

Abstract
Molecular dynamics simulations using physics-based atomistic force fields have been increasingly used to characterize the heterogeneous structural ensembles of intrinsically disordered proteins (IDPs). To evaluate the accuracy of latest atomistic explicit-solvent force fields in modelling larger IDPs with non-trivial structural features, we focus on the 61-residue N-terminal transactivation domain (TAD) of tumor suppressor p53, an important protein in cancer biology that has been extensively studied and abundant experimental data is available for evaluation of simulated ensembles. We performed extensive replica exchange with solute tempering simulations, in excess of 1.0 μs/replica, to generate disordered structural ensembles of p53-TAD using six latest explicit solvent protein force fields. Multiple local and long-range structural properties, including chain dimension, residual secondary structures, and transient long-range contacts, were analyzed and compared against available experimental data. The results show that IDPs such as p53-TAD remains highly challenging for atomistic simulations due to conformational complexity and difficulty in achieving adequate convergence. Structural ensembles of p53-TAD generated using various force fields differ significantly from each other. The a99SB-disp force field demonstrates the best agreement with experimental data at all levels, and proves to be suitable for simulating unbound p53-TAD and how its conformational properties may be modulated by phosphorylation and other cellular signals or cancer-associated mutations. Feasibility of such detailed structural characterization is a key step towards establishing the sequence-disordered ensemble-function-disease relationship of p53 and other biologically important IDPs.
Graphical Abstract
Introduction
Lacking well-defined three-dimensional structures under physiological conditions, intrinsically disordered proteins (IDPs) are functional proteins that deviate from the traditional protein structure-function paradigm1–10. Sequence analysis have shown that nearly one half of eukaryotic proteins contain disordered regions whose lengths are least 30 amino acids11, which implies that many proteins may rely on intrinsic disorder to perform biological functions. Indeed, IDPs could specifically interact with many cellular targets under different conditions, thus playing critical roles in cellular signaling and regulation5, 7, 8, 11–13. Missense mutation of IDPs, changes in IDP concentration, or aggregation of IDPs are frequently associated with human diseases, including cancers, cardiovascular diseases, diabetes, and neurodegenerative diseases11, 13–16. Thus, there is a great need to characterize the detailed structural and dynamic properties of IDPs in order to better understand the functional mechanisms of IDPs in many biological processes.
The highly dynamic and heterogeneous conformations of IDPs pose great challenges in their experimental characterization. They must be described using ensembles of diverse structures that does not lend themselves to description using traditional methods that are geared toward describing a coherent set of similar structures. For a disordered protein state, only ensemble-averaged properties can be measured in most cases17–21, which alone are insufficient to uniquely define the heterogeneous ensemble17, 22, 23. In contrast, molecular dynamics (MD) simulations using physics-based atomistic force fields could provide high temporal and spatial resolution information for the system of interest, thus offering a powerful tool in characterizing the detailed structures of IDPs. Properly validated using experimental data, the simulated ensemble could provide the much needed basis for establishing how intrinsic conformational disorder mediates protein function and how such functional mechanism may be altered in diseases.
Nevertheless, challenges still exist in atomistic simulations of IDPs. First of all, IDPs usually are more expanded than folded proteins with the same number of residues, requiring larger explicit solvent simulation boxes and significantly increasing the computational cost. Efficient enhanced sampling techniques24–27 and/or implicit treatment of solvent28–31 are often needed to reduce the computational cost of adequate sampling of the large conformational space of IDPs. However, a more fundamental challenge lies in the accuracy of current protein force fields, since the quality of force field ultimately determines the reliability of simulated ensembles. The free energy landscape of an IDP often displays a multitude of comparably shallow minima32, 33, and accurate description of relative prevalence of these important sub-states has pushed the limit of protein force fields. In fact, the need for accurate simulation of IDPs has prompted intensive efforts towards developing improved atomistic force fields28, 34–42.
It has been shown that many modern atomistic force fields could describe folded proteins and some small IDPs quite well. For example, 12 structurally distinct proteins have been shown43 to reversibly fold into their native states using the CHARMM22* force field39. By systematic comparison against 524 NMR measurements on very short peptides (≤ 4 residues) and a folded protein, ubiquitin44, ff99sb-ildn-phi45 or ff99sb-ildn-NMR46 force fields were found to yield errors that are comparable to the experimental uncertainty. In another benchmark study47, both ff99SB*-ILDN42, 48, 49 and CHARMM22*39 were found to reproduce experimental data on folded proteins and small IDPs (10–15 residues) quite well. As for slightly larger IDPs, for instance, the 24-residue arginine/serine (RS) peptide, CHARMM 22*39 was observed to agree best with experimental measurement50. However, it has also been increasingly recognized that most explicit solvent atomistic force fields often generate overly compact conformations for the unfolded state of proteins and larger IDPs37, 38, 51. Several strategies then have been proposed to alleviate this problem. For example, a new water force field, TIP4P-D37, has been developed, where water dispersion interaction was increased by approximately 50% compared with the TIP3P water force field52. Another approach was to scale up the Lennard-Jones (LJ) interactions between protein and water, such as in the Amber ff03ws force field38. More recently, the CHARMM36m35 and a99SB-disp34 force fields were developed based on extensive simulations of tens of folded and disordered proteins and peptides, demonstrating impressive levels of accuracy in simulating both folded and intrinsically disordered proteins.
These exciting developments in protein force field optimization have now paved the way for broader application of atomistic simulations to larger and more complex IDPs of biological and biomedical interest. In this work, we focus on the N-terminal transactivation domain of tumor suppressor p53 (p53-TAD, residues 1–61), one of the most frequently mutated proteins in human cancers53, 54. The stability and activity of p53 are tightly controlled by its interactions with key regulators, such as the E3 ubiquitin ligase MDM2 and transcriptional coactivator CBP/p30055. These interactions are mediated through the intrinsically disordered TAD domain of p53, which can specifically recognize these targets and often gain stable secondary and tertiary structures upon binding. Under normal cellular conditions, p53-TAD binds more tightly to MDM2, leading to polyubiquitination and degradation of p5356, 57. Under prolonged genotoxic stress, p53-TAD accumulates phosphorylation at multiple sites, which reduces binding affinity to MDM2 and enhances binding to the CBP/p300, resulting in stabilization and activation of p5358–62. So far, the molecular mechanisms of how phosphorylation modulates binding affinity to MDM2 and CBP/p300 is not obvious. The effects of phosphorylation on binding is typically understood by examining how it may provide additional interaction sites or disrupt the existing binding interface in the complex state. However, available structures of p53-TAD complexes63–66 suggest that most phosphorylation sites are outside of the binding interface. Instead, the effects of p53-TAD phosphorylation are likely achieved through modulation of the unbound state, where the disordered ensemble is poised to respond sensitive to posttranslational modifications and mutations. Altered level of partially formed secondary structures and/or tertiary contacts may lead to different folding conformational entropy cost upon specific binding and modulate the binding affinity67, 68. Therefore, the disordered ensemble of p53-TAD is likely a central conduit of p53 regulation, and its detailed characterization will be critical for establishing the sequence-disordered ensemble-function-disease relationship of p53.
As a key step towards detailed characterization of the structural ensembles of p53-TAD using MD simulations, one needs to carefully examine if the current protein force field could accurately recapitulate key structural features revealed by available experimental measurements. Due to its biological and biomedical significance, p53-TAD has been extensively characterized using several experimental techniques69–74, including NMR, single-molecule FRET (smFRET), and time-resolved FRET (TR-FRET). These studies have reveal that p53-TAD contains non-trivial structural features, including multiple residual helical elements69, 72, 74 and transient long-range contact formation73, 75. The conformational complexity and availability of large amount of experimental data also makes p53-TAD an ideal model system for evaluating the quality of current protein force fields for simulating of increasingly complex IDPs of biological interest.
In the present work, we evaluate six of the latest protein force fields including CHARMM36m35 (herein termed c36m), CHARMM36m35 with an alternative water force field35 (herein termed c36mw), CHARMM22*39, 76, 77 (herein termed c22*), ff99SB-ILDN48, 49, ff99SB-ILDN49 with TIP4P-D water force field37 (herein termed ff99SB-ILDN-TIP4P-D), and a99SB-disp34. To obtain sufficient sampling of relevant conformational space for the 61-residue p53-TAD peptide, we deployed the replica exchange with solute tempering (REST2) enhanced sampling technique27, 78, which allows tempering on only the region of interest (e.g., protein) and dramatically reduce the computational cost of replica exchange types of simulations in explicit solvent79–82. Two independent REST2 simulations were performed starting from distinct conformations for each force field examined, to allow rigorous evaluation of the convergence of simulated ensembles. The aggregated simulation time is over 192 μs. A wide range of local and long-range structural features of p53-TAD were carefully analyzed and compared directly against available experimental measurements.
Methods
Simulation details
The p53-TAD domain (residues 1–61: MEEPQ SDPSV EPPLS QETFS DLWKL LPENN VLSPL PSQAM DDLML SPDDI EQWFT EDPGP D) was studied in this work. The N- and C- termini were capped with an acetyl group and N-methyl amide, respectively. Six latest protein force fields were examined, including c36m35, c36mw35, c22*39, 76, 77, ff99SB-ILDN48, 49, ff99SB-ILDN-TIP4P-D37, 49, and a99SB-disp34. For each force field, two independent REST2 simulations were performed, one starting from a highly helical state (i.e., control run) and the other one starting from an extended state (i.e., folding run). This allows rigorous evaluation of simulation convergence. The protein was simulated in a truncated octahedron box containing ~24,000 water molecules. The shortest distance between two opposite hexagons was ~ 9.8 nm. 14 Na+ were added to neutralize the system.
All simulations were carried out using GROMACS 5.1.483, 84 patched with PLUMED 2.3.081, 85, 86. The initial conformation was first energy minimized using a steepest descent algorithm for 6000 steps. NVT simulation at 298 K was then performed for 100 ps, following by 1 ns of NPT simulation at 298 K and 1 atm, during which the position of protein heavy atoms was restrained using harmonic potentials with force constants of 1000 kJ/mol/nm2. The system was then equilibrated under the same NPT condition for another 1 ns, removing all restraints on the protein. From this NPT simulation, the mean volume of the simulation box was calculated, and one conformation was selected with its volume closest to the mean volume. This conformation was then used as the initial structure for the production NVT run at 298 K. Only the protein was subjected to tempering, which was achieved by scaling the solute-solute and solute-solvent interactions by λ and , respectively. 16 replicas were used in all cases, with λ values set to 1.00, 0.97, 0.93, 0.90, 0.87, 0.84, 0.81, 0.79, 0.76, 0.73, 0.71, 0.68, 0.66, 0.64, 0.62 and 0.60, respectively. They correspond to 16 exponentially spaced effective temperatures of the protein, ranging from 298 K to 500 K. Replica exchange was attempted every 2 ps, and the averaged exchange acceptance ratio was ~25%. Each simulation lasted for ~1 μs/replica. In simulations using c36m35, c36mw35, or c22*39, 76, 77 force field, van der Waals interactions were truncated at 1.2 nm, with the force smoothly switched to zero starting from 1.0 nm and the neighboring list updated every 10 steps. When ff99SB-ILDN49, ff99SB-ILDN-TIP4P-D, or a99SB-disp34 was used, the van der Waals interactions were cut off at 1.0 nm with the neighboring list updated every 5 steps. For all simulations, long-range electrostatic interactions were treated using the Particle Mesh Ewald (PME) method87. Lengths of all bonds involving hydrogen atoms were constrained using the LINCS algorithm88, which allows for an integration time step of 2 fs.
Analysis
By examining the evolution of secondary structure and radius of gyration of p53-TAD, the first 300 ns trajectories of each REST2 simulation were excluded from all analysis to remove the initial condition dependence. Simulated structure ensembles were constructed by collecting conformations sampled in the replica of λ = 1.00. Unless otherwise specified, all structural analysis were performed using the GROMACS toolset83, 84 and in-house scripts. Chemical shifts of simulated structures were calculated using SHIFTX289, and the ensemble-averaged values were compared with experimental data90. For secondary chemical shift analysis, random coil values91, 92 were subtracted from both calculated and experimental90 chemical shifts. For conformational clustering analysis, trajectories from independent control and folding simulations were combined. For such a large and highly dynamic system, the root-mean-square deviation (RMSD) in the Cartesian coordinate space is not an optimal distance metric. Therefore, featurization was first performed using the DRID algorithm, distribution of reciprocal of interatomic distances93, implemented in MSMBuilder 3.6.194, with only Cα atoms selected in this calculation. K-means clustering was then performed in the DRID space. Different numbers of total clusters were tested, including 50, 100, 500, 1000, 5000, and 10000. Final results were reported for 1000 total clusters, because smaller values would result in large clustering error, while larger values do not significantly decrease the clustering error, i.e., sum of squared distances of samples to their closest cluster center (see Fig. S1).
The paramagnetic relaxation enhancement (PRE) effects of four sites (D7, E28, A39 and D61) were calculated for simulated ensembles, and directly compared with previously published experimental results71. The ratios of peak intensities of oxidized (paramagnetic) and reduced (diamagnetic) resonances were calculated according to where 95,96 Here r is the distance between spin label and residue; K is 1.23×10−32 cm6s−2; and <> indicates ensemble averaging. Consistent with experimental conditions71, Larmor frequency ωH is 600 MHz; the correlation time τc is 3.3 ns; the intrinsic R2 relaxation rate is 16 s−1; and the INEPT evolution time t is set to 9.8 ms. Since our simulations didn’t include the paramagnetic spin labels used in the actual NMR measurement71, Cα-Cα distances were used to approximate the electron-proton distances.
Results and Discussion
Evaluation of self-consistency of simulated ensembles
The p53-TAD peptide provides an exceptional model system to understand the sequence-disordered ensemble-function-disease relationship of IDPs. In order to reliably simulate the disordered ensembles of p53-TAD using de novo simulations, the first, and probably one of the most important steps is to generate well-converged simulations. Here, the convergence of our REST2 simulations was mainly assessed by comparing the structural ensembles generated from the independent control and folding runs (see Methods). As shown in Fig. 1, the overall dimension of p53-TAD (indicated by the radius of gyration, Rg) appears well-converged in c22*, ff99SB-ILDN, ff99SB-ILDN-TIP4P-D, and a99SB-disp simulations. Similar observations could be made for the distributions of end-to-end distance (Fig. S2). Acceptable convergence at the level of secondary structure is also found for the REST2 simulations using these four force fields (Fig. 2). This was further supported by comparing the results calculated from different segments of each trajectory, demonstrating the self-consistency of each individual simulation (Fig. S3 and S4).
Figure 1.

Probability distributions of Rg of p53-TAD calculated from independent control (red) and folding (green) simulations, for all six force fields examined in this work. The corresponding ensemble averaged values are indicated using vertical bars on the x-axis.
Figure 2.

Residue helicity profiles of p53-TAD calculated from independent control (red) and folding (green) simulations using six force fields.
However, it seems challenging to obtain convergence for c36m and c36mw simulations, as reflected by the distributions of Rg (Fig. 1), end-to-end distance (Fig. S2) and residue helicity profiles (Fig. 2). For example, as shown in Fig. 2, residues 10–25 barely unfolded in the control simulations (where initial structures were highly helical), while it remained full disordered in the folding simulations (where initial structures were highly extended). As a result, REST2 simulations as configured were not able to generate converged ensembles when these two force fields were used to study p53-TAD. We note that both sets of simulations appeared to reach steady states, where distributions for individual control or folding REST2 runs stopped changing (Fig. S3 and S4). This again highlights the importance of using independent runs with drastically different initial conformations to rigorously evaluate convergence in IDP simulations. Two factors may contribute to slow convergence of c36m and c36mw simulations. The free energy barrier separating helical and unfolded coil states may be higher in these two force fields such that longer helix-coil transition timescales may have significantly slowed down the convergence of REST2 simulations. On the other hand, the apparent difficulty of achieving convergence with REST2 simulations using c36m and c36mw may also be due to imperfect compensation between the scaled solute-solute and solute-solvent interaction energies, which will hinder the sampling efficiency of REST type of simulations78. To further understand this issue, we extracted the last conformations at the λ = 1.00 condition from the four REST2 simulations to initiate four multi-μs MD simulations at 298 K. After about another 1 μs, the seemingly long-lived helical states for residues 10–25 from control REST2 simulations became unfolded in both c36m and c36mw force folds; yet partial helix refolding was not observed in any of these 7 μs simulations (see Fig. S5). This suggests that lack of convergence of REST2 simulations in c36m and c36mw is likely attributed to the force field characteristics instead of the REST2 protocol. Due to a lack of convergence, the quality of c36m and c36mw will not be reliably evaluated based on the current REST2 and constant temperature simulations. Therefore, we will mainly focus on evaluating c22*, ff99SB-ILDN, ff99SB-ILDN-TIP4P-D, and a99SB-disp in the remainder of this study, where well-converged structural ensembles are available from REST2 simulations.
Overall dimension of p53-TAD varies significantly among different force fields
Previous NMR study has shown that under normal cellular conditions, p53-TAD could interact simultaneously with HDM2 (the human homolog of MDM2) and CBP domains, thus forming ternary complexes59. The fate of these ternary complexes can be regulated by many cellular events, such as phosphorylation of p53-TAD, thus affecting the stability and activity of p53. According to this model, the compactness of p53-TAD may have a consequence in its availability to these regulatory proteins and in coordinating various interactions, as proposed previously75. Therefore, in order to study the “structure”-function relationship of p53-TAD using de novo simulations, it is very important to model its overall dimension correctly. Here we examine if these six force fields could describe the level of compaction of p53-TAD. The results show that chain dimensions of simulated p53-TAD ensembles depend strongly on the force field used. For instance, simulations using c22* and ff99SB-ILDN yield significantly more compact ensembles compared to those generated by ff99SB-ILDN-TIP4P-D and a99SB-disp simulations (Fig. 1 and Fig. S2). The ensemble averaged Rg values were 1.26, 1.33, 2.61, and 2.60 nm in c22*, ff99SB-ILDN, ff99SB-ILDN-TIP4P-D and a99SB-disp simulations, respectively. This is not surprising as both of the latter two force fields have been parameterized specifically to overcome the over compaction bias of earlier force fields34, 37. Despite a lack of convergence, scaling up protein-water interactions in the c36mw force field clearly leads to more expanded structural ensembles, which appears more in-line with results from ff99SB-ILDN-TIP4P-D and a99SB-disp simulations (Fig. 1 and S2). The Stokes radius of a slightly longer p53-TAD peptide (residues 1–73) was found to be 2.38 nm based on size exclusion chromatography71. Therefore, both c22* and ff99SB-ILDN, and likely c36m, overestimate the compaction of p53-TAD.
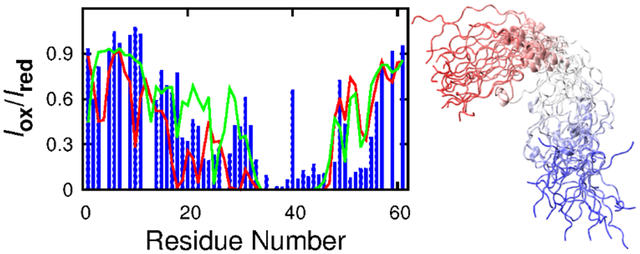
We further assess the ability of these force fields to correctly describe the overall compactness of p53-TAD as well as transient long-range ordering by comparing the back-calculated PRE effects with experimental measurements. PRE measurement provides ensemble-averaged distance information between the paramagnetic spin label at a specific residue location and all protons throughout the rest of the protein; it provides unparallelly rich information to examine the presence (and absence) of transient long-range ordering of the simulated ensembles. In Fig. 3, we compare the back-calculated PRE effects with experimental results on labelling at residues 7 and 61, where the terminal locations of the spin label provide particularly useful information on the peptide chain dimension. Quantitative comparison of experimental and back-calculated PRE effects is summarized in Table 1. These results clearly confirm that the simulated ensemble generated using c22* and ff99SB-ILDN are overly compact, leading to global over-estimation of PRE effects (Fig. 3). As for c36m and c36mw simulations, although not converged, better agreement with experimental PRE measurements appear to be achieved compared to the older c22* force field (Fig. S6 and Table 1), demonstrating their improvement in describing the overall dimension of IDPs. Moreover, ff99SB-ILDN-TIP4P-D and a99SB-disp simulations are able to reliably capture the chain dimension and long-range ordering of p53-TAD, generating conformational ensembles with minimal over-estimation of PRE effects (Fig. 3 and Table 1).
Figure 3.

Calculated (lines) and experimental (grey bars) PRE effects induced by paramagnetic spin labelling at residues 7 (top row) and 61 (bottom row). Red and green traces are calculated from independent control and folding simulations, respectively.
Table 1.
Root-mean-square (RMS) error of PRE effects of simulated ensembles in comparison with experimental measurements.
| c22* | ff99SB-ILDN | ff99SB-ILDN-TIP4P-D | a99SB-disp | c36m | 36mw | ||
|---|---|---|---|---|---|---|---|
| Residue 7 | Control | 0.68 | 0.71 | 0.35 | 0.29 | 0.38 | 0.21 |
| Folding | 0.69 | 0.70 | 0.23 | 0.19 | 0.50 | 0.35 | |
| Residue 28 | Control | 0.36 | 0.42 | 0.31 | 0.32 | 0.27 | 0.34 |
| Folding | 0.34 | 0.39 | 0.24 | 0.34 | 0.35 | 0.43 | |
| Residue 39 | Control | 0.48 | 0.50 | 0.28 | 0.31 | 0.36 | 0.32 |
| Folding | 0.49 | 0.48 | 0.26 | 0.24 | 0.37 | 0.31 | |
| Residue 61 | Control | 0.37 | 0.55 | 0.23 | 0.21 | 0.25 | 0.31 |
| Folding | 0.34 | 0.60 | 0.26 | 0.22 | 0.32 | 0.34 | |
| Overall RMS error | 0.49 | 0.56 | 0.27 | 0.27 | 0.36 | 0.33 | |
Ensemble-averaged distances between several pairs of residues of p53-TAD have also been determined previously using smFRET and TR-FRET measurement70 (see Figs. 4 and S7). For the distances between residues 1 and 17 (Fig. 4 top row), results from c22* and Amber ff99SB-ILDN simulations agreed well with experimental values, while ff99SB-ILDN-TIP4P-D and a99SB-disp appeared to slightly overestimate this distance. For another residue pair, 14 and 30 (Fig. 4 middle row), all four force fields showed reasonable agreement with the experimental data. However, caution needs to be taken to interpret the absolute distance estimation from smFRET and TR-FRET experiments, since fluorescence properties and energy transfer also depends on environment and orientation of the dye97. Therefore, the distances between these two residue pairs calculated from all simulations didn’t seem to qualitatively different from experimental measurement. On the other hand, the distance between residues 10 and 56 was significantly underestimated in c22* and ff99SB-ILDN simulations, while better agreement was observed for ff99SB-ILDN-TIP4P-D and a99SB-disp force fields (Fig. 4 bottom row). Since these two residues are close to N- and C-termini, respectively, in the primary sequence, it may be used to approximate the end-to-end distance and thus overall dimension of the peptide. The results again suggest that c22* and ff99SB-ILDN-derived structures are too collapsed and that ff99SB-ILDN-TIP4P-D and a99SB-disp can faithfully predict the chain dimension of p53-TAD.
Figure 4.

Probability distributions of distances between three pairs of residues, 1 and 17 (top row), 14 and 30 (middle row), and 10 and 56 (bottom row), calculated from independent control (red) and folding (green) simulations using four force fields. The corresponding ensemble averages were indicated as vertical bars. The vertical black line indicated experimental values from smFRET and TR-FRET measurements70.
Dependence of p53-TAD secondary structure on force field
The level of secondary structure of IDPs is often finely tuned by their sequences, which is critical for signaling fidelity8. Many disease mutations in IDPs have been predicted to alter secondary structure with higher probabilities than functionally neutral mutations15, which may perturb the protein interaction network and lead to mis-regulation. Multiple partial helices have also been identified in p53-TAD69, 74 and suggested to play roles in mediating p53’s interactions with key regulators including MDM2 and CBP/p30055. Cancer associated p53-TAD mutant K24N has been shown to reduce the helicity of residues 18–2774. Therefore, it’s very important to evaluate how well current force fields can capture residual secondary structures. Like chain dimension, secondary structure content of p53-TAD also depends strongly on the chosen force field. The helical content in c22* simulations seemed to be the highest among these four force fields, where three segments (residues 15–25, 35–45 and 45–55) constantly showed helical propensity of at least 40% (Fig. 2). In contrast, there was almost no residual helical structure in both control and folding simulations using ff99SB-ILDN-TIP4P-D force field (Fig. 2). The residual helicities for the other two force fields, ff99SB-ILDN and a99SB-disp, were in between these two extremes. Previous NMR studies have shown that residues 17–29 were ~10% helical, and two turns were assigned to the regions of 40–44 and 48–52, respectively69, 74. Nonetheless, continual sequential dNN NOEs and negative secondary Hα chemical shifts were found in the latter two regions69, implying that partial helical elements may exist. Results from a99SB-disp force field appear the most consistent with these experimental observations (see Fig. 2).
To better quantify the level of agreement of simulated ensembles with NMR results, we calculated the secondary chemical shifts of Cα and C’ atoms, which are sensitive to the secondary structure. The profiles of secondary chemical shift are shown in Figs. 5 and 6, and quantitative comparison between simulated and experimental results is summarized in Table 2. These results demonstrate that secondary chemical shifts of Cα and C’ atoms from a99SB-disp simulations agreed the best with experimental measurements, suggesting that this force field could properly describe the secondary structure propensity of p53-TAD. Note that SHIFTX2 could predict chemical shifts for folded proteins with RMS errors of 0.4412 and 0.5330 ppm for Cα and C’ atoms, respectively89. Therefore, according to Table 2, results from ff99SB-ILDN force field also seem in reasonable agreement with experimental values. In contrast, the content of helical structure is significantly underestimated by ff99SB-ILDN-TIP4P-D and over-estimated by c22* (Figs. 5 and 6).
Figure 5.

Secondary chemical shift analysis for Cα atoms in p53-TAD. Calculations were performed using independent control (red) and folding (green) simulations for five force fields. Experimental results were obtained from previously published work90.
Figure 6.

Secondary chemical shift analysis for C’ atoms in p53-TAD. Calculations were performed using independent control (red) and folding (green) simulations for five force fields. Experimental results were obtained from previously published work90.
Table 2.
RMS error of Cα and C’ chemical shifts for simulated ensembles in comparison with experimental values. The values within the SHIFTX2 prediction errors were shown in italics.
| c22* | ff99SB-ILDN | ff99SB-ILDN-TIP4P-D | a99SB-disp | ||
|---|---|---|---|---|---|
| Cα (ppm) | Control | 1.429 | 0.734 | 0.479 | 0.554 |
| Folding | 0.665 | 0.375 | 0.473 | 0.367 | |
| C’ (ppm) | Control | 0.824 | 0.481 | 0.591 | 0.417 |
| Folding | 0.544 | 0.376 | 0.578 | 0.345 | |
We note a curious observation regarding the propensity of forming 310 helices from multiple fore fields. As summarized in Fig. S8, the 310 helical content in simulations using three CHARMM force fields was low, and significantly lower than the corresponding α-helical content (Fig. 2). This can be directly attributed to the CMAP term used in CHARMM force fields, which was designed to rebalance the propensities of forming α, 310 and π helices76. In contrast, the Amber family of force fields appears to generate significant 310 helices. The only exception was ff99SB-ILDN-TIP4P-D, but this may be a direct result of increased water dispersion in TIP4P-D water force field that destabilizes both α (e.g., Fig. 2) and 310 helices. As shown in Fig. S8, the 310 helical propensity is comparable to the α-helical content for residues 20–25 in both ff99SB-ILDN and a99SB-disp simulations. Similar observations have also been reported previously for ff99SB*-ILDN42, 48, 49, a force field modified from ff99SB-ILDN by rebalancing the backbone helix-coil propensity, where the probability of forming 310 helix is higher in ff99SB*-ILDN simulations than in those using other force fields (see Figure 2 in reference50). Nevertheless, it is not clear how such an apparent systematic bias may affect IDP simulations using these force fields.
Detection of transient long-range contact formation using different force fields
Transient long-range interactions in IDPs are known to have important functional implication98–100. For example, previous NMR study has shown that loss of transient long-range interactions in α-synuclein, an IDP associated with Parkinson’s disease, could lead to completely unfolded conformations that aggregate more rapidly100. Such transient long-range interactions have also been found in p53-TAD73, 75, and they can be modulated both locally and remotely by binding of regulatory proteins or post-translational modification73. Thus, in order to obtain reliable simulated structural ensembles of p53-TAD, the force field also needs to be able to capture such important structural features. Experimentally, PRE in combination with site-specific spin labelling provides a powerful tool in detecting transient long-range contact formation for IDPs75, 101–103. The increase in relaxation rate of a proton is proportional to 1/r6, where r is the distance between proton and the unpaired electron in a spin label. Because of this 1/r6 dependence, PRE effect is suitable for identifying transient long-range contacts and insensitive to conformations with large proton-electron distance. Since PRE measurement could detect contact formation between unpaired electron and all amide protons in a protein, it provides valuable information for globally validating our simulation ensembles. Experimental PRE measurement75 found that residue 28 could become close to many residues ranging from 16 to 55 (see Fig. 7 top row). This feature could be captured by all simulations, although c22* and ff99SB-ILDN simulations seemed to underestimate the distances to both N- and C-termini, and a99SB-disp may overestimate the distances between residue 28 and residues 45–55 (Fig. 7 top row). Similar observations could also be found for PRE induced by spin labelling at residue 39 (Fig. 7 bottom row). Notably, a99SB-disp and ff99SB-ILDN-TIP4P-D force fields could even recapitulate some fine features in the experimental PRE profiles (Figs. 3 and 7). Taken together, transient long-range interactions from ff99SB-ILDN-TIP4P-D and a99SB-disp simulations agreed the best with experimental PRE (Table 1).
Figure 7.

Calculated (lines) and experimental (grey bars) PRE effects induced by paramagnetic spin labelling at residues 28 (top row) and 39 (bottom row). Red and green traces are calculated from independent control and folding simulations, respectively.
The presence of long-range contacts can be more directly and systematically examined using the contact probability map. As shown in Fig. 8, c22* and ff99SB-ILDN force fields generate significantly more transient interactions than ff99SB-ILDN-TIP4P-D and a99SB-disp force fields, consistent with the above observation that they may over-estimate the compactness of p53-TAD (e.g., Fig. 1). Nevertheless, many weakly populated tertiary contacts were also constantly observed in both ff99SB-ILDN-TIP4P-D and a99SB-disp simulations (Fig. 8), implying that the structural ensemble is highly heterogeneous. This can be illustrated by conformational clustering analysis shown in Figs. 9 and S9–S11, where no major cluster with over 2% occupancy was identified (see Methods for details). Since many of these long-range contacts involve the regions important for binding to MDM2 or CBP domains (e.g., residues 16–26, 48–55, see Fig. 8), in the future it would be interesting to study how these transient long-range contacts are modulated by mutation or post-translational modification, thus shedding light on the molecular mechanisms of p53 regulation.
Figure 8.

Contact probability maps calculated from independent control (top left) and folding (bottom right) simulations using four force fields. A contact is considered formed if the minimum distance between heavy atoms of two residues is less than or equal to 0.42 nm. Local and intermediate range contacts between residues separated by ≤ 4 residues were not shown for clarity.
Figure 9.

Centroid structures and populations of the top 12 most populated clusters for the structural ensemble derived from a99SB-disp control and folding simulations. The p53-TAD peptide is shown in Cartoon, with the color changing from red (at N-terminus) to blue (at C-terminus).
Conclusions
Accurate description of various local and long-range structural features of large, complex IDPs using empirical force fields is crucial for successful application of de novo simulations to characterize the disordered structure ensembles of IDPs and understand their sequence-disordered ensemble-function-disease relationships. We have explored the 61-residue p53-TAD as an exceptional model system to assess the ability of six latest protein force fields to describe non-trivial structural features within the disordered ensemble. Structural ensembles generated using μs-timescale REST2 enhanced sampling simulations were carefully analyzed and compared to a wide-range of experimental measurements derived from NMR, smFRET, and TR-FRET experiments. The results show that complex IDPs like p53-TAD remain very challenging for atomistic explicit solvent simulations, not only in achieving adequate level of convergence but also in proper description of local and long-range structural properties. Results from different force fields can vary significantly. Both c36m and c36mw fail to generate converged ensembles despite multiple μs simulation timescales. c22* generated overly compact structural ensembles, and residual helicity is also slightly over-estimated. Similar problem of over-compactness was also observed in ff99SB-ILDN simulations, but the helical propensity agreed well NMR measurement. More expanded structures can be obtained by combining ff99SB-ILDN force field with the TIP4P-D water force field; this seems to be achieved at the expense of over-destabilizing protein-protein interactions, leading to significant under-estimation of residual secondary structures in p53-TAD. Encouragingly, the latest a99SB-disp force field seems capable of faithfully recapitulating virtually all experimental measurements examined, including the overall chain dimension, residual secondary structures, and transient long-range ordering. As such, we anticipate that atomistic simulations using a99SB-disp will provide a viable approach for understanding how the disordered state of p53-TAD may be modulated by multi-site phosphorylations and/or many cancer-associated mutations in biology and cancers. Such structural insights will pave the way for establishing the sequence-disordered ensemble-function-disease relationships of p53-TAD and other regulatory IDPs.
Supplementary Material
Acknowledgements
This work is supported by the National Institutes of Health (GM114300). The computing was performed on the Pikes cluster housed in the Massachusetts Green High-Performance Computing Center (MGHPCC).
REFERENCES
- 1.Wright PE; Dyson HJ, Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 1999, 293 (2), 321–331. [DOI] [PubMed] [Google Scholar]
- 2.Dunker AK; Brown CJ; Lawson JD; Iakoucheva LM; Obradovic Z, Intrinsic disorder and protein function. Biochemistry 2002, 41 (21), 6573–82. [DOI] [PubMed] [Google Scholar]
- 3.Tompa P, Intrinsically unstructured proteins. Trends Biochem Sci 2002, 27 (10), 527–533. [DOI] [PubMed] [Google Scholar]
- 4.Dyson HJ; Wright PE, Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Bio 2005, 6 (3), 197–208. [DOI] [PubMed] [Google Scholar]
- 5.Uversky VN; Oldfield CJ; Dunker AK, Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit 2005, 18 (5), 343–384. [DOI] [PubMed] [Google Scholar]
- 6.Dunker AK; Silman I; Uversky VN; Sussman JL, Function and structure of inherently disordered proteins. Curr Opin Struc Biol 2008, 18 (6), 756–764. [DOI] [PubMed] [Google Scholar]
- 7.Smock RG; Gierasch LM, Sending Signals Dynamically. Science 2009, 324 (5924), 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright PE; Dyson HJ, Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Bio 2015, 16 (1), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Click TH; Ganguly D; Chen J, Intrinsically disordered proteins in a physics-based world. Int J Mol Sci 2010, 11 (12), 5292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teilum K; Olsen JG; Kragelund BB, Functional aspects of protein flexibility. Cellular and molecular life sciences : CMLS 2009, 66 (14), 2231–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iakoucheva LM; Brown CJ; Lawson JD; Obradovic Z; Dunker AK, Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol 2002, 323 (3), 573–84. [DOI] [PubMed] [Google Scholar]
- 12.Mitrea DM; Kriwacki RW, Regulated unfolding of proteins in signaling. Febs Lett 2013, 587 (8), 1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babu MM; van der Lee R; de Groot NS; Gsponer J, Intrinsically disordered proteins: regulation and disease. Curr Opin Struct Biol 2011, 21 (3), 432–40. [DOI] [PubMed] [Google Scholar]
- 14.Lu HC; Chung SS; Fornili A; Fraternali F, Anatomy of protein disorder, flexibility and disease-related mutations. Frontiers in molecular biosciences 2015, 2, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vacic V; Markwick PR; Oldfield CJ; Zhao X; Haynes C; Uversky VN; Iakoucheva LM, Disease-associated mutations disrupt functionally important regions of intrinsic protein disorder. PLoS Comput Biol 2012, 8 (10), e1002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uversky VN; Dave V; Iakoucheva LM; Malaney P; Metallo SJ; Pathak RR; Joerger AC, Pathological Unfoldomics of Uncontrolled Chaos: Intrinsically Disordered Proteins and Human Diseases. Chemical reviews 2014, 114 (13), 6844–6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JH, Towards the physical basis of how intrinsic disorder mediates protein function. Archives of biochemistry and biophysics 2012, 524 (2), 123–131. [DOI] [PubMed] [Google Scholar]
- 18.Kruschel D; Zagrovic B, Conformational averaging in structural biology: issues, challenges and computational solutions. Molecular BioSystems 2009, 5 (12), 1606–1616. [DOI] [PubMed] [Google Scholar]
- 19.Fisher CK; Stultz CM, Constructing ensembles for intrinsically disordered proteins. Curr Opin Struc Biol 2011, 21 (3), 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuler B; Eaton WA, Protein folding studied by single-molecule FRET. Curr Opin Struc Biol 2008, 18 (1), 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eliezer D, Biophysical characterization of intrinsically disordered proteins. Curr Opin Struc Biol 2009, 19 (1), 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brucale M; Schuler B; Samori B, Single-Molecule Studies of Intrinsically Disordered Proteins. Chemical reviews 2014, 114 (6), 3281–3317. [DOI] [PubMed] [Google Scholar]
- 23.Ganguly D; Chen J, Structural Interpretation of Paramagnetic Relaxation Enhancement-Derived Distances for Disordered Protein States. J Mol Biol 2009, 390 (3), 467–477. [DOI] [PubMed] [Google Scholar]
- 24.Zhang WH; Chen JH, Accelerate Sampling in Atomistic Energy Landscapes Using Topology-Based Coarse-Grained Models. J Chem Theory Comput 2014, 10 (3), 918–923. [DOI] [PubMed] [Google Scholar]
- 25.Moritsugu K; Terada T; Kidera A, Scalable free energy calculation of proteins via multiscale essential sampling. J. Chem. Phys 2010, 133 (22). [DOI] [PubMed] [Google Scholar]
- 26.Sugita Y; Okamoto Y, Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 1999, 314 (1–2), 141–151. [Google Scholar]
- 27.Liu P; Kim B; Friesner RA; Berne BJ, Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (39), 13749–13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KH; Chen JH, Optimization of the GBMV2 implicit solvent force field for accurate simulation of protein conformational equilibria. Journal of computational chemistry 2017, 38 (16), 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitalis A; Pappu RV, ABSINTH: A New Continuum Solvation Model for Simulations of Polypeptides in Aqueous Solutions. Journal of computational chemistry 2009, 30 (5), 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das RK; Pappu RV, Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc Natl Acad Sci U S A 2013, 110 (33), 13392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao AH; Crick SL; Vitalis A; Chicoine CL; Pappu RV, Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (18), 8183–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berlow RB; Dyson HJ; Wright PE, Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J Mol Biol 2018, 430 (16), 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Habchi J; Tompa P; Longhi S; Uversky VN, Introducing Protein Intrinsic Disorder. Chemical reviews 2014, 114 (13), 6561–6588. [DOI] [PubMed] [Google Scholar]
- 34.Robustelli P; Piana S; Shaw DE, Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (21), E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr., CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature methods 2017, 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robertson MJ; Tirado-Rives J; Jorgensen WL, Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J Chem Theory Comput 2015, 11 (7), 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piana S; Donchev AG; Robustelli P; Shaw DE, Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J Phys Chem B 2015, 119 (16), 5113–5123. [DOI] [PubMed] [Google Scholar]
- 38.Best RB; Zheng W; Mittal J, Balanced Protein–Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J Chem Theory Comput 2014, 10 (11), 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piana S; Lindorff-Larsen K; Shaw DE, How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys J 2011, 100 (9), L47–L49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nerenberg PS; Jo B; So C; Tripathy A; Head-Gordon T, Optimizing Solute-Water van der Waals Interactions To Reproduce Solvation Free Energies. J Phys Chem B 2012, 116 (15), 4524–4534. [DOI] [PubMed] [Google Scholar]
- 41.Choi JM; Pappu RV, Improvements to the ABSINTH Force Field for Proteins Based on Experimentally Derived Amino Acid Specific Backbone Conformational Statistics. J Chem Theory Comput 2019, 15 (2), 1367–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Best RB; Hummer G, Optimized Molecular Dynamics Force Fields Applied to the Helix-Coil Transition of Polypeptides. J Phys Chem B 2009, 113 (26), 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindorff-Larsen K; Piana S; Dror RO; Shaw DE, How Fast-Folding Proteins Fold. Science 2011, 334 (6055), 517–520. [DOI] [PubMed] [Google Scholar]
- 44.Beauchamp KA; Lin YS; Das R; Pande VS, Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements. J Chem Theory Comput 2012, 8 (4), 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nerenberg PS; Head-Gordon T, Optimizing Protein-Solvent Force Fields to Reproduce Intrinsic Conformational Preferences of Model Peptides. J Chem Theory Comput 2011, 7 (4), 1220–30. [DOI] [PubMed] [Google Scholar]
- 46.Li DW; Bruschweiler R, NMR-Based Protein Potentials. Angew Chem Int Edit 2010, 49 (38), 6778–6780. [DOI] [PubMed] [Google Scholar]
- 47.Lindorff-Larsen K; Maragakis P; Piana S; Eastwood MP; Dror RO; Shaw DE, Systematic Validation of Protein Force Fields against Experimental Data. PloS one 2012, 7 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C, Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65 (3), 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE, Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78 (8), 1950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rauscher S; Gapsys V; Gajda MJ; Zweckstetter M; de Groot BL; Grubmuller H, Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. J Chem Theory Comput 2015, 11 (11), 5513–24. [DOI] [PubMed] [Google Scholar]
- 51.Piana S; Klepeis JL; Shaw DE, Assessing the accuracy of physical models used in protein-folding simulations: quantitative evidence from long molecular dynamics simulations. Curr Opin Struc Biol 2014, 24, 98–105. [DOI] [PubMed] [Google Scholar]
- 52.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79 (2), 926–935. [Google Scholar]
- 53.Muller PAJ; Vousden KH, Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25 (3), 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kandoth C; McLellan MD; Vandin F; Ye K; Niu BF; Lu C; Xie MC; Zhang QY; McMichael JF; Wyczalkowski MA; Leiserson MDM; Miller CA; Welch JS; Walter MJ; Wendl MC; Ley TJ; Wilson RK; Raphael BJ; Ding L, Mutational landscape and significance across 12 major cancer types. Nature 2013, 502 (7471), 333–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris SL; Levine AJ, The p53 pathway: positive and negative feedback loops. Oncogene 2005, 24 (17), 2899–2908. [DOI] [PubMed] [Google Scholar]
- 56.Grossman SR; Deato ME; Brignone C; Chan HM; Kung AL; Tagami H; Nakatani Y; Livingston DM, Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003, 300 (5617), 342–344. [DOI] [PubMed] [Google Scholar]
- 57.Brooks CL; Gu W, p53 ubiquitination: Mdm2 and beyond. Molecular cell 2006, 21 (3), 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kruse JP; Gu W, Modes of p53 Regulation. Cell 2009, 137 (4), 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferreon JC; Lee CW; Arai M; Martinez-Yamout MA; Dyson HJ; Wright PE, Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (16), 6591–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teufel DP; Bycroft M; Fersht AR, Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28 (20), 2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee CW; Ferreon JC; Ferreon ACM; Arai M; Wright PE, Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (45), 19290–19295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levine AJ; Hu W; Feng Z, The P53 pathway: what questions remain to be explored? Cell death and differentiation 2006, 13 (6), 1027–1036. [DOI] [PubMed] [Google Scholar]
- 63.Kussie PH; Gorina S; Marechal V; Elenbaas B; Moreau J; Levine AJ; Pavletich NP, Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274 (5289), 948–953. [DOI] [PubMed] [Google Scholar]
- 64.Lee CW; Martinez-Yamout MA; Dyson HJ; Wright PE, Structure of the p53 Transactivation Domain in Complex with the Nuclear Receptor Coactivator Binding Domain of CREB Binding Protein. Biochemistry 2010, 49 (46), 9964–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee CW; Arai M; Martinez-Yamout MA; Dyson HJ; Wright PE, Mapping the Interactions of the p53 Transactivation Domain with the KIX Domain of CBP. Biochemistry 2009, 48 (10), 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Lello P; Jenkins LMM; Jones TN; Nguyen BD; Hara T; Yamaguchi H; Dikeakos JD; Appella E; Legault P; Omichinski JG, Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Molecular cell 2006, 22 (6), 731–740. [DOI] [PubMed] [Google Scholar]
- 67.Ganguly D; Chen J, Atomistic details of the disordered states of KID and pKID. Implications in coupled binding and folding. Journal of the American Chemical Society 2009, 131 (14), 5214–5223. [DOI] [PubMed] [Google Scholar]
- 68.Liu XR; Chen JL; Chen JH, Residual Structure Accelerates Binding of Intrinsically Disordered ACTR by Promoting Efficient Folding upon Encounter. J Mol Biol 2019, 431 (2), 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee H; Mok KH; Muhandiram R; Park KH; Suk JE; Kim DH; Chang J; Sung YC; Choi KY; Han KH, Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem 2000, 275 (38), 29426–29432. [DOI] [PubMed] [Google Scholar]
- 70.Huang F; Rajagopalan S; Settanni G; Marsh RJ; Armoogum DA; Nicolaou N; Bain AJ; Lerner E; Haas E; Ying L, Multiple conformations of full-length p53 detected with single-molecule fluorescence resonance energy transfer. Proceedings of the National Academy of sciences 2009, 106 (49), 20758–20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lowry DF; Stancik A; Shrestha RM; Daughdrill GW, Modeling the accessible conformations of the intrinsically unstructured transactivation domain of p53. Proteins: Structure, Function, and Bioinformatics 2008, 71 (2), 587–598. [DOI] [PubMed] [Google Scholar]
- 72.Wells M; Tidow H; Rutherford TJ; Markwick P; Jensen MR; Mylonas E; Svergun DI; Blackledge M; Fersht AR, Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (15), 5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lum JK; Neuweiler H; Fersht AR, Long-Range Modulation of Chain Motions within the Intrinsically Disordered Transactivation Domain of Tumor Suppressor p53. Journal of the American Chemical Society 2012, 134 (3), 1617–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhan YA; Wu HW; Powell AT; Daughdrill GW; Ytreberg FM, Impact of the K24N mutation on the transactivation domain of p53 and its binding to murine double-minute clone 2. Proteins 2013, 81 (10), 1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vise P; Baral B; Stancik A; Lowry DF; Daughdrill GW, Identifying long-range structure in the intrinsically unstructured transactivation domain of p53. Proteins 2007, 67 (3), 526–530. [DOI] [PubMed] [Google Scholar]
- 76.MacKerell AD; Feig M; Brooks CL, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. Journal of computational chemistry 2004, 25 (11), 1400–1415. [DOI] [PubMed] [Google Scholar]
- 77.MacKerell AD; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiorkiewicz-Kuczera J; Yin D; Karplus M, All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 1998, 102 (18), 3586–3616. [DOI] [PubMed] [Google Scholar]
- 78.Wang LL; Friesner RA; Berne BJ, Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2). J Phys Chem B 2011, 115 (30), 9431–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pang X; Zhou HX, Disorder-to-Order Transition of an Active-Site Loop Mediates the Allosteric Activation of Sortase A. Biophys J 2015, 109 (8), 1706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang K; Garcia AE, Acceleration of Lateral Equilibration in Mixed Lipid Bilayers Using Replica Exchange with Solute Tempering. J Chem Theory Comput 2014, 10 (10), 4264–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Terakawa T; Kameda T; Takada S, On Easy Implementation of a Variant of the Replica Exchange with Solute Tempering in GROMACS. Journal of computational chemistry 2011, 32 (7), 1228–1234. [DOI] [PubMed] [Google Scholar]
- 82.Stirnemann G; Sterpone F, Recovering Protein Thermal Stability Using All-Atom Hamiltonian Replica-Exchange Simulations in Explicit Solvent. J Chem Theory Comput 2015, 11 (12), 5573–5577. [DOI] [PubMed] [Google Scholar]
- 83.Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar]
- 84.Pall S; Abraham MJ; Kutzner C; Hess B; Lindahl E, Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. Lect Notes Comput Sc 2015, 8759, 3–27. [Google Scholar]
- 85.Tribello GA; Bonomi M; Branduardi D; Camilloni C; Bussi G, PLUMED 2: New feathers for an old bird. Comput Phys Commun 2014, 185 (2), 604–613. [Google Scholar]
- 86.Bussi G, Hamiltonian replica exchange in GROMACS: a flexible implementation. Mol Phys 2014, 112 (3–4), 379–384. [Google Scholar]
- 87.Darden T; York D; Pedersen L, Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. The Journal of chemical physics 1993, 98 (12), 10089–10092. [Google Scholar]
- 88.Hess B; Bekker H; Berendsen HJC; Fraaije JGEM, LINCS: A linear constraint solver for molecular simulations. Journal of computational chemistry 1997, 18 (12), 1463–1472. [Google Scholar]
- 89.Han B; Liu YF; Ginzinger SW; Wishart DS, SHIFTX2: significantly improved protein chemical shift prediction. Journal of biomolecular NMR 2011, 50 (1), 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wong TS; Rajagopalan S; Freund SM; Rutherford TJ; Andreeva A; Townsley FM; Petrovich M; Fersht AR, Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic acids research 2009, 37 (20), 6765–6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wishart DS; Nip AM, Protein chemical shift analysis: a practical guide. Biochem Cell Biol 1998, 76 (2–3), 153–163. [DOI] [PubMed] [Google Scholar]
- 92.Wishart DS; Bigam CG; Holm A; Hodges RS; Sykes BD, H-1, C-13 and N-15 Random Coil Nmr Chemical-Shifts of the Common Amino-Acids .1. Investigations of Nearest-Neighbor Effects. Journal of biomolecular NMR 1995, 5 (1), 67–81. [DOI] [PubMed] [Google Scholar]
- 93.Zhou T; Caflisch A, Distribution of Reciprocal of Interatomic Distances: A Fast Structural Metric. J Chem Theory Comput 2012, 8 (8), 2930–2937. [DOI] [PubMed] [Google Scholar]
- 94.Beauchamp KA; Bowman GR; Lane TJ; Maibaum L; Haque IS; Pande VS, MSMBuilder2: Modeling Conformational Dynamics on the Picosecond to Millisecond Scale. J Chem Theory Comput 2011, 7 (10), 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Battiste JL; Wagner G, Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 2000, 39 (18), 5355–5365. [DOI] [PubMed] [Google Scholar]
- 96.Ganguly D; Chen J, Modulation of the Disordered Conformational Ensembles of the p53 Transactivation Domain by Cancer-Associated Mutations. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roy R; Hohng S; Ha T, A practical guide to single-molecule FRET. Nature methods 2008, 5 (6), 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krois AS; Dyson HJ; Wright PE, Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (48), E11302–E11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ranjan P; Kumar A, Perturbation in Long-Range Contacts Modulates the Kinetics of Amyloid Formation in alpha-Synuclein Familial Mutants. Acs Chem Neurosci 2017, 8 (10), 2235–2246. [DOI] [PubMed] [Google Scholar]
- 100.Bertoncini CW; Jung YS; Fernandez CO; Hoyer W; Griesinger C; Jovin TM; Zweckstetter M, Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (5), 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Clore GM; Iwahara J, Theory, Practice, and Applications of Paramagnetic Relaxation Enhancement for the Characterization of Transient Low-Population States of Biological Macromolecules and Their Complexes. Chemical reviews 2009, 109 (9), 4108–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gillespie JR; Shortle D, Characterization of long-range structure in the denatured state of staphylococcal nuclease .1. Paramagnetic relaxation enhancement by nitroxide spin labels. J Mol Biol 1997, 268 (1), 158–169. [DOI] [PubMed] [Google Scholar]
- 103.Clore GM; Tang C; Iwahara J, Elucidating transient macromolecular interactions using paramagnetic relaxation enhancement. Curr Opin Struc Biol 2007, 17 (5), 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.