

Abstract
Noncoding RNA sequences, including long noncoding RNAs, small nucleolar RNAs, and untranslated mRNA regions, accomplish many of their diverse functions through direct interactions with RNA-binding proteins (RBPs). Recent efforts have identified hundreds of new RBPs that lack known RNA-binding domains, thus underscoring the complexity and diversity of RNA-protein complexes. Recent progress has expanded the number of methods for studying RNA-protein interactions in two general categories: approaches that characterize proteins bound to an RNA of interest (RNA-centric), and those that examine RNAs bound to a protein of interest (protein-centric). Each method has unique strengths and limitations, which makes it important to select optimal approaches for the biological question being addressed. Here we review methods for the study of RNA-protein interactions, with a focus on their suitability for specific applications.
RNA and proteins are interconnected biomolecules that can promote each other’s life cycles and functions through physical interactions1. The coding sequence of mRNA carries the instructions for protein synthesis and some regulatory sequences, and the untranslated regions of mRNA influence the fate of the encoded protein by regulating its protein translation, localization, and interactions with other proteins2. Proteins, in turn, can bind and modulate RNA expression and function from RNA synthesis to degradation3. RNA-protein interactions are key to cellular homeostasis, and perturbations of RNA-RBP interactions can lead to cellular dysfunction and disease4,5. Recent work has substantially expanded the number of putative RNA-protein associations in eukaryotes, underscoring the need for a versatile array of methods to identify and characterize their interactions6,7.
Methods for studying the physical interactions between RNA and protein can be classified by the type of molecule they start with. RNA-centric methods start with an RNA of interest and are used to study proteins that associate with that RNA. Protein-centric methods, in contrast, start with a protein of interest and focus on the RNAs that bind it. Recent progress has expanded the number of both RNA-centric and protein-centric methods. Each currently available method has particular advantages and drawbacks, and thus method selection must be tailored to the relevant biological question. This review provides a selective overview of a subset of these methods and hopefully will assist scientists in their selection of optimal methods to address a particular research question. While there are several methods in each area, the review is focused on recent methods that have demonstrated substantial conceptual and technical advances.
Expanding the RNA-binding proteome
Canonical RBPs are defined by the presence of RNA-binding domains, such as the hnRNP K homology domain and the RNA-recognition motif8; however, recent efforts have identified novel RBPs with no annotated RNA-binding domains9. Thus, it is not possible to use protein sequence and structural information alone to predict whether an individual protein is indeed an RBP. Direct experimentation is required to generate a census of RBPs in the cell. Experimental methods to identify RBPs in cells use UV cross-linking to create a covalent bond between RNA and protein. Oligo(dT) capture has been used after cross-linking to isolate poly(A)-binding proteins for proteomic identification10. These methods are limited to identification of the RNA-binding proteome of poly(A) RNA. Recently, Chen and colleagues developed click-chemistry-assisted RNA interactome capture (CARIC), which uses metabolic labeling of RNAs with an alkynyl uridine analog to enable RNA capture independent of polyadenylation11. UV-based methods for studying the RNA-binding proteome have been applied to several cell types in species ranging from Caenorhabditis elegans to humans12,13. From these studies, a large number of RBPs have been discovered, suggesting that approximately 5% of the human proteome consists of RBPs10. The application of polyadenylation-agnostic methods such as CARIC in more cell types is likely to further expand the known repertoire of RBPs. Among the newly discovered RBPs are several metabolic enzymes such as adenylate kinase and fatty acid synthase14. It remains unknown how RNA binding affects the primary function of these metabolic enzymes. For example, how does the identified RNA-protein interaction affect the given RNA involved? How does it affect the metabolic function of the protein? These types of questions can be addressed with complementary RNA-centric and protein-centric methods.
RNA-centric methods: discovering RBPs bound to RNAs of interest
RNA is bound to protein throughout its life cycle. The changing medley of RNA-protein interactions is critical for cellular function, and is restructured on the basis of subcellular location and environmental stimuli3. These dynamic interactions are often transient, which makes it a challenge to identify the most important proteins bound to a given RNA. Broadly speaking, these methods can be classified into two categories (Fig. 1). In vitro methods commonly are used to study RNAs and proteins outside the context of an intact cell. In vivo approaches are used to investigate RNA-protein interactions in the cellular environment and are subdivided according to whether cross-linking is used. Each in vitro and in vivo RNA-centric method has particular strengths and drawbacks, which makes it important to select a method tailored to the biological question being addressed.
Fig. 1 |. Schematic representation of RNA-centric methods.
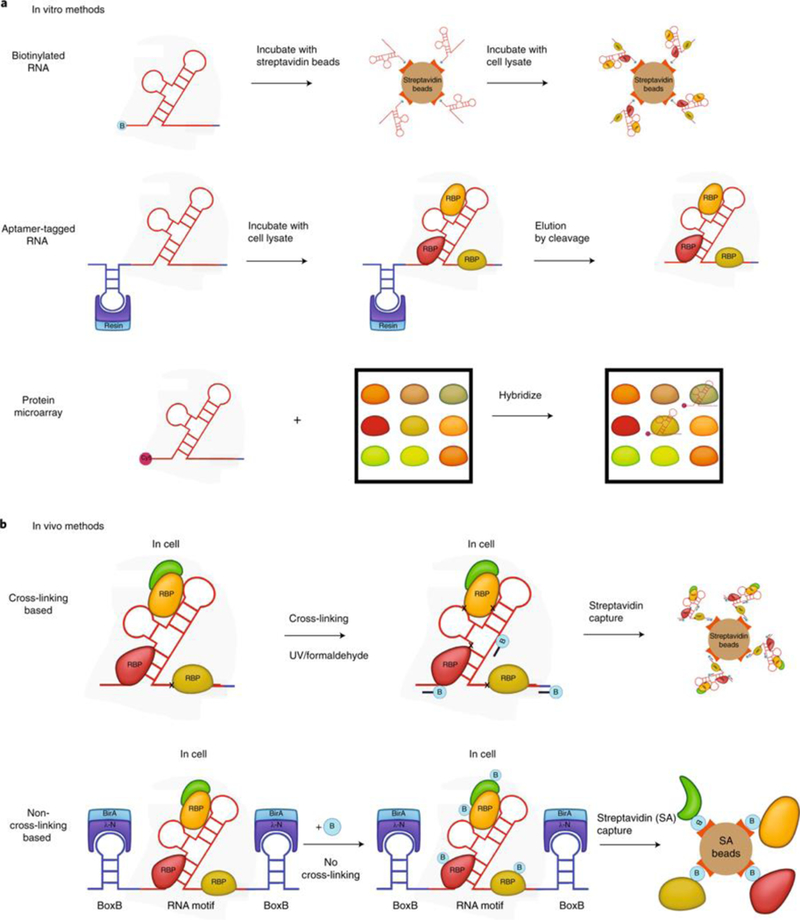
a, In vitro methods. Top, schematic of end-biotinylated-RNA pulldown. RNA is synthesized with biotin at the 5′ or 3′ end and combined with streptavidin. Recombinant or cellular-extract proteins bind to RNA. After being washed, the beads are boiled to elute and identify RNA-bound proteins. Middle, schematic of aptamer-tagged-RNA capture (S1, Cys4) methods. The RNA of interest is in vitro-transcribed with an RNA tag (blue). The RNA tag binds RNA (red) to a resin support. Proteins in the cellular extract bind to RNA. After washing steps, RNA complex is eluted with imidazole for Cys4 or biotin for the S1 aptamer method. Bottom, schematic of a protein microarray. RNA is in vitro-transcribed with Cy5. The RNA is then added to a human protein microarray spotted with −9,400 proteins. After washing steps, fluorescence is used to detect and quantitate RNA bound on spotted proteins on the microarray. b, Top, schematic of in vivo cross-linking methods. Cross-linking-based methods use either UV (RAP, PAIR, MS2-BioTRAP, TRIP) or formaldehyde cross-linking (CHART, ChIRP). Biotinylated oligonucleotide probes are hybridized to the RNA of interest, and the RNA and cross-linked proteins are purified for downstream analysis. Bottom, schematic of an in vivo non-cross-linking method (RaPID). BoxB RNA stem loops (blue) flank the RNA sequence of interest (red). RaPID (LN-HA-BirA*) fusion protein binding to BoxB sites leads to biotinylation of proteins associated with the inserted RNA sequence in living cells grown in biotin-containing media. Streptavidin beads are used to capture biotinylated protein.
In vitro methods.
In vitro methods commonly use in vitro-transcribed (IVT) RNA that contains a tag that binds to resin (Fig. 1a). After the IVT RNA is bound to resin, cellular extract is added, and subsequently washing steps are carried out to define the RBPs bound to the IVT RNA bait15.The speed and ease of these in vitro methods are balanced by several disadvantages. IVT RNA might not have the same modifications or structure that a given RNA has in cells. Likewise, if recombinant proteins are used, they may lack post-translational modifications that influence RNA association, and the use of high concentrations of protein may also promote nonspecific associations. The use of cellular extracts as a protein source may overcome some of these challenges, but it may also bias an experiment toward the detection of abundant proteins. A particular advantage of in vitro methods is the possibility of mutagenesis studies designed to identify the specific RNA nucleotides and protein amino acids involved in the binding of a given RNA-protein pair.
For in vitro studies, the simplest method of RNA tagging for pull-down is 5′ or 3′ RNA biotinylation. End-labeled biotinylated RNA is bound to streptavidin beads16, then cellular extract is added and beads are subsequently washed. With this method, there is no elution of RNA, and the beads are boiled in SDS for proteomic analysis16. An alternative approach is to add RNA aptamer tags to the RNA of interest17,18. Numerous tags have been developed for in vitro methods, including several recently developed methods15. Stoecklin and colleagues used the S1 aptamer tag, which binds to streptavidin beads, thus enabling biotin to be used later to competitively bind streptavidin and elute the S1-aptamer-tagged RNA19. Doudna and colleagues leveraged Cys4 endoribonuclease, which binds to a Cys4 hairpin loop20. Using imidazole, the nuclease can cleave off the hairpin loop and liberate the RBP-bound RNA (Fig. 1a)20. Cellular proteins can bind nonspecifically to resin. Elution of RBP-bound RNA (rather than all protein) off resin reduces background by excluding nonspecific resin-bound proteins from downstream analysis.
An alternative in vitro approach is to use IVT RNA labeled with Cy5 dye and hybridize it to a protein microarray containing approximately 9,400 recombinant human proteins (Human ProtoArray)21. Proteins that capture the Cy5 RNA are detected via fluorescence reading (Fig. 1a). The protein microarray method does not require cellular extracts and enables the discovery of direct RNA-protein interactions22. However, the method is limited by the folding and post-translational modifications of spotted proteins and may be potentially distorted by artificial concentrations. As a category of methods, in vitro methods overall are particularly useful for characterizing the binding of specific known RNA-protein interactions.
In vivo methods with cross-linking.
Researchers can use protein-RNA cross-linking to identify in vivo interactions by purifying the RNA under denaturing conditions that remove noncovalent interactions, and subsequently extracting only the cross-linked proteins for identification. Formaldehyde is a small, bifunctional cross-linker that readily permeates cells and cross-links macromolecules within 2 A, including protein-protein, protein-DNA, and protein-RNA complexes, creating a reversible covalent linkage23. UV light cross-links protein to nucleic acid at zero distance, and in an irreversible, covalent bond24,25. Each method of cross-linking presents distinct advantages and disadvantages. Because UV light is a zero-distance cross-linker and because it does not cross-link protein-protein interactions with an efficiency anywhere near that of formaldehyde, it is a more specific cross-linker than formaldehyde. However, the efficiency of UV cross-linking is lower24. Both UV and formaldehyde cross-linking have biases. UV cross-linking has a slight uridine preference26, and double-stranded RNA is known to be poorly cross-linked27. With respect to proteins, the efficiency of UV cross-linking varies by amino acid28. The structure and surface area of the protein-RNA interaction are other factors that are likely to affect UV cross-linking efficiency, but the current understanding of cross-linking efficiency remains too incomplete to allow quantitative predictions. We expect further research to make progress on this essential problem. In contrast, with formaldehyde cross-linking, strongly nucleophilic lysine residues are preferentially cross-linked29. Formaldehyde promotes cross-linking between proteins in addition to protein-RNA interactions, and thus it is difficult to distinguish proteins that contact RNA directly from those that are complexed with directly bound proteins. The biases and the low efficiency of both of these cross-linking modalities necessitate high input cell numbers, ranging from 108 to 109 cells, to maximize the capture of RNA-protein interactions30–32.
Several in vivo methods use either UV or formaldehyde crosslinking (Fig. 1b). Methods that use UV cross-linking include RNA affinity purification (RAP)33,34, peptide-nucleic-acid-assisted identification of RBPs (PAIR)35, MS2 in vivo biotin-tagged RAP (MS2-BioTRAP)36, and tandem RNA isolation procedure (TRIP)37 (Table 1). Although they share a common UV cross-linking approach, these methods differ in experimental setup. RAP uses long, 120-oligo-nucleotide probes to pull down RNA-RBP complexes after crosslinking and has been used to study noncoding RNAs such as Xist and FIRRE32,34. PAIR uses peptide nucleic acid probes with cell-penetrating peptides to gain entry into cells and hybridize to RNA, after which the RNA is purified, along with bound RBPs35. MS2-BioTRAP uses the interaction between MS2 hairpin loop and MS2 coat protein to tether protein to RNA38. Both MS2 hairpin RNA and MS2 coat protein are expressed in the same cell and form a stable complex, enabling the fusion MS2 coat protein to be used as a handle to purify the MS2-containing RNA after UV cross-linking36. The ectopic expression of MS2-tagged RNA might not reflect physiological levels of RNA, which can potentially impair the accuracy of downstream proteomic analysis. TRIP is used to study polyadenylated RNA and uses a dual purification: poly(A) RNA is purified first, and then biotinylated antisense oligonucleotides (ASOs) are used to hybridize with the RNA of interest in the poly(A) mixture by base pairing, after which the RNA-ASO complex is purified with streptavidin beads37. Chromatin isolation by RNA purification (ChIRP)30,39 and capture hybridization analysis of RNA targets (CHART)31, in contrast, use formaldehyde to cross-link RNA to proteins. CHART requires an additional RNase H assay to identify accessible sites for probes, whereas ChIRP does not require prior knowledge of RNA accessibility and uses shorter, 20-mer oligonucleotide probes30,40. These biotinylated probes hybridize with RNA in the cell and are captured with streptavidin beads for proteomic analysis. These UV and formaldehyde cross-linking methods allow the RNA-centric study of RNA-protein interactions in vivo.
Table 1 |.
Comparison of RNA-centric methods
| Method | Application | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Biotinylated RNA | SMN mRNA | Strong binding of biotin-end- labeled RNA with streptavidin beads | In vitro; potentially biased toward abundant proteins in cell extracts | 16 |
| S1 aptamer | ARE motif | Easy elution of RBP complex from streptavidin beads with biotin | In vitro; potentially biased toward abundant proteins | 19 |
| Cys4 | Pre-miRNA | Elution of RBP complex with imidazole | In vitro; potentially biased toward abundant proteins | 20 |
| Protein microarray | TINCR, SNORD50 | No cellular extract required; no MS required | In vitro; limited to direct interactions with proteins spotted on microarray | 21,22 |
| RAP | Xist, FIRRE noncoding RNA | In vivo; high specificity with UV cross-linking and long oligonucleotide probes (120 nt) | High input cell numbers | 33 |
| TRIP | p27 mRNA, CEP-1 mRNA | In vivo; high specificity with UV cross-linking | Two capture steps with poly(A) and biotinylated ASO capture decrease efficiency | 37 |
| PAIR | ANK mRNA | In vivo; high specificity with UV cross-linking | Cost and effort for production of peptide nucleic acid | 35 |
| MS2-BioTRAP | IRES | In vivo; high specificity with UV cross-linking | Requires MS2 conjugation to RNA, transfection/infection of RNA and labeler protein, and high input cell numbers | 36 |
| CHART | Xist, MALAT1, NEAT1 | In vivo | Additional RNase H step to identify accessible sites for probes; high input cell numbers | 31,52 |
| ChIRP | TERC, Xist | In vivo; no prior knowledge of RNA accessibility required for probe design | Short probes may pull down similar sequence fragments; high input cell numbers | 30,39 |
| RaPID | ZIKV-host protein interactome; 3′ untranslated region motifs | In vivo; low number of cells required; direct labeling of protein | Requires BoxB link to RNA; short sequence limits; transfection/infection of RNA and labeler protein | 44 |
In vivo methods without cross-linking.
Proximity proteomics has recently been applied for the RNA-centric study of RNA-protein interactions in living cells without the use of any form of crosslinking. ‘Promiscuous’ biotin ligases, predominantly used to study protein-protein interactions41,42, convert biotin to a reactive bio-tin-5-AMP intermediate, which is released from the enzyme to covalently label any nearby exposed lysine amino acid residue43. Because the intracellular environment is reducing, biotin-5-AMP is quenched and becomes unreactive within a distance of 20 nm of its point of release41. Hence, proteins that are within a distance of 20 nm will be labeled with biotin preferentially compared with other proteins in the cell. The RNA-protein interaction detection (RaPID) method allows one to use this spatial detection constraint to detect RBPs bound to RNA by tagging an RNA of interest with a BoxB aptamer to recruit a fusion protein of λ-N and a promiscuous biotin ligase44. The biotin sprayer binds the BoxB motif through its λ-N domain and labels proteins proximal to its bound RNA (Fig. 1b). Because 20 nm corresponds to ~66 nt of linear RNA, placing BoxB aptamers both 5′ and 3′ of an RNA sequence of interest should enable the study of RNAs up to 132 nt long with this approach, and the structured nature of some RNAs might permit the study of substantially longer sequences. This approach relies on direct labeling of RBPs and does not require cross-linking or purification of the RNA45. Although there are benefits to this approach, including speed, cost, the low number of cells needed, and ease of use, there are downsides as well. First, the BoxB site needs to be proximal to the RNA sequence of interest, and thus the ‘bait’ RNA has to be expressed artificially, such as by plasmid transfection. Additionally, RNA can fold into complex structures, so the positioning of the BoxB aptamer needs to be carefully considered for longer RNA species. Given these limitations, including the 20-nm limit for the ‘reach’ of this method, RaPID might be best for studies of shorter (≤132 nt) RNA motifs.
Proteomic analysis.
Most RNA-centric approaches use quantitative mass spectrometry (MS) to discover RBPs bound to RNA. At a practical level, MS techniques can be divided between those that use labeling and those that do not46. Labeling methods can involve the differential use of stable isotope labels or chemical tagging of proteins in samples and controls. Hence, the ratio of labeled peptides can be used to obtain enrichment scores and identify true binding partners46. Labeling MS methods, such as SILAC and iTRAQ, are particularly useful with formaldehyde cross-linking or an in vitro method, as these approaches have a lower specificity, and results could be contaminated by proteins bound nonspecifically47. However, labeling methods require more technical expertise in MS data analysis and are more expensive. Label-free MS identifies proteins in both samples and controls; the challenge is in distinguishing true binding partners from nonspecific proteins. Analytical tools such as SAINT can be used with spectral count data from nonquantitative MS to effectively score the probability of a bona fide RBP-RNA interaction48. The use of more than two replicates each for samples and controls is advisable to increase stringency and avoid false positives in nonquantitative MS analysis. For negative controls, we recommend an RNA derived by scrambling the sequence of the RNA of interest. Scrambled sequences have the same length and nucleotide composition but a different primary, and therefore secondary, structure of RNA. Positive controls where the RNA has known binding partners can be included in experiments designed to validate both the experimental setup and proteomic analysis. In methods that require the purification of cellular RNA (CHART, RAP, etc.), verifying the isolated RNA by sequencing will increase confidence that proteomic analysis has indeed found interactions with the RNA of the interest. Proteomic analysis requires multiple steps from raw data acquisition to mapping and differential analysis49. There are a variety of open access and proprietary software tools for this, with attendant advantages and disadvantages50,51. It is critical to keep track of and report the tools and settings used for analysis.
RNA-centric method selection
Given the diversity of methods available, which one is optimal for a given application? Three factors can help to guide the selection of an appropriate RNA-centric method.
The first relates to the choice between an in vitro or in vivo approach. In vitro approaches are particularly powerful for determining which nucleotides and amino acids contribute to known RNA-protein interactions. In vivo methods are helpful in studies that rely on the cellular environment, where the localization of RNAs and proteins may vary from organelle to organelle (nucleus, cytoplasm, endoplasmic reticulum, etc.). Thus, in vivo methods may be best for discovering and studying RNA-protein interactions when subcellular localization, RNA and protein modifications, or a dynamic range of local protein concentrations are a factor.
The second consideration relates to RNA abundance. The copy number of the RNA of interest is critical for the detection of RNA-protein interactions. The higher the RNA copy number, the fewer cells are required in order for RNA-protein interactions to be detected in vivo. For example, the noncoding RNAs MALAT1 and NEAT1 have high expression, and thus fewer cells are needed to obtain their RBP interactome52. Many in vivo methods that use cross-linking to detect RBP interactions, however, require 100 million to 1 billion cells30,33,52. Thus, the effort and cost required to scale up experiments to study low-copy-number RNA can be over-whelming. In such a setting, the better alternative might be to use in vitro methods to study RNA-protein interactions.
The third factor involves the strength of the studied RNA- protein interaction, which especially influences the choice of cross-linking approach. Experimentally measured RNA-protein dissociation constants vary widely, ranging from high nanomolar to picomolar concentrations53. Cross-linking modalities differ in efficiency, and in general, the weaker the interaction between RNA and protein, the less likely it will be captured with UV cross-linking as opposed to formaldehyde-cross-linking-based methods. Thus, more cells might be needed to capture RNA-protein interactions in methods that use UV light versus formaldehyde for cross-linking. Furthermore, with cross-linking methods, the RNA needs to be purified with the use of oligonucleotide capture probes before bound RBPs can be isolated. The efficiency of the oligonucleotide capture further decreases the efficiency of RNA-protein interaction capture, and this necessitates higher input cell numbers. Taken together (Fig. 1), these considerations can help to guide the selection of an appropriate RNA-centric methodology.
Protein-centric methods: characterizing RNAs that bind a protein of interest
Protein-centric methods start with a protein of interest and characterize its interaction with RNA. Commonly, these approaches either directly purify the protein to find associated RNAs or use selective chemical modification of RNA in a way that relies on its association with the protein of interest. The overwhelming majority of studies that identify RNAs bound to a given protein do so by purifying the protein of interest. The most common approach in this case is to make use of the long-known fact that protein will chemically cross-link to nucleic acid in vivo when hit by UV light at approximately 254 nm (refs. 54,55). The use of 254-nm induced cross-linking played an important role in the initial identification of RBPs56. Almost all amino acids cross-link (D, E, N, and Q being the exceptions)57. Although RNA-protein cross-linking is generally thought to proceed through an initial excitation of the nucleobase, it should be noted that UVC light also induces some protein-protein cross-linking58–60. Methods that involve UV cross-linking followed by purification of the protein of interest and identification of bound RNAs are broadly termed cross-linking immunoprecipitation (CLIP)61 methods, with those that use high-throughput sequencing (HITS) forming the CLIP-seq family of methods62. Methods that use a similar protocol but an alternative cross-linker are also sometimes referred to as CLIP methods27.
A common difficulty with CLIP-seq is immunopurifying enough cross-linked RNA, which can become a problem with poor crosslinking efficiency, low RNA-ribonucleoprotein complex abundance, poor antibodies, inefficient library preparation, or combinations thereof. Unfortunately, there is no universally accepted answer to the question of how much purified cross-linked RNA is enough for CLIP. Practically, a common approach includes CLIP followed by an evaluation of whether cross-linked RNA can be visualized by dye or 32P labeling. Another common tack is to see whether a given CLIP-seq effort generates satisfactory libraries. CLIP-seq is covered in greater detail below.
If sufficient UV cross-linked complexes cannot be purified, then options diverge: if (1) indirect interactions are tolerable and (2) binding sites within RNAs do not need to be determined, then the standard method is RIP-seq63,64. RIP-seq may be conceptualized as RNA-seq after protein purification, or CLIP-seq without the removal of non-cross-linked RNAs. Essentially, immunopurification is carried out under nondenaturing conditions that are intended to preserve cellular complexes, thereby removing the need for cross-linking. RIP-seq can also provide RNA binding sequence locations if the RNase digestion is optimized63. The conventional wisdom is that CLIP has a higher signal-to-noise ratio than RIP, as might be expected from the removal of noncovalent interactions, and as supported by the often vanishingly small noepitope CLIP-seq control datasets. However, it is conceivable that RIP might have a higher signal-to-noise ratio for proteins with very poor cross-linking.
In the case that either indirect interactions are not tolerable or RIP-seq is not satisfactory, there are alternative cross-linking reagents. PAR-CLIP65, which uses 4-thiouridine and/or 5-thioguanine as a nucleotide analog, is advantageous in cases where UV light is not penetrating deep enough into the sample. However, we note that PAR-CLIP results have generally been similar to those of regular cross-linking66,67. Investigators choosing a method for their protein of interest may consult ref. 10, which identifies proteins that can be cross-linked to RNA either by 254-nm UV light or by nucleotide analog, and compares their relative efficiencies.
Recently, formaldehyde was used as a cross-linking reagent for CLIP on a double-stranded RNA-binding protein27, which are generally thought to UV cross-link poorly. Methylene blue has been used to cross-link double-stranded RNA to RBPs68, but this method has not yet been widely applied in CLIP. Many compounds known to crosslink RNA to protein, such as diepoxybutane69, 2-iminothiolane70, and DTT71, have not been studied as reagents for CLIP, and it is likely that many more uncharacterized cross-linking compounds exist. Regardless of whether standard UV cross-linking or alternative methods are used, methods that rely on protein purification for protein-centric RNA studies constitute a mainstay of the field, foremost among which are the quickly expanding varieties of CLIP.
CLIP-seq
A vast number of alternative CLIP-seq protocols have been published. We present a subway-map view of how these protocols progress from immunoprecipitation to PCR amplification in Fig. 2 (also see Supplementary Table 1). Some steps, such as initial dephosphorylation of RNA, ligation to the 3′ end of RNA, and reverse transcription, are universal, whereas others are method specific. In the original CLIP-seq protocol, 5’ and 3’ adaptors are ligated to purified RNA, and reverse transcriptase has to proceed through the crosslinked nucleotide62. Reverse transcription through the cross-linked nucleotide is 10–25% efficient for SuperScript IV72, but manganese appears to increase this rate73. CLIP variants 1 (ref. 67) and 2 (refs. 74,75) streamline the original protocol by doing both ligations on-bead. The RNA processing steps in the CLIP protocol CRAC (UV crosslinking and analysis of cDNA) are the same as in variant 1, but are preceded by a denaturing purification76. iCLIP removes the 5′ adaptor ligation to RNA, replacing it with a circularization of the cDNA77. eCLIP replaces the 5′ adaptor ligation with a 3′ cDNA ligation78, and monitored eCLIP uses both a 5′ ligation and a 3′ cDNA ligation72. irCLIP is similar to iCLIP but makes use of a biotinylated, fluorescent 3′ DNA adaptor79. BrdU-CLIP uses a nucleotide analog in reverse transcription to separate cDNA from unreacted reverse-transcription primer80. The recent GoldCLIP method is a shortened iCLIP protocol that removes the protein gel step and, like CRAC, includes an on-bead denaturation81. To date, eCLIP has probably produced the largest number of datasets, owing to its use it in the ENCODE project.
Fig. 2 |. Subway map of CLIP protocols, from immunoprecipitation to PCR.
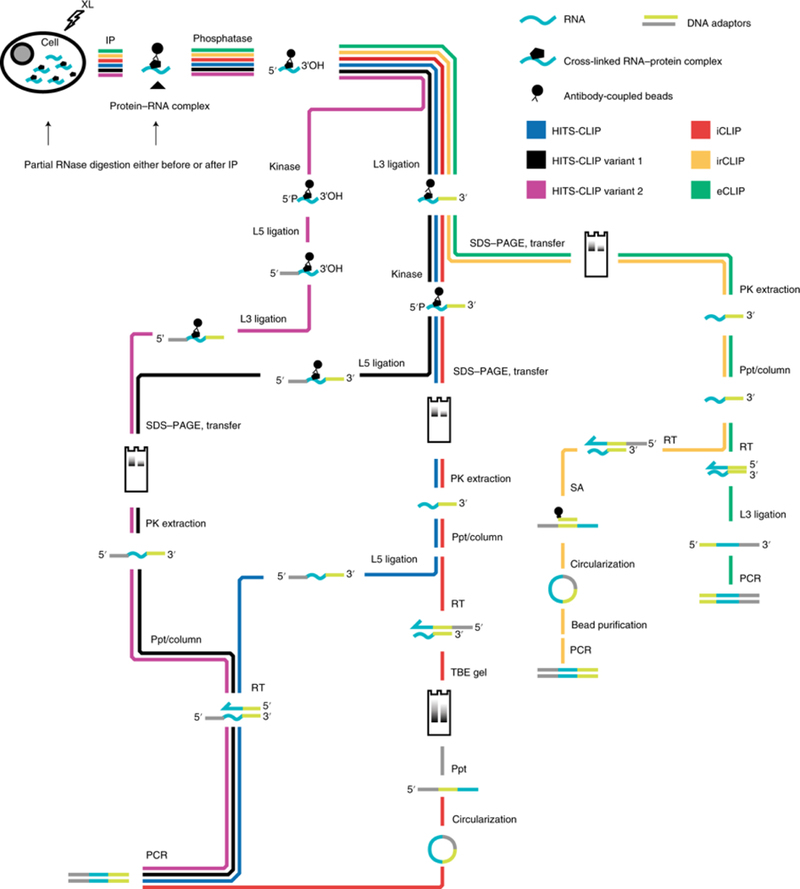
The chart highlights steps of various representative CLIP-seq protocols. Not all steps are included. XL, UV cross-link; IP, immunopurification; phosphatase, removal of 3′ phosphate; kinase, addition of 5′ phosphate; RT, reverse transcription; L3, 3′ adaptor ligation to RNA or DNA; L5, 5′ adaptor ligation; PK extraction, proteinase K extraction from nitrocellulose membrane; Ppt/column, alcohol precipitation or column cleanup of nucleic acid; TBE, Tris-borate-EDTA; SA, streptavidin.
There are currently no published data that would allow an estimation of the overall efficiency of any CLIP-seq method, which would require quantification of the number of input cellular complexes and the total library complexity at the end. In fact, it is rare for CLIP-seq methods to quantify the efficiency of any step. Investigators looking to begin CLIP with an optimal method face some confusion, as there is little in the way of true quantitative comparisons. Some advantages to each method are noted in Table 2. Fluorescent DNA adaptors (as in irCLIP79) may be particularly helpful for investigators initiating CLIP studies, as they do not interfere with any method and provide a way to track sample throughout library preparation, which can reveal where sample may be lost. The technical end point of CLIP-seq method evolution would be the sequencing of bound RNAs directly, which would bypass nearly every step in Fig. 2, but direct RNA sequencing82 has not yet been coupled to CLIP-seq. An intrinsic challenge to all CLIP-seq methods that rely on antibody immunoprecipitation is the requirement for high-quality antibodies. Although this can be overcome by the insertion of epitope tags, concerns about the physiological expression of tagged proteins and the possibility of the tag itself affecting RNA binding are limitations. CLIP-seq methods, however, continue to evolve, and represent an active area of protein-centric method innovation.
Table 2 |.
Features of selected CLIP methods
| Method | Advantages | Disadvantages | References |
|---|---|---|---|
| HITS-CLIP/PAR-CLIP | Well-established method; can identify interactions at the 3′ end of RNA | Long protocol; UV cross-linking may be poor; reverse transcriptase must bypass the cross-linked nucleotide | 62,65 |
| iCLIP | Well-established method; reverse transcriptase does not need to bypass the cross-linked nucleotide | Long protocol; UV cross-linking may be poor; interactions near the 3′ end of an RNA may be unidentifiable because reverse transcriptase stops at the cross-linked nucleotide | 77 |
| eCLIP | Avoids the circularization step of iCLIP, which can be an unreliable reaction | Long protocol; UV cross-linking may be poor; must ligate a single-stranded DNA adaptor to single-stranded cDNA | 78 |
| irCLIP | Uses a fluorescent adaptor to visualize cross-linked RNA at each step; efficient | Long protocol; UV cross-linking may be poor; interactions near the 3′ end of an RNA may be unidentifiable because reverse transcriptase stops at the cross-linked nucleotide | 79 |
| HITS-CLIP variants 1 and 2 | Short protocol; increased reliability because both ligations are carried out on-bead | UV cross-linking may be poor; reverse transcriptase must bypass the cross-linked nucleotide | 67,74–76 |
| GoldCLIP | No gel purification step; short protocol | UV cross-linking may be poor; must express a fusion protein; interactions near the 3′ end of an RNA may be unidentifiable because reverse transcriptase stops at the cross-linked nucleotide | 81 |
| fCLIP | Replaces UV cross-linking with formaldehyde, which may have a higher efficiency for proteins binding double-stranded RNA | Long protocol; formaldehyde cross-linking for CLIP is not as well understood as standard UV cross-linking | 27 |
| BrdU-CLIP | BrdU in reverse transcription removes ‘empty adaptor’ reads that can clutter HITS data | Long protocol; UV cross-linking may be poor; interactions near the 3′ end of an RNA may be unidentifiable because reverse transcriptase stops at the cross-linked nucleotide | 80 |
| TRIBE | No need to purify the protein of interest; no dependence on UV cross-linking; RBP interaction may occur anywhere in RNA | Less extensive examples of effective use of the method | 90 |
| RNA tagging | No need to purify the protein of interest; no dependence on UV cross-linking; straightforward protocol | Has not been demonstrated outside Saccharomyces cerevisiae; might not work on RBPs distant from the 3′ end of RNA | 91 |
Advantages and disadvantages of different methods of identifying RNAs bound to a protein of interest. Only a selected subset of CLIP methods is included, and many other excellent protocols exist.
CLIP-seq analysis.
There is no universal standard for CLIP-seq analysis, and new pipelines continue to emerge. This might be due to variation in study aims and in how background is defined. Table 3 summarizes some of the different analytic approaches. Several features of CLIP data may influence the analytic path taken. In chromatin immunoprecipitation (ChIP)-seq, the amount of nucleic acid (DNA) is fixed at two copies, whereas the abundance of transcription factors varies. In CLIP-seq, the abundance of both RNAs and RNA-binding proteins varies over orders of magnitude. At some frequency, all RNAs contact all proteins, and high abundances of RNA or protein may make low-affinity interactions common enough to be easily detected by CLIP As a result, the identification of an interaction between a specific RNA and protein as having occurred by, say, the clustering of cross-link-induced mutations does not by itself necessarily provide robust insight into its frequency or physiological relevance.
Table 3 |.
Approaches used to interpret CLIP-seq data
| Study | Approach | References |
|---|---|---|
| Nova | CLIP data are evaluated on the basis of enrichment relative to a control immunopurification. | 62 |
| Ago | Calculates the expected number of reads per RNA from RNA abundance and randomizes their positions to determine the odds of a peak as high as that observed. | 94 |
| eCLIP | CLIP data are evaluated on the basis of enrichment relative to RNA from the input lysate run on an SDS-PAGE gel and transferred to a nitrocellulose membrane. | 78 |
| FOX2 | Reads are scrambled randomly in transcript to calculate the odds of the observed peak height. This differs from the method used for Ago above94 in that the reads are drawn from the CLIP data, not RNA abundance. As a result, this approach identifies a locational bias within a given RNA, not affinity for the RNA molecule of interest, although the latter is indirectly tested by the requirement for having enough reads to identify a locational bias. | 95 |
| Puf2p | Raw peak height cutoff, enrichment relative to local CLIP signal, and enrichment over expectation from RNA abundance. This method is a combination of a local signal pileup analysis like that of FOX295, enrichment relative to RNA abundance like that of Ago94, and a third raw peak height requirement. | 67 |
| FBF | Compares each of the different approaches available at the time of its development: local pileup analysis (similar to FOX2 above95), enrichment over RNA abundance (similar to Ago94), enrichment over a control immunopurification (similar to Nova62), CIMS96, and CITS80. Enrichment over a control immunopurification was found to most accurately reflect known biology. | 97 |
| Nova, Ago | Frequency of cross-link-induced mutation pileup versus random permutations. Termed CIMS. The notes on FOX2 apply to this approach as well. | 96 |
| hnRNPC, TIA1, Rbfox | Frequency of reverse-transcription termination positions versus random permutations. Termed CITS80. The notes on FOX2 apply to this approach as well. | 80, 98,99 |
| Many | Frequency of PAR-CLIP-specific mutations. We refer the reader to a more specialized review of CLIP-seq analysis tools100. | 75, 101, 102 |
A non-exhaustive list of selected approaches is noted. This table presents examples of the different concepts of CLIP-seq analysis, rather than comparing the numerous implementations of those approaches.
Quantitation of CLIP-seq data is one current area of analytic challenge. If PCR duplicates are removed, each read in a CLIP library should, in principle, correspond to a single cross-linked protein-RNA complex isolated from a cell. That is, barring technical artifacts, each sequenced DNA fragment must have been transcribed from a single cross-linked RNA fragment purified from the cell. If the CLIP library-preparation protocol has approximately the same efficiency for nearly all RNA fragments, as is commonly assumed, then each stack of reads in a region per all reads corresponds to that region’s proportion of all cross-linked complexes. As the reads-per-million value represents the read count per all reads multiplied by 1 million, reads-per-million in CLIP-seq data represents the frequency of cross-links at a given RNA out of all crosslinks. Because this frequency includes a cross-linking efficiency factor (the proportion of RNA-protein complexes that react with UV light to form a covalent bond) that is dependent on the RNA and may vary considerably, it might not always be accurate to treat cross-link frequencies as true RNA-protein interaction frequencies. The currently unpredictable variability of cross-linking efficiency for different RNA sequences introduces a second complexity in CLIP-seq compared with ChIP-seq. The number of reads at a region in a sequenced CLIP library is equal to the number of cross-links at that region multiplied by a long string of multiplicative efficiency factors, one for each step in library construction. That is, there is some number of cross-linked RNA-protein complexes containing a given RNA present in the lysate input to a CLIP experiment, and at each subsequent step of library construction only a fraction of molecules is correctly processed. As a result, CLIP signal tends to vary on a log scale between replicates, and, as with RNA-seq data83, a conversion to a log scale is frequently helpful.
A large number of CLIP analysis programs have been published (Table 3); however, none has as yet been universally adopted as a standard, and it is not uncommon for a method to be used in only a handful of studies. This is likely due to the fact that an experienced bioinformatician can write a script to implement any of the approaches in Table 3 with a similar amount of labor as it would take to adapt an existing tool, thereby avoiding giving up both control of and detailed understanding of the analytic pipeline. CLIP analysis programs are likely to become more user-friendly with time, but meanwhile, multipurpose software libraries to deal with HITS data have also become more extensively functional and easier to adapt to situations like CLIP. For example, the combination of Python libraries HTSeq84, NumPy85, SciPy86, statsmodels87, and pandas88 (to name a few) provides an extremely powerful framework for CLIP analysis, and Jupyter89 notebooks create an interactive environment for programming analysis suites. It is possible that no complete CLIP analysis package will become standard, although a consensus may form regarding well-written, especially fast algorithms for specialized CLIP-related tasks. At present, it probably remains optimal for CLIP analysis to involve mostly bespoke code that uses multiple forms of read count and mutation analysis, although programs such as those referenced in Table 3 may be satisfactory for many experiments.
Methods not requiring protein purification
Methods to identify the RNA targets of an RBP without purifying the RBP are relatively new, and currently rely on two different chemical modifications of RNA. In the first, TRIBE90, the RBP of interest is fused to the enzyme ADAR, which deaminates nearby adenosines, after which deaminated bases are subsequently identified by sequencing. In the second, RNA tagging91, the RBP of interest is fused to the enzyme poly(U) polymerase, which adds poly(U) tails to bound RNAs. Tails are subsequently identified by sequencing of the 3′ ends of RNA. The coupling of RBPs to peroxidase tags has also been used to identify RNAs in specific subcellular compartments; this method has not yet been used to identify direct RNA targets92. Far more enzymatic modifications of RNA are possible, and we expect this field to see rapid growth as more and better methods are found. Especially exciting are the possibility of multiple distinct chemical marks being made by separate RBPs for studies of combinatorial regulation, and the combination of chemical modification with RBP purifications to study the locations of RBP-RNA interactions.
Conclusion
The technical and conceptual advances in methods for studying RNA-protein interactions have shed light on complex and critical RNA-protein interactions in cells. There are areas where further innovation could spur the accessibility of these methods to researchers. Cross-linking is the crux of several methods used to identify RBPs and to define RNAs bound to protein. Current UV and formaldehyde cross-linking approaches are inefficient, and better cross-linking methods could capture RNA-protein interactions efficiently with fewer cells. Advances in orthogonal areas could also generate novel methods to study RNA-protein interactions. The discovery of RNA-specific Cas proteins could be adapted to probe RNA-protein interactions in cells93. Fusion of these Cas proteins with enzymes could label either RNA or protein at specific spots along an RNA. With potentially exciting new tools on the horizon, it is important for researchers to be aware of the strengths and limitations of different methods. In virtually all cases, orthogonal methods are essential to validate results based on a single method. For example, RBPs discovered via an RNA-centric method should be validated by the complementary protein-centric method (CLIP, etc.) for confident identification of bona fide RNA-protein interactions. Taken together, the expanding arsenal of both RNA-centric and protein-centric methods for the study of RNA-protein interactions will accelerate progress in this expanding area of biology.
Supplementary Material
Acknowledgements
We thank members of the Khavari lab for helpful discussions and apologize to colleagues whose work was not cited because of the space limitations of this review. This work was supported by grant 1F32AR072504 to D.F.P., by a USVA Merit Review grant, and by NIAMS/NIH grants AR45192 and AR49737 to P.A.K.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41592-019-0330-1.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zhu X Seeing the yin and yang in cell biology. Mol. Biol. Cell 21, 3827–3828 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayr C Regulation by 3′-untranslated regions. Annu. Rev. Genet. 51, 171–194 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Moore MJ From birth to death: the complex lives of eukaryotic mRNAs. Science 309, 1514–1518 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Allerson CR, Cazzola M & Rouault TA Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia-cataract syndrome. J. Biol. Chem. 274, 26439–26447 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Batista PJ & Chang HY Long noncoding RNAs: cellular address codes in development and disease. Cell 152, 1298–1307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urdaneta EC et al. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. bioRxiv Preprint at https://www.biorxiv.org/content/10.1101/333385v1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trendel J et al. The human RNA-binding proteome and its dynamics during translational arrest. Cell 176, 391–403 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Re A, Joshi T, Kulberkyte E, Morris Q & Workman CT RNA-protein interactions: an overview. In RNA Sequence, Structure and Function: Computational and Bioinformatic Methods (eds. Gorodkin J & Ruzzo WL) 491–521 (Humana Press, Totowa, NJ, 2014). [DOI] [PubMed] [Google Scholar]
- 9.Hentze MW, Castello A, Schwarzl T & Preiss T A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Castello A et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406 (2012).Comprehensive poly(A) RNA-protein interactome capture performed in HeLa cells. [DOI] [PubMed] [Google Scholar]
- 11.Huang R, Han M, Meng L & Chen X Transcriptome-wide discovery of coding and noncoding RNA-binding proteins. Proc. Natl Acad. Sci. USA 115, E3879–E3887 (2018).Utilization of RNA metabolic labeling to comprehensively capture the RNA-protein interactome independent of RNA polyadenylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matia-Gonzalez AM, Laing EE & Gerber AP Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 22, 1027–1033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castello A et al. Comprehensive identification of RNA-binding domains in human cells. Mol. Cell 63, 696–710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castello A, Hentze MW & Preiss T Metabolic enzymes enjoying new partnerships as RNA-binding proteins. Trends Endocrinol. Metab. 26, 746–757 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faoro C & Ataide SF Ribonomic approaches to study the RNA-binding proteome. FEBS Lett. 588, 3649–3664 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Zheng X et al. Detecting RNA-protein interaction using end-labeled biotinylated RNA oligonucleotides and immunoblotting In RNA-Protein Complexes and Interactions (ed. Lin R-J) 35–44 (Humana Press, New York, 2016). [DOI] [PubMed] [Google Scholar]
- 17.Hartmuth K, Vornlocher H-P & Luhrmann R Tobramycin affinity tag purification of spliceosomes In mRNA Processing and Metabolism (ed. Schoenberg DR) 47–64 (Humana Press, Totowa, NJ, 2004). [DOI] [PubMed] [Google Scholar]
- 18.Hogg JR & Collins K RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 13, 868–880 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leppek K & Stoecklin G An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 42, e13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HY et al. RNA-protein analysis using a conditional CRISPR nuclease. Proc. Natl Acad. Sci. USA 110, 5416–5421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kretz M et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493, 231–235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siprashvili Z et al. The noncoding RNAs SN0RD50A and SN0RD50B bind K-Ras and are recurrently deleted in human cancer. Nat. Genet. 48, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutherland BW, Toews J & Kast J Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J. Mass Spectrom. 43, 699–715 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Li X, Song J & Yi C Genome-wide mapping of cellular protein-RNA interactions enabled by chemical crosslinking. Genomics Proteomics Bioinformatics 12, 72–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beckmann BM RNA interactome capture in yeast. Methods 118–119, 82–92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugimoto Y et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol. 13, R67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim B & Kim VN fCLIP-seq for transcriptomic footprinting of dsRNA-binding proteins: lessons from DROSHA. Methods 152, 3–11 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Meisenheimer KM & Koch TH Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 32, 101–140 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Hoffman EA, Frey BL, Smith LM & Auble DT Formaldehyde crosslinking: a tool for the study of chromatin complexes. J. Biol. Chem. 290, 26404–26411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu C, Qu K, Zhong FL, Artandi SE & Chang HY Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 44, 667–678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon MD et al. The genomic binding sites of a noncoding RNA. Proc. Natl Acad. Sci. USA 108, 20497–20502 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McHugh CA et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McHugh CA & Guttman M RAP-MS: a method to identify proteins that interact directly with a specific RNA molecule in cells. Methods Mol. Biol. 1649, 473–488 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Hacisuleyman E et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 21, 198–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng F et al. A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nat. Protoc. 1, 920–927 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Tsai BP, Wang X, Huang L & Waterman ML Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol. Cell. Proteomics 10, M110.007385 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matia-Gonzalez AM, Iadevaia V & Gerber AP A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 118–119, 93–100 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Parrott AM et al. RNA aptamers for the MS2 bacteriophage coat protein and the wild-type RNA operator have similar solution behaviour. Nucleic Acids Res. 28, 489–497 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu C et al. Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simon MD Capture hybridization analysis of RNA targets (CHART). Curr. Protoc. Mol. Biol. 101, 21.25.1–21.25.16 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kim DI et al. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl Acad. Sci. USA 111, E2453–E2461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim DI et al. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–1196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roux KJ, Kim DI, Raida M & Burke B A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramanathan M et al. RNA-protein interaction detection in living cells. Nat. Methods 15, 207–212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramanathan M & Khavari P RNA-protein interaction detection (RaPID). Protocol Exchange 10.1038/protex.2018.003 (2018). [DOI] [Google Scholar]
- 46.Bantscheff M, Schirle M, Sweetman G, Rick J & Kuster B Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 (2007).Comprehensive analysis of mass spectrometry methods used in proteomics. [DOI] [PubMed] [Google Scholar]
- 47.Butter F, Scheibe M, Morl M & Mann M Unbiased RNA-protein interaction screen by quantitative proteomics. Proc. Natl Acad. Sci. USA 106, 10626–10631 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mellacheruvu D et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai T-H, Wang M & Ressom HW Preprocessing and analysis of LC-MS-based proteomic data. Methods Mol. Biol. 1362, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Röst HL et al. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748 (2016).This paper presents a common platform with ready-made workflows allowing users to perform reproducible mass spectrometry analysis with ease. [DOI] [PubMed] [Google Scholar]
- 51.Cox J et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011). [DOI] [PubMed] [Google Scholar]
- 52.West JA et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 55, 791–802 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang X, Li H, Huang Y & Liu S The dataset for protein-RNA binding affinity. Protein Sci. 22, 1808–1811 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith KC & Aplin RT A mixed photoproduct of uracil and cysteine (5-S-cysteine-6-hydrouracil). A possible model for the in vivo cross-linking of deoxyribonucleic acid and protein by ultraviolet light. Biochemistry 5, 2125–2130 (1966). [DOI] [PubMed] [Google Scholar]
- 55.Goddard J, Streeter D, Weber C & Gordon MP Studies on the inactivation of tobacco mosaic virus by ultraviolet light. Photochem. Photobiol. 5, 213–222 (1966). [DOI] [PubMed] [Google Scholar]
- 56.Choi YD & Dreyfuss G Isolation of the heterogeneous nuclear RNA-ribonucleoprotein complex (hnRNP): a unique supramolecular assembly. Proc. Natl Acad. Sci. USA 81, 7471–7475 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kramer K et al. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 11, 1064–1070 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwartz JC, Wang X, Podell ER & Cech TR RNA seeds higher-order assembly of FUS protein. Cell Rep. 5, 918–925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tundup S, Akhter Y, Thiagarajan D & Hasnain SE Clusters of PE and PPE genes of Mycobacterium tuberculosis are organized in operons: evidence that PE Rv2431c is co-transcribed with PPE Rv2430c and their gene products interact with each other. FEBS Lett. 580, 1285–1293 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Itri F et al. Identification of novel direct protein-protein interactions by irradiating living cells with femtosecond UV laser pulses. Biochem. Biophys. Res. Commun. 492, 67–73 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Ule J et al. CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Licatalosi DD et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469 (2008).First paper to apply high-throughput sequencing to a CLIP method. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicholson CO, Friedersdorf M & Keene JD Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA 23, 32–46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tenenbaum SA, Carson CC, Lager PJ & Keene JD Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl Acad. Sci. USA 97, 14085–14090 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hafner M et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kishore S et al. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods 8, 559–564 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Porter DF, Koh YY, VanVeller B, Raines RT & Wickens M Target selection by natural and redesigned PUF proteins. Proc. Natl Acad. Sci. USA 112, 15868–15873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu ZR, Wilkie AM, Clemens MJ & Smith CW Detection of double-stranded RNA-protein interactions by methylene blue-mediated photo-crosslinking. RNA 2, 611–621 (1996). [PMC free article] [PubMed] [Google Scholar]
- 69.Baumert HG, Skold SE & Kurland CG RNA-protein neighbourhoods of the ribosome obtained by crosslinking. Eur. J. Biochem. 89, 353–359 (1978). [DOI] [PubMed] [Google Scholar]
- 70.Wower I, Wower J, Meinke M & Brimacombe R The use of 2-iminothiolane as an RNA-protein cross-linking agent in Escherichia coli ribosomes, and the localisation on 23S RNA of sites cross-linked to proteins L4, L6, L21, L23, L27 and L29. Nucleic Acids Res. 9, 4285–4302 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaman U et al. Dithiothreitol (DTT) acts as a specific, UV-inducible cross-linker in elucidation of protein-RNA interactions. Mol. Cell. Proteomics 14, 3196–3210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hocq R, Paternina J, Alasseur Q, Genovesio A & Le Hir H Monitored eCLIP: high accuracy mapping of RNA-protein interactions. Nucleic Acids Res. 46, 11553–11565 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Nostrand EL, Shishkin AA, Pratt GA, Nguyen TB & Yeo GW Variation in single-nucleotide sensitivity of eCLIP derived from reverse transcription conditions. Methods 126, 29–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Creamer TJ et al. Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet. 7, e1002329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benhalevy D, McFarland HL, Sarshad AA & Hafner M PAR-CLIP and streamlined small RNA cDNA library preparation protocol for the identification of RNA binding protein target sites. Methods 118–119, 41–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Granneman S, Kudla G, Petfalski E & Tollervey D Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc. Natl Acad. Sci. USA 106, 9613–9618 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Konig J et al. iCLIP—transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J. Vis. Exp. 2011, 2638 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016).This paper introduced the eCLIP method, which has produced the largest number of CLIP datasets so far. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zarnegar BJ et al. irCLIP platform for efficient characterization of protein-RNA interactions. Nat. Methods 13, 489–492 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weyn-Vanhentenryck SM et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 6, 1139–1152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gu J et al. GoldCLIP: gel-omitted ligation-dependent CLIP. Genomics Proteomics Bioinformatics 16, 136–143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garalde DR et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Yip SH, Wang P, Kocher JA, Sham PC & Wang J Linnorm: improved statistical analysis for single cell RNA-seq expression data. Nucleic Acids Res. 45, e179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van der Walt S, Colbert SC & Varoquaux G The NumPy array: a structure for efficient numerical computation. Comput. Sci. Eng. 13, 22–30 (2011). [Google Scholar]
- 86.Oliphant TE Python for scientific computing. Comput. Sci. Eng. 9, 10–20 (2007). [Google Scholar]
- 87.Seabold S & Perktold J Statsmodels: econometric and statistical modeling with Python In Proc. 9th Python in Science Conference (SciPy 2010) (eds. van der Walt S & Millman J) 57–61 (LuLu Press, Morrisville, NC, 2010). [Google Scholar]
- 88.McKinney W Data structures for statistical computing in Python In Proc. 9th Python in Science Conference (SciPy 2010) (eds. van der Walt S, & Millman J) 57–61 (LuLu Press, Morrisville, NC, 2010). [Google Scholar]
- 89.Perez F & Granger BE IPython: a system for interactive scientific computing. Comput. Sci. Eng. 9, 21–29 (2007). [Google Scholar]
- 90.McMahon AC et al. TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell 165, 742–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lapointe CP, Wilinski D, Saunders HAJ & Wickens M Protein-RNA networks revealed through covalent RNA marks. Nat. Methods 12, 1163–1170 (2015).First paper to identify RNAs bound to a protein of interest by selective enzymatic modification of bound RNAs followed by high-throughput sequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaewsapsak P, Shechner DM, Mallard W, Rinn JL & Ting AY Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 6, e29224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cox DBT et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chi SW, Zang JB, Mele A & Darnell RB Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460, 479–486 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yeo GW et al. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 16, 130–137 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang C & Darnell RB Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 29, 607–614 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Prasad A et al. The PUF binding landscape in metazoan germ cells. RNA 22, 1026–1043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.König J et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 17, 909–915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang Z et al. iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol. 8, e1000530 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De S & Gorospe M Bioinformatic tools for analysis of CLIP ribonucleoprotein data. Wiley Interdiscip. Rev. RNA 8, e1404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Corcoran DL et al. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 12, R79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sievers C, Schlumpf T, Sawarkar R, Comoglio F & Paro R Mixture models and wavelet transforms reveal high confidence RNA-protein interaction sites in M0V10 PAR-CLIP data. Nucleic Acids Res. 40, e160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.