

Abstract
Tumor-necrosis factor (TNF) and its superfamily members are pleiotropic cytokines. Activation of TNF can lead to distinct cellular outcomes including inflammation, cell survival, and different forms of cell death, such as apoptosis and necrosis in a context-dependent manner. However, our understanding of what determines the versatile functions of TNF is far from complete. Here, we examined the molecular mechanisms that distinguish the forms of cell death induced by Eiger (Egr), the sole homolog of TNF in Drosophila. We show that expression of Egr in the developing Drosophila eye simultaneously induces apoptosis and apoptosis-independent developmental defects indicated by cellular disorganization, both of which rely on the c-Jun N-terminal kinase (JNK) signaling activity. Intriguingly, when effector caspases DrICE and Dcp-1 are defective or inhibited, expression of Egr triggers necrosis which is characterized by loss of cell membrane integrity, translucent cytoplasm, and aggregation of cellular organelles. Moreover, such Egr-induced necrosis depends on the catalytic activity of the initiator caspase Dronc and the input from JNK signaling but is independent of their roles in apoptosis. Further mosaic analysis with mutants of scribble (scrib), an evolutionarily conserved tumor suppressor gene regulating cell polarity, suggests that Egr/JNK-mediated apoptosis and necrosis establish a two-layered defense system to inhibit the oncogenic growth of scrib mutant cells. Together, we have identified caspase- and JNK-dependent mechanisms underlying Egr-induced apoptosis versus necrosis and their fail-safe roles in tumor suppression in an intact organism in vivo.
Subject terms: Necroptosis, Disease model
Introduction
Apoptosis is a major form of programmed cell death critical for development and damage response1. The key factors driving apoptosis are caspases, a family of cysteine proteases. These apoptotic caspases can further group into initiator and effector caspases2. The initiator caspases, once activated, cleave and activate the effector caspases leading to apoptosis. Unlike apoptosis, necrosis has long been considered as an uncontrolled form of cell death. However, recent studies have revealed that certain types of necrosis are molecularly regulated3,4. Intriguingly, our growing understanding of apoptosis and regulated necrosis has unveiled multiple molecular interplays between them5. One example of such molecules is the tumor-necrosis factor (TNF) superfamily, a group of cytokines which was initially discovered because of its antitumor activity6. Binding of TNF to their receptors promotes recruitment of various death-inducing protein complexes depending on the context7. A key component common in these complexes is caspase-8, an initiator caspase. Once activated, caspase-8 cleaves and activates effector caspases, such as caspase-3 and −7, to trigger apoptosis8. Notably, caspase-8 also cleaves and inactivates two receptor-interacting serine/threonine-protein kinases (RIPKs), RIPK1, and RIPK3, which mediate activation of necroptosis, a form of regulated necrosis9–12. Therefore, when Caspase-8 is deficient or inhibited, activation of TNF induces necrosis via RIPK1 and PIPK3.
In addition to cell death, functions of TNF family members have been revealed in immunity, inflammation, cell survival, and proliferation6. Many of these functions are mediated by pleiotropic molecules, such as the Nuclear Factor-κB (NF-κB) transcription factor13, the c-Jun N-terminal kinase (JNK), and the p38 mitogen-activated protein kinase (p38-MAPK)14,15. It is therefore not surprising that dysregulated TNF signaling is associated with numerous pathological conditions including cancer16,17. However, our understanding of the mechanisms that determine distinct outcomes of TNF signaling is still far from complete.
Eiger (Egr) is the sole Drosophila homolog of TNF18,19. Wengen20 and, more recently, Grindelwald21 have been identified as its receptors. Similar to mammalian TNFs, Egr plays multiple roles in regulating cell death, host defense, tissue growth, and regeneration in a context-dependent manner22–24. Notably, Egr exerts its functions mainly through activation of JNK. For example, expression of Egr under the control of an eye-specific driver GMR (GMR > egr) activates JNK leading to cell death and small adult eyes18,19,25. Intriguingly, GMR > egr-induced cell death may be nonapoptotic because its eye ablation phenotype cannot be suppressed by P35 (ref.18), an inhibitor of the effector caspases DrICE and Dcp-1 (ref.26). However, what seems to be a paradox, GMR > egr-induced small eyes can be suppressed by inhibition of the initiator caspase Dronc19,25. It is therefore not yet clear whether and how Egr can induce both apoptotic and nonapoptotic cell death.
Here, we report that Egr primarily induces JNK-dependent apoptosis and, simultaneously, apoptosis-independent cellular disorganization when it is expressed in the Drosophila eye. However, when apoptosis is blocked by inhibition of the effector caspases DrICE and Dcp-1, expression of Egr induces necrosis instead. Intriguingly, the initiator caspase Dronc is required for both Egr-induced apoptosis and necrosis, but at different protein levels. Its catalytic activity cooperates with an apoptosis-independent role of JNK signaling to activate necrosis. Furthermore, under pathological conditions, e.g., in cells mutant for scribble (scrib) which is a tumor suppressor gene regulating cell polarity, activation of Egr-JNK establishes a two-layered defense mechanism with apoptosis and, if apoptosis fails, necrosis in place to prevent oncogenic tissue overgrowth. Therefore, Drosophila models can be employed to dissect the molecular interplays between apoptosis and necrosis in vivo.
Results
The cleaved Dcp-1 antibody is a specific marker for activated effector caspases DrICE and Dcp-1 in Drosophila
To determine whether GMR > Egr induces apoptosis or nonapoptotic cell death, we first sought to identify a marker specifically recognizing activated effector caspases, e.g., the cleaved DrICE and Dcp-1, in Drosophila because antibodies recognizing the cleaved human caspase-3 are not specific to these proteins27. A recently developed cleaved Dcp-1 (Asp216) antibody (referred to as cDcp1) from Cell Signaling Technology is a polyclonal antibody raised against the large 22 kDa fragment of cleaved Dcp-1. Although this antibody has been increasingly used to label apoptosis in Drosophila28, a detailed characterization of its specificity has not been performed. To address this, we used GMR-hid, a transgene activating apoptosis in the eye29. In late 3rd instar larval eye disks, compared with wild type (Fig. 2e), GMR-hid induces two waves of apoptotic cells as indicated by TUNEL, an assay detecting DNA fragmentation therefore labeling apoptotic cells (Fig. 1a)30. The cDcp1 antibodies recognize these two apoptotic waves (Fig. 1b). Interestingly, these apoptotic signals persist in dcp-1 null mutants (Fig. 1c, d). It suggests that cleaved Dcp-1 is not the only protein recognized by cDcp1. In addition to Dcp-1, DrICE is another major effector caspase in somatic tissues31,32. Indeed, GMR-hid-induced apoptosis is almost completely lost in drICE null mutants (Fig. 1e). Because DrICE and Dcp-1 share similar sequences cleaved by Dronc27,33, it is possible that cDcp1 also recognizes the cleaved DrICE. Consistent with this idea, cDcp1 detects a relatively low level of proteins, presumably the cleaved Dcp-1, in drICE mutants (Fig. 1f). In contrast, the cDcp1- and TUNEL-signals are lost in dcp-1; drICE double mutants (Fig. 1g, h). Therefore, cDcp1 recognizes both cleaved Dcp-1 and cleaved DrICE.
Fig. 2. GMR > egr induces apoptosis via hid in the Drosophila eye.

a–d Adult eye images. Compared with wild type (a), expression of egr under the control of GMR (GMR-GAL4 UAS-egr, GMR > egr) induces a strong eye ablation phenotype (b). This phenotype is completely suppressed by an RNAi knockdown of egr (egrRNAi, c) or expression of puckered (puc, d), a negative regulator of JNK. Late 3rd instar larval eye disks labeled with cDcp1 (e–h, j) or cDcp1 and GFP (i–i′′). Compared with wild type (e), GMR > egr induces massive apoptosis indicated by cDcp1 labeling (f). This apoptosis is suppressed in dronc null mutants (g) or by expression of a GMR-p35 transgene (h). In GMR > egr disks with hid mutant clones marked by lack of GFP (i), apoptosis is blocked in the clones (highlighted by yellow dotted lines in i′ and i′′ which are enlarged images of the outlined area in i). In contrast, rpr mutants (rpr87/XR38a, combination of a deletion and a null mutant of rpr) do not suppress GMR > egr-induced apoptosis (j). k–n Adult eye images. hid mutant clones (k), expression of a RING domain-deleted, therefore stabilized, form of Diap1 (GMR-BIR, l) or dronc null mutants (pharate adults were dissected out of the pupal cases, m) strongly suppress GMR > egr-induced eye ablation phenotype. In contrast, rpr mutants (rpr87/XR38a) do not suppress the small eyes induced by GMR > egr (n)
Fig. 1. The cDcp1 antibody recognizes cleaved DrICE and Dcp-1 in Drosophila.
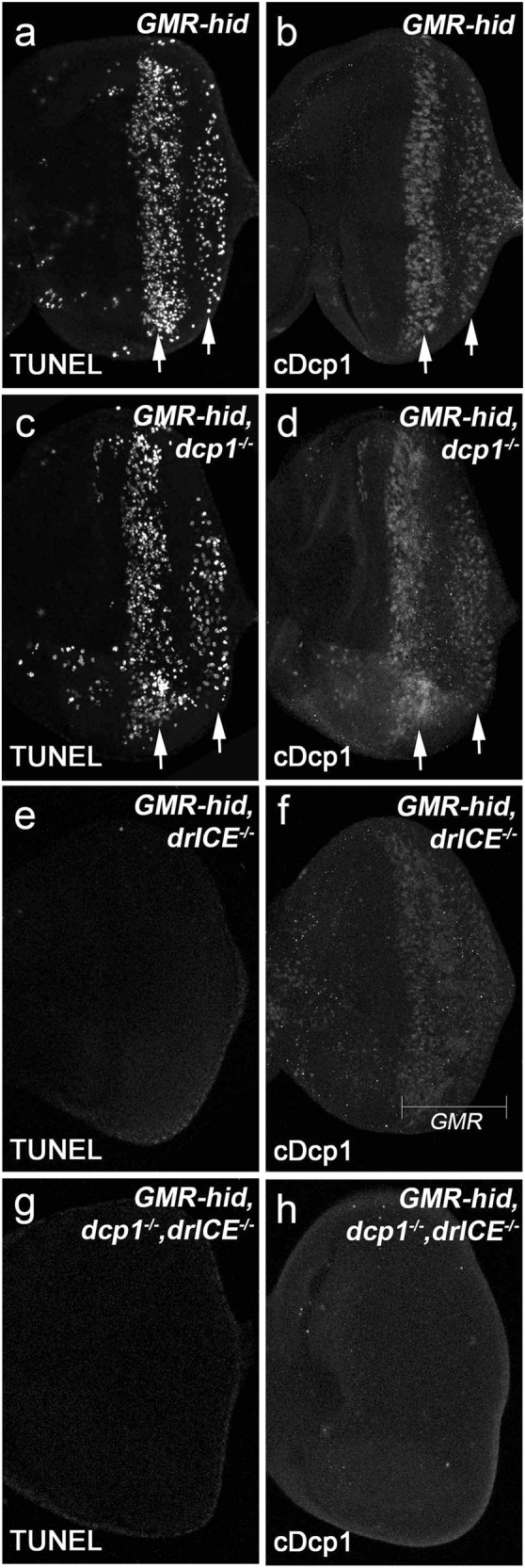
Late 3rd instar larval eye disks labeled with either the TUNEL assay (a, c, e, g) or the cDcp1 antibodies (b, d, f, h). Expression of hid under the control of GMR (GMR-hid) induces two apoptotic waives indicated by either TUNEL (a, arrows) or cDcp1 (b, arrows) staining. GMR-hid-induced two apoptotic waves are recognized by both TUNEL (c) and cDcp1 (d) in dcp-1 null mutants. GMR-hid-induced apoptosis is blocked in drICE null mutants as indicated by lack of TUNEL labeling (e). However, cDcp1 recognizes a low level of signals in the whole GMR domain in the same genetic background (f). No GMR-hid-induced signals were detected by either TUNEL (g) or cDcp1 (h) in drICE and dcp-1 double null mutants
Expression of Eiger induces strong apoptosis through the canonical apoptosis pathway and the pro-apoptotic gene hid
It has been reported that GMR > egr activates JNK resulting in an eye ablation phenotype (compare 2b to 2a)18,19, which can be suppressed by RNAi knockdown of egr (Fig. 2c) or expression of puckered (puc) (Fig. 2d), a negative regulator of JNK34,35. To assess whether GMR > egr induces apoptosis, we used the cDcp1 antibody. Compared with wild type (Fig. 2e), a strong wave of cDcp1-labeling was observed in GMR > egr disks (Fig. 2f). To confirm it is apoptosis, we performed genetic analyses on the key components in the apoptosis pathway. Loss of Dronc, the major initiator caspase mediating apoptosis in Drosophila36, or expression of P35, an inhibitor of activated DrICE and Dcp-1 (ref.26), completely blocks the cDcp1 signals in GMR > egr (Fig. 2g, h). Consistently, GMR > egr-induced eye ablation phenotype is suppressed in dronc null mutants (compare Fig. 2m to 2b). Moreover, expression of a RING domain-deleted (therefore stabilized) form of Diap1 (BIR), an apoptosis inhibitor acting upstream of Dronc37, also suppresses GMR > egr small eyes (compare Fig. 2l to 2b). Therefore, GMR > egr triggers massive apoptosis in the developing Drosophila eye.
To identify which proapoptotic genes mediate GMR > egr-induced apoptosis, we examined expression of hid and rpr by using their reporters38. Compared with the control, expression of hid, but not rpr, significantly increases in GMR > egr (Supplementary Fig. S1). Moreover, loss of rpr in rpr87/XR38, a combination of a deletion39 and a null mutant of rpr (ref.40), does not affect GMR > egr-induced apoptosis (Fig. 2j). In contrast, such apoptosis is lost in hid mutant clones (Fig. 2i–i′′). Consistent with these results, the hid mutant clones, but not rpr mutants, suppress GMR > egr-induced small eyes (Fig. 2k, n). Taken together, expression of Egr activates apoptosis through the proapoptotic gene hid in the developing Drosophila eye.
Expression of Eiger induces developmental defects independent of apoptosis
Notably, unlike expression of puc (Fig. 2d), loss of dronc or expression of Diap1 (GMR > BIR) does not completely restore the GMR > egr eyes back to normal. These eyes show glassy appearance suggesting defects in their ommatidial patterning (Fig. 2l, m). In contrast, expression of a dominant negative form of bsk (bskDN, bsk = Drosophila JNK) or hemizygous mutants of Tak1 (Tak12527, null mutant41), an upstream kinase of JNK, almost completely rescue GMR > egr-induced adult eye defects including the glassy eye appearance (Fig. 3a, b). Therefore, GMR > egr also induces apoptosis-independent, but JNK-dependent, developmental defects (Fig. 3c).
Fig. 3. GMR > egr induces nonapoptotic, but JNK-dependent, cellular disorganization in the Drosophila eye.
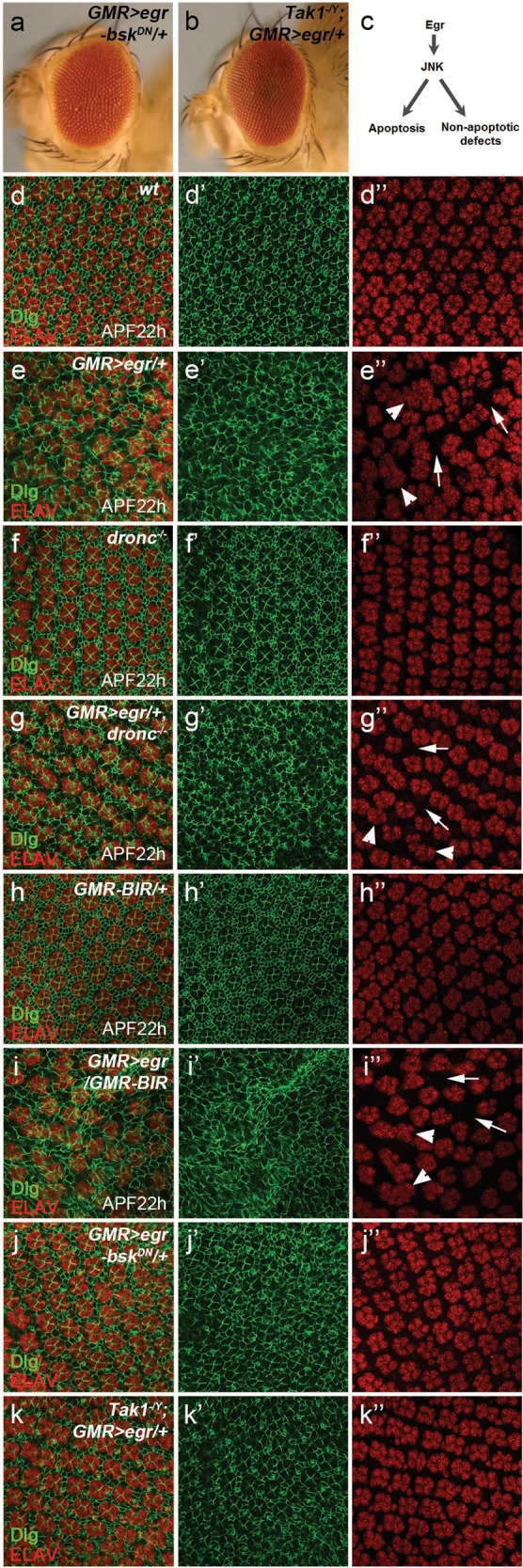
a, b Adult eye images. Expression of a dominant negative form of bsk (bskDN, bsk = Drosophila JNK) (a) or a hemizygous mutant of Tak1 (Tak1-/Y, b), an upstream kinase of JNK, almost completely suppresses GMR > egr-induced eye ablation phenotype. (c) A diagram showing that JNK signaling induced by Egr can lead to both apoptosis and nonapoptotic defects in the developing eye. APF22h pupal eye disks labeled with a cellular membrane maker Dlg (green in d–k and d′–k′) and a neuronal marker ELAV (red in d–k and d′′–k′′). In wild-type disks (d–d′′), ommatidia (each is composed of eight photoreceptor neurons), as indicated by ELAV, and interommatidial cells, as indicated by Dlg, are well-patterned. In contrast, defective cellular organization was observed in GMR > egr disks (e–e′′). Examples of these defects such as ommatidial fusion (arrowheads in e′′, g′′, i′′) and increased interommatidial spacing (arrows in e′′, g′′, i′′) are highlighted. dronc null mutants (f–f′′, dronc−/−) or expression of a stabilized form of Diap1 (h–h′′, GMR-BIR) neither alter the ommatidial patterning in wild-type eye disks (compare f–f′′ and h–h′′ to d–d′′) nor suppress the irregular ommatidial organization in GMR > egr disks (compare g–g′′ and i–i′′ to e–e′′). In contrast, expression of a dominant-negative form of JNK (bskDN, j–j′′) or a hemizygous mutant of Tak1 (k–k′′) strongly suppresses the cellular disorganization induced by GMR > egr
To further characterize this nonapoptotic developmental defects, we examined cellular organization in the pupal eye disks, which are composed of well-patterned ommatidia (Fig. 3d, d′′, ELAV labeling) and interommatidial cells (Fig. 3d, d′). At 25 °C, developmental apoptosis occurs at 28 h after pupal formation (APF28h) to remove extra interommatidial cells in eye disks (Supplementary Fig. S2)42,43. In contrast, no apoptosis is observed at APF22h (Fig. 4a, a′). We therefore focused our analysis of GMR > egr on the stage of APF22h to avoid developmental apoptosis. Compared with wild-type eye disks in which ommatidia and interommatidial cells are well-patterned (Fig. 3d–d′′), GMR > egr induces disorganization of these cells (Fig. 3e–e′′) indicated by both increased interommatidial spacing (Fig. 3e′′, arrows) and ommatidial fusion (Fig. 3e′′, arrowheads). Such defects persist when apoptosis is inhibited in dronc mutants (Fig. 3g-g′′) or by expression of Diap1 (Fig. 3i–i′′). As controls, ommatidial organization is not affected by loss of dronc (Fig. 3f–f′′) or expression of Diap1 (Fig. 3h–h′′) alone. Moreover, GMR > egr-induced ommatidial organization defects are strongly suppressed by expression of bskDN (Fig. 3j–j′′) or in hemizygous mutants of Tak1 (Fig. 3k–k′′). Altogether, these data suggest that expression of egr induces both apoptosis and nonapoptotic, but JNK-dependent, cellular disorganization in the developing eye (Fig. 3c).
Fig. 4. Nonapoptotic cell death is induced in GMR > egr when effector caspases DrICE and Dcp-1 are inhibited.
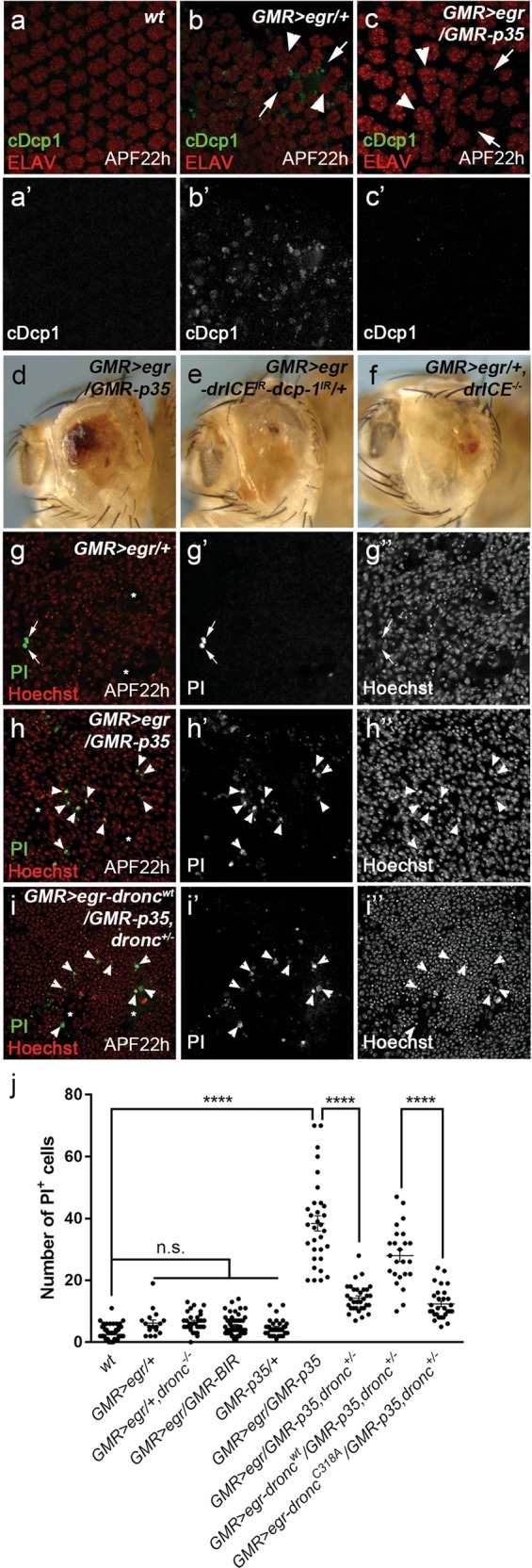
APF22h pupal disks labeled with cDcp1 (green in a, b, c and gray in a′, b′, c′) and ELAV (red in a, b, c), a neuronal marker. At this stage, no apoptotic cells were detected in wild-type disks (cDcp1, a, a′). Ommatidia are also well-patterned (ELAV, a). In contrast, strong apoptosis was detected at APF22h in GMR > egr eye disks (cDcp1, b and b′). Cellular disorganization indicated by increased interommatidial spacing (arrows, b) and ommatidial fusion (arrowheads, b) was observed. Although GMR > egr-induced apoptosis is almost completely blocked by expression of P35 (cDcp1, c and c′), the irregular ommatidial organization (c, arrows and arrowheads) is not suppressed. d–f Adult eye images. Expression of P35 (GMR-p35, d), RNAi knockdown of drICE and dcp-1 (e), or drICE null mutants (f) do not or only slightly suppress GMR > egr-induced eye ablation phenotype (compare 4d, e, f to 2b). APF22h pupal eye disks labeled with Propidium Iodide (PI, green in g, h, i and gray in g′, h′, i′) and Hoechst (red in g, h, i and gray in g′′, h′′, i′′). In GMR > egr disks, PI detects a background level of signals (arrows, g–g′′) which often do not co-localize with the Hoechst labeling, a nucleus marker. In contrast, expression of P35 (GMR > egr/GMR-p35) results in a strong increase of PI-positive nuclei, majority of which are also Hoechst-positive (arrowheads, h–h′′). Suppression of these PI signals in dronc heterozygous mutants can be reversed by expression of a wild-type dronc transgene (arrowheads, i–i′′). Asterisks indicate irregular cellular spacing caused by expression of Egr in the corresponding eye disks. j Quantification of PI-positive cell numbers in APF22h pupal eye disks of various genetic backgrounds as indicated. One-way ANOVA with Bonferroni multiple comparison test was used to compute p-values. Asterisks indicate statistically significant changes (**** p< 0.0001). A background low level of PI-labeling was observed in both wild type and GMR > egr disks. This low PI-labeling in GMR > egr is not increased in dronc mutants or by expression of a stabilized form of Diap-1 (GMR-BIR). The PI-labeling is also low in GMR-p35 disks. In contrast, strong PI-labeling was observed in GMR > egr/GMR-p35 disks. This PI-labeling is largely suppressed in dronc heterozygous mutants (GMR > egr/GMR-p35, dronc+/−). In this background, further expression of a wild-type form of Dronc (GMR > egr-droncwt/GMR-p35, dronc+/−), but not a catalytic site-mutated form of Dronc (GMR > egr-droncC318A/GMR-p35, dronc+/−), is sufficient to restore the PI signals
Inhibition of effector caspases in GMR > egr induces necrosis
One intriguing observation is that, unlike dronc mutants or expression of Diap1, expression of P35, an inhibitor of effector caspases, cannot or only slightly suppresses the GMR > egr-induced eye ablation phenotype (compare Fig. 4d to Fig. 2b, quantified in Fig. 6f)18,19,44. One possible explanation is that P35 may not be sufficient to block GMR > egr-induced apoptosis because it is a pseudo-substrate of effector caspases26,45. We examined this possibility. Compared with wild type (Fig. 4a, a′), GMR > egr induces massive apoptosis and defective ommatidial patterning in pupal eye disks at APF22h (Fig. 4b, b′). Expression of P35 is sufficient to block apoptosis (Fig. 4c, c′). However, unlike dronc mutants or expression of Diap1, expression of P35 in GMR > egr still results in small adult eyes (compare Fig. 4d to Fig. 2l, m). This is not due to any artefacts induced by P35 itself because double RNAi knockdown of drICE and dcp-1 or drICE mutants do not suppress, or even enhance, GMR > egr-induced eye ablation phenotype (Fig. 4e, f). These RNAi lines and mutants are functional because they can suppress GMR-hid-induced apoptosis (Supplementary Fig. S3). These suggest that inhibition of effector caspases, particularly DrICE, in GMR > egr may induce another form of cell death while it suppresses apoptosis.
Fig. 6. GMR > egr induces Dronc-dependent necrosis when DrICE and Dcp-1 are inhibited.

a, a′ APF22h pupal eye disks labeled with cDcp1 (green in d and gray in d′) and ELAV (red in d). Loss of one copy of dronc does not or only slightly suppress GMR > egr-induced apoptosis. b–e Adult eye images. Although loss of one copy of dronc only slightly inhibits GMR > egr-induced small eye phenotype (compare 6b to 2b), it strongly suppresses the eye ablation phenotype induced by GMR > egr/GMR-p35 (compare 6c to 4d). This suppression is neutralized by expression of a wild-type form of Dronc (d), but not a catalytic site-mutated form of Dronc (e). f, g Adult eye images. Expression of Dronc and P35 in dronc heterozygous mutants does not reduce the eye size although it causes an eye pigmentation defect (f). Expression of a catalytic site-mutated form of Dronc does not result in any eye defects (g). h Quantification of the average adult eye size (mean ± SD) of various genetic backgrounds as indicated. One-way ANOVA with Bonferroni multiple comparison test was used to compute p-values. Asterisks indicate statistically significant changes (*P < 0.05 or ****P < 0.0001). Suppression of GMR > egr by expression of P35 is not statistically significant (n.s.). Heterozygous dronc mutants only weakly suppress GMR > egr-induced small eyes (GMR > egr/+, dronc+/−). But they strongly suppress GMR > egr/GMR-p35-induced eye ablation phenotype (GMR > egr/GMR-p35, dronc+/−). In this background, further expression of a wild-type form of Dronc (GMR > egr-droncwt/GMR-p35, dronc+/−), but not a catalytic site-mutated form of Dronc (GMR > egr-droncC318A/GMR-p35, dronc+/−), is sufficient to restore the eye ablation phenotype
To further characterize this nonapoptotic cell death, we performed labeling with propidium iodide (PI), a dye enters a cell and binds to its DNA when the cell membrane is disrupted. Because loss of membrane integrity is a hallmark of necrosis, PI has been used to label necrotic cells (Supplementary Fig. S4a,b)46,47. Interestingly, no PI-positive cells were detected in late 3rd instar larval GMR > egr disks with or without expression of P35 (Supplementary Fig. S4c, d). We then examined a later pupal stage (APF22h). In GMR > egr, a very low level of PI-labeling was observed (Fig. 4g, g′). Notably, most of these PI signals are unspecific because they do not co-localize with a DNA marker Hoechest (Fig. 4g, g′′, arrows). In contrast, expression of P35 strongly induces PI-labeling in GMR > egr disks (Fig. 4h, h′). Importantly, majority of these PI signals co-localize with Hoechest-positive nuclei (Fig. 4h, h′′, arrowheads) suggesting that they specifically label membrane-compromised cells. As a control, expression of P35 alone does not induce PI-labeling (Fig. 4j). Altogether, these data suggest that Egr may induce necrosis when apoptosis is blocked. To further confirm this, we performed the Transmission Electron Microscopy (TEM) analysis on GMR > egr/GMR-p35 pupal eye disks. Compared with the wild-type control (Fig. 5a), cells with typical apoptotic features48 such as high-electron-density chromatin condensation and apoptotic bodies were frequently observed in GMR > egr disks (Fig. 5b, b′). In contrast, cells with typical necrotic features were observed in GMR > egr/GMR-p35 disks (Fig. 5c-c′′). These cells have translucent cytoplasm, mal-shaped or unidentifiable nuclei, and aggregation of cellular organelles such as endoplasmic reticulum, which are characteristics of necrotic cell death48,49. Therefore, necrosis is induced in GMR > egr/GMR-p35 pupal eye disks.
Fig. 5. Nonapoptotic cell death in GMR > egr/GMR-p35 shows morphological features of necrosis.

TEM images of wt (a), GMR > egr/ + (b, b′) and GMR > egr/GMR-p35 (c–c′′) disks at APF22h. b′ is an enlarged image for the outlined area in b. c′, c′′ are enlarged images for the outlined areas in c. Compared with the wild-type eye disk cell which has a large nucleus (yellow arrow, a), apoptotic features such as high-electron-density chromatin condensation (yellow arrows, b) and apoptotic bodies (dark aggregates in b and b′) are frequently observed in GMR > egr disks (b). Expression of P35 in GMR > egr (c), however, induces necrotic cell features such as translucent cytoplasm, mal-shaped (arrow, c′′) or unidentifiable nuclei (c′), and aggregation of endoplasmic reticulum and other cellular organelles (arrowheads, c′ and c′′). Asterisk indicates a phagolysosome
The initiator caspase Dronc mediates Eiger-induced necrosis
To determine how necrosis is activated when effector caspases are inhibited, we first examined PI-labeling in GMR > egr disks in a background of dronc mutants or with expression of the stabilized Diap1 (GMR-BIR). This is because both of them suppress GMR > egr-induced apoptosis upstream of effector caspases (Fig. 2l, m). Only a background level of PI-labeling was detected under these conditions (Fig. 4j). Strikingly, loss of one copy of dronc strongly suppresses the PI-labeling induced in GMR > egr/GMR-p35 (Fig. 4j). However, it only weakly affects GMR > egr-induced apoptosis (Fig. 6a, a′) hence the eye ablation phenotype (Fig. 6b, h). Consistent with these observations, dronc heterozygous mutants strongly suppress the GMR > egr/GMR-p35 small eyes (Fig. 6c, h). Therefore, Dronc is a key component mediating necrosis when GMR > egr-induced apoptosis is inhibited.
It is known that Dronc, the initiator caspase, cleaves its downstream effector caspases, e.g., DrICE and Dcp-1, to activate apoptosis in Drosophila33,50. We thus examined whether the catalytic activity of Dronc is required for necrosis induced in GMR > egr/GMR-p35. To do this, either a wild-type Dronc (droncwt) or a catalytic site-mutated form of Dronc (droncC318A) transgene was used51. As expected, expression of the wild-type Dronc in GMR > egr/GMR-p35; dronc+/− animals restores PI-positive necrosis (Fig. 4i–i′′, quantified in 4j) and, consequently, the eye ablation phenotype (Fig. 6d, h). In contrast, expression of droncC318A in the same background does not have such effects (Figs. 4i and 6e, h). As controls, expression of droncwt or droncC318A in a similar background without expression of egr does not result in small eyes (Fig. 6f, g). Notably, an eye pigmentation defect was observed when droncwt is overexpressed (Fig. 6f), consistent with what has been previously reported33,50. Altogether, these data suggest that the catalytic activity of Dronc is required for Egr-induced necrosis.
JNK signaling is required for Eiger- and Dronc-induced necrosis
Unlike GMR > egr, the eye ablation phenotype induced by expression of hid, e.g., GMR-hid, can be suppressed by expression of P35 (ref.26) or reduction of DrICE and Dcp-1 (Supplementary Fig. S3). Therefore, factor(s) other than inhibition of apoptosis is required for induction of necrosis in GMR > egr/GMR-p35. Because Egr activates the JNK signaling upstream of hid-mediated apoptosis, we examined whether there is a nonapoptotic input from JNK contributing to induction of necrosis when apoptosis is blocked. Hypomorphic mutants of bsk, hep, MKK4 and Tak1, genes encoding various kinases in the JNK pathway52, were used. We observed that heterozygosity of these mutants can only moderately suppress the GMR > egr-induced eye ablation phenotype (compare Fig. 7a–d to Fig. 2b). In contrast, except hep1 (Fig. 7f), heterozygosity of bsk1, MKK4G680 or Tak12 mutants strongly suppresses the GMR > egr/GMR-p35 small eyes (compare Fig. 7e, g, h to Fig. 2b). Moreover, we observed that overexpression of Dronc in eye disks, i.e., GMR > droncwt, induces a moderate level of apoptosis (Supplementary Fig. S5a–c). It activates necrosis when apoptosis is blocked (compare Fig. 7j to 7i). In contrast, expression of the catalytic site-mutated form of Dronc does not induce necrosis (Fig. 7k). Because JNK is activated at a basal level in photoreceptor neurons during eye development53, we examined whether JNK is required for Dronc-induced necrosis. Although JNK activity is not further enhanced by overexpression of Dronc (Supplementary Fig. S5d–i), loss of one copy of bsk or Tak1, as well as the Tak1 null mutants, suppress necrosis in the GMR > droncwt/GMR-p35 disks (Fig. 7l, m). Therefore, in addition to its roles in apoptosis and development, JNK signaling also contributes to induction of necrosis (Fig. 7n).
Fig. 7. JNK signaling contributes to Eiger- and Dronc-induced necrosis when apoptosis is blocked.

a–h Adult eye images. Heterozygous mutants of bsk1 (a), hep1 (b), MKK4G680 (c) and Tak12 (d) can only weakly or moderately inhibit GMR > egr-induced eye ablation phenotype (compared with Fig. 2b). In contrast, GMR > egr/GMR-p35-induced small eyes are strongly suppressed by heterozygous mutants of bsk1(e), MKK4G680 (g) and Tak12 (h), but not hep1(f) (compared with Fig. 4d). i–l Late 3rd instar larval eye disks labeled with Propidium Iodide (PI). Compared with expression of a wild-type form of Dronc (GMR > droncwt/+, i), co-expression of Dronc and P35 (GMR > dronc/GMR-p35) induces PI-positive necrosis (arrowhead, j). In contrast, PI-labeling is not observed when a catalytic site-mutated form of Dronc is expressed instead (GMR > droncC318A/GMR-p35, k). Loss of one copy of bsk (bsk+/−) strongly suppresses necrosis induced in GMR > droncwt/GMR-p35 (l). m Quantification of PI-positive cell numbers in late 3rd instar larval eye disks of various genetic backgrounds as indicated. One-way ANOVA with Bonferroni multiple comparison test was used to compute p-values. Asterisks indicate statistically significant changes (**** p< 0.0001). Loss of one copy of Tak1 (Tak1+/−) or a Tak1 null mutant (Tak1−/−) strongly suppresses necrosis induced in GMR > droncwt/GMR-p35. n A diagram showing comparable molecular mechanisms of regulated necrosis in Drosophila and mammals. The Drosophila TNF (Egr), similar to its mammalian counterparts, has multiple context-dependent functions including induction of necrosis when apoptosis is blocked. In mammals, necrosis can occur when inhibition of caspase-8 on RIPK1 and RIPK3 is removed. JNK contributes to both apoptosis and necrosis. While in Drosophila, effector caspases DrICE and Dcp-1 inhibit Egr-induced necrosis. Once this inhibition is removed, the initiator caspase Dronc can activate necrosis with an additional input(s) from JNK signaling. Key factors that mediate this necrosis downstream of caspases and JNK are currently unknown (indicated by the question mark). Moreover, energy metabolism regulators have been implicated in regulation of Egr/TNF-induced signaling responses although their exact roles remain to be determined (see “Discussion”)
Egr/JNK-mediated necrosis restricts the oncogenic potential of scrib mutant cells
To further determine roles of Egr/JNK-mediated necrosis under pathophysiological conditions, we examined mutants of scrib, a well-characterized tumor suppressor gene regulating the epithelial cell polarity54–56. Loss of scrib leads to Egr-mediated activation of JNK (Supplementary Fig. S6)57,58. Although animals entirely mutant for scrib die at the larval stage with massive tissue overgrowth54, scrib mutant cells (scrib−/− clones) in an otherwise wild-type tissue are eliminated via JNK-mediated apoptosis59. Consistent with these reports, we observed small scrib mutant clones in the wild-type larval eye disks (Fig. 8a). Interestingly, when p35 is expressed in scrib−/− clones to inhibit apoptosis (scrib−/−-p35 clones), these clones show no signs of overgrowth and their average size is close to the wild-type control (Fig. 8b, g). Therefore, apoptosis is not the only mechanism that restricts the oncogenic potential of scrib mutant cells. Compared with the scrib−/− clones (Fig. 8a′), strong PI-labeling was detected in the scrib−/−-p35 clones (Fig. 8b′). This suggests that necrosis is induced in these clones. In support of this, compared with adults with scrib−/− clones (Fig. 8d), over 90% of survived adults (n = 44) with scrib−/−-p35 clones have eyes with necrotic patches (Fig. 8e). We then examined whether JNK is required for necrosis induced in scrib−/−-p35 clones. Expression of bskDN in the scrib−/−-p35 clones (scrib−/−-p35-bskDN clones) completely suppresses the PI-labeling (Fig. 8c′). This results in massively overgrown clones which occupy most of the entire eye disk in all samples we have examined (n > 50, Fig. 8c, g). Consequently, these animals are pupal lethal (Fig. 8f). Collectively, these data suggest that, similar to what we observed in the GMR > egr/GMR-p35 disks, Egr/JNK-mediated necrosis is activated in scrib mutant cells when apoptosis is deficient. Importantly, in this situation, necrosis constrains the oncogenic tissue overgrowth.
Fig. 8. Egr/JNK-mediated necrosis restricts oncogenic growth of scrib mutant cells.

a–c′ Late 3rd instar larval eye disks with GFP-positive scrib mutant (scrib−/−, a), scrib−/−-p35 (b) or scrib−/−-p35-bskDN (c) clones in an otherwise wild-type background. These disks are labeled with Propidium Iodide (PI, red in a–c and gray in a′–c′). Compared with scrib mutant clones (a, a′), PI-positive necrosis is induced in scrib−/−-p35 clones (arrows, b, b′). This necrosis is suppressed by inhibition of JNK via expression of bskDN (c, c′). d, e Adult eye images. scrib mutant clones only cause a mild rough-eye phenotype (d). In contrast, necrotic patches (arrow, e) are observed in over 90% of scrib−/−-p35 mosaic eyes (n = 44). f scrib−/−-p35-bskDN mosaic animals are pupal lethal. g Quantification of the clone/disk size ratio in late 3rd instar larval eye disks with various genetic backgrounds as indicated. The ratio is a comparison of the total clone size in each eye disk to the full disk size. One-way ANOVA with Bonferroni multiple comparison test was used to compute p-values. Asterisks indicate statistically significant changes (****P < 0.0001). Compared with wild-type clones which occupy an average of 40% of the whole eye disk, scrib mutant (scrib−/−) clones are much smaller with an average of 8% coverage on the disk. Expression of P35 in scrib−/− clones (scrib−/−-p35) moderately increases their sizes leading to an average disk coverage of 28%. Further expression of bskDN in these clones results in their massive overgrowth and increases the clone/disk ratio to an average of 78%
Discussion
Our findings in this study suggest an analogy between Drosophila and mammals in regulation of TNF-induced apoptosis and necrosis (Fig. 7n). Caspases play critical roles in these processes. In mammals, caspase-8, an initiator caspase, exerts a permissive role on TNF-induced apoptosis but an inhibitory role on necrosis. Here, we report that both apoptosis and necrosis induced by Egr, the Drosophila TNF, depend on the initiator caspase Dronc. Loss of one copy of dronc blocks necrosis but not apoptosis suggesting that different levels of Dronc are required for apoptosis versus necrosis. In contrast to Dronc, effector caspases DrICE and Dcp-1, particularly DrICE, appear to inhibit Egr-induced necrosis. Such inhibition can be released by either expression of the inhibitor P35 or reduction of DrICE and Dcp-1. However, inhibition of DrICE and Dcp-1 alone is not sufficient to trigger a switch from apoptosis to necrosis. It requires a further input from JNK signaling. Interestingly, the mammalian JNK has been implicated in promoting TNF-induced and Caspase-8-mediated apoptosis60. It can also contribute to necrosis but the underlying mechanisms remain elusive61,62. Therefore, JNK may have an evolutionarily conserved role in connecting activation of apoptosis and, if apoptosis fails, necrosis. Such interlinked apoptosis and necrosis may have pathophysiological implications because many human diseases including cancers are frequently associated with both apoptosis and necrosis63,64. In this study, we observed that Egr/JNK-mediated apoptosis and necrosis act as a two-layered defense system to restrict the oncogenic potential of scrib, a well-established tumor suppressor gene. It is therefore interesting to see whether apoptosis and necrosis cooperate to prevent tumorigenesis in mammals.
Another intriguing observation in our study is that expression of the full-length wild-type Dronc is sufficient to induce necrosis when apoptosis is blocked. Although the full-length Dronc is enzymatically inactive under physiological conditions, its overexpression triggers a moderate level of apoptosis (Supplementary Fig. S5b), which is probably due to spontaneous activation of the full-length Dronc50. Suppression of this apoptosis by P35 results in a switch from apoptosis to necrosis (Fig. 7j). Interestingly, although expression of the proapoptotic genes such as hid activates Dronc and apoptosis33,50, it does not seem to trigger necrosis when apoptosis is blocked. This suggests that activation of Dronc alone is not sufficient to induce necrosis. It has recently been reported that the subcellular localization of Dronc determines its apoptotic versus nonapoptotic functions65,66. Therefore, overexpression of Dronc may allow its protein level in a particular subcellular domain to exceed a threshold thus trigger necrosis. This may not occur in GMR-hid because it activates but does not induce expression of Dronc (Supplementary Fig. S5i). Notably, the catalytic activity of Dronc is required for both Dronc- and Egr-induced necrosis. Hence, identification of the nonapoptotic substrates of Dronc may provide critical information to understand how Dronc mediates necrosis.
What are other factors that coordinate the inputs from caspases and JNK to differentiate apoptosis versus necrosis? This is an important question that remains largely open. In mammals, caspase-8 cleaves and inactivates RIPK1 and RIPK3 therefore inhibits TNF-induced necroptosis67,68. However, no homologs of RIPK1 and RIPK3 have been reported in Drosophila. Notably, RIPK3 mediates TNF-induced necrosis at least partially via its role in regulation of energy metabolism and reactive oxygen species (ROS) production11. The concept of metabolic checkpoints has been proposed to describe the complex roles of metabolism in regulation of cell death including necrosis69. Intriguingly, a recent study in Drosophila also revealed roles of the energy metabolism pathways in regulation of GMR > egr-induced eye ablation phenotype44. As we show in this study, expression of Egr can lead to distinct outcomes including apoptosis, nonapoptotic developmental defects and necrosis in a context dependent manner (Fig. 7n). It is therefore interesting to see whether the metabolism pathways regulate Egr-induced necrosis and, if yes, how they interact with caspases and JNK in this process. Furthermore, necrosis-like cell death in Drosophila has also been reported in the stressed larval brains49, the developmental male testes70,71 and female ovaries72. In these forms of necrosis, effector caspases and the catalytic activity of Dronc are not required. Hence, future research is needed to understand how different types of necrosis are regulated at the molecular level. What′s more, a series of intracellular events specific to necrosis, such as mitochondrial dysfunction, ATP depletion and increased cytosolic Ca2+, appear to be conserved in multiple organisms73. Therefore, studies on various necrosis models in Drosophila will likely provide insights into understanding the molecular regulation of necrosis.
Material and methods
Drosophila genetics
Genetic crosses for all experiments were reared at 25 °C. GMR-GAL4 UAS-egr (GMR > egr)19, GMR-hid10 (ref.29), dcp-1Prev1 (ref.74), drICEΔ1 (ref.31), droncI29 (ref.36), rpr87 (ref.40), XR38 (ref.39), hid05014 (ref.29), GMR-BIR37, GMR-p35 (ref.26), hid20-10-lacZ38, rprXRE-lacZ38, sev > GluLc46, MKK4G680 (ref. 75), UAS-droncwt and UAS-droncC318A (ref.50), UAS-bskDN (on Chr.III) (ref.76), scrib1 and scrib2 (refs.77,78) were as described. bsk1, hep1, Tak12527, Tak12 and UAS-bskDN (on Chr.X) were obtained from the Bloomington stock center. UAS-drICERNAi and UAS-dcp-1RNAi were obtained from the NIG-Fly stock center. UAS-egrRNAi (108814) was obtained from the VDRC stock center.
Mosaic analysis
For mosaic analysis with hid mutant clones, larvae of the following genotype were analyzed at the late 3rd instar larval stage: ey-FLP/+; GMR > egr/+; FRT80B/hid05014 FRT80B. For mosaic analyses with wild-type control, scrib mutant (scrib−/−), scrib−/−-p35 or scrib−/−-p35-bakDN clones, the following genotypes were used: (1) ey-FLP/+; act > y+> GAL4 UAS-GFP/+; FRT82B/FRT82B tub-GAL80; (2) ey-FLP/+; act > y+> GAL4 UAS-GFP/+; FRT82B scrib1/FRT82B tub-GAL80; (3) ey-FLP/+; act > y+> GAL4 UAS-GFP/UAS-p35; FRT82B scrib1/FRT82B tub-GAL80; (4) ey-FLP/+; act > y+> GAL4 UAS-GFP/UAS-p35; FRT82B scrib1 UAS-bakDN/FRT82B tub-GAL80. Similar mosaic analyses with scrib2 mutants showed comparable results.
Immunohistochemistry
Pupal or larval disks were dissected, fixed (with 4% paraformaldehyde for 30 min at room temperature), and then labeled with antibodies using standard protocols as described30. Primary antibodies used were rabbit anti-cDcp1 (the cleaved Dcp-1 antibody, 1:500, Cell Signaling), rabbit anti-phospho-JNK (pJNK, 1:200, Calbiochem), rat anti-ELAV, mouse anti-Dlg, mouse anti-β-Gal and mouse anti-MMP1 (all 1:50, DHSB). To generate the rabbit anti-Dronc antibody, the full-length dronc cDNA was amplified by PCR with primers F-5′CGGGATCCATGCAGCCGCCGGAGCT3′ and R-5′CGGAATTCCTATTCGTTGAAAAACCCGGGATT3′. It was then subcloned into BamHI and EcoRI sites in pET-28a (Novagen) to produce a His-tagged recombinant protein. The purified recombinant protein was then inject into rabbits to generate a polyclonal antibody. The antisera were subsequently affinity purified. Secondary antibodies were goat Fab fragments conjugated to Alex488, 555, or 647 (all 1:1000) from Molecular Probes.
Propidium Iodidum (PI), Hoechst labeling and TUNEL
For PI and Hoechst double labeling on pupal disks, freshly dissected disks were incubated in dark with 4 µM PI (Sigma-Aldrich) and 16 µM Hoechst (ThermoFisher) in Schneider's media for 1 h at room temperature. The disks were then fixed with 4% paraformaldehyde for 20 min at room temperature followed by gentle washes as previously described47. For PI labeling alone, larval or pupal eye disks were incubated with 15 µM PI for 10 min at room temperature followed by fixation (with 4% paraformaldehyde for 20 min at room temperature) and gentle washes as previously described46. PI labeling can also be observed and scored without fixation. For TUNEL (terminal deoxynucleotide transferase-mediated dUTP end labeling), dissected and fixed (with 4% paraformaldehyde for 20 min at room temperature) disks were incubated in 100 mM Na-citrate with 0.1% Triton X-100 for 30 min at 65 °C, followed by detection of dying cells using an in situ cell death detection kit (Roche)30.
Imaging and statistical analysis
Fluorescent eye disk images were taken with either a Zeiss or Leica confocal microscope. Adult fly eye images were taken using a Zeiss stereomicroscope equipped with an AxioCam ICC1 camera. For statistical analysis of PI-labeling (Fig. 4j and 7m), at least 20 pupal or larval disks from each genotype were used for counting the number of PI-positive cells. For quantification of adult eye size (Fig. 6h), the “histogram” function in Adobe Photoshop CS was used to measure ten representative adult eyes of each genotype. For quantification of the clone/disk size ratio in late 3rd instar mosaic eye disks (Fig. 8g), the sizes of GFP-positive clones and the whole eye disk were measured using the “histogram” function in Adobe Photoshop CS before the ratio is calculated for each disk. Ten representative mosaic disks of each genotype were used. For all quantifications, the statistical significance was evaluated through a one-way ANOVA with Bonferroni multiple comparison test using GraphPad Prism.
Transmission electron microscopy (TEM)
Freshly dissected pupal eye–brain complexes were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 45 min followed by a secondary fixation of 1% Osmium Tetroxide for 1 h at room temperature. The samples were then washed with the buffer (10 × 5 min) and dehydrated in ascending concentrations of ethanol before they were embedded in epoxy resin. Sections (90 nm) were prepared and stained with uranyl acetate and lead citrate followed by examination using a JEOL 1200EX electron microscope fitted with a tungsten filament. Images were acquired through a GATAN MultiScan camera.
Supplementary information
Acknowledgements
We would like to thank Paul Stanley and Theresa Morris at the Centre for Electron Microscopy in the University of Birmingham for their assistance on TEM. The confocal imaging was carried out with support of the Birmingham Advanced Light Microscopy (BALM) facility. We thank Kai Liu for advice on PI-labeling. We are grateful to Konrad Basler, Andreas Bergmann, Lei Liu, Pascal Meier, Mirka Uhlirova, the Bloomington Stock Center, the NIG-Fly stock center in Japan, the VDRC stock center in Vienna and the Developmental Studies Hybridoma Bank (DSHB) in Iowa for fly stocks and reagents. M.L. is supported by the China Scholarship Council (CSC)-Birmingham joint PhD program. Y.F. is supported by Grant BB/M010880/1 from the Biotechnology and Biological Sciences Research Council (BBSRC) UK and the Marie Curie Career Integration Grant (CIG) 630846 from the European Union's Seventh Framework Program (FP7).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by E. Baehrecke
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41419-019-1862-0).
References
- 1.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- 3.Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat. Rev. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 5.Vanden Berghe, T., Kaiser, W. J., Bertrand, M. J. M. & Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell Oncol.2 (2015). [DOI] [PMC free article] [PubMed]
- 6.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 2015;15:362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 8.Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta. 2011;1813:558–563. doi: 10.1016/j.bbamcr.2011.01.026. [DOI] [PubMed] [Google Scholar]
- 9.Cho Y, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He SD, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 11.Zhang DW, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 14.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 15.Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014;26:237–245. doi: 10.1016/j.smim.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sedger LM, McDermott MF. TNF and TNF-receptors: from mediators of cell death and inflammation to therapeutic giants—past, present and future. Cytokine Growth Factor Rev. 2014;25:453–472. doi: 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 17.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igaki T, et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21:3009–3018. doi: 10.1093/emboj/cdf306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno E, Yan M, Basler K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 2002;12:1263–1268. doi: 10.1016/S0960-9822(02)00954-5. [DOI] [PubMed] [Google Scholar]
- 20.Kanda H, Igaki T, Kanuka H, Yagi T, Miura M. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J. Biol. Chem. 2002;277:28372–28375. doi: 10.1074/jbc.C200324200. [DOI] [PubMed] [Google Scholar]
- 21.Andersen DS, et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature. 2015;522:482–486. doi: 10.1038/nature14298. [DOI] [PubMed] [Google Scholar]
- 22.Igaki T, Miura M. The Drosophila T. N. F. ortholog Eiger: emerging physiological roles and evolution of the TNF system. Semin. Immunol. 2014;26:267–274. doi: 10.1016/j.smim.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Agrawal N, et al. The Drosophila TNF Eiger is an adipokine that acts on insulin-producing cells to mediate nutrient response. Cell Metab. 2016;23:675–684. doi: 10.1016/j.cmet.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Fogarty CE, et al. Extracellular reactive oxygen species drive apoptosis-induced proliferation via drosophila macrophages. Curr. Biol. 2016;26:575–584. doi: 10.1016/j.cub.2015.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma X, et al. NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res. 2012;22:425–431. doi: 10.1038/cr.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- 27.Fan Y, Bergmann A. The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 2010;17:534–539. doi: 10.1038/cdd.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkissian T, Timmons A, Arya R, Abdelwahid E, White K. Detecting apoptosis in Drosophila tissues and cells. Methods. 2014;68:89–96. doi: 10.1016/j.ymeth.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- 30.Fan Y, Bergmann A. Multiple mechanisms modulate distinct cellular susceptibilities toward apoptosis in the developing Drosophila eye. Dev. Cell. 2014;30:48–60. doi: 10.1016/j.devcel.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muro I, et al. The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development. 2006;133:3305–3315. doi: 10.1242/dev.02495. [DOI] [PubMed] [Google Scholar]
- 32.Xu D, et al. The effector caspases drICE and dcp-1 have partially overlapping functions in the apoptotic pathway in Drosophila. Cell Death Differ. 2006;13:1697–1706. doi: 10.1038/sj.cdd.4401920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawkins CJ, et al. The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J. Biol. Chem. 2000;275:27084–27093. doi: 10.1074/jbc.M000869200. [DOI] [PubMed] [Google Scholar]
- 34.McEwen DG, Peifer M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development. 2005;132:3935–3946. doi: 10.1242/dev.01949. [DOI] [PubMed] [Google Scholar]
- 35.Narasimamurthy R, et al. Structure-function analysis of Eiger, the Drosophila TNF homolog. Cell Res. 2009;19:392–394. doi: 10.1038/cr.2009.16. [DOI] [PubMed] [Google Scholar]
- 36.Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–2134. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- 38.Fan Y, et al. Dual roles of Drosophila p53 in cell death and cell differentiation. Cell Death Differ. 2010;17:912–921. doi: 10.1038/cdd.2009.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson C, Carney GE, Taylor BJ, White K. reaper is required for neuroblast apoptosis during Drosophila development. Development. 2002;129:1467–1476. doi: 10.1242/dev.129.6.1467. [DOI] [PubMed] [Google Scholar]
- 40.Moon NS, et al. E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet. 2008;4:e1000153. doi: 10.1371/journal.pgen.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stronach B, Perrimon N. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 2002;16:377–387. doi: 10.1101/gad.953002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cagan RL, Ready DF. The emergence of order in the Drosophila pupal retina. Dev. Biol. 1989;136:346–362. doi: 10.1016/0012-1606(89)90261-3. [DOI] [PubMed] [Google Scholar]
- 43.Wolff, T. & Ready, D. F. Pattern formation in the Drosophila retina. Vol. II (Cold Spring Harbor Laboratory Press, 1993).
- 44.Kanda H, Igaki T, Okano H, Miura M. Conserved metabolic energy production pathways govern Eiger/TNF-induced nonapoptotic cell death. Proc. Natl Acad. Sci. USA. 2011;108:18977–18982. doi: 10.1073/pnas.1103242108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riedl SJ, Renatus M, Snipas SJ, Salvesen GS. Mechanism-based inactivation of caspases by the apoptotic suppressor p35. Biochemistry. 2001;40:13274–13280. doi: 10.1021/bi010574w. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y, Hou L, Li Y, Ni J, Liu L. Neuronal necrosis and spreading death in a Drosophila genetic model. Cell Death Dis. 2013;4:e723. doi: 10.1038/cddis.2013.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Obata F, et al. Induction of rapid and selective cell necrosis in Drosophila using Bacillus thuringiensis Cry toxin and its silkworm receptor. BMC Biol. 2015;13:48. doi: 10.1186/s12915-015-0160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 49.Kuang C, et al. A novel fizzy/Cdc20-dependent mechanism suppresses necrosis in neural stem cells. Developmet. 2014;141:1453–1464. doi: 10.1242/dev.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meier P, Silke J, Leevers SJ, Evan GI. The Drosophila caspase DRONC is regulated by DIAP1. Embo J. 2000;19:598–611. doi: 10.1093/emboj/19.4.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chew SK, et al. The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev. Cell. 2004;7:897–907. doi: 10.1016/j.devcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 52.Igaki T. Correcting developmental errors by apoptosis: lessons from Drosophila JNK signaling. Apoptosis. 2009;14:1021–1028. doi: 10.1007/s10495-009-0361-7. [DOI] [PubMed] [Google Scholar]
- 53.Willsey, H. R. et al. Localized JNK signaling regulates organ size during development. Elife5 (2016). [DOI] [PMC free article] [PubMed]
- 54.Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–116. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- 55.Bergstralh DT, St Johnston D. Epithelial cell polarity: what flies can teach us about cancer. Essays Biochem. 2012;53:129–140. doi: 10.1042/bse0530129. [DOI] [PubMed] [Google Scholar]
- 56.Elsum I, Yates L, Humbert PO, Richardson HE. The Scribble-Dlg-Lgl polarity module in development and cancer: from flies to man. Essays Biochem. 2012;53:141–168. doi: 10.1042/bse0530141. [DOI] [PubMed] [Google Scholar]
- 57.Cordero JB, et al. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell. 2010;18:999–1011. doi: 10.1016/j.devcel.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell. 2009;16:458–465. doi: 10.1016/j.devcel.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–5779. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 61.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr., Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004;18:2905–2915. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sosna J, et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol. Life Sci. 2014;71:331–348. doi: 10.1007/s00018-013-1381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 64.Najafov A, Chen H, Yuan J. Necroptosis and Cancer. Trends Cancer. 2017;3:294–301. doi: 10.1016/j.trecan.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang Y, Neuman SD, Bashirullah A. Tango7 regulates cortical activity of caspases during reaper-triggered changes in tissue elasticity. Nat. Commun. 2017;8:603. doi: 10.1038/s41467-017-00693-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amcheslavsky A, et al. Plasma membrane localization of apoptotic caspases for non-apoptotic functions. Dev. Cell. 2018;45:450–464. doi: 10.1016/j.devcel.2018.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng SS, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 69.Green DR, Galluzzi L, Kroemer G. Metabolic control of cell death. Science. 2014;345:1466–+. doi: 10.1126/science.1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yacobi-Sharon K, Namdar Y, Arama E. Alternative germ cell death pathway in Drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev. Cell. 2013;25:29–42. doi: 10.1016/j.devcel.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 71.Napoletano Francesco, Gibert Benjamin, Yacobi-Sharon Keren, Vincent Stéphane, Favrot Clémentine, Mehlen Patrick, Girard Victor, Teil Margaux, Chatelain Gilles, Walter Ludivine, Arama Eli, Mollereau Bertrand. p53-dependent programmed necrosis controls germ cell homeostasis during spermatogenesis. PLOS Genetics. 2017;13(9):e1007024. doi: 10.1371/journal.pgen.1007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bass BP, et al. Cell-autonomous requirement for DNaseII in nonapoptotic cell death. Cell Death Differ. 2009;16:1362–1371. doi: 10.1038/cdd.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 74.Laundrie B, et al. Germline cell death is inhibited by P-element insertions disrupting the dcp-1/pita nested gene pair in Drosophila. Genetics. 2003;165:1881–1888. doi: 10.1093/genetics/165.4.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geuking P, Narasimamurthy R, Lemaitre B, Basler K, Leulier F. A non-redundant role for Drosophila Mkk4 and hemipterous/Mkk7 in TAK1-mediated activation of JNK. PloS ONE. 2009;4:e7709. doi: 10.1371/journal.pone.0007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uhlirova M, Bohmann D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 2006;25:5294–5304. doi: 10.1038/sj.emboj.7601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perez, E., Lindblad, J. L. & Bergmann, A. Tumor-promoting function of apoptotic caspases by an amplification loop involving ROS, macrophages and JNK in Drosophila. Elife6, e26747 (2017). [DOI] [PMC free article] [PubMed]
- 78.Zeitler J, Hsu CP, Dionne H, Bilder D. Domains controlling cell polarity and proliferation in the Drosophila tumor suppressor Scribble. J. Cell Biol. 2004;167:1137–1146. doi: 10.1083/jcb.200407158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.