

Bacteria adapt to infections by evolving variants that are more fit and persistent. These recalcitrant variants are typically observed in chronic infections. However, it is unclear when and why these variants evolve. To address these questions, we used a porcine chronic wound model to study the evolutionary dynamics of Pseudomonas aeruginosa in a mixed-strain infection. We isolated hyperbiofilm variants that persisted early in the infection. Interstrain interactions were also observed, where adapted variants acquired CRISPR-mediated immunity to phages. We show that when initiating infection, P. aeruginosa experiences strong positive selection for hyperbiofilm phenotypes produced by mutants of a single chemosensory system, the Wsp pathway. We predict that hyperbiofilm variants are early adaptations to infection and that interstrain interactions may influence bacterial burden and infection outcomes.
KEYWORDS: CRISPR-Cas, Pseudomonas aeruginosa, RSCV, Wsp, bacteriophages, biofilms, chronic infection, cyclic di-GMP, evolution, exopolysaccharide
ABSTRACT
Opportunistic pathogens establishing new infections experience strong selection to adapt, often favoring mutants that persist. Capturing this initial dynamic is critical for identifying the first adaptations that drive pathogenesis. Here we used a porcine full-thickness burn wound model of chronic infection to study the evolutionary dynamics of diverse Pseudomonas aeruginosa infections. Wounds were infected with a mixed community of six P. aeruginosa strains, including the model PA14 strain (PA14-1), and biopsies taken at 3, 14, and 28 days postinfection. Hyperbiofilm-forming rugose small-colony variants (RSCVs) were the earliest and predominant phenotypic variant. These variants were detected on day 3 and persisted, with the majority evolved from PA14-1. Whole-genome sequencing of PA14-1 RSCV isolates revealed driver mutations exclusively in the wsp pathway, conferring hyperbiofilm phenotypes. Several of the wsp mutant RSCVs also acquired CRISPR-Cas adaptive immunity to prophages isolated from the P. aeruginosa wound isolate (B23-2) that was also present in the inoculum. These observations emphasize the importance of interstrain dynamics and the role of lysogenic phages in the survival of an invading pathogen. Rather than being a side effect of chronicity, the rapid rise of RSCVs in wounds is evidence of positive selection on the Wsp chemosensory system to produce mutants with elevated biofilm formation capacity. We predict that RSCVs provide a level of phenotypic diversity to the infecting bacterial community and are common, early adaptations during infections. This would likely have significant consequences for clinical outcomes.
INTRODUCTION
Chronic infections that persist despite extensive treatment are often attributed to biofilms, which are communities of adhered microorganisms encased in an extracellular polymeric substance (EPS) (1). Complicating chronic infections is the high likelihood that bacterial populations adaptively evolve, producing novel subpopulations with persistent phenotypes and increased fitness. When opportunistic pathogens leave an environmental reservoir to colonize a host, there are potentially many beneficial mutations available for selection in this environment. Defining this new fitness landscape and the evolutionary dynamics of pathogen adaptation to the host environment is critical to understanding infection pathology, as the emergence and selection of adapted variants in an infection are often associated with worsening clinical outcomes (2). Adapted variants are typically identified later in the infection, when eradication has become increasingly difficult. By monitoring the rise of adapted genotypes within pathogen populations, as the infection becomes established, we can understand the environment in which the pathogen adapts and predict the fitness constraints as a result of the adaptation. This may enable prediction of evolutionary pathways that could be targeted to improve treatment efficacy (3, 4).
Of interest to chronic infections are the emergence of rugose small-colony variants (RSCVs), which are associated with persistence (5). RSCVs are characterized by high fitness in biofilms, and they have been isolated from chronic infections (6, 7) and in vitro-grown biofilm experiments (8, 9). This suggests that there is strong selection for ecological diversification in both in vivo- and in vitro-grown biofilms. One of the most studied bacterial adaptive responses to chronic infection is that of Pseudomonas aeruginosa to the cystic fibrosis (CF) lung (10). CF patients exhibit airway abnormalities, where P. aeruginosa biofilms commonly colonize the mucus lining, and establish persistent pulmonary infections (11, 12). RSCVs are isolated from up to 50% of P. aeruginosa-positive CF sputum samples (6, 7). When isolated, RSCV frequencies range widely between patient samples, from 0.1 to 100% of the total population (7, 13). However, in comparison to the chronic pulmonary infections of CF patients, little is understood regarding P. aeruginosa adaptation in other common chronic infections, such as chronic wounds. Furthermore, understanding the selective forces that drive the emergence and frequency of RSCVs is important because of their association with hyperbiofilm-forming phenotypes (14, 15), increased tolerance to antimicrobials (13), and enhanced resistance to immunity (5).
The RSCV phenotype is commonly caused by mutations in pathways that lead to elevated cyclic diguanylate monophosphate (c-di-GMP) (2). c-di-GMP is a messenger molecule that signals the transition from planktonic to biofilm lifestyle in many bacteria (16). In P. aeruginosa, increased c-di-GMP, among many responses, leads to overproduction of exopolysaccharides, Psl and Pel, and matrix proteins (14, 17, 18). RSCVs with driver mutations in the Wsp (wrinkly spreader) pathway, originally identified in Pseudomonas fluorescens (19, 20), are commonly isolated from in vitro-grown biofilms, where approximately 70% of isolated P. aeruginosa RSCVs can be complemented by wspF in trans (17). Mutations in the Wsp pathway have been implicated in P. aeruginosa evolution in CF patients (21, 22). However, their importance in relation to other identified adaptive mutations and frequency in other chronic infections is unclear.
Despite our understanding of the divergent phenotypes of evolved variants, it is currently unclear which mutations and pathways experience selection in an infection, what new niches become occupied, and how these adaptations enable pathogen survival. It is often not feasible to monitor bacterial evolution from the onset of infection, as it is difficult to understand microbial dynamics in a clinical setting due to patient care, sampling difficulties, and cost. This has similarly proven challenging to monitor in a research setting, as there are few chronic infection models that mimic what is observed clinically. Furthermore, mutations also exist in the context of the larger microbial community. Chronic infection models typically address the adaptive traits of only a single founding clone, but susceptible individuals are continually exposed to different strains of opportunistic pathogens, particularly in the case of environmental organisms like P. aeruginosa. To address these challenges, we used a porcine full-thickness thermal injury wound model, which closely reflects human clinical chronic wounds (23–27), and is considered a general model for studying chronic infections established by bacterial biofilms (23, 25–27). Here, these wounds were inoculated with a coinfection of six P. aeruginosa strains, and we determine which strains persist, which produce adaptive colony variants, and identify the genetic and physiological pathways of adaptive evolution.
RESULTS
P. aeruginosa strains PA14-1 and PAO1-B11 were predominant in a mixed-strain chronic burn wound infection.
To determine the relative fitness of different P. aeruginosa strains and how the population evolves during chronic infection, we infected porcine full-thickness burn wounds with an inoculum consisting of approximately equal (but not identical) numbers of six different P. aeruginosa strains. Wounds were infected with two model strains (PA14-1 and PAO1-B11), three clinical isolates (B23-2, CF18-1, and S54485-1), and a water isolate (MSH10-2) (Table 1). Each strain had a unique nucleotide barcode introduced at the neutral Tn7 site (Table 1). These strains share similar growth kinetics (see Fig. S1A and B in the supplemental material) and biofilm formation capacity (Fig. S1C). Wound biopsies were taken 3, 14, and 28 days postinfection (dpi) for bacterial quantification.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype and/or characteristic(s) | Reference or source |
|---|---|---|
|
P. aeruginosa strains used in the infectiona |
||
| PA14-1 | Barcode CAAAAGGACA; Gent | |
| PAO1-B11 | Barcode GTGTCGTGGG; Gent | |
| B23-2 | Wound isolate; barcode GCCTATTGTG; Gent | Lubbock, TX |
| CF18-1 | Nonmucoid CF isolate; barcode GTTACGTCAA; Gent | 60 |
| MSH10-2 | Water isolate; barcode TATCAGATTT; Gent | 60 |
| S54485-1 | UTIb isolate; barcode TTAAACTAGG; Gent | 60 |
| P. aeruginosa strains | ||
| PA14 | ||
| PA14ΔwspF | Clean wspF deletion | |
| PAO1 | ||
| PAO1ΔwspF | Clean wspF deletion (JJH356) | |
| PA14-1attB::lacZ | lacZ from miniCTX-lacZ introduced at the attB site | This study |
| RSCV-2 wt-wspA | Variant wspA replaced by the wild-type allele | This study |
| RSCV-1 wt-wspA | Variant wspA replaced by the wild-type allele | This study |
| RSCV-40 wt-wspA | Variant wspA replaced by the wild-type allele | This study |
| E. coli strains | ||
| NEB5-α | NEBc | |
| S17 | NEB | |
| Plasmids | ||
| pEX18Ap | Gene deletion vector | 47 |
| pEX18Ap::wt-wspA | wspA-complementing construct | This study |
| pCdrA::gfp | CdrA promoter fused to gfp; Carb | 31 |
| pMH487 | Empty vector for pCdrA::gfp; Carb | 31 |
| pJN2133 | PA2133 cloned into pJN105; Gent | 32 |
| pHERD20T | Empty vector; Carb | 56 |
| pHERD2133 | PA2133 from pJN2133 cloned into pHEDR20T; Carb | This study |
| miniCTX-lacZ | Tet | 58 |
All strains used in the infection are resistant to gentamicin. Barcodes are located at the Tn7 site on the genome.
UTI, urinary tract infection.
NEB, New England Biolabs.
Phenotypic profile of P. aeruginosa strains used in the infection. (A) Metabolic activity of the six P. aeruginosa strains assayed for 16h at 37°C using a Biolog system. (B) Area under the curve (AUC) of the Biolog kinetic plot depicted in panel A. **, P value of <0.01. (C) Biofilms of the six P. aeruginosa strains were grown for 24 h at 37°C in a 96-well plate. Biomass levels were quantified by crystal violet staining. Data are depicted as mean ± SD (n = 3). (D) The frequency of each strain in the starting inoculum was determined by sequencing the strain-specific barcodes at the Tn7 site. The proportion of each barcode was expressed as a percentage of the total sequence counts. Download FIG S1, PDF file, 0.1 MB (128.8KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Colony-forming unit (CFU) counts revealed that wounds remained colonized with approximately 105 bacteria up to 28 dpi (Fig. 1A). To quantify the proportion of each strain over time, genomic DNA was isolated from biopsy tissue samples, and amplicons spanning the barcode were sequenced. As early as 3 dpi, PA14-1 and PAO1-B11 became the predominant strains in the infection, outcompeting the four other strains by orders of magnitude (Fig. 1B). To test whether PA14-1 and PAO1-B11 dominated in the infection due to host factors or due to interstrain competition, the six ancestor strains were competed together in planktonic culture in vitro, at similar starting frequencies used to inoculate the porcine wounds. Under these conditions, after 24 h, the wound isolate B23-2 outcompeted the other five strains (Fig. 1C). After 3 days, B23-2 remained the predominant strain, with PAO1-B11 the only other strain to be consistently detected, at approximately 2% of the population (Fig. 1C). This indicates that the predominance of PA14-1 and PAO1-B11 in the wounds was likely due to in vivo-specific factors (see Discussion).
FIG 1.

P. aeruginosa burden in a mixed-strain chronic burn wound infection. (A) Biopsies were taken from wounds at 3, 14, and 28 dpi. Biopsy specimens were homogenized and plated for CFU/gram, each plated in triplicate, with a minimum of four biopsy specimens taken from each wound. Significance was determined using a one-way ANOVA. ns, not significant; *, P value of <0.05. (B) Genomic DNA was isolated from homogenized tissue, and the strain-specific barcodes at the Tn7 site were sequenced. A minimum of four biopsy specimens from each wound was sequenced. Significance was determined using a one-way ANOVA: a, P value of <0.05 compared to PA14-1; b, P value of <0.05 compared to PAO1-B11. (C) The six ancestor strains were competed in planktonic culture in vitro for 12 days. Three replicates were performed at each time point. For panels B and C, the proportion of strain barcodes were expressed as a percentage of the total sequence reads to determine the relative frequency of each strain.
RSCVs were selected for during porcine chronic burn wound infections.
To identify functionally distinct mutants within the infections, we used colony morphology as an indicator (28, 29). Homogenized biopsy specimens were grown on Vogel-Bonner minimal medium supplemented with Congo red and brilliant blue dyes (VBMM). RSCVs were isolated from all three time points, with two subpopulations observed; one that had a pink, rugose phenotype and a second that had an orange, textured phenotype (Fig. 2A). The RSCV phenotype was stable across four passages on selective and nonselective growth media, consistent with the hypothesis that these variants were due to stable genetic mutations. In comparison, RSCVs were not isolated from the in vitro planktonic competition.
FIG 2.

P. aeruginosa RSCVs isolated from porcine chronic burn wound infections. (A) Representative colony morphologies of RSCVs isolated from homogenized porcine burn wound tissue. RSCVs were plated on VBMM, and the colony morphology was compared to the ancestor strain (labeled). Bars, 2 mm. (B) Frequency of the total RSCV population isolated at each time point expressed as a percentage of the total P. aeruginosa population. Data are presented as mean ± standard deviation (SD). Significance was determined using a one-way ANOVA: **, P value of <0.01; ****, P value of <0.0001 compared to 14 dpi. (C) Frequency of the ancestor strain that the RSCVs evolved from, expressed as a percentage of the total RSCV subpopulation. Data presented as mean ± SD. Significance was determined using a Student’s t test: **, P value of <0.01; ***, P value of <0.001.
The total RSCV abundance in the wounds was quantified as a percentage of the total P. aeruginosa burden. RSCV frequency was low 3 dpi (0.02% ± 0.12%), peaked at 14 dpi, at approximately 2% (2.15% ± 5.25%), and declined at 28 dpi (0.19% ± 0.65%) (Fig. 2B). Despite their low prevalence, their rapid rise to detectable frequency allowed us to quantify the selective pressure acting on RSCVs in the wound, across a range of possible starting frequencies, according to equation 1. The selective coefficient (s) was >0.1, demonstrating that RSCVs were under strong positive selection (Table 2).
TABLE 2.
Relative fitness, expressed as selection coefficient (s), of RSCVs from porcine burn wounds inferred for potential starting frequencies
| Starting frequency (RSCV/ancestor) |
s (mean ± SD) at the following days postinfection: |
||
|---|---|---|---|
| 3 | 14 | 28 | |
| 1:105 | 0.585 ± 0.464 | 0.441 ± 0.126 | 0.18 ± 0.055 |
| 1:106 | 0.618 ± 0.579 | 0.565 ± 0.177 | 0.229 ± 0.068 |
| 1:107 | 0.651 ± 0.710 | 0.689 ± 0.238 | 0.278 ± 0.098 |
| 1:108 | 0.684 ± 0.849 | 0.813 ± 0.304 | 0.327 ± 0.134 |
RSCVs were detected only from the model strains PA14-1 (the pink phenotype [Fig. 2A]) and PAO1-B11 (the orange phenotype [Fig. 2A]). RSCVs derived from PA14-1 were isolated across all time points, whereas PAO1-B11 RSCVs were isolated from 14 and 28 dpi. At the later two time points, PA14-1 RSCVs remained the predominant subpopulation (Fig. 2C).
PA14-1 RSCVs contained driver mutations exclusively within the wsp pathway.
As PA14-1 RSCVs were the predominant evolved phenotype, we focused on this subpopulation for the remainder of this study. Whole-genome sequencing was performed on 27 randomly selected PA14-1 RSCVs to identify the mutation(s) accounting for the RSCV phenotype.
We identified putative driver mutations exclusively in the wsp cluster, specifically, an in-frame, 42-bp deletion (Δ285−298 aa) in wspA, and a frameshift 5-bp deletion (V154fs) in wspF (Table 3). RSCVs with the wspA mutation were predominant across all time points, and wspF mutants were identified only at 14 dpi. Using RSCV-2 as a representative wspA mutant, we replaced the variant wspA with the wild-type allele using two-step allelic recombination to rescue the deletion. This resulted in the RSCV colony phenotype reverting to the wild-type phenotype (Fig. 3), demonstrating that the wspA Δ285-298 mutation was responsible for the RSCV phenotype.
TABLE 3.
Mutations identified in PA14-1 RSCVs
| Sample |
Driver mutation |
Secondary mutation |
|||||
|---|---|---|---|---|---|---|---|
| Day | RSCV no. | Wound no. | Gene | Mutationa | Frequency (%) | Gene | Mutation |
| 3 | 1 | 1 | wspA | 285-298del | pslO-pslE | Δ14,299 bp | |
| 2 | 2 | wspA | 285-298del | ||||
| pslO-pslE | Δ14,299 bp | ||||||
| 4 | 1 | wspA | 285-298del | PA14_13130/PA14_13140 | TNN→TGC | ||
| fabI/ppiD | TTC→TCC | ||||||
| Day 3 summary | wspA | 285-298del | 100 | ||||
| 14 | 3 | 4 | wspA | 285-298del | |||
| 6 | 4 | wspA | 285-298del | ||||
| 7 | 4 | wspA | 285-298del | ||||
| 8 | 4 | wspA | 285-298del | ||||
| 9 | 4 | wspA | 285-298del | ||||
| 10 | 4 | wspA | 285-298del | ||||
| 12 | 4 | wspA | 285-298del | CRISPR–Cas1/hpb | +60bp | ||
| 13 | 1 | wspA | 285-298del | Glutamyl-tRNA reductase | L71L (CTG→TTG) | ||
| 14 | 4 | wspA | 285-298del | fabI/ppiD | TTC→TCC | ||
| 16 | 4 | wspA | 285-298del | ||||
| 17 | 4 | wspA | 285-298del | ||||
| 20 | 4 | wspF | V154fs | ||||
| 24 | 1 | wspA | 285-298del | ||||
| 27 | 1 | wspA | 285-298del | ||||
| 28 | 2 | wspA | 285-298del | ||||
| 36 | 3 | wspF | V154fs | ||||
| 37 | 3 | wspA | 285-298del | ||||
| 38 | 3 | wspF | V154fs | ||||
| 86 | 3 | wspA | 285-298del | CRISPR–Cas1/hp | +60bp | ||
| Day 14 summary | wspA | 285-298del | 84.2 | ||||
| wspF | V154fs | 15.8 | |||||
| 28 | G→TT 585bp | ||||||
| wspD | CT→AG 588-589bp | ||||||
| 40 | 1 | wspA | 285-298del | S197S (TCG→TCA) | |||
| PA14_54090 | A248V (GCG→GTG) | ||||||
| 42 | 3 | wspA | 285-298del | ||||
| 43 | 3 | wspA | 285-298del | ||||
| 45 | 3 | wspA | 285-298del | ||||
| 87 | 2 | wspA | 285-298del | ||||
| Day 28 summary | wspA | 285-298del | 100 | ||||
Deleted amino acid residues are indicated. del, deletion; fs, frameshift.
hp, hypothetical protein.
FIG 3.
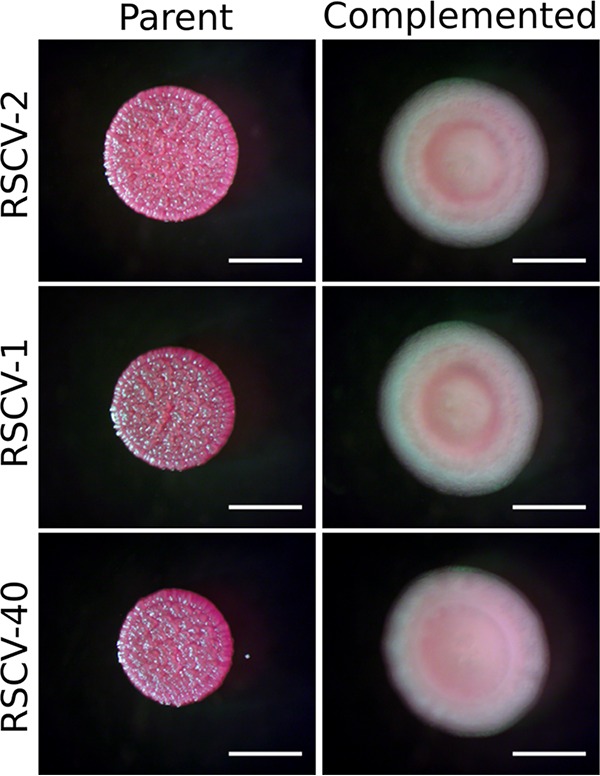
Complementation of the wspA Δ285-298 mutation. wspA was complemented in representative RSCVs by replacing wspA Δ285-298 with the wild-type allele on the genome. RSCV-2 was selected as a representative RSCV with the wspA driver mutation alone. RSCV-1 and RSCV-40 have the wspA driver mutation as well as ΔpslE-pslO and wspD secondary mutations, respectively. Parent and complemented RSCVs (labeled) were grown on VBMM, and colony morphologies were assessed. Bars, 2 mm.
Some of the RSCVs also possessed secondary mutations (Table 3), possibly demonstrating further adaptation in the wound. Two wspA RSCVs from 3 dpi (Table 3; RSCV-1 and RSCV-4) underwent a 14,299-bp deletion that removed the remaining psl operon. PA14 naturally lacks Psl, since pslA-pslD are absent (30). In these two isolates, the remaining genes of the psl operon, pslE-pslO were deleted. In the RSCV-1 background, complementation of wspA reverted the RSCV colony phenotype to wild type (Fig. 3), indicating that deletion of the remaining psl operon did not influence the RSCV phenotype.
Evidence of additional adaptive mutations in the wsp cluster was detected on 28 dpi. RSCV-40, in addition to having the wspA Δ285-298 driver mutation, had three separate mutations in wspD which led to an early stop codon. However, as the mutations occurred at the wspD 3′ end, the WspD N terminus may still be expressed and functional (Table 3). In this isolate, complementation of wspA reverted the RSCV colony phenotype to wild type (Fig. 3), indicating that the wspD mutations did not influence the RSCV colony phenotype. However, this further points toward the strong selective pressure on the Wsp pathway in the chronic infection.
We were also interested in identifying how the PA14-1 non-RSCV population adapted to the infection and whether this population acquired mutations that did not result in divergent colony phenotypes. When we sequenced randomly selected PA14-1 non-RSCV isolates (23 total), relatively few had acquired chromosomal mutations (see Supplementary Results in Text S1 in the supplemental material; Table S1). These isolates had similar levels of biofilm formation and metabolic kinetics compared to the ancestor strain (see Supplementary Results in Text S1; Fig. S2), suggesting that the emergence of RSCVs was the main source of phenotypic diversification in the wounds.
Supplemental Materials and Methods and Results. Download Text S1, PDF file, 0.2 MB (175.1KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The PA14-1 non-RSCV population in the porcine wounds behave similarly to the ancestor strain. (A) Five non-RSCV colonies were randomly selected from each wound. The ancestor of each colony was identified and expressed as a percentage of the total number of colonies isolated from that time point. (B) Biofilms (24 h) were grown in 96-well plates, and biomass levels were quantified by crystal violet staining. Biomass levels are expressed as a percentage relative to PA14-1, which was set at 100%. ns indicates no significant difference. (C) AUC of Biolog kinetic curves. AUC are expressed as a percentage relative to PA14-1, which was set at 100%. Gray scale in panels B and C indicate different wounds. (D to F) Kinetic Biolog curves of representative non-RSCV PA14-1 isolates from each time point (labeled). Data are presented as mean ± SD (n = 3). *, P value of <0.05; **, P value of <0.01; ***, P value of <0.001. Download FIG S2, PDF file, 0.4 MB (395.4KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mutations in non-RSCV PA14-1 population. Download Table S1, PDF file, 0.1 MB (56.3KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RSCVs isolated from wounds had elevated intracellular c-di-GMP levels.
As all PA14-1 RSCVs acquired driver mutations in the wsp cluster, we predicted that both the wspA and wspF mutations led to overproduction of c-di-GMP, resulting in the RSCV phenotype. To assess whether c-di-GMP levels were elevated, the pCdrA::gfp plasmid (31) was introduced into representative PA14-1 RSCVs. In this plasmid, the cdrA promoter, which is under c-di-GMP regulation, is fused to a promoterless gfp (31). As expected, the representative PA14-1 RSCVs showed increased levels of green fluorescence compared to the ancestor PA14-1, indicating that the RSCVs had elevated c-di-GMP levels (Fig. 4A). There were no differences in fluorescent signal between the RSCVs, regardless of the driver mutation or the presence of secondary mutations, indicating that the c-di-GMP levels were similar across the representative PA14-1 RSCVs (Fig. 4A). Introduction of a plasmid encoding the phosphodiesterase (PDE; enzyme that degrades c-di-GMP [16]) PA2133, under an arabinose-inducible promoter, into the representative PA14-1 RSCVs reverted the colony morphology to the ancestral type (Fig. 4B). The empty vector did not influence the RSCV colony morphology (Fig. 4B). Collectively, this indicates that elevated c-di-GMP levels were responsible for the RSCV colony phenotype, as has been observed in P. aeruginosa wspF mutants (32).
FIG 4.
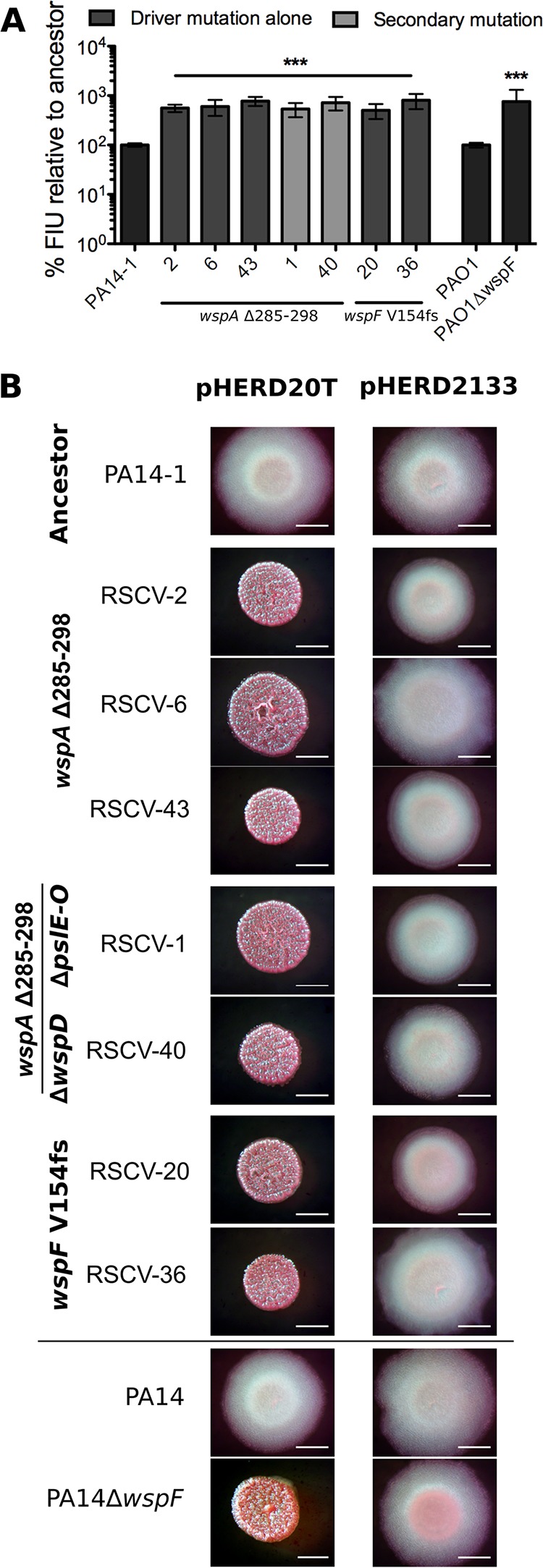
PA14-1 RSCVs have elevated intracellular levels of c-di-GMP which are responsible for the RSCV phenotype. (A) Green fluorescence was measured in representative RSCVs with the c-di-GMP reporter plasmid pCdrA::gfp. Increased GFP signal correlates to increased intracellular c-di-GMP levels. Fluorescence intensity units (FIU) of RSCVs were determined relative to the ancestor strain, which was set at 100%. PAO1ΔwspF and its isogenic parent PAO1 were used as controls. Data are presented as mean ± SD (n = 3). Significance was determined using a one-way ANOVA: ***, P value of <0.001 compared to the ancestor strain. (B) Colony morphology of representative RSCVs grown on VBMM plus 0.1% arabinose. pHERD20T is the empty vector. pHERD2133 has the PDE PA2133 cloned under an arabinose-inducible promoter. PA14ΔwspF and its isogenic parent, PA14, were used as controls. For both assays, representative RSCVs were selected. wspA mutants from each time point, RSCV-2, RSCV-6, and RSCV-43, were selected. Representative wspA mutants with secondary mutations were selected. RSCV-1 has the remaining psl operon deleted, and RSCV-40 has the additional wspD mutations. Representative wspF mutants, RSCV-20 and RSCV-36, were also selected. Bars, 2 mm.
Wsp-dependent RSCVs were more fit than the ancestor.
RSCV colony morphology and elevated c-di-GMP are often associated with increased biofilm formation capacity. To investigate this possibility, biofilms were grown in microtiter plates for 24 h and stained with crystal violet. All 27 PA14-1 RSCVs displayed increased biofilm formation capacity compared to the ancestor PA14-1 (Fig. 5A). This increased biofilm formation was not due to elevated metabolic activity, as PA14-1 RSCVs had similar growth kinetics, except for an extended lag phase (Fig. S3).
FIG 5.
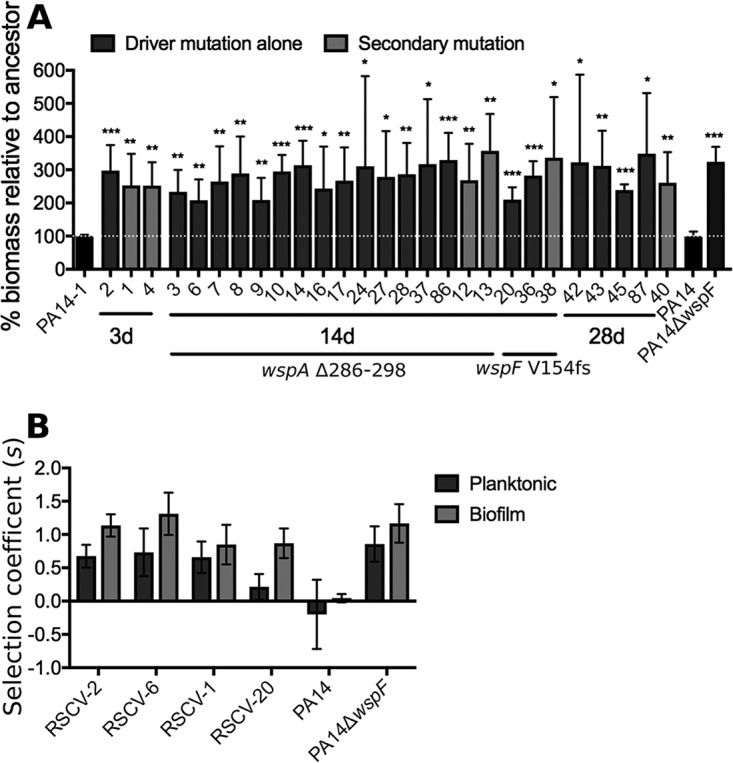
PA14-1 RSCVs have increased biofilm formation and fitness relative to the ancestor PA14-1. (A) PA14-1 RSCVs were grown in 96-well plates for 24 h. Biofilm biomass was stained and quantified by crystal violet. Biofilm biomass levels were expressed as a percentage relative to the ancestor strain, which was set at 100% for each replicate. Data are presented as mean plus SD (n = 3). Significance was determined using a one-way ANOVA: *, P value of <0.05; **, P value of <0.01; ***, P value of <0.001. (B) Fitness of representative PA14-1 RSCVs relative to the ancestor PA14-1. PA14ΔwspF and its isogenic parent PA14, compared to PA14-1, were used as controls. Strains were grown for 48 h as either a planktonic culture or biofilm, and selection of the RSCVs relative to PA14-1attB::lacZ was determined by calculating the selection rate according to equation 1. Data are presented as mean ± SD (n = 5).
PA14-1 RSCVs from the porcine wounds have an extended lag phase compared to the ancestor strain. (A) AUC of Biolog metabolic kinetic curves of all sequenced PA14-1 RSCVs, expressed as a percentage relative to PA14-1. PA14ΔwspF and its parent PA14 were used for comparison. *, P value of <0.01; #, P value of <0.001 compared to the ancestor strain. (B to E) Kinetic Biolog curves of representative PA14-1 RSCVs (labeled). Data are presented as mean ± SD (n = 3). Download FIG S3, PDF file, 0.3 MB (335.6KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To determine the fitness of PA14-1 RSCVs relative to the ancestor strain in vitro, representative RSCVs were grown together with the ancestor PA14-1 tagged with lacZ at the neutral attB site (PA14-1attB::lacZ). Strains were grown either in planktonic culture or in a biofilm assay (33). The number of CFUs of each strain and the selection coefficient (s) were determined after 2 days as a measurement of fitness. All RSCVs tested were more fit than the ancestor under both modes of growth (Fig. 5B). Fitness in biofilm tended to be greater than in planktonic conditions, but these differences were not significant. The driver mutation did not appear to influence the selection rate, as RSCVs with either the wspA Δ285-298 or wspF V154fs had comparable fitness levels (Fig. 5B; RSCV-2 and RSCV-6 compared to RSCV-20). Furthermore, RSCV-1 (ΔpslE-pslO) had similar fitness levels compared to RSCV-2 and RSCV-6 (Fig. 5B). This suggests that loss of the remaining psl operon does not lead to an increased fitness advantage over the RSCV phenotype alone under these laboratory conditions.
In vivo acquisition of immunity to viral infection through CRISPR expansion.
Two of the PA14-1 RSCV isolates, RSCV-12 and RSCV-38, acquired a 60-bp insertion at the clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated proteins (Cas) locus (Table 3). Both sequences inserted at the intergenic region (−1549/+271) between PA14_33350 (RS13600) and PA14_33370 (RS13605) at the genomic position 2,937,205. The last 28 bp of the inserted sequences were identical between the two isolates and aligned to the repetitive elements in the PA14 CRISPR array (CRISPR2 [34]) (Fig. 6A). However, the first 32 bp differed, indicative of CRISPR spacer sequences (Fig. 6A) that are specific to infective mobile genetic elements. A BLAST search of the inserted CRISPR spacer against the ancestor strains aligned to a prophage sequence in the B23-2 genome assembly, indicating potential phage infection across strains (see Supplementary Results in Text S1; Fig. S4).
FIG 6.

PA14-1 isolates RSCV-12 and RSCV-38 are resistant to infection by phage isolated from B23-2. (A) The 60-bp insertion sequence in the CRISPR array of RSCV-12 and RSCV-38. Prophages were isolated from B23-2, and plaque assays were performed to determine the level of phage infection for representative RSCV isolates (B) and the ancestor P. aeruginosa strains (C). RSCV-6 and RSCV-12 both have the same driver wspA mutation, while RSCV-36 and RSCV-38 have the same driver wspF mutation. The presence (+) or absence (−) of the CRISPR insertion is indicated. The driver mutation for each RSCV is labeled. Data are presented as mean plus SD (n = 4). Significance was determined using a Student’s t test: *, P value of <0.05.
Contig sequence from B23-2 with the protospacer sequence. There is currently no annotated sequence for B23. Therefore, the CRISPR spacer sequences were aligned against the contig sequences of the ancestor B23-2, which was sequenced along with the representative RSCVs. Protospacer for the CRISPR spacer in RSCV-12 (contig 107) (A) and RSCV-38 (contig 95) (B) were identified in two separate contigs (bold text). For the protospacer sequence in contig 95 (B), there was one base pair mismatch when comparing the CRISPR spacer to the protospacer (base pair that is not bold). After each protospacer, there is a conserved GG PAM motif (underlined) which is required for type 1-F CRISPR-Cas families which P. aeruginosa PA14 possesses. A BLAST search of the contig sequences identified CRISPR spacers in P. aeruginosa strains (Table S2) aligned to other protospacers in contig 107 (A), indicated by the red and blue text. Download FIG S4, PDF file, 1.6 MB (1.7MB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We therefore predicted that RSCV-12 and RSCV-38 would both be resistant to phages isolated from B23-2 due to CRISPR-Cas adaptive immunity. To test this, we grew B23-2 in mitomycin C and harvested the phage-enriched supernatant. P. aeruginosa strains were incubated with the phage lysate, and plaque assays were performed. RSCV-12 and RSCV-38 isolates were resistant to phage infection, with no plaques observed for RSCV-38, and only a single plaque in one replicate for RSCV-12 (Fig. 6B). Infection of RSCV-6 and RSCV-36 (CRISPR−), which have identical wsp driver mutations as RSCV-12 and RSCV-38 (CRISPR+), respectively, had similar levels of phage sensitivity compared to the ancestor PA14-1 (Fig. 6B). This indicates that the acquired CRISPR spacers in RSCV-12 and RSCV-38 produced immunity to phages isolated from B23-2. Each of the ancestor strains was assessed for phage susceptibility, and as expected, B23-2 was resistant to infection, whereas the remaining strains showed various levels of phage sensitivity (CF18-1 > MSH10-2, S54485-1 > PA14-1, PAO1-B11; Fig. 6C).
DISCUSSION
Here we describe the rapid evolution of adaptive P. aeruginosa mutants with conspicuous colony phenotypes arising in a clinically relevant model of chronic infection. There is a consensus in the field that evolved variants arise in an infection as a consequence of adaptation over extended periods of time. However, we isolated hyperbiofilm-forming RSCVs from early stages of infection, which suggests that variants may evolve by positive selection more rapidly than originally appreciated. Therefore, contrary to current understanding, RSCVs may be a common, early adaptation during infections, and the selective pressures driving RSCV evolution may persist throughout these infections.
Interestingly, the competitive fitness of the six different P. aeruginosa strains differed in vivo and in vitro. In the wounds, both PA14-1 and PAO1-B11 outcompeted the remaining four strains, while in planktonic culture, B23-2 predominated (Fig. 1). This suggests that in vivo environmental factors may be responsible for driving the P. aeruginosa strain dynamics in the wound. We predict that in vitro, the prophage from B23-2 may be induced and responsible for driving strain competition and dynamics in planktonic culture. It would be interesting to explore this further and to determine, if so, why these dynamics might be dampened in vivo. This highlights the notion that fitness in vitro does not necessarily correlate to fitness in vivo and stresses the importance for using appropriate models when studying in vivo systems.
Because of their competitive superiority in the wound, RSCVs were recovered only from PA14-1 and PAO1-B11 (Fig. 2C). P. aeruginosa RSCVs were highly adaptive in the infection and in laboratory conditions (Table 2 and Fig. 5B). The estimated selection coefficients determined in vivo (s = 0.180 to 0.813; Table 2) were up to five times greater than those identified in the Lenski long-term evolution lines (35), pointing toward the strong positive selection experienced by RSCVs in the wounds. In addition, similar to our results with PA14-1, Burkholderia cenocepacia variants containing wsp mutations isolated from an in vitro biofilm evolution assay had high selection coefficients (36). Together, these results suggest that wsp mutants experience significant positive selection both in vivo and in vitro and are broadly adaptive across different environments, where the biofilm lifestyle predominates.
Despite these strong selection coefficients, RSCVs remained at relatively low frequencies throughout the infection (Fig. 2B). This implies that RSCVs may be highly favorable when rare in the wounds, but less favorable when abundant, a process referred to as negative frequency-dependent selection. Negative frequency-dependent selection has been observed for evolved rugose variants of P. fluorescens (37–39). Niche competition (37, 38) and division of labor (39) with the ancestor strain drove the evolution of P. fluorescens rugose variants from static planktonic and colony growth, respectively. In both cases, diversification of the population was maintained by negative frequency-dependent selection (37–39). In the wound infections, the low-frequency RSCVs could facilitate the ancestor strain in colonizing and establishing biofilms in the wound by producing more EPS. This elevated EPS may enable persistence in an environment constantly exposed to stressors, including fluctuating antibiotics and immune defense (5). However, this may be a shortsighted evolutionary strategy for an opportunistic pathogen, like P. aeruginosa, with a significant environmental reservoir. The mutations in wsp are rarely reverted and constrain some of the phenotypic plasticity required for survival in fluctuating environments. Accordingly, we predict that in a more homogeneous fitness landscape, RSCVs at low frequencies could be enriched. As an example supporting this hypothesis, higher RSCV frequencies have been observed in CF patients under prolonged exposure to antimicrobials, particularly aerosolized antibiotics (6, 10).
Every sequenced PA14-1 RSCV acquired a driver mutation in the wsp cluster, demonstrating that in chronic wounds, the Wsp pathway specifically undergoes selection, and that wsp mutants may be more fit than other c-di-GMP-regulating pathways that confer the RSCV phenotype. The wspA Δ285-298 was the most common driver mutation and was isolated early in the infection (Table 3). There are two potential explanations for the rapid rise of this single wspA mutant in the infection. The first is that it may have been present in the initial inoculum at undetected levels. The second is that this region may be hypermutable owing to a direct repeat located at either side of the deletion (see Fig. S5A). We are currently unable to discern between these two scenarios; however, it is significant that the population rapidly diversifies in the wound due to strong positive selection of adaptive phenotypes provided by wsp mutations.
WspA deletions. (A) The 42-bp deletion in wspA is located between direct repeats. The gene sequence of PA14 wspA is shown. The 42-bp deletion (853-894bp) in the wspA mutants is underlined. Either side of this sequence is a direct repeat indicated in bold text. (B) Alignment of WspA deletions. Deletions in homologous regions of wspA have been observed in in vitro-evolved RSCVs; a 286-307aa deletion in P. aeruginosa PAO1 (MJK8), a 284-311aa deletion in P. fluorescence Pfl01 and a 307-313aa deletion in Burkholderia cenocepacia HI2424. The different domains of WspA were determined from Pfam analysis from the Pseudomonas Genome Database and the Burkholderia Genome Database. Domains are colored as follows: purple for the ligand binding domain or the four-helix bundle domain, green for the HAMP or linker domain, and blue for the MCP signaling domain. The region of the deletion in each protein is indicated in red. Download FIG S5, PDF file, 2.0 MB (2MB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supporting the second theory is the observation that mutations in this wspA region also appear to be under selection to produce RSCVs in laboratory biofilms (Fig. S5B). The P. aeruginosa PAO1 RSCV isolate MJK8, which evolved during biofilm growth in a tube reactor (14), has an in-frame 66-bp deletion (Δ286–307aa) in the same region as the wspA Δ285-298 isolated here (17). P. fluorescens Pfl01, when grown as a colony biofilm, evolved RSCVs with driver mutations in wspC, wspA, and wspE (39). One of the wspA mutations was an in-frame 84-bp deletion (Δ284-311aa), again occurring in the homologous region (39). Finally, B. cenocepacia HI2424 RSCVs with wspA and wspE mutations were isolated from a biofilm bead evolution experiment (36). The majority of mutations identified were nonsynonymous single nucleotide polymorphisms (SNPs); however, one of the wspA mutations was an in-frame 21-bp deletion (Δ307-313aa), again in the homologous region (36) (Fig. S5B). Homology modeling of PA14 WspA suggests that the Δ285-298 occurs opposite the predicted methylation site (see Supplementary Results in Text S1; Fig. S6). We therefore predict that small deletions in the homologous region could alter how WspA is methylated/demethylated and ultimately lead to constitutive signaling and autoinduction of the Wsp pathway.
A 14-aa deletion in WspA is predicted to occur opposite the methylation site. (A) Schematic of WspA. The different domains of WspA were determined from the Pseudomonas Genome Database Pfam analysis. LBD, ligand binding domain or the four-helix bundle domain (3-182aa); HAMP, linker domain (213-261aa); SD, MCP signaling domain (348-505aa). The region of the 14aa deletion is indicated in red (285-298aa). (B) The WspA cytoplasmic domain amino acid sequence. The domains are indicated by the same colors as in panel A. The signaling domain contains two additional features, the kinase interacting subdomain, or “tip” domain in dark blue (382-420aa) and the predicted methylation site in navy blue (492-501aa). Both the heptad registers (reg) and the heptad number (h#) are labeled, with consecutive heptads indicated in alternating black and gray text. (C) Homology model of PA14 WspA modeled against the T. maritime MCP (PDB 3JA6) generated using SWISS-MODEL. Colors correspond to the domains indicated in panel B. The model spans 250-541aa of WspA. Download FIG S6, PDF file, 0.4 MB (433.3KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In addition to driver mutations, some PA14-1 RSCVs also gained secondary mutations. Of particular interest was the Δ14,299 bp, which deleted the remaining psl operon (Table 3; RSCV-1 and RSCV-4). We predicted that this deletion might lead to increased fitness of the RSCVs, over the RSCV driver mutation alone. However, deletion of the remaining psl operon did not provide additional fitness benefits under the conditions tested. Therefore, the remaining psl operon (pslE-pslO) in PA14 may play a role outside Psl synthesis, which may have a fitness cost in the wound environment.
Additional secondary mutations of interest were the 60-bp insertions in the CRISPR-Cas array of RSCV-12 and RSCV-38 (Table 3 and Fig. 6A), which encoded resistance to phage(s) isolated from B23-2 (Fig. 6B). It has only recently been confirmed that the P. aeruginosa type I-F CRISPR-Cas system provides adaptive immunity to phages with a target protospacer (40). This is dependent on the presence of the correct protospacer adjacent motif (PAM) in the mobile genetic element (40). In support of this, both protospacers contain the type I-F CRISPR-Cas-specific GG PAM (Fig. S4). Furthermore, B23-2 contig 107 contained two additional protospacers to which CRISPR spacers in P. aeruginosa have been reported (Table S2 and Fig. S4A). Of interest was the observation that PA14 already contains a CRISPR spacer identical to a protospacer in contig 107 (Table S2 and Fig. S4A), possibly indicating that PA14 had already been exposed to the prophage in B23-2. However, the ancestral PA14-1 was still sensitive to infection (Fig. 6B and C), presumably due to the incorrect PAM (Fig. S4A). This highlights the importance of insertion of the correct CRISPR spacer in mediating phage immunity. This is only the second report of CRISPR-Cas acquired immunity in P. aeruginosa strains (40), and to our knowledge, this is the first report of CRISPR-Cas adaptive immunity acquired in an infection.
CRISPR spacers in P. aeruginosa strains with a protospacer aligning to contig 107 in B23-2. Download Table S2, PDF file, 0.01 MB (15.6KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Our data indicate that P. aeruginosa experiences strong selective forces in chronic infections, from interstrain phage predation to increased biofilm or aggregate production, and in response rapidly evolve during the initial stages of infection. Parallel evolution of the wsp pathway was observed across all time points, suggesting that wsp mutants are early, highly beneficial adaptations to infection. This also indicates that the Wsp system is a potent pathway under selection in chronic infections to produce hyperbiofilm variants. This is despite the availability of other c-di-GMP-regulating pathways to produce RSCVs, both in vivo and in vitro (18, 41–45). We predict that RSCVs produced through the wsp pathway may be an adaptation common to chronic infections, and developing therapies that target the RSCV subpopulation or prevent their emergence could be transferrable across these infections.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Bacterial strains and plasmids used in this study are detailed in Table 1. Complementation constructs were made using Gibson Assembly (New England Biolabs [NEB]) (46). Primers used to create the constructs are detailed in Table S3 in the supplemental material. Constructs were incorporated into the P. aeruginosa genome using two-step allelic recombination as previously described (47).
Primers used in this study. Download Table S3, PDF file, 0.02 MB (16.1KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Porcine full-thickness chronic burn wound model.
Swine were housed and studied according to the Ohio State University IACUC-approved protocols. Porcine full-thickness chronic burn wounds were established as previously described (23). Briefly, two pigs were subjected to thermal injury, giving six full-thickness burns per pig and covered with impermeable wound dressings. Wounds were infected 3 days after injury with equal amounts of six different P. aeruginosa strains to achieve a final 250-μl inoculum at 108 bacteria (1.6 × 107 each for a total of 1 × 108), which was spread over the wounds and allowed to air dry before the wound dressing was reapplied. Wounds were infected with P. aeruginosa strains PA14-1, PAO1-B11, B23-2, CF18-1 (GenBank identifier [ID] NZ_KI519281), MSH10-2 (GenBank ID NZ_KE138672), and S54485-1 (GenBank ID NZ_KI519256). Prior to infection, each strain had been tagged with a unique barcode at the Tn7 site on the genome (see Supplementary Methods in Text S1 in the supplemental material; Table 1).
Wound healing was monitored 3, 14, and 28 days postinfection. At each time point, four to eight 8-mm punch biopsies were taken from two wounds on each pig (four wounds for each time point). Biopsy specimens were homogenized in 1 ml phosphate-buffered saline (PBS) and plated on Pseudomonas isolation agar (PIA) supplemented with 100 μg/ml gentamicin for CFU/gram tissue. To screen for the emergence of adapted P. aeruginosa variants, homogenized tissue was also plated onto adjusted Vogel-Bonner minimal medium (VBMM) (see Supplementary Methods in Text S1) supplemented with 100 μg/ml gentamicin. Colony morphology variants were passaged onto PIA followed by two rounds on Luria agar (LA), before being plated back onto VBMM (without antibiotics) to confirm that the variant phenotype was a result of a stable mutation. Confirmed colony variants were stored at −80°C.
The selection of RSCVs in the wound was determined by calculating the selection coefficient (s) according to equation (1) (48)
| (1) |
where Tx is day x and M and W are the number of mutant and wild-type cells, respectively, at day x and 0.
Sequencing and analysis.
To determine the frequency of each strain across the infection, strain-specific barcodes were amplified and given Illumina sequencing adapters as detailed in Supplementary Methods in Text S1. Library sequencing pools were sequenced on NextSeq and MiniSeq High Output SE75 runs at the Petit Institute Molecular Evolution Core Facility at Georgia Institute of Technology. Analysis script is available on GitHub (https://github.com/glew8/Barcode_Sequencing). Briefly, FastQC 0.11.7 and MultiQC v1.5 were used to confirm sufficient sequencing quality (49, 50). Cutadapt 1.13 was used to select sequences with the insert sequence and parse reads containing each barcode, which were counted using egrep (51).
To identify the ancestor strain that the isolated RSCVs evolved from, colony PCRs were performed using ancestor strain-specific primers. Forward primers contained the unique barcode used to tag each ancestor strain at the Tn7 site. Primers are indicated in Table S3.
To identify mutations, genomic DNA was isolated from colony variants using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s protocol. Clonal DNA was sequenced on the Illumina NextSeq 500 at the University of Pittsburgh Microbial Genomics Sequencing center using a modified protocol for library prep using the Illumina Nextera kit (52). 2x151bp sequencing reads for selected isolates were trimmed and quality filtered using Trimmomatic v0.36 (settings: LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:70) (53). The reads passing quality filtering were then used for variant calling with the open-source program breseq v0.30.0 using default settings (54).
Planktonic competition.
To compete the six ancestor strains in vitro, strains were grown independently overnight in supplemented M9 medium (42.2 mM Na2HPO4, 22 mM KH2PO4, 21.7 mM NaCl, 18.7 mM NH4Cl, 0.1 mM CaCl2, 1 mM MgSO4, and 11.1 mM glucose, supplemented with 20 ml/liter minimum essential medium [MEM] essential amino acids, 10 ml/liter MEM nonessential amino acids, and 1 ml each of trace mineral solutions A, B, and C [Corning catalog no. 25021-3Cl] [55]). Replicate tubes containing 5 ml supplemented M9 medium were inoculated with 50 μl of each overnight culture. Each day, for 12 days (∼80 generations total), 50 μl of culture was transferred to a new tube filled with 5 ml supplemented M9 medium. Populations were frozen on days 1, 3, 4, 6, 7, 9, 10, and 12. At each time point, genomic DNA was isolated and sequenced as described above for mutation calling. Three replicates were sequenced at each time point. All sequencing reads from isolates are deposited in NCBI SRA under Biosample accession numbers SAMN11956547 to SAMN11956595.
c-di-GMP assays.
c-di-GMP reporter plasmid pCdrA::gfp and empty vector pMH487 (31) were electroporated into selected isolates. Overnight cultures were diluted to an optical density at 600 nm (OD600) of 0.1, and the cell pellets were resuspended in PBS. One hundred microliters were transferred to a polystyrene black 96-well plate (Corning). Cells were measured on a SpectraMax i3 plate reader (Molecular Devices) for OD600 and GFP fluorescence. Fluorescence was measured as fluorescence intensity units (FIU) using an excitation of 485 nm and emission of 535 nm. Four biological replicates, each with three technical replicates, were performed. The FIU values were normalized to OD, and the average autofluorescence from the empty vector control was subtracted for each strain. The FIU of RSCVs was determined relative to the ancestor strain, which was set at 100%.
The P. aeruginosa PDE PA2133 was shuttled from pJN2133 (32) as an EcoRI-XbaI fragment into pHERD20T (56), forming the new plasmid pHERD2133. This was electroporated into selected isolates. Colony morphology was assayed on VBMM with 0.1% arabinose.
Biofilm assays.
Microtiter crystal violet assays were performed as previously described (57). Briefly, overnight cultures were diluted 1:10 into fresh media, and 100 μl was transferred into a 96-well round-bottom polyvinyl chloride (PVC) plate (Corning) and incubated for 24 h at 37°C in a humidified chamber. Biofilms were stained with 120 μl of 0.1% crystal violet for 30 min at room temperature. Crystal violet was extracted with 150 μl of 100% ethanol for 30 min at room temperature. One hundred microliters was transferred to a new plate, and OD590 was measured on a SpectraMax i3 plate reader (Molecular Devices). Three biological replicates, each with two technical replicates, were performed.
Fitness assays.
The ancestor strain PA14-1 was tagged with lacZ at the attB site to generate the strain PA14-1attB::lacZ. Briefly, miniCTX-lacZ (58) was conjugated into PA14-1. The vector backbone was removed by electroporating pFLP2 (59), which was subsequently cured by growth on LA supplemented with 10% sucrose. Colonies were screened for tetracycline and carbenicillin sensitivity and blue selection on 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal).
Overnight cultures were grown in 5 ml tryptic soy broth (TSB) (30 g/liter TSB). For each competition, five replicate tubes were inoculated with 25 μl of overnight culture of both the RSCV and the marked ancestor (PA14-1attB::lacZ). Cultures were inoculated into tubes containing 5 ml of TSB either with a 7-mm polystyrene bead (biofilm) or without the bead (planktonic). After 24 h, 50 μl of culture was transferred to a second tube containing fresh TSB for the planktonic cultures, and for the biofilm cultures, the biofilm-coated polystyrene bead was transferred to a second tube containing an uninoculated bead. After a second round of 24-h growth, all competitions were diluted and plated for CFU counts onto tryptic soy agar plates solidified with 1.5% agar and supplemented with 80 mg/ml X-Gal. In order to assess biofilm CFU, the beads were sonicated in PBS with a probe sonicator for 10 s at 30% power, and the resulting supernatant was used for plating. The selection coefficient (s) was calculated according to equation 1.
Prophage isolation and plaque assay.
To isolate prophages from B23-2, an overnight culture of B23-2 was diluted 1:100 and incubated for 30 min at 37°C with shaking at 200 rpm. Mitomycin C (0.5 μg/ml) was added to the culture, and the OD600 was measured. The culture was reincubated, and the OD600 was measured every hour. When the OD600 began to decrease, the cells were pelleted by centrifugation, and the supernatant was filter sterilized and stored at 4°C.
To determine the level of susceptibility of P. aeruginosa strains to the bacteriophage(s) isolated from B23-2, 100 to 200 μl of mid-log P. aeruginosa culture was incubated with 100-μl serial dilutions of bacteriophage lysate for 15 min at 37°C. The infection was added to 5 ml molten soft agar (LB solidified with 0.7% agar) supplemented with 10 mM CaCl2 and MgSO4. This was then poured over solidified hard agar (LB solidified with 1.5% agar), allowed to solidify, and incubated overnight. The number of resulting plaques was then counted, and the number of PFU/ml was determined.
Statistical analysis.
Data are presented as mean ± standard deviation (SD). To determine whether data conformed to a normal distribution, a Shapiro-Wilk test was performed. All of the data sets were normally distributed except for the PFU/ml data (Fig. 6). For these data sets, means were compared using the nonparametric t test. All other comparisons were made using a one-way analysis of variance (ANOVA) with a Tukey’s post-hoc test and Student’s t test. Analyses were performed using GraphPad Prism v.5 (GraphPad Software). Statistical significance was determined using a P value of <0.05.
Data availability.
The reference sequences for variant calling were acquired from NCBI’s RefSeq database (NC_002516.2 for PAO1, NC_008463.1 for PA14). All sequencing reads from isolates are deposited in NCBI SRA under Bioproject number PRJNA491911 and Biosample accession numbers SAMN10101410 to SAMN10101459.
ACKNOWLEDGMENTS
We thank Michael Kann for his help with the planktonic and biofilm fitness assays.
A CFF Postdoctoral Research Fellowship (GLOAG17F0) supported E.S.G. V.S.C. and C.W.M. were supported by NIH grants (R01GM110444 and U01AI124302). D.J.W. was supported by NIH grants (R01AI077628, R01AI097511 and R01AI134895).
E.S.G. and C.W.M. performed the experimental work. J.S.H. performed the colony PCR. S.B.C. infected and sampled the porcine burn wounds. D.S. generated the sequence library. M.W. and G.R.L. quantified the strain frequency in the wounds. A.S.-L. and C.W.M. quantified the strain frequencies from planktonic growth. E.S.G., C.W.M., M.W., V.S.C., and D.J.W. conceptualized the project and wrote the manuscript.
Footnotes
Citation Gloag ES, Marshall CW, Snyder D, Lewin GR, Harris JS, Santos-Lopez A, Chaney SB, Whiteley M, Cooper VS, Wozniak DJ. 2019. Pseudomonas aeruginosa interstrain dynamics and selection of hyperbiofilm mutants during a chronic infection. mBio 10:e01698-19. https://doi.org/10.1128/mBio.01698-19.
Contributor Information
Frederick M. Ausubel, Mass General Hospital.
Stephen Lory, Harvard Medical School.
Paul Turner, Yale University.
REFERENCES
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.Evans TJ. 2015. Small colony variants of Pseudomonas aeruginosa in chronic bacterial infection of the lung in cystic fibrosis. Future Microbiol 10:231–239. doi: 10.2217/fmb.14.107. [DOI] [PubMed] [Google Scholar]
- 3.Lässig M, Mustonen V, Walczak AM. 2017. Predicting evolution. Nat Ecol Evol 1:0077. doi: 10.1038/s41559-017-0077. [DOI] [PubMed] [Google Scholar]
- 4.Lind PA, Libby E, Herzog J, Rainey PB. 2019. Predicting mutational routes to new adaptive phenotypes. Elife 8:e38822. doi: 10.7554/eLife.38822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pestrak MJ, Chaney SB, Eggleston HC, Dellos-Nolan S, Dixit S, Mathew-Steiner SS, Roy S, Parsek MR, Sen CK, Wozniak DJ. 2018. Pseudomonas aeruginosa rugose small-colony variants evade host clearance, are hyper-inflammatory, and persist in multiple host environments. PLoS Pathog 14:e1006842. doi: 10.1371/journal.ppat.1006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haussler S, Tummler B, Weissbrodt H, Rohde M, Steinmetz I. 1999. Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin Infect Dis 29:621–625. doi: 10.1086/598644. [DOI] [PubMed] [Google Scholar]
- 7.Thomassen MJ, Demko CA, Boxerbaum B, Stern RC, Kuchenbrod PJ. 1979. Multiple isolates of Pseudomonas aeruginosa with differing antimicrobial susceptibility patterns from patients with cystic fibrosis. J Infect Dis 140:873–880. doi: 10.1093/infdis/140.6.873. [DOI] [PubMed] [Google Scholar]
- 8.Boles BR, Thoendel M, Singh PK. 2004. Self-generated diversity produces “insurance effects” in biofilm communities. Proc Natl Acad Sci U S A 101:16630–16635. doi: 10.1073/pnas.0407460101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flynn KM, Dowell G, Johnson TM, Koestler BJ, Waters CM, Cooper VS. 2016. Evolution of ecological diversity in biofilms of Pseudomonas aeruginosa by altered cyclic diguanylate signaling. J Bacteriol 198:2608–2618. doi: 10.1128/jb.00048-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogardt M, Heesemann J. 2010. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol 300:557–562. doi: 10.1016/j.ijmm.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 11.Lam J, Chan R, Lam K, Costerton JW. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect Immun 28:546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. 2000. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- 13.Drenkard E, Ausubel FM. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740–743. doi: 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- 14.Kirisits MJ, Prost L, Starkey M, Parsek MR. 2005. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microbiol 71:4809–4821. doi: 10.1128/AEM.71.8.4809-4821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma L, Jackson KD, Landry RM, Parsek MR, Wozniak DJ. 2006. Analysis of Pseudomonas aeruginosa conditional psl variants reveals roles for the psl polysaccharide in adhesion and maintaining biofilm structure postattachment. J Bacteriol 188:8213–8221. doi: 10.1128/JB.01202-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hengge R. 2009. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 17.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, Wozniak DJ, Harwood CS, Parsek MR. 2009. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pu M, Sheng L, Song S, Gong T, Wood TK. 2018. Serine hydroxymethyltransferase ShrA (PA2444) controls rugose small-colony variant formation in Pseudomonas aeruginosa. Front Microbiol 9:315. doi: 10.3389/fmicb.2018.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goymer P, Kahn SG, Malone JG, Gehrig SM, Spiers AJ, Rainey PB. 2006. Adaptive divergence in experimental populations of Pseudomonas fluorescens. II. Role of the GGDEF regulator WspR in evolution and development of the wrinkly spreader phenotype. Genetics 173:515–526. doi: 10.1534/genetics.106.055863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spiers AJ, Kahn SG, Bohannon J, Travisano M, Rainey PB. 2002. Adaptive divergence in experimental populations of Pseudomonas fluorescens. I. Genetic and phenotypic bases of wrinkly spreader fitness. Genetics 161:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marvig RL, Sommer LM, Molin S, Johansen HK. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet 47:57. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- 22.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy S, Elgharably H, Sinha M, Ganesh K, Chaney S, Mann E, Miller C, Khanna S, Bergdall VK, Powell HM, Cook CH, Gordillo GM, Wozniak DJ, Sen CK. 2014. Mixed-species biofilm compromises wound healing by disrupting epidermal barrier function. J Pathol 233:331–343. doi: 10.1002/path.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaney SB, Ganesh K, Mathew-Steiner S, Stromberg P, Roy S, Sen CK, Wozniak DJ. 2017. Histopathological comparisons of Staphylococcus aureus and Pseudomonas aeruginosa experimental infected porcine burn wounds. Wound Repair Regen 25:541–549. doi: 10.1111/wrr.12527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Summerfield A, Meurens F, Ricklin ME. 2015. The immunology of the porcine skin and its value as a model for human skin. Mol Immunol 66:14–21. doi: 10.1016/j.molimm.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan TP, Eaglstein WH, Davis SC, Mertz P. 2001. The pig as a model for human wound healing. Wound Repair Regen 9:66–76. doi: 10.1046/j.1524-475x.2001.00066.x. [DOI] [PubMed] [Google Scholar]
- 27.Abdullahi A, Amini-Nik S, Jeschke MG. 2014. Animal models in burn research. Cell Mol Life Sci 71:3241–3255. doi: 10.1007/s00018-014-1612-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Branda SS, Vik A, Friedman L, Kolter R. 2005. Biofilms: the matrix revisited. Trends Microbiol 13:20–26. doi: 10.1016/j.tim.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Haussler S, Fuqua C. 2013. Biofilms 2012: new discoveries and significant wrinkles in a dynamic field. J Bacteriol 195:2947–2958. doi: 10.1128/JB.00239-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman L, Kolter R. 2004. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol 186:4457–4465. doi: 10.1128/JB.186.14.4457-4465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rybtke MT, Borlee BR, Murakami K, Irie Y, Hentzer M, Nielsen TE, Givskov M, Parsek MR, Tolker-Nielsen T. 2012. Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosa. Appl Environ Microbiol 78:5060–5069. doi: 10.1128/AEM.00414-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hickman JW, Tifrea DF, Harwood CS. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A 102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poltak SR, Cooper VS. 2011. Ecological succession in long-term experimentally evolved biofilms produces synergistic communities. ISME J 5:369–378. doi: 10.1038/ismej.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cady K, White A, Hammond J, Abendroth M, Karthikeyan R, Lalitha P, Zegans M, O’Toole G. 2011. Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 157:430–437. doi: 10.1099/mic.0.045732-0. [DOI] [PubMed] [Google Scholar]
- 35.Khan AI, Dinh DM, Schneider D, Lenski RE, Cooper TF. 2011. Negative epistasis between beneficial mutations in an evolving bacterial population. Science 332:1193–1196. doi: 10.1126/science.1203801. [DOI] [PubMed] [Google Scholar]
- 36.Cooper VS, Staples RK, Traverse CC, Ellis CN. 2014. Parallel evolution of small colony variants in Burkholderia cenocepacia biofilms. Genomics 104:447–452. doi: 10.1016/j.ygeno.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Rainey PB, Rainey K. 2003. Evolution of cooperation and conflict in experimental bacterial populations. Nature 425:72. doi: 10.1038/nature01906. [DOI] [PubMed] [Google Scholar]
- 38.Rainey PB, Travisano M. 1998. Adaptive radiation in a heterogeneous environment. Nature 394:69. doi: 10.1038/27900. [DOI] [PubMed] [Google Scholar]
- 39.Kim W, Levy SB, Foster KR. 2016. Rapid radiation in bacteria leads to a division of labour. Nat Commun 7:10508. doi: 10.1038/ncomms10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O’Toole GA. 2012. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J Bacteriol 194:5728–5738. doi: 10.1128/JB.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ueda A, Wood TK. 2009. Connecting quorum sensing, c-di-GMP, pel polysaccharide, and biofilm formation in Pseudomonas aeruginosa through tyrosine phosphatase TpbA (PA3885). PLoS Pathog 5:e1000483. doi: 10.1371/journal.ppat.1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malone JG, Jaeger T, Spangler C, Ritz D, Spang A, Arrieumerlou C, Kaever V, Landmann R, Jenal U. 2010. YfiBNR mediates cyclic-di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog 6:e1000804. doi: 10.1371/journal.ppat.1000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giddens SR, Jackson RW, Moon CD, Jacobs MA, Zhang XX, Gehrig SM, Rainey PB. 2007. Mutational activation of niche-specific genes provides insight into regulatory networks and bacterial function in a complex environment. Proc Natl Acad Sci U S A 104:18247–18252. doi: 10.1073/pnas.0706739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones CJ, Newsom D, Kelly B, Irie Y, Jennings LK, Xu B, Limoli DH, Harrison JJ, Parsek MR, White P, Wozniak DJ. 2014. ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Pathog 10:e1003984. doi: 10.1371/journal.ppat.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irie Y, Starkey M, Edwards AN, Wozniak DJ, Romeo T, Parsek MR. 2010. Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol Microbiol 78:158–172. doi: 10.1111/j.1365-2958.2010.07320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 47.Choi K-H, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5:30. doi: 10.1186/1471-2180-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper VS. 2018. Experimental evolution as a high-throughput screen for genetic adaptations. mSphere 3:e00121-18. doi: 10.1128/mSphere.00121-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom. [Google Scholar]
- 50.Ewels P, Magnusson M, Lundin S, Käller M. 2016. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 52.Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. 2015. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10:e0128036. doi: 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq, p 165–188. In Sun L, Shou W (ed), Engineering and analyzing multicellular systems: methods and protocols. Springer, Berlin, Germany. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santos-Lopez A, Marshall CW, Scribner MR, Snyder D, Cooper VS. 2019. Biofilm-dependent evolutionary pathways to antibiotic resistance. bioRxiv 10.1101/581611. [DOI] [PMC free article] [PubMed]
- 56.Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74:7422–7426. doi: 10.1128/AEM.01369-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Toole GA. 2011. Microtiter dish biofilm formation assay. J Vis Exp 47:2437. doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 59.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 60.Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, Miyada CG, Lory S. 2003. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:8484–8489. doi: 10.1073/pnas.0832438100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotypic profile of P. aeruginosa strains used in the infection. (A) Metabolic activity of the six P. aeruginosa strains assayed for 16h at 37°C using a Biolog system. (B) Area under the curve (AUC) of the Biolog kinetic plot depicted in panel A. **, P value of <0.01. (C) Biofilms of the six P. aeruginosa strains were grown for 24 h at 37°C in a 96-well plate. Biomass levels were quantified by crystal violet staining. Data are depicted as mean ± SD (n = 3). (D) The frequency of each strain in the starting inoculum was determined by sequencing the strain-specific barcodes at the Tn7 site. The proportion of each barcode was expressed as a percentage of the total sequence counts. Download FIG S1, PDF file, 0.1 MB (128.8KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental Materials and Methods and Results. Download Text S1, PDF file, 0.2 MB (175.1KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The PA14-1 non-RSCV population in the porcine wounds behave similarly to the ancestor strain. (A) Five non-RSCV colonies were randomly selected from each wound. The ancestor of each colony was identified and expressed as a percentage of the total number of colonies isolated from that time point. (B) Biofilms (24 h) were grown in 96-well plates, and biomass levels were quantified by crystal violet staining. Biomass levels are expressed as a percentage relative to PA14-1, which was set at 100%. ns indicates no significant difference. (C) AUC of Biolog kinetic curves. AUC are expressed as a percentage relative to PA14-1, which was set at 100%. Gray scale in panels B and C indicate different wounds. (D to F) Kinetic Biolog curves of representative non-RSCV PA14-1 isolates from each time point (labeled). Data are presented as mean ± SD (n = 3). *, P value of <0.05; **, P value of <0.01; ***, P value of <0.001. Download FIG S2, PDF file, 0.4 MB (395.4KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mutations in non-RSCV PA14-1 population. Download Table S1, PDF file, 0.1 MB (56.3KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PA14-1 RSCVs from the porcine wounds have an extended lag phase compared to the ancestor strain. (A) AUC of Biolog metabolic kinetic curves of all sequenced PA14-1 RSCVs, expressed as a percentage relative to PA14-1. PA14ΔwspF and its parent PA14 were used for comparison. *, P value of <0.01; #, P value of <0.001 compared to the ancestor strain. (B to E) Kinetic Biolog curves of representative PA14-1 RSCVs (labeled). Data are presented as mean ± SD (n = 3). Download FIG S3, PDF file, 0.3 MB (335.6KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Contig sequence from B23-2 with the protospacer sequence. There is currently no annotated sequence for B23. Therefore, the CRISPR spacer sequences were aligned against the contig sequences of the ancestor B23-2, which was sequenced along with the representative RSCVs. Protospacer for the CRISPR spacer in RSCV-12 (contig 107) (A) and RSCV-38 (contig 95) (B) were identified in two separate contigs (bold text). For the protospacer sequence in contig 95 (B), there was one base pair mismatch when comparing the CRISPR spacer to the protospacer (base pair that is not bold). After each protospacer, there is a conserved GG PAM motif (underlined) which is required for type 1-F CRISPR-Cas families which P. aeruginosa PA14 possesses. A BLAST search of the contig sequences identified CRISPR spacers in P. aeruginosa strains (Table S2) aligned to other protospacers in contig 107 (A), indicated by the red and blue text. Download FIG S4, PDF file, 1.6 MB (1.7MB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
WspA deletions. (A) The 42-bp deletion in wspA is located between direct repeats. The gene sequence of PA14 wspA is shown. The 42-bp deletion (853-894bp) in the wspA mutants is underlined. Either side of this sequence is a direct repeat indicated in bold text. (B) Alignment of WspA deletions. Deletions in homologous regions of wspA have been observed in in vitro-evolved RSCVs; a 286-307aa deletion in P. aeruginosa PAO1 (MJK8), a 284-311aa deletion in P. fluorescence Pfl01 and a 307-313aa deletion in Burkholderia cenocepacia HI2424. The different domains of WspA were determined from Pfam analysis from the Pseudomonas Genome Database and the Burkholderia Genome Database. Domains are colored as follows: purple for the ligand binding domain or the four-helix bundle domain, green for the HAMP or linker domain, and blue for the MCP signaling domain. The region of the deletion in each protein is indicated in red. Download FIG S5, PDF file, 2.0 MB (2MB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A 14-aa deletion in WspA is predicted to occur opposite the methylation site. (A) Schematic of WspA. The different domains of WspA were determined from the Pseudomonas Genome Database Pfam analysis. LBD, ligand binding domain or the four-helix bundle domain (3-182aa); HAMP, linker domain (213-261aa); SD, MCP signaling domain (348-505aa). The region of the 14aa deletion is indicated in red (285-298aa). (B) The WspA cytoplasmic domain amino acid sequence. The domains are indicated by the same colors as in panel A. The signaling domain contains two additional features, the kinase interacting subdomain, or “tip” domain in dark blue (382-420aa) and the predicted methylation site in navy blue (492-501aa). Both the heptad registers (reg) and the heptad number (h#) are labeled, with consecutive heptads indicated in alternating black and gray text. (C) Homology model of PA14 WspA modeled against the T. maritime MCP (PDB 3JA6) generated using SWISS-MODEL. Colors correspond to the domains indicated in panel B. The model spans 250-541aa of WspA. Download FIG S6, PDF file, 0.4 MB (433.3KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CRISPR spacers in P. aeruginosa strains with a protospacer aligning to contig 107 in B23-2. Download Table S2, PDF file, 0.01 MB (15.6KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S3, PDF file, 0.02 MB (16.1KB, pdf) .
Copyright © 2019 Gloag et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The reference sequences for variant calling were acquired from NCBI’s RefSeq database (NC_002516.2 for PAO1, NC_008463.1 for PA14). All sequencing reads from isolates are deposited in NCBI SRA under Bioproject number PRJNA491911 and Biosample accession numbers SAMN10101410 to SAMN10101459.