; 1HNMR (400 MHz, DMSO-d6): δ 9.33 (s, 1H,–NH D2O exchangeable), δ 8.74–8.72 (d, J = 8.8 Hz, 2H, phthalazine), δ 8.31–8.28 (d, J = 8.8 Hz, 2H, phthalazine), δ 8.14–8.11 (d, J = 8.6 Hz, 2H, Ar-H), δ 8.09–8.06 (d, J = 8.6 Hz, 2H, Ar-H), δ 7.49–7.47 (d, J = 7.6 Hz, 1H, Ar-H), δ 7.47–7.44 (d, J = 8.6 Hz, 2H, Ar-H), δ 6.55–6.53 (d, J = 8.6 Hz, 1H, Ar-H), δ 3.89 (s, 2H, CH2), δ 2.19 (s, 3H, CH3); 13CNMR (100 MHz, DMSO) : δ 173.4, 162.4, 151.0, 139.4, 138.4, 133.1, 132.1, 128.9, 128.6, 128.4, 126.9, 126.9, 125.9, 123.3, 123.15, 121.9, 119.4, 115.7, 112.2, 75.4, 20.9, FT-IR (υ max, cm−1) 3414 (NH), 3034 (CH aromatic), 2976 (CH aliphatic); MS (Mwt.: 375.85): m/z 375.94 (M+, 1.09%), 349.94 (4.65%), 267.15 (100.00%); Anal. Calcd for C22H18ClN3O: C, 70.30; H, 4.83; N, 11.18; Found: C, 70.43; H, 4.88; N, 11.32.
Supplementary Material
Acknowledgements
The authors are grateful to The National Cancer Institute, Maryland, USA for performing anticancer activity. TMI would like to acknowledge Prof. Frank Boeckler (University of Tuebingen, Germany) for granting access to XBScore and to some functions of MOE.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1. Zhang YS, Liu Y, Chen D, et al. Synthesis and antitumor activities of novel 1,4-disubstituted phthalazine derivatives. Eur. J. Med. Chem 2010;45:3504–10. [DOI] [PubMed] [Google Scholar]
- 2. Imramovský A, Jorda R, Pauk K, et al. Substituted 2-hydroxy-N-(arylalkyl)benzamides induce apoptosis in cancer cell lines. Eur J Med Chem 2013;68:253–9. [DOI] [PubMed] [Google Scholar]
- 3. Renehan AG, Booth C, Potten CS. What is apoptosis, and why is it important? BMJ 2001;322:1536–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cortes E, Ravandi F, O'Brien S, Kantarjian H. Targeted therapies in hematology and their impact on patient care: chronic and acute myeloid leukemia. Semin Hematol 2013;50:271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scott EN, Meinhardt G, Jacques C, et al. Vatalanib: the clinical development of a tyrosine kinase inhibitor of angiogenesis in solid tumours. Expert Opin Investig Drugs 2007;16:367–79. [DOI] [PubMed] [Google Scholar]
- 7. Hasinoff BB, Pate D. The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro . Toxicol Appl Pharm 2010;249:132–9. [DOI] [PubMed] [Google Scholar]
- 8. Ricci MS, Kim SH, Ogi K, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007;12:66–80. [DOI] [PubMed] [Google Scholar]
- 9. Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006;66:11851–8. [DOI] [PubMed] [Google Scholar]
- 10. Zhang W, Konopleva M, Ruvolo VR, et al. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia 2008;22:808–18. [DOI] [PubMed] [Google Scholar]
- 11. Chuma M, Terashita K, Sakamoto N. New molecularly targeted therapies against advanced hepatocellular carcinoma: From molecular pathogenesis to clinical trials and future directions. Hepatol Res 2015;45:E1–11. [DOI] [PubMed] [Google Scholar]
- 12. Blanc J, Geney R, Menet C. Type II kinase inhibitors: an opportunity in cancer for rational design. Anticancer Agents Med Chem 2013;13:731–47. [DOI] [PubMed] [Google Scholar]
- 13. Sodani K, Patel A, Anreddy N, et al. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem Pharmacol 2014;89:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garuti L, Roberti M, Bottegoni G. Small molecule aurora kinases inhibitors. Curr Med Chem 2009;16:1949–63. [DOI] [PubMed] [Google Scholar]
- 15. Shallal HM, Russu WA. Discovery, synthesis, and investigation of the antitumor activity of novel piperazinylpyrimidine derivatives. Eur J Med Chem 2011;46:2043–57. [DOI] [PubMed] [Google Scholar]
- 16. Rice KD, Kim MH, Bussenius J, et al. Pyrazolopyrimidines as dual Akt/p70S6K inhibitors. Bioorg Med Chem Lett 2012;22:2693–7. [DOI] [PubMed] [Google Scholar]
- 17. Pollard JR, Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J Med Chem 2009;52:2629–51. [DOI] [PubMed] [Google Scholar]
- 18. Musumeci F, Radi M, Brullo C, Schenone S. Vascular endothelial growth factor (VEGF) receptors: drugs and new inhibitors. J Med Chem 2012;55:10797–822. [DOI] [PubMed] [Google Scholar]
- 19. Abouzid KM, Khalil N, Ahmed E. Synthesis and evaluation of anti-proliferative activity of 1,4-disubstituted phthalazines. Med Chem Res 2012;21:3288–93. [Google Scholar]
- 20. Sammond DM, Veal JM, Nolte RT, et al. Discovery of a novel and potent series of dianilinopyrimidineurea and urea isostere inhibitors of VEGFR2 tyrosine kinase. Bioorg Med Chem Lett 2005;15:3519–23. [DOI] [PubMed] [Google Scholar]
- 21. Stout DM, Matier WL, Barcelon-Yang C, et al. Synthesis and antiarrhythmic and parasympatholytic properties of substituted phenols. 3. Modifications to the linkage region (region 3). J Med Chem 1985;28:295–8. [DOI] [PubMed] [Google Scholar]
- 22. Nakatsu MN, Sainson RCA, Aoto JN, et al. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1. Microvas Res 2003;66:102–12. [DOI] [PubMed] [Google Scholar]
- 23. Park H-J, Zhang Y, Georgescu SP, et al. Human umbilical vein endothelial cells and human dermal microvascular endothelial cells offer new insights into the relationship between lipid metabolism and angiogenesis. Stem Cell Rev 2006;2:93–101. [DOI] [PubMed] [Google Scholar]
- 24. Alley MC, Monks A, Hursey ML, et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 1988;48:589–601. [PubMed] [Google Scholar]
- 25. Grever MR, Chabner BA. The National Cancer Institute: cancer drug discovery and development program. Semin Oncol 1992;19:622–38. [PubMed] [Google Scholar]
- 26. Jiang BH, Jiang G, Zheng JZ, et al. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth 2001;12:363–9. [PubMed] [Google Scholar]
- 27. Behl C, Ziegler C. Cell aging: molecular mechanisms and implications for disease. 1st ed Berlin Heidelberg: Springer-Verlag; 2014. [Google Scholar]
- 28. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilcken R, Zimmermann MO, Lange A, et al. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem 2013;56:1363–88. [DOI] [PubMed] [Google Scholar]
- 30. Wilcken R, Zimmermann MO, Lange A, et al. Addressing methionine in molecular design through directed sulfur-halogen bonds. J Chem Theory Comput 2011;7:2307–15. [DOI] [PubMed] [Google Scholar]
- 31. Zimmermann MO, Lange A, Boeckler FM. Evaluating the potential of halogen bonding in molecular design: automated scaffold decoration using the new scoring function XBScore. J Chem Inf Model 2015;55:687–99. [DOI] [PubMed] [Google Scholar]
- 32. Smolyar NN, Yutilov YM. Cyclotransformation in the series of fused 5-nitropyridin-2(1H)-ones. Russ J Org Chem 2008;44:274–81. [Google Scholar]
- 33.Gabriel S, Neumann A. Umlagerung von Phtalidderivatenin AbkÖmmlinge des α γ - Diketohydrindens. Chemische Berichte 1893;26:708–9. [Google Scholar]
- 34. Zhang QR, Xue DQ, He P, et al. Synthesis and antimicrobial activities of novel 1,2,4-triazolo [3,4-a] phthalazine derivatives. Bioorg Med Chem Letters 2014;24:1236–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.