

Abstract
Antibiotic resistant organisms (AROs) are a major concern to public health worldwide. While antibiotics have been naturally produced by environmental bacteria for millions of years, modern wide-spread use of antibiotics have enriched resistance mechanisms in human-impacted bacterial environments. Antibiotic resistance genes (ARGs) continue to emerge and spread rapidly. To combat the global threat of antibiotic resistance, researchers must develop methods to rapidly characterize AROs and ARGs; monitor their spread across space and time; and identify novel ARGs and resistance pathways. We review how high-throughput sequencing-based methods can be combined with classic culture-based assays to characterize, monitor, and track AROs and ARGs. Then, we evaluate genomic and metagenomic methods for identifying ARGs and biosynthetic pathways for novel antibiotics from genomic datasets. Together, these genomic analyses can improve surveillance and prediction of emerging resistance threats and accelerate the development of new antibiotic therapies to combat resistance.
Keywords: genomics, antibiotics, resistance, bacteria, bioinformatics, computation biology, pharmacoresistance, infectious disease
Introduction
Since Paul Ehrlich’s discovery that arsphenamine could cure syphilis-infected rabbits and Alexander Fleming’s serendipitous discovery of the natural product penicillin, antibiotics have been among the most successful chemotherapies in the history of medicine(1). In addition to directly treating infectious diseases, a leading cause of morbidity and mortality for most of human history, antibiotics enable treatments like invasive surgery, organ transplantation, and cancer chemotherapy, which would otherwise be unsafe due to the high risk of infection(2). The emergence of widespread antibiotic resistance significantly compromises these advancements. The WHO described antibiotic resistance as the single greatest challenge in infectious disease today, threatening high and low gross domestic product countries alike(3). One analysis of data from the European Union estimated that in 2015 there were over 600,000 infections and 33,000 deaths attributable to antibiotic-resistant organisms (AROs), figures that have increased by nearly 2.5-fold compared to similar estimates from 2007(4).
Although human use of antibiotics began in the 20th century, antibiotics have been naturally produced by environmental bacteria for millions of years(3). Because antibiotic production appears to be a natural feature of bacterial communities, it should not be surprising that defense mechanisms against those compounds appear as an evolutionary consequence(5). Sequencing of DNA preserved in ancient environmental samples have detected diverse antibiotic resistance genes (ARGs) in metagenomes of 30,000-year-old permafrost sediments and bacteria recovered from a cave isolated from human contact for four million years(6, 7). Similarly, sequencing of the host-associated microbiomes of previously uncontacted Amerindians detected numerous ARGs against modern antibiotics(8). The collection of resistance genes in a given microbiome is known as the resistome (Table 1). Worryingly, longitudinal analyses of resistomes have suggested that human use of antibiotics have influenced their composition(3, 9).
Table 1.
Glossary of terms.
| Term | Definition |
|---|---|
| Antibiotic resistance gene (ARG) | A gene that confers an organism the ability to resist an antibiotic treatment that would otherwise affect it |
| Antibiotic resistant organism (ARO) | An organism that is not controlled or killed by an antibiotic treatment that would otherwise affect it |
| Biosynthetic gene cluster (BGC) | A physically clustered group of two or more genes in a particular genome that together encode a biosynthetic pathway for the production of a specialized metabolite |
| Commensal | A microbe that coexists with a host without detriment or obvious benefit to the host |
| Contig | Contiguous DNA sequence assembled from shorter sequencing reads |
| Homology | In the context of genomics, the existence of a shared ancestry between two genes |
| Horizontal gene transfer | The transfer of genes between microbes that are not in a parent-offspring relationship. Usually occurs through transformation, conjugation, or transduction |
| Isolate | A pure strain of microbe which has been separated from a mixed microbial culture |
| Microbiome | The collective community of microbial genomes in a given environment or sample |
| Next-generation sequencing (NGS) | A collection of DNA sequencing technologies that allow for rapid generation of sequencing data that is orders of magnitude larger in volume than Sanger sequencing. Includes technologies like Illumina, Roche 454, and Ion Torrent sequencing |
| Open reading frame (ORF) | A sequence of DNA that is translated into protein |
| Pathogen | An infectious agent whose presence results in disease or damage to the host |
| Profile Hidden Markov model (pHMM) | A statistical model where hidden states emit observed sequences with a defined probability. Widely used for genome annotation, where protein/gene families are modeled as “states” and the primary amino acid/nucleotide sequence is the observed sequence |
| Resistome | The collection of resistance genes in a given environment or sample. This includes intrinsic, acquired, proto-, and cryptic resistance genes. |
| Proto-resistance gene | A gene that has the potential to evolve into a resistance element |
| Cryptic resistance gene | A resistance gene that is embedded in the bacterial chromosome, but does not obviously confer resistance due to low expression |
| Strain | A genetic variant or subtype of a microbial species |
| Whole genome sequencing (WGS) | The complete sequencing of genes from an organism |
| Whole metagenome sequencing (WMS) | The complete sequencing of genes from all organisms in a given environment or sample |
Selective pressure from wide-spread use of antibiotics in agricultural and clinical settings have enriched natural resistance mechanisms in human-impacted bacterial environments. Analysis of soil samples from 1940 to present-day revealed increased abundance of ARGs to a wide range of antibiotics, and clinical resistance to every class of antibiotics has been observed, sometimes within the same year as deployment(3, 9). This is further highlighted by the rapid global spread of plasmid-borne resistance determinants like mcr1, blaKPC, blaNDM-1, and qnr, which provide resistance to antibiotics of last-resort(5). However, the clinical focus on antibiotic resistance in human pathogens has historically led to an under-sampling and underestimate of the diverse array of resistance determinants that exist in environmental bacteria, which may be exchanged with pathogenic hosts through horizontal gene transfer(10–13). Therefore, the study of antibiotic resistance has recently expanded from examination of isolated, cultivatable and easily-cultured pathogens to include metagenomic approaches that capture resistomes of microbial communities across diverse habitats (10, 14, 15). These genomic assays are ideally combined with functional and culture-based assays to improve surveillance and prediction of emerging resistance threats(16, 17). Further, by characterizing the vast biosynthetic capacity of the microbiome, we can accelerate the development of new antibiotic therapies to combat resistance(18, 19).
In this review, we provide an overview of how high-throughput sequencing-based methods can be combined with classic culture-based assays to improve diagnosis and treatment of infectious disease. We examine how these new methods are used to characterize, monitor, and track resistance determinants. Next, we critically evaluate the genomic and metagenomic methods that have been developed for identifying ARGs and biosynthetic pathways for novel antibiotics from genomic datasets, and highlight exciting new methods that have utilized sequencing technologies to explore genomic diversity beyond what nature can provide.
Using genomics to better characterize and identify AROs and ARGs
Surveying the presence and abundance of AROs is frequently done through culture-based methods, where microbial colonies are isolated and identified by phenotypic characteristics, such as morphology, size, and pigmentation, or by chemical profile using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Figure 1A,B)(20). After identification, phenotypic antibiotic susceptibility of isolates can be determined by disk diffusion, antibiotic gradient, or broth micro-dilution to determine minimum inhibitory concentrations (Figure 1B)(21). Together, culture-based methods allow for characterization of isolates but are limited in speed and scale(22). Because of this, screening a large number and variety of isolates from both patients and environmental settings can be a tedious and expensive process(22). Recent studies in research and diagnostic microbiology pair culture-based methods with next-generation sequencing (NGS) (Figure 1B). Advances in NGS throughput have enabled relatively inexpensive and fast whole genome sequencing (WGS) of isolates, permitting WGS to be used more readily in both research and diagnostic microbiology(23). WGS analyses allow for accurate identification of isolate strain phylogeny, ARGs, and epidemiological traits, which is critical when tracking outbreaks(16, 24) (Figure 1B).
Figure 1.
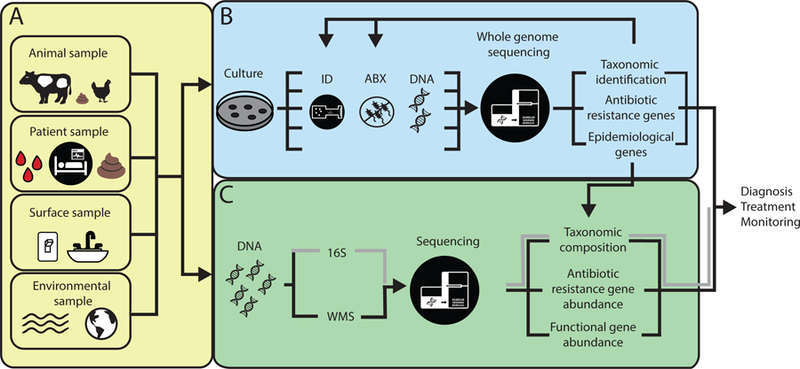
Biological and environmental samples can be interrogated for ARO and ARG enrichment using whole genome sequencing (WGS) and metagenomics analyses. A) Samples can be collected from diverse origin, including biological samples such as tissue, blood, or fecal samples from animal or patient samples. Environmental samples can be collected directly from built environment surfaces or natural environments such as soil and water sources. B) In culture-based methods, isolates are extracted from samples and processed individually to determine species identification and antibiotic susceptibility. After DNA extraction, each isolate can be sequenced using WGS. WGS analyses can determine taxonomic identification, antibiotic resistance genotype, and epidemiological genes. These characteristics can be compared to culture-based results, creating genotype-phenotypic characterization of isolates and improved characterization of species and resistome. WGS analyses can also be used to improve sequence based searches for metagenomics analyses. C) In metagenomics analyses, genomic DNA is extracted directly from a sample, then 16S or whole metagenomics sequencing is done. After 16S amplification and sequencing, taxonomic composition is determined. After WMS, taxonomic composition, antibiotic resistance gene abundance, and functional gene abundance can be analyzed. All of these data can be combined to improve diagnosis, treatment, and ARO and ARG surveillance.
The future of monitoring and combating the presence of AROs and ARGs lies in successfully integrating and optimizing culture-based methods with WGS analyses to rapidly identify, characterize, and track emerging pathogens. Diverse microbial species and strains can have very different pathological patterns, and accurate identification of strain and characterization of resistance profiles is critical when monitoring and treating pathogens in humans(24, 25). For example, Klebsiella is a genus of Gram-negative bacteria that is commonly found in both environmental and human-associated samples. Klebsiella pneumoniae is well known for being an opportunistic human pathogen associated with nosocomial infections and worldwide antibiotic resistance(26). The related species Klebsiella variicola, identified in 2003, was considered less pathogenic; however, recent studies suggest that 2–10% of K. pneumoniae strains isolated from infections are actually misidentifications of K. variicola, indicating that K. variicola is more prevalent in infections than initially thought(26–28). Further, there is an increased mortality in patients infected with K. variicola compared to patients with K. pneumoniae, and members of the two species have different resistance profiles(24, 27, 28). By integrating culture-based and WGS analyses, a recent study successfully reclassified commonly misidentified K. variicola isolates and better characterized phylogeny and resistance profiles of this emerging pathogen(24). This study uncovered many high-risk antibiotic resistance genes in K. variicola genomes from a variety of geographic locations(24). Further, by using WGS to investigate key virulence factors, this study demonstrated that genomic changes in K. variicola pilus genes were associated with uropathogenicity in a mouse model of urinary tract infections(24). Altogether, recent studies of K. variicola highlight the importance of accurate identification and characterization of emerging pathogens and how culture-based and WGS analyses can be integrated to gain a better understanding of emerging pathogens(24, 26–28).
Using genomics to better track outbreaks and monitor reservoirs of AROs and ARGs
Beyond identification and characterization of AROs, it is critical to track and predict antibiotic resistant outbreaks and reservoirs of AROs and ARGs(16, 17, 29). To do this, studies must be able to accurately identify the relatedness and antibiotic resistance profiles of isolates. A recent study in Australia used WGS and culture-based methods to track an emerging pathogen, vanA-possessing vancomycin-resistant Enterococcus faecium (VREfm)(16). Prior to 2015, vanA VREfm was rare in Australia but has subsequently had a dramatic increase with 44% of the VREfm isolates identified as vanA in 2016(16). WGS of VREfm isolates was used to determine the phylogenetic relationship of this emerging pathogen in Australia. Using core genome single nucleotide polymorphism (SNP) diversity, this study found 26 of the isolates from 24 patients in the outbreak were from the same strain; 17 of those isolates could not be differentiated at the core genome level; and the remaining 9 isolates differed from the main cluster by 1–6 SNPs(16). This resolution allowed the study to track the emergence of colonization and clinical infection from vanA VREfm(16). In another study, clonal and strain tracking was used to determine the spread of a multidrug-resistant Sphingomonas koreensis outbreak in the United States(17). Sphinogomonas species are common in built and aquatic environments but have been implicated in a range of infections, including pneumonia, meningitis, catheter-associated bloodstream infections, and wound infections(17). This study investigated 6 infections of Sphingomonas over a 6-month period at the US National Institutes of Health (NIH). Core genome single nucleotide variants (SNVs) were used to identify and track strains across patients and clinical water sources(17). Four of the S. koreensis patient isolates had high genomic similarity to each other (>99.9% average nucleotide identity (ANI)) and to environmental S. koreensis isolates from water and faucet samples collected from inside patient rooms (>99.7% ANI)(17). This genomic similarity suggested that there was a common environmental reservoir for this pathogen that had persisted in the hospital infrastructure over time(17). Further, banked S. koreensis isolates from 2006 also showed high similarity to isolates from the 2016 outbreak (>99.8% ANI), suggesting this reservoir had persisted for a decade(17). These studies demonstrate the capabilities of WGS in tracking individual strains in outbreaks.
Beyond tracking the strains across an outbreak, WGS analyses can be used to determine the effectiveness of mediation strategies to prevent future ARO outbreaks. One such study tracked a large Klebsiella pneumoniae carbapenemase (KPC)-producing E. coli outbreak to determine the impact of ARO decontamination intervention after an outbreak. KPC-producing E. coli are uncommon and the emergence of an outbreak with carbapenem resistance is a particular concern, because of the extremely limited treatment options for such infections(29). In 2015, a sudden increase of KPC-producing E. coli colonization was detected in patient fecal samples at the Central Manchester University Hospital NHS Foundation Trust (United Kingdom), spurring a genomic epidemiological study on isolates from patients and the environment. While environmental samples were taken from sinks, drains, toilets, sluices, and other high-touch sites, all carbapenem-resistant Enterobacteriaceae (CRE) cultures were associated with wastewater sites(29). WGS analyses indicated that the outbreak was due to the spread of a KPC-producing E. coli clone among both patients and the environment(29). A decontamination intervention was performed, and CRE incidence declined after the wards were closed and the plumbing replaced(29). However, shortly after the intervention, wastewater sites were rapidly recolonized again by the KPC-producing clone, suggesting the importance of developing better containment and elimination strategies(29). Together, these studies demonstrate how WGS can be used as a tool to better characterize emerging pathogens, monitor reservoirs of AROs and ARGs, and evaluate success of reservoir decontamination interventions within hospitals.
While WGS continues to become quicker and less expensive, the technology is more expensive and computationally intensive than other clinical methods to identify and characterize pathogens(30). Sequencing costs can vary from $100-$500 a genome based on a number of factors including type of sequencing, genome size, depth of sequencing, and number of samples processed(31, 32). Further, a maintained bioinformatics pipeline is essential to store, process and interpret WGS for clinical relevance. This pipeline comes with associated costs and additional necessary expertise. Because of these costs, WGS currently does not replace the need for other clinical methods for individual patient diagnosis and is best applied when characterizing emerging pathogens or outbreaks of antibiotic resistant organisms.
Using metagenomics to improve identification of AROs and clinical treatment
Culture-based methods provide valuable insights to AROs colonization and allow for WGS investigation into ARO strains across outbreaks. However, these methods often restrict studies to a few genes of interest or to easily cultured organisms, precluding studies on the emergence and spread of AROs and ARGs in diverse and complex uncultured microbial communities. As ARGs can spread from environmental organisms to human pathogens, it is essential to survey and characterize environmental microbes alongside human pathogens(10). Uncultured microbial communities can be interrogated using metagenomics, wherein genomic DNA (gDNA) is extracted directly from a sample without the need to first culture microorganisms (Figure 1C). After DNA extraction, there are two prominent methods to characterize whole microbial communities: 16S sequencing and whole metagenome sequencing (WMS). 16S metagenomic studies amplify and sequence a small variable segment of the 16S RNA gene. The 16S rRNA gene is highly conserved and present in almost all bacteria. From 16S sequences, it is possible to infer microbial taxonomic presence and relative abundance from a sample. In WMS, all genomic DNA is fragmented and sequenced directly after extraction without amplification. By comparing the fragmented shotgun reads to databases of known functional and resistance genes, it is possible to determine relative abundance of taxa and known functional and resistance genes(33–35) (Figure 1C). Both metagenomic methods are relatively quick and high throughput compared to culture-based methods(3). Further, longitudinal metagenomic approaches are well suited for addressing questions of burden and accumulation of antibiotic resistance. Samples taken from the same patient or population across treatment will allow researchers to directly address how resistance has changed and determine strategies to minimize the emergence and transmission of resistance. Thus, metagenomic analyses can be a useful tool to monitor and characterize AROs and ARGs in areas where culture-based methods are insufficient or inconvenient.
While culture-based methods are the standard by which to diagnose and confirm infectious agents in clinical microbiology, many samples entering clinical laboratories return negative despite being subjected to a battery of conventional laboratory tests(25, 36). In cases where a causative agent of infection is not identified, metagenomic analyses can be used to rapidly identify potential pathogens(25). For example, hundreds of pathogens have been associated with infectious encephalitis, and in as many as 60% of encephalitis cases, conventional laboratory assays fail to detect a causative agent(37). Accurate and rapid identification of the infectious agent is critical as the main alternative diagnosis is immune-mediated encephalitis, which requires immune suppression for treatment(37). In one striking case, an HIV-infected patient was admitted to a hospital with lethally diffuse brain lesions(25). The patient’s blood and cerebrospinal fluid tests were negative for common microbial infections. Unbiased pathogen detection by metagenomic sequencing of cerebrospinal fluid found sequences mapping to Toxoplasma gondii. After identification, the patient was immediately treated with azithromycin plus co-trimoxazole for toxoplasmosis encephalitis and showed significant short-term improvement, but for complex reasons was lost to follow-up(25). T. gondii, while a known neurological opportunistic pathogen, is difficult to culture and not easily detected by microscopic examination. PCR assays for detection typically require an a priori suspicion of T. gondii and are not readily available in many regions(25). In this case and many others like it, WMS analyses have the capability to directly and rapidly identify pathogens presence, improving diagnosis and treatment of patients.
Metagenomics can also be used to rapidly monitor dysbiotic states of the microbiome that may induce or be correlated with disease and disease progression. For example, early detection and intervention is critical for treating liver cirrhosis(38). Early interventions can avoid or delay patient decompensation, but chronic liver disease is often asymptomatic in early cases, and diagnosis for cirrhosis is typically done after liver biopsy, an invasive procedure that is not applicable to all patients and can lead to complications of its own(38). However, metagenomic studies have found a direct relationship between gut dysbiosis and the progression of cirrhosis in patients(38). In these cases, metagenomic analyses can be used to determine the progression of liver cirrhosis by monitoring the depletion of species and gene richness in stool samples. This has the potentially to improve detection and treatment across the course of disease progression(38).
There are also limitations to metagenomic analyses from clinical samples that must be considered when evaluating metagenomic analyses as a tool in clinical diagnosis. While metagenomic analyses may have higher sensitivity in pathogen detection and identification than culture-based methods(25, 36), it has yet to be determined if metagenomic analyses have higher sensitivity than other molecular-based diagnosis methods such as pathogen-specific PCR(25). Molecular-based methods have been used successfully and rapidly in many clinical cases; however, their clinical sensitivity can be highly variable and they are more prone to false positives due to contamination or non-causative agents(39). Further, in human samples, the amount of human genomic DNA often vastly outnumbers the amount of microbial genomic DNA(40). This disparity can greatly influence the read depth necessary to detect microbial presence. Tools are being developed that selectively deplete eukaryotic DNA, allowing enhanced interrogation of microbial presence(40). Metagenomics also shares the same sequencing and computation costs discussed for WGS. Because of these costs and limitations, metagenomics is applied in patients after an infectious agent cannot be identified by other means and in human samples where there is a known microbial presence.
Using metagenomics to monitor environmental and biological reservoirs of antibiotic resistance
The spread of antibiotic resistant infectious agents is of rising global concern. One specific example of this is blaNDM-1, which can confer resistance to a wide range of β-lactam antibiotics, including carbapenems, a class of last-resort antibiotics(41). blaNDM-1 was initially discovered in 2008 with a suspected origin of India(41). Since then, different bacterial hosts, such as Klebsiella, and E. coli, etc. containing blaNDM-1 have been associated with outbreaks in hospitals across a wide geographic area, including all continents except South America and Antarctica(41). Further, blaNDM-1 has been identified in drinking water samples in the Indian subcontinent(42). While blaNDM-1 is not unique in its clinical relevance, it stands as a concerning example of how ARGs can disseminate rapidly in the environment to compromise disease treatment in humans. A metagenomic characterization study found blaNDM-1 in hospital wastewater, indicating wastewaters as a hotspot for ARGs(43). An important aspect of understanding and combating the rising prevalence of antibiotic resistance is to survey and monitor environmental reservoirs for infectious agents and accumulation of ARGs. The human microbiome is regularly exposed and influenced by environmental bacteria(44, 45). These environmental bacteria could act as a reservoir of ARGs, which may be exchanged with pathogens that in turn could infect humans(10). To best predict the emergence and abundance of AROs and ARGs, researchers must characterize and predict the spread of environment AROs and ARGs and their influence on human pathogens. While many environments can act as reservoirs of infectious agents, host-associated commensals and water reservoirs have recently been highlighted as particular important to monitor(13, 46–48). Metagenomic analyses allow for long-term and rapid reservoir monitoring that can then be directed and verified using culture-based methods.
Domesticated animals pose a potentially dangerous biological reservoir for AROs and ARGs, because of the high risk and relative ease of zoonotic transmission to humans(49, 50). Antibiotics at both therapeutic and non-therapeutic concentrations are used extensively in livestock (e.g., poultry, cattle, swine), with total annual volumes far exceeding human antibiotic use(51). Remnant antibiotic compounds and their metabolites are excreted in livestock manure, litter, and wastewater(47, 51, 52). Metagenomic surveillance of animal waste products show an enrichment of ARGs in environmental bacteria associated with these animal reservoirs(51, 52). In 2015, the emergence of the first plasmid-mediated colistin resistance gene, mcr-1, was found from a surveillance project of commensal E. coli in pigs in China(13). Colistin is a last-resort antibiotic, and before the discovery of mcr-1, colistin resistance had only been observed through chromosomal mutations(13). A plasmid-mediated resistance gene allows the gene to be easily maintained in populations and transfer rapidly into pathogenic strains(13). Since the discovery of mcr-1, it has been found in animal and food sources and in community-acquired infections(53). Monitoring ARG enrichment in animal reservoirs may help predict emerging human pathogens. However, the magnitude by which AROs or ARGs in animal reservoirs contribute to human pathogens and infections remains unclear, and further studies are required to provide quantitative assessments of these risks.
Water supplies are home to a wide diversity of microbes, and can become contaminated by known human pathogens that have been categorized as serious or urgent clinical antimicrobial resistant threats(29, 46, 48). Given that many acquired ARGs are harbored on conjugative plasmids that can self-mobilize between diverse bacterial species, water environments represent a reservoir where ARGs can exchange between benign bacteria and human pathogens(48). Wastewaters receive diverse discharges with a high presence and abundance of microbial organisms(54, 55). These discharges include antibiotic remnants from use in humans and animals, which may can increase selection pressures on ARGs native to water microbial communities (Figure 2). Although wastewater is treated to eliminate coliform bacteria, treatments may be ineffective at destroying ARGs, resulting in ARG enrichment in surviving wastewater microbes(55–57). Further, functional metagenomic studies (methods described below) have identified increases in phenotypically resistant clones recovered from samples downstream of wastewater treatment plants, suggesting that wastewater treatment and disposal increases the reservoir of resistance mechanisms in the environment(58). One recent study used metagenomics to better understand ARO and ARG abundance in the Chaobia River, a major source of drinking water, agricultural irrigation, and waste in China(59). Metagenomic analyses demonstrated that the Chaobia River has elevated ARG abundance compared to other areas in the region and to areas without human interaction such as deep-sea and Antarctic sediment. The dominant ARGs found were associated with antibiotics that had been heavily used in human medicine, veterinary therapy, and livestock growth(59). This increase in ARGs in major water sources may have a direct impact on millions of humans through an increase in AROs and ARGs in human drinking supplies (Figure 2). Metagenomic analyses of drinking water across 25 cities on 3 continents shows a wide diversity of ARG types in human drinking supplies with the majority of samples having higher ARG abundance than environmental sediment and soil samples(60). Altogether these studies illustrate the ubiquitous presence of ARO and ARGs in water supplies and the importance of understanding the origin and spread of ARGs in drinking water. Unless monitored and mediated, ARO and ARG environmental reservoirs can contribute to a vicious cycle that spreads AROs and ARGs over time and vast distances (Figure 2)(54).
Figure 2.
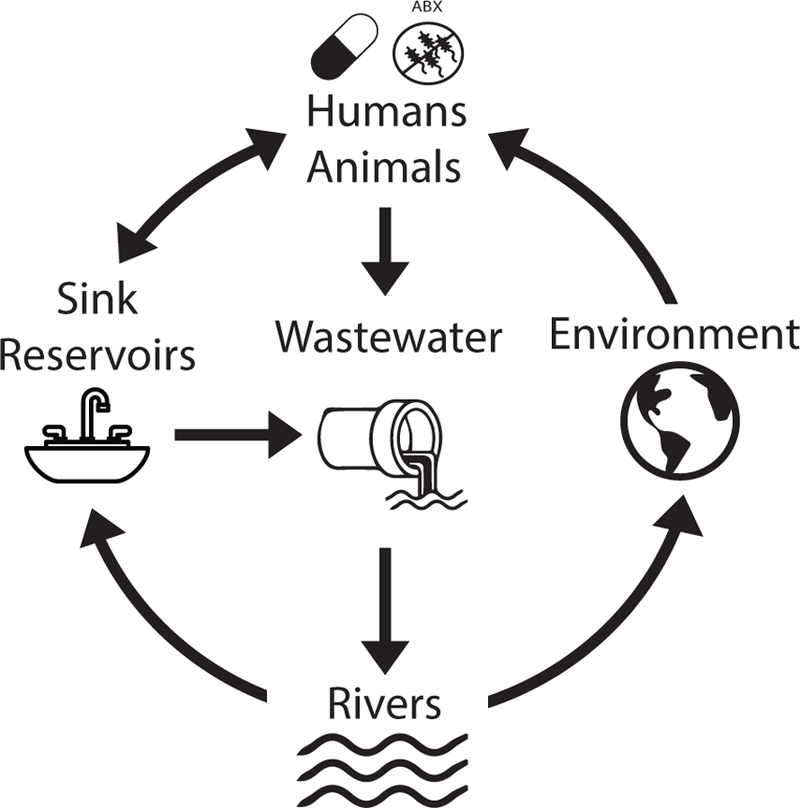
Antibiotic remnants from antibiotic use in humans and animals increases antibiotic concentration and ARO and ARG abundance in wastewaters and in sink reservoirs in hospitals due to human shedding. After treatment, wastewater progresses to reservoirs such as rivers, resulting in ARG enrichment in downstream agricultural and human water sources. From rivers, ARG enrichment may be passed on to household water supplies, resulting in an ARG enrichment in sinks due to increased ARO and ARG abundance in source water. ARG enrichment in rivers also results in increased ARG abundance in sediment and other environmental water sources. This cycle can result in ARG enrichment in human pathogens and persistent environmental reservoirs of AROs and ARGs.
Discovery of Novel Antibiotics and Resistance Mechanisms
The discovery of novel antibiotic lead compounds and resistance mechanisms has accelerated with the advancements in sequencing technology(19, 61–63). Concurrent with these advancements, databases containing assembled genomes, annotated open reading frames (ORFs), and metagenomes have expanded, which allows sequencing to serve as an emerging method for surveillance of pathogens and ARGs(64–67). However, direct observation of these pathogens or ARGs is only part of the clinically-relevant utility that microbial sequencing has been able to provide. This section will discuss various methods that have been used to discover new antibiotics and resistance mechanisms, or provide novel insights into previously characterized mechanisms.
Sequence-based methods
One common strategy of looking for ARGs is to use the primary nucleotide or amino acid sequence of known or putative ARGs(24, 56, 59, 60, 68). These methods most commonly work by aligning a query sequence to known ARGs or markers from a curated database(64–67). Sequence-based methods can be performed rapidly and in a massively parallel manner compared to culture-based methods of resistance determination(3). Further, as new resistance mechanisms are discovered, existing datasets can be re-screened for new ARGs. However, choosing the right analysis depends on the type of sample collected and requires some expertise in bioinformatics to process and interpret the data.
Analysis of isolates
The analysis of bacterial isolates by WGS currently proceeds primarily by highly-multiplexed short-read sequencing technologies (e.g., Illumina). The reads can then be mapped to a reference genome or a database using tools like Bowtie2(69). This method is advantageous because it is faster and less computationally demanding than de novo genome assembly (discussed below)(70). However, it relies on the completeness of the reference database to which the query reads are mapped. If an organism is underrepresented in the reference database, or if an ARG is novel or a distant homolog, then it will not be detected(71). Further, because of the short reads (<500 bp) used in most WGS studies, some reads may spuriously align to sequences from different genes due to repetitive DNA elements and local sequence homology(72). Another consequence of short read length is the loss of genomic context surrounding a particular ARG. This is of particular concern for antibiotic resistance research, because it diminishes the ability to detect transmission of ARGs through synteny with mobile genetic elements like plasmids and transposons.
Assembly of genomes from short reads into contiguous fragments (contigs) without the aid of a reference genome is referred to as de novo assembly. A recent review discusses popular genome assemblers(30) The ability to analyze large contigs overcomes many of the problems of read mapping. Using assembled contigs, ARGs can be identified in the context of their surrounding genomic content, and genomes from organisms that share low sequence identity with reference sequences can be analyzed. There are limitations to this method, however. Assembly from reads is computationally demanding, requires high sequence coverage compared to read mapping, and its completeness and accuracy can be compromised by sequencing errors and repetitive DNA elements(35, 70). One exciting advance in this area has been the development of long-read sequencing technologies like SMRT (Single-Molecule Real-Time) and Nanopore Sequencing(73, 74). Instead of the 50–150bp reads typically produced by technologies like Illumina, these methods produce reads of 10kb to over 100kb(73, 74). One advantage of these longer reads for studying antibiotic resistance of bacterial isolates is that they greatly simplify the issue of de novo assembly and can even resolve a complete bacterial genome (i.e. a single contig)(75). Perhaps the most exciting advancement of these technologies is that they produce data much faster than short-read technologies (in as little as a few minutes compared to several days)(76). Nanopore sequencing in particular excels at producing data rapidly and without the need for expensive and sophisticated equipment, making it an ideal platform for point-of-care deployment. As proof of concept, Bradley et al. demonstrated that antibiotic resistance profiles of Staphylococcus aureus could be accurately predicted using Nanopore technology in as little as 7 hours(77).
Analysis of metagenomes
The analysis of microbial communities by whole metagenome shotgun sequencing (WMS) is more complicated than isolate WGS analysis. Because data from shotgun metagenomics contains information from an unknown number of organisms with varying amounts of sequence coverage and sequencing depth, de novo assembly is not a trivial task(78). A recent review compares popular metagenomic assemblers{}(79). }{Once a genome is assembled from WMS, it can be annotated and analyzed in the same manner as an isolate genome by WGS. However, de novo assembly from short-read metagenomes remains a challenge, and plasmids (which commonly harbor ARGs) are difficult to link to their host since they replicate separately from the host bacterial chromosome (80). Similar to isolate genomes, long-read sequencing greatly simplifies de novo assembly of metagenomes. In addition to improving assemblies, long-read sequencing can better resolve plasmids from metagenomes, and simultaneous measurement of DNA methylation patterns can link plasmids to their host(80). The rapid turnaround of long-read technologies have also made clinical analysis of metagenomes more feasible. Using Nanopore sequencing, Břinda et al. were able to accurately predict antibiotic susceptibility for Streptococcus pneumonaie from sputum samples within minutes, without the need for time-intensive culturing steps(76).
Read-mapping can also be applied to metagenomic data but this quickly becomes computationally demanding as metagenomic sequencing depth and reference database size increases. Further, short read-length can lead to false positive identifications due to local sequence homology(35). To overcome these problems, the ShortBRED (Short, Better Representative Extract Dataset) method was developed for assembly-free identification of ARGs in metagenomes(35). ShortBRED builds short peptide sequences (markers) that are unique to a specific protein family, which are then used as a reference to map metagenomic reads. This method has been used to quickly and accurately quantify the resistome of organisms from numerous environments(62, 63). While most work has focused on ARGs, ShortBRED has the potential to identify and quantify any group of functionally-related proteins, for example, those involved in biosynthesis of natural products.
Search strategies
Concurrent with the proliferation of NGS technologies, the development of annotated databases of nucleotide or amino acid sequences of ARGs has allowed for the rapid screening of genomic or metagenomic datasets for resistance. Query sequences, whether from contigs or reads, can be aligned to sequences from known ARGs which can used to identify resistance mechanisms directly from sequences using pairwise sequence alignment tools like the Basic Local Alignment Search Tool (BLAST) or Bowtie2(69, 81).
One of the first databases for ARGs is the Antibiotic Resistance Gene Database (ARDB)(64) (http://ardb.cbcb.umd.edu/), which is a manually curated database representing >20,000 ARGs. Unfortunately, this database is not currently maintained, with the last update occurring in 2009. One actively updated database is the Comprehensive Antibiotic Resistance Database (CARD)(65) (https://card.mcmaster.ca/), which is also a manually curated database of known resistance determinants and their associated antibiotics, organized by gene ontology. CARD also offers the Resistance Gene Identifier (RGI), which is a tool with both web-based and downloadable versions for large-scale analyses(65). In addition to these general databases, several databases are maintained for specific classes of antibiotics, of note the Bush–Jacoby β-lactamase Lahey database and the Lactamase Engineering Database (LacED)(66, 67). One suite of tools, ResFinder and PointFinder (https://cge.cbs.dtu.dk/services/ResFinder/), combines the ADRB with numerous specialized databases to create a robust set of acquired ARGs and sequence variants known to confer resistance which can be searched against(82).
A challenge with alignment-based search methods is the risk of false negatives. Because reference databases are biased towards human pathogens and culturable bacteria, the chance of missing genes of interest when mining metagenomic datasets is very high(71). One solution is to employ profile hidden Markov models (pHMMs). pHMMs are statistical models trained on sequences with a known function that can be used to identify remote homologues and novel sequences with similar function but low pairwise identity(83). Because of this ability, they have been a popular choice for discovery of novel resistance and biosynthetic genes. Resfams (http://www.dantaslab.org/resfams) is a curated database of pHMMs that was trained using protein sequences from the CARD, LacED, and Bush–Jacoby β-lactamase databases(71). To better capture resistance profiles from uncultured bacteria, Resfams was also trained on ARGs obtained from culture-independent functional selections from soil and human commensal microbiotas. Compared to sequence alignment, an HMM-based approach leads to significantly more sensitive and less biased screening of environmental metagenomes. When searching soil and human gut microbiomes, BLAST searches using CARD and ADRB were unable to identify 64% of the ARGs identified with Resfams(71). Using a gold-standard set of annotated genes from 95 multi-drug resistant soil isolates (reported in Forsberg et al, 2012(10)), analysis with Resfams did not identify a single ARG that was not predicted by extensive manual annotation of the sequences(71). This sensitivity and specificity demonstrate the ability of HMM-based approaches to accurately screen ARGs in genomes of underrepresented organisms.
Diverse environmental microbiomes and the bioactive molecules they produce represent a rich source of potential lead compounds for novel antibiotics, which may be identified using similar methods to those used for metagenomic ARG characterization. Similar to ARG identification, BLAST-based searches may not capture biosynthetic gene clusters (BGCs) present in organisms that are underrepresented in databases. To address this, the antibiotics and secondary metabolite analysis shell (antiSMASH) was developed, which integrates a pHMM-based method to identify potential BGCs(84). Also, antiSMASH implements many other tools, such as domain analysis, active site prediction, and chemical structure prediction. AntiSMASH is able to rapidly identify and characterize many different types of BGCs, including the important polyketide synthase and non-ribosomal peptide synthetase families. Using this pipeline, researchers were able to identify over 3000 small-molecule BGCs in 752 metagenomes from the NIH Human Microbiome Project(85). As proof of concept, the authors purified and characterized a predicted thiopeptide (an antibiotic class that was shown to be widely distributed in the microbiomes of human commensals) and demonstrated its antibacterial activity against numerous potential human pathogens, including Staphylococcus aureus, Enterococcus faecalis, Corynebacterium aurimucosum, and Gardnerella vaginalis(85).
Sequence-based methods are high-throughput compared to culture-based methods, and enable analysis of organisms that are recalcitrant to culturing. This allows for rapid and reasonably accurate screening of genomic or metagenomic datasets for functions of interest. However, large-scale analyses still require access to sophisticated sequencing equipment, massive computational resources, and expertise in bioinformatics, making their deployment to the clinic more difficult. This is a rapidly changing area, and the development of long-read sequencing technologies represents a step towards practical point-of-care deployment(76, 77). Further, software tools are continually being improved to be accessible for a broad user base, as well as lightweight enough to run on an average laptop computer(76, 77). Many of the software tools discussed thus far are available free and open-source, which increases accessibility outside of large academic centers.
Functional metagenomics
Because sequence-based methods ultimately rely on comparisons to prior information, they are limited in their ability to identify entirely novel mechanisms. Functional metagenomics is a unique method for mining functions of interest from metagenomes in an unbiased, culture-independent way(10, 86–88). The basic functional metagenomic workflow involves extraction of metagenomic DNA from a community of interest (e.g., soil, human or animal gut) cloning fragments into an expression vector, creating the functional metagenomic library. The library is then transformed into a heterologous host which can be screened for a function of interest in a high-throughput manner(87, 89). The gene or genes responsible for the function can be identified by sequencing the selected plasmids. This method outperforms solely sequence-based methods at discovering novel genes because the selection is independent of prior knowledge about a mechanism or representation in a reference database(8, 10, 62, 87–89). More importantly, it allows for the identification of functions from organisms that would be otherwise extremely difficult to culture. This section will discuss cases where this method has been successfully employed to discover novel ARGs and bioactive compounds.
In the case of antibiotic resistance discovery, simply plating a functional metagenomic library on media containing antibiotics at concentrations that inhibit the selection host can select for potential novel ARGs. Several studies have used functional metagenomics to discover thousands of novel ARGs from metagenomes of diverse phylogenetic backgrounds and geographic locations(8, 10, 14). Recently a comprehensive screen of functional metagenomic libraries was able to identify a new family of enzymes that conferred resistance to tetracycline antibiotics by chemical inactivation(89). This significantly expanded the class of tetracycline-inactivating enzymes. In fact, prior to this the only biochemically-characterized enzyme in this class was Tet(X), whose encoding gene (tet(X)) was isolated from a plasmid from the human commensal Bacteroides fragilis. These new enzymes (Tet(47)-Tet(55)), dubbed the tetracycline destructases, shared at most 24.4% amino acid identity to Tet(X), demonstrating the power of functional metagenomics to identify genes that share function but with low sequence identity to known genes. Comparative genomic analysis of tet(49)-tet(55) revealed another tetracycline destructase in the genome of the pathogen Legionella longbeachae, which was also shown to provide resistance when expressed in a heterologous host as well as the parent pathogenic host(89, 90).
Identification of novel clinically-relevant ARGs is ideally followed by investigations of their substrate specificity and sequence-structure-function relationships, as well as to screen for inhibitors which can enable re-deployment of the targeted antibiotics. For example, x-ray crystallographic structural determination of the new soil-derived tetracycline destructases showed that while they share little primary sequence identity with Tet(X), they exhibit a similar overall three-dimensional architecture, though with a few key differences (90). The authors hypothesized that the shared structural information could be used in rational design of tetracycline destructase inhibitors, analogous to the commonly used β-lactamase inhibitors(91). This led to the identification of anhydrotetracycline as a potential lead compound for tetracycline destructase inhibition. Further, the co-crystal structure of Tet(50) with anhydrotetracycline revealed a unique binding mode where the enzyme is locked in an inactive conformation. This powerful structure-function investigation led to the conclusion that anhydrotetracycline could be coupled with tetracyclines to inhibit this class of enzymes, which was validated in vitro and against model pathogens expressing tetracycline destructases(90). Recently these same authors described a novel synthesis scheme for generating semi-synthetic anhydrotetracycline derivatives with broader inhibitor potency and enzyme specificity, including rescue of next-generation tetracyclines against soil-, pathogen-, and human gut-derived tetracycline destructases(92). This functional approach to ARG discovery provided a unique, proactive opportunity to discover new therapeutic compounds to counteract an emerging threat before it explodes into the wider population, which is particularly significant given the recent identification of Tet(X) in a number of multidrug resistant pathogens(93), and the even more recent clinical approvals of next-generation tetracycline antibiotics – eravacyline and omadacycline(94, 95).
Screening for hits in a functional metagenomic library is most convenient when the desired outcome is gain-of-function required for survival of the transformed host bacterium. Antibiotic resistance is a natural application of this method, but other studies have employed clever strategies to leverage functional metagenomics to instead discover production of novel biomolecules, including antibiotics(19). One such strategy utilizes a double-agar method to screen for production of antibiotic compounds(19). In this method, the heterologous host containing the library is plated on agar, where it is allowed to incubate for enough time to produce antibiotic compounds. Next, the plates are overlaid with top agar containing an indicator strain. A functional metagenomic clone that produces an antibiotic is identified by inhibition of growth of the indicator strain. This method was used to identify 12 clones from a library representing ~12.25 GB of DNA from a gut microbiome that were able to inhibit the growth of E. coli. Sequencing of the clones identified the colicin V gene cluster, which was responsible for production of the colicin V bacteriocin(19).
While the studies discussed above demonstrate the power of functional metagenomics, it is not without its limitations. One major challenge is that it relies on expression of genes outside of their native hosts. Differences have been observed when expressing the same metagenomic library in different hosts(96, 97). This challenge may be addressed by selecting a phylogenetically diverse group of hosts for expression of a functional metagenomic library, though this increases the cost and reduces the throughput of the system(96, 97). Conversely, just because a clone produces a selected phenotype of interest (e.g., antibiotic resistance) in the heterologous host, especially during forced over-expression, it does not mean that it produces the same phenotype in the original host, or indeed that it is even expressed in that native context. Accordingly, ARGs discovered from functional metagenomic selections are better categorized as illuminating functional resistome potential, which may contribute to horizontal ARG spread, rather than describing the true resistance phenotypes of the microbial members of the studied community. Culture-based phenotyping still remains the only guaranteed way to directly assess resistance of specific microbes to specific antibiotics.
Using massively parallel sequencing and synthetic biology as a tool
Advances in NGS technology have not only made searching for interesting, but uncharacterized genes much faster and easier, but have opened the door to exciting new technologies that allow for better characterization of known genes or mechanisms. Methods that combine bioinformatic, microbiologic, and synthetic biology represent exciting new approaches to discover new antibiotic compounds or new insights into drug resistance.
One example of this is in the use of bioinformatics to improve access to the biosynthetic capacity of metagenomic libraries. Because BGCs often share common sequence motifs, PCR amplification of these motifs from a metagenomic sample using degenerate primers followed by amplicon sequencing allows profiling of the biosynthetic capacity of that sample(18). To facilitate this, the environmental Surveyor of Natural Product Diversity (eSNaPD, available at http://esnapd2.rockefeller.edu/) was developed. eSNaPD is a bioinformatics platform designed for analysis of amplicons in the context of natural product biosynthesis. Using sequence tags from known BGCs, eSNaPD phylogenetically organizes amplicon sequences based on BGC content and predicted chemical output, highlighting novel or divergent biosynthetic pathways. Samples predicted to contain these novel or divergent pathways can then be prioritized for functional metagenomic library construction, expression, and screening. Recently, this method was successfully used to discover malacidins, a previously uncharacterized class of calcium-dependent antibiotics from 2,000 unique soil samples(61). One extracted malacidin was shown to inhibit growth of a wide range of Gram-positive bacteria, including multidrug-resistant pathogens(61).
The previous example demonstrates how bioinformatics can be combined with metagenomic sequencing to explore the rich diversity of natural products that microbial communities can offer. Perhaps an even more interesting application of sequencing technologies has been to pursue diversity that is not represented in nature. The development of deep mutational scanning represents one step in that direction(98–100). Deep mutational scanning is an extension of the classic experiment where a researcher measures how specific mutations affect the function of a protein of interest, but instead of examining a handful of mutations, the researcher can simultaneously observe the effects of thousands of different protein variants simultaneously. Briefly, a mutant library is made using multiplex PCR and primers designed with random codons that amplify the target gene with every possible amino acid substitution at every possible location in the gene. Next, the library is cloned into an appropriate host, expressed, and selected for a function of interest. The cells with active protein variants are enriched and the cells with inactive variants are depleted, which is quantified by deep sequencing of the library from selected cells and unselected (control) cells(99). By creating an unbiased mutant library, deep mutational scanning can reveal intrinsic protein properties, unexpected observations, and insights into the evolution of that protein(98). Recently, deep mutational scanning was applied to the NDM-1 carbapenemase, where it was used to unveil the sequence requirements for catalysis of various β-Lactam antibiotics(100). Interestingly, the authors found that the residues required for hydrolysis of penicillins and cephalosporins are a subset of those required for carbapenem hydrolysis. This suggests that the broad substrate profile of NDM-1 could have evolved from selective pressure exclusively from carbapenems. Further, the results imply that if an NDM-1 inhibitor is developed, it should be used in combination with carbapenems because the stricter sequence requirements for carbapenem hydrolysis reduces the number of substitutions possible that may confer inhibitor resistance(100).
Conclusion
The evolution and spread of antibiotic resistance are a pressing global concern. The only way to successfully combat the rise of antibiotic resistance is through a comprehensive approach that identifies, characterizes, and tracks emerging and evolving pathogens by integrating culture-based methods, genomics, and metagenomics; and by developing experimental and computational tools to identify mechanisms of resistance and develop new antibiotics to combat the evolution of antibiotic resistance.
Figure 3.
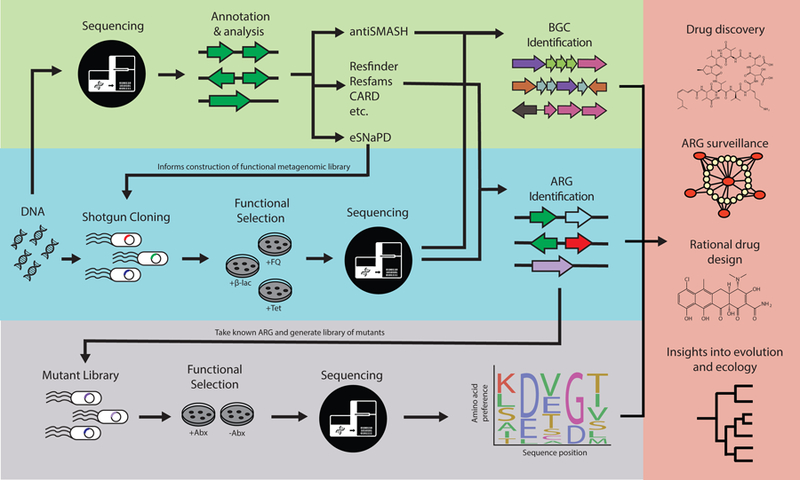
The application of NGS to bacterial isolates and metagenomes has accelerated drug development by enabling the discovery of novel antibiotics and informing the rational design of drugs that reverse resistance mechanisms. Genomic data can enable intelligent surveillance methods for tracking the origin and spread of ARGs, as well as insights into the evolutionary context behind emergence of a particular resistance mechanism. This can be done through sequence-based computational tools (top) or function-based methods, such as functional metagenomics methods (middle). Note the vertical arrows depicting how sequence- and function-based methods can be combined to make inferences about a particular resistance mechanism, such as through deep mutational scanning (bottom).
Acknowledgements
We would like to acknowledge Alaric D’Souza, Robert F. Potter, Manish Boolchandani, and Drew Schwartz for providing feedback on this paper.
Funding: This work was supported in part by awards to G.D. through the National Institute of Allergy and Infectious Diseases (NIAID: https://www.niaid.nih.gov/), the Eunice Kennedy Shriver National Institute of Child Health & Human Development (https://www.nichd.nih.gov/), and the National Center for Complementary and Integrative Health (https://nccih.nih.gov/) of the National Institutes of Health (NIH) under award numbers R01AI123394, R01HD092414, and R01AT009741, respectively.
Footnotes
Conflict of Interest: The authors declared no competing interests for this work.
Publisher's Disclaimer: Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
References
- (1).Aminov RI A brief history of the antibiotic era: lessons learned and challenges for the future. Front Microbiol 1, 134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Brown ED & Wright GD Antibacterial drug discovery in the resistance era. Nature 529, 336–43 (2016). [DOI] [PubMed] [Google Scholar]
- (3).Adu-Oppong B, Gasparrini AJ & Dantas G Genomic and functional techniques to mine the microbiome for novel antimicrobials and antimicrobial resistance genes. Ann N Y Acad Sci 1388, 42–58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cassini A et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis 19, 56–66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Crofts TS, Gasparrini AJ & Dantas G Next-generation approaches to understand and combat the antibiotic resistome. Nat Rev Microbiol 15, 422–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).D’Costa VM et al. Antibiotic resistance is ancient. Nature 477, 457–61 (2011). [DOI] [PubMed] [Google Scholar]
- (7).Bhullar K et al. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 7, e34953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Clemente JC et al. The microbiome of uncontacted Amerindians. Sci Adv 1, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Knapp CW, Dolfing J, Ehlert PA & Graham DW Evidence of increasing antibiotic resistance gene abundances in archived soils since 1940. Environ Sci Technol 44, 580–7 (2010). [DOI] [PubMed] [Google Scholar]
- (10).Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO & Dantas G The shared antibiotic resistome of soil bacteria and human pathogens. Science 337, 1107–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Poirel L, Rodriguez-Martinez JM, Mammeri H, Liard A & Nordmann P Origin of plasmid-mediated quinolone resistance determinant QnrA. Antimicrob Agents Chemother 49, 3523–5 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang R et al. The global distribution and spread of the mobilized colistin resistance gene mcr-1. Nat Commun 9, 1179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu YY et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 16, 161–8 (2016). [DOI] [PubMed] [Google Scholar]
- (14).Forsberg KJ et al. Bacterial phylogeny structures soil resistomes across habitats. Nature 509, 612–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Finley RL et al. The scourge of antibiotic resistance: the important role of the environment. Clin Infect Dis 57, 704–10 (2013). [DOI] [PubMed] [Google Scholar]
- (16).Douglas AP et al. Utilizing genomic analyses to investigate the first outbreak of vanA vancomycin-resistant Enterococcus in Australia with emergence of daptomycin non-susceptibility. J Med Microbiol, (2019). [DOI] [PubMed]
- (17).Johnson RC et al. Investigation of a Cluster of Sphingomonas koreensis Infections. N Engl J Med 379, 2529–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Katz M, Hover BM & Brady SF Culture-independent discovery of natural products from soil metagenomes. J Ind Microbiol Biotechnol 43, 129–41 (2016). [DOI] [PubMed] [Google Scholar]
- (19).Cohen LJ, Han S, Huang YH & Brady SF Identification of the Colicin V Bacteriocin Gene Cluster by Functional Screening of a Human Microbiome Metagenomic Library. ACS Infect Dis 4, 27–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wieser A, Schneider L, Jung J & Schubert S MALDI-TOF MS in microbiological diagnostics-identification of microorganisms and beyond (mini review). Appl Microbiol Biotechnol 93, 965–74 (2012). [DOI] [PubMed] [Google Scholar]
- (21).Jorgensen JH & Ferraro MJ Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin Infect Dis 49, 1749–55 (2009). [DOI] [PubMed] [Google Scholar]
- (22).Didelot X, Bowden R, Wilson DJ, Peto TEA & Crook DW Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13, 601–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Koser CU et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog 8, e1002824 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Potter RF et al. Population Structure, Antibiotic Resistance, and Uropathogenicity of Klebsiella variicola. MBio 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hu Z et al. Metagenomic next-generation sequencing as a diagnostic tool for toxoplasmic encephalitis. Ann Clin Microbiol Antimicrob 17, 45 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rodrigues C, Passet V, Rakotondrasoa A & Brisse S Identification of Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella variicola and Related Phylogroups by MALDI-TOF Mass Spectrometry. Front Microbiol 9, 3000 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lu Y, Feng Y, McNally A & Zong Z Occurrence of colistin-resistant hypervirulent Klebsiella variicola. J Antimicrob Chemother 73, 3001–4 (2018). [DOI] [PubMed] [Google Scholar]
- (28).Maatallah M et al. Klebsiella variicola is a frequent cause of bloodstream infection in the stockholm area, and associated with higher mortality compared to K. pneumoniae. PLoS One 9, e113539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Decraene V et al. A Large, Refractory Nosocomial Outbreak of Klebsiella pneumoniae Carbapenemase-Producing Escherichia coli Demonstrates Carbapenemase Gene Outbreaks Involving Sink Sites Require Novel Approaches to Infection Control. Antimicrob Agents Chemother 62, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Boolchandani M, D’Souza A & Dantas G Sequencing-based methods and resources to study antimicrobial resistance. Nature Reviews Genetics, (2019). [DOI] [PMC free article] [PubMed]
- (31).In: Applications of Clinical Microbial Next-Generation Sequencing: Report on an American Academy of Microbiology Colloquium held in Washington, DC, in April 2015 (Washington (DC: ), 2016). [PubMed] [Google Scholar]
- (32).Quainoo S et al. Whole-Genome Sequencing of Bacterial Pathogens: the Future of Nosocomial Outbreak Analysis. Clin Microbiol Rev 30, 1015–63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Truong DT et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 12, 902–3 (2015). [DOI] [PubMed] [Google Scholar]
- (34).Abubucker S et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8, e1002358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G & Huttenhower C High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLoS Comput Biol 11, e1004557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Guo LY et al. Detection of pediatric bacterial meningitis pathogens from cerebrospinal fluid by next-generation sequencing technology. J Infect, (2018). [DOI] [PubMed]
- (37).Boucher A et al. Epidemiology of infectious encephalitis causes in 2016. Med Mal Infect 47, 221–35 (2017). [DOI] [PubMed] [Google Scholar]
- (38).Shao L, Ling Z, Chen D, Liu Y, Yang F & Li L Disorganized Gut Microbiome Contributed to Liver Cirrhosis Progression: A Meta-Omics-Based Study. Front Microbiol 9, 3166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Opota O, Jaton K & Greub G Microbial diagnosis of bloodstream infection: towards molecular diagnosis directly from blood. Clin Microbiol Infect 21, 323–31 (2015). [DOI] [PubMed] [Google Scholar]
- (40).Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R & Zengler K Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 6, 42 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Johnson AP & Woodford N Global spread of antibiotic resistance: the example of New Delhi metallo-beta-lactamase (NDM)-mediated carbapenem resistance. J Med Microbiol 62, 499–513 (2013). [DOI] [PubMed] [Google Scholar]
- (42).Walsh TR, Weeks J, Livermore DM & Toleman MA Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis 11, 355–62 (2011). [DOI] [PubMed] [Google Scholar]
- (43).Ng C et al. Characterization of Metagenomes in Urban Aquatic Compartments Reveals High Prevalence of Clinically Relevant Antibiotic Resistance Genes in Wastewaters. Front Microbiol 8, 2200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lax S et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lax S & Gilbert JA Hospital-associated microbiota and implications for nosocomial infections. Trends Mol Med 21, 427–32 (2015). [DOI] [PubMed] [Google Scholar]
- (46).Vaz-Moreira I, Nunes OC & Manaia CM Diversity and antibiotic resistance in Pseudomonas spp. from drinking water. Sci Total Environ 426, 366–74 (2012). [DOI] [PubMed] [Google Scholar]
- (47).Wen X et al. Occurrence and contamination profiles of antibiotic resistance genes from swine manure to receiving environments in Guangdong Province southern China. Ecotoxicol Environ Saf 173, 96–102 (2019). [DOI] [PubMed] [Google Scholar]
- (48).Vaz-Moreira I, Nunes OC & Manaia CM Bacterial diversity and antibiotic resistance in water habitats: searching the links with the human microbiome. FEMS Microbiol Rev 38, 761–78 (2014). [DOI] [PubMed] [Google Scholar]
- (49).Ruiz-Fons F A Review of the Current Status of Relevant Zoonotic Pathogens in Wild Swine (Sus scrofa) Populations: Changes Modulating the Risk of Transmission to Humans. Transbound Emerg Dis 64, 68–88 (2017). [DOI] [PubMed] [Google Scholar]
- (50).Richard M et al. Factors determining human-to-human transmissibility of zoonotic pathogens via contact. Curr Opin Virol 22, 7–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhu YG et al. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc Natl Acad Sci U S A 110, 3435–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Xie WY, Yang XP, Li Q, Wu LH, Shen QR & Zhao FJ Changes in antibiotic concentrations and antibiotic resistome during commercial composting of animal manures. Environ Pollut 219, 182–90 (2016). [DOI] [PubMed] [Google Scholar]
- (53).Ruan Z et al. Emergence of a ST2570 Klebsiella pneumoniae isolate carrying mcr-1 and blaCTX-M-14 recovered from a bloodstream infection in China. Clin Microbiol Infect, (2019). [DOI] [PubMed]
- (54).Reddy B & Dubey SK River Ganges water as reservoir of microbes with antibiotic and metal ion resistance genes: High throughput metagenomic approach. Environ Pollut 246, 443–51 (2019). [DOI] [PubMed] [Google Scholar]
- (55).Fernandes T, Vaz-Moreira I & Manaia CM Neighbor urban wastewater treatment plants display distinct profiles of bacterial community and antibiotic resistance genes. Environ Sci Pollut Res Int, (2019). [DOI] [PubMed]
- (56).Guo J, Li J, Chen H, Bond PL & Yuan Z Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res 123, 468–78 (2017). [DOI] [PubMed] [Google Scholar]
- (57).Czekalski N et al. Inactivation of Antibiotic Resistant Bacteria and Resistance Genes by Ozone: From Laboratory Experiments to Full-Scale Wastewater Treatment. Environ Sci Technol 50, 11862–71 (2016). [DOI] [PubMed] [Google Scholar]
- (58).Amos GC, Zhang L, Hawkey PM, Gaze WH & Wellington EM Functional metagenomic analysis reveals rivers are a reservoir for diverse antibiotic resistance genes. Vet Microbiol 171, 441–7 (2014). [DOI] [PubMed] [Google Scholar]
- (59).Chen H, Bai X, Jing L, Chen R & Teng Y Characterization of antibiotic resistance genes in the sediments of an urban river revealed by comparative metagenomics analysis. Sci Total Environ 653, 1513–21 (2019). [DOI] [PubMed] [Google Scholar]
- (60).Ma L et al. Catalogue of antibiotic resistome and host-tracking in drinking water deciphered by a large scale survey. Microbiome 5, 154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hover BM et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat Microbiol 3, 415–22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Pehrsson EC et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature 533, 212–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Gibson MK et al. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat Microbiol 1, 16024 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Liu B & Pop M ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res 37, D443–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Jia B et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45, D566–D73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Bush K & Jacoby GA Updated functional classification of beta-lactamases. Antimicrob Agents Chemother 54, 969–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Thai QK, Bos F & Pleiss J The Lactamase Engineering Database: a critical survey of TEM sequences in public databases. BMC Genomics 10, 390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Zankari E et al. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J Antimicrob Chemother 68, 771–7 (2013). [DOI] [PubMed] [Google Scholar]
- (69).Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Lin Y, Li J, Shen H, Zhang L, Papasian CJ & Deng HW Comparative studies of de novo assembly tools for next-generation sequencing technologies. Bioinformatics 27, 2031–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Gibson MK, Forsberg KJ & Dantas G Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J 9, 207–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Bocanegra-Ibarias P et al. Molecular and microbiological report of a hospital outbreak of NDM-1-carrying Enterobacteriaceae in Mexico. PLoS One 12, e0179651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Eid J et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–8 (2009). [DOI] [PubMed] [Google Scholar]
- (74).Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S & Bayley H Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol 4, 265–70 (2009). [DOI] [PubMed] [Google Scholar]
- (75).Liao YC, Lin SH & Lin HH Completing bacterial genome assemblies: strategy and performance comparisons. Sci Rep 5, 8747 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Brinda K et al. Lineage calling can identify antibiotic resistant clones within minutes. bioRxiv, (2018).
- (77).Bradley P et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun 6, 10063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Ghurye JS, Cepeda-Espinoza V & Pop M Metagenomic Assembly: Overview, Challenges and Applications. Yale J Biol Med 89, 353–62 (2016). [PMC free article] [PubMed] [Google Scholar]
- (79).van der Walt AJ, van Goethem MW, Ramond JB, Makhalanyane TP, Reva O & Cowan DA Assembling metagenomes, one community at a time. BMC Genomics 18, 521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Beaulaurier J et al. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat Biotechnol 36, 61–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J Mol Biol 215, 403–10 (1990). [DOI] [PubMed] [Google Scholar]
- (82).Zankari E et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67, 2640–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Eddy SR Profile hidden Markov models. Bioinformatics 14, 755–63 (1998). [DOI] [PubMed] [Google Scholar]
- (84).Blin K et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res 45, W36–W41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Donia MS et al. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158, 1402–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Rondon MR et al. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66, 2541–7 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Pehrsson EC, Forsberg KJ, Gibson MK, Ahmadi S & Dantas G Novel resistance functions uncovered using functional metagenomic investigations of resistance reservoirs. Front Microbiol 4, 145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Moore AM et al. Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes. PLoS One 8, e78822 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Forsberg KJ, Patel S, Wencewicz TA & Dantas G The Tetracycline Destructases: A Novel Family of Tetracycline-Inactivating Enzymes. Chem Biol 22, 888–97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Park J et al. Plasticity, dynamics, and inhibition of emerging tetracycline resistance enzymes. Nat Chem Biol 13, 730–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Drawz SM & Bonomo RA Three decades of beta-lactamase inhibitors. Clin Microbiol Rev 23, 160–201 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Markley JL et al. Semisynthetic Analogues of Anhydrotetracycline as Inhibitors of Tetracycline Destructase Enzymes. ACS Infect Dis, (2019). [DOI] [PMC free article] [PubMed]
- (93).Leski TA et al. Multidrug-resistant tet(X)-containing hospital isolates in Sierra Leone. Int J Antimicrob Agents 42, 83–6 (2013). [DOI] [PubMed] [Google Scholar]
- (94).Zhanel GG et al. Review of Eravacycline, a Novel Fluorocycline Antibacterial Agent. Drugs 76, 567–88 (2016). [DOI] [PubMed] [Google Scholar]
- (95).Tanaka SK, Steenbergen J & Villano S Discovery, pharmacology, and clinical profile of omadacycline, a novel aminomethylcycline antibiotic. Bioorg Med Chem 24, 6409–19 (2016). [DOI] [PubMed] [Google Scholar]
- (96).Martinez A et al. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol 70, 2452–63 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Craig JW, Chang FY, Kim JH, Obiajulu SC & Brady SF Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl Environ Microbiol 76, 1633–41 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Fowler DM & Fields S Deep mutational scanning: a new style of protein science. Nat Methods 11, 801–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Fowler DM, Stephany JJ & Fields S Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat Protoc 9, 2267–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Sun Z, Hu L, Sankaran B, Prasad BVV & Palzkill T Differential active site requirements for NDM-1 beta-lactamase hydrolysis of carbapenem versus penicillin and cephalosporin antibiotics. Nat Commun 9, 4524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]