

Abstract
Background and Purpose
Attention training reverses the neurodegeneration and memory loss promoted by infusion of amyloid‐β (Aβ) peptide in rats and increases the density of α7 nicotinic ACh receptors (α7nAChRs) in brain areas related to memory. Hence, we aimed to assess the role of α7nAChRs in the memory recovery promoted by attention training.
Experimental Approach
C57Bl/6 mice were chronically infused with Aβ, Aβ plus the α7 antagonist methyllycaconitine (MLA), or MLA alone. Control animals were infused with vehicle. Animals were subjected weekly to the active avoidance shuttle box for 4 weeks (attention training). The brain and serum were collected for biochemical and histological analysis.
Key Results
Aβ caused cognitive impairment, which was reversed by the weekly training, whereas Aβ + MLA also promoted memory loss but with no reversal with weekly training. MLA alone also promoted memory loss but with only partial reversal with the training. Animals infused with Aβ alone showed senile plaques in hippocampus, no change in BDNF levels in cortex, hippocampus, and serum, but increased AChE activity in cortex and hippocampus. Co‐treatment with MLA increased AChE activity and senile plaque deposition in hippocampus as well as reducing BDNF in hippocampus and serum, suggesting a lack of α7nAChR function leads to a loss of neuroprotection mechanisms.
Conclusions and Implications
The α7nAChR has a determinant role in memory recovery and brain resilience in the presence of neurodegeneration promoted by Aβ peptide. These data support further studies concerning these receptors as pharmacological targets for future therapies.
Abbreviations
- AD
Alzheimer's disease
- Aβ
amyloid‐β
- BDNF
brain‐derived neurotrophic factor
- CAR
conditioned avoidance response
- MLA
methyllycaconitine
- α7nAChR
α7 nicotinic ACh receptor
What is already known
Cholinergic system modulates memory, and its function can be influenced by an individual's lifestyle.
Strategies like attention training can reverse memory loss promoted by neurodegeneration, forming a cognitive reserve.
What this study adds
Antagonism of α7nAChRs prevented neuroplasticity, increased senile plaques, and decreased cognitive function.
What is the clinical significance
It is possible that α7 agonists and anticholinesterasic agents together with complementary strategies could be used to maintain cognitive reserve.
1. INTRODUCTION
Alzheimer's disease (AD) is still the principal concern all over the world regarding the ageing process, as it is the main cause of gradual enhancement in cognitive disability (Lane, Hardy, & Schott, 2018). It is well known that AD has a multifactorial aetiology resulting in progressive and irreversible dementia. Most of the patients (99%) present the sporadic form of the disease, with a late onset (beginning after 65 years of age) characterized by an increase in amyloid‐β (Aβ) accumulation, leading to a great number of senile plaques, formation of neurofibrillary tangles, and inflammation (Ballard et al., 2011; Masters et al., 2015; Thal, Walter, Saido, & Fändrich, 2015; Scheltens et al., 2016; Lane et al., 2018). A small proportion of patients (1%), however, present the familial, most severe form of the disease, with mutations in genes related to the processing of Aβ peptide and early onset—around 45 years of age (Wattmo & Wallin, 2017). As a consequence for both forms, the neuronal loss and synaptic disruption impair learning and memory, cognitive capacity, and ability to deal with daily living skills (Dorostkar, Zou, Blazquez‐Llorca, & Herms, 2015; Wattmo & Wallin, 2017). Whitehouse et al. (1982) showed that AD patients presented 75% loss in cholinergic neurons of the nucleus basalis of Meynert, indicating that a deficiency in function of the cholinergic system is critical for the development of the disease. After that, the loss of cholinergic neurons and the consequent reduction in choline transportation and in the density of ACh receptors were also reported in many studies. Post‐mortem examinations have shown a significant reduction in the activity of choline acetyltransferase, the enzyme that synthesizes ACh, and an increase in AChE, which degrades the neurotransmitter ACh (Duan et al., 2014; García‐Ayllón, Silveyra, & Sáez‐Valero, 2008; Sivaprakasam, 2006). Therefore, until today, the pharmacological strategy to delay the progression of the disease in the initial or intermediate phases is the use of AChE inhibitors (rivastigmine, galantamine, and donepezil; Kulshreshtha & Piplani, 2016; Waite, 2015), leading to an increase in cholinergic system function and LTP maintenance. Besides, several studies point to the involvement of α7 nicotinic ACh receptor (α7nAChR) as a promising therapeutic target in the management of AD (Hernandez & Dineley, 2012). α7nAChR is a homopentameric neuronal receptor permeable to calcium ions and is one of the most frequently found ACh receptors in the CNS. Its ionotropic/metabotropic dual action has allowed the receptor's implication in memory modulation and anti‐inflammatory and neuroprotective mechanisms (Corradi & Bouzat, 2016; Foucault‐Fruchard et al., 2017; Picciotto, Higley, & Mineur, 2012). On the other hand, some studies have shown that the pharmacological blockade of α7nAChRs with the antagonist methyllycaconitine (MLA) produced cognitive deficits in mice (Addy, Nakajama, & Levin, 2003; Andriambeloson, Huyard, Poiraud, & Wagner, 2014). MLA is an alkaloid extracted from the seeds of Delphinium brawnii. It is a highly selective inhibitor of the well‐known α7nAChR antagonist α‐bungarotoxin in vertebrate brain with affinity in concentrations as low as the nanomolar range (Davies et al., 1999; Turek, Kang, Campbell, Arneric, & Sullivan, 1995; Alkondon, Pereira, Wonnacott & Albuquerque, 1992).
There is, as yet, no efficient strategy to treat neuronal and memory loss during the development of the disease. However, many studies have shown that retaining cognitive function during the ageing process depends on the formation of cognitive and structural reserves (Balthazar, Schöwe, Cipolli, Buck, & Viel, 2018; Baraldi et al., 2013; Grant, Dennis, & Li, 2014; Lavrencic et al., 2018; Stern, 2012). This could lead to the development of improved brain resilience against future injuries. These reserves are built through physical, social, motor, sensorial, and cognitive activities and can be key to separate preserved memory during normal ageing and the initial stages of dementia.
In a previous study, our research team showed that rats chronically infused with Aβ peptide and subsequently submitted to attention training had an improvement in memory. In addition, an increase in α7nAChRs in areas related to attention, memory, and emotions was observed (Viel, Caetano, Albuquerque, Araujo, & Buck, 2012). In this way, it was suggested that attention training improved cognitive reserves and helped reverse memory loss by mechanisms dependent on α7nAChRs. The aim of this study was to assess the role of α7nAChR in memory recovery produced by sustained attention training following chronic infusion of Aβ peptide.
2. METHODS
2.1. Animal welfare and ethical statement
A total of 77 male C57Bl/6 mice (2 months old) were provided by the National Institute of Pharmacology Facility, Federal University of São Paulo. This number of animals was necessary as we had to subject the animals to the active avoidance apparatus in order to select those who had the ability to learn and memorize the task (Viel et al., 2008).
The animals were kept in ventilated cages (Alesco, Brazil) at a controlled room temperature (22–24°C) and humidity (55–65%), on a 12‐hr light/dark cycle with food and water available ad libitum. All care procedures were strictly performed according to the guidelines for animal experimentation as stipulated in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication Number 86–23, Bethesda, MD), and all procedures were approved by the Ethics Committee on Experimental Research of the Institute of Biomedical Sciences, University of São Paulo, Brazil (#164, Sheet 143, Book 02). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. The authors declare that every effort was made to minimize the number of animals used and their level of suffering.
2.2. Experimental protocols and design
2.2.1. Assessment of aversive‐related memory response and sustained attention
Evaluation of memory evocation and neurostimulation were performed using an active avoidance apparatus that produces alert and sustained attention in animals (Viel et al., 2008), based on a protocol previously used by our group (Amaral et al., 2010; Lemos et al., 2010). The trials were performed in a two‐way shuttle box (Ugo Basile, Comerio, Italy) consisting of two compartments accessible to each other by a hole in the dividing wall. Each animal was placed individually and then acclimatized to the shuttle‐box apparatus for 5 min before each session. The animal was then subjected to 50 avoidance conditioning trials (acquisition test), with a pause of 5 min after the 25th trial. Each trial consisted of a 2‐s conditioned stimulus, that is, a buzzer (70 dB, 760 Hz) and a light. If the animal did not cross to the other side during this period, a scramble shock of 0.2 mA for 4 s was delivered through the floor grid (unconditioned stimulus). Each trial was separated by fixed intertrial intervals (20 s), during which animals were left inside the apparatus. Animals were subjected to this protocol for three consecutive days in order to ensure that all animals in the experiment had the same level of acquired memory. During this period, the number of conditioned avoidance responses (CARs), in which the animals moved to the other compartment of the shuttle box before the beginning of the unconditioned stimulus, was recorded. Those that reached a satisfactory learning performance—that is, those achieving CAR rates between 30% and 70% (Amaral et al., 2010; Viel et al., 2008)—were selected to be subjected to the surgical procedures (described below). So, from the 77 animals initially selected for the experiment, 57 were included in the study. Weekly repetition of the procedure was used to improve sustained attention and promote reversal of memory loss, as described previously (Viel et al., 2012). All tests were carried out during the light phase (9:00 a.m.–3:00 p.m.). While the animal was in the equipment, the operator only observed and appropriately saved data. The equipment does not permit any operator interference, but any sign of freezing (immobilization of animal) during the protocol was sufficient to make the operator stop the equipment and take the animal out. Animals that presented this behaviour accounted for those with less than 30% CAR rates. The experimental design is shown in the timeline below (Figure 1).
Figure 1.
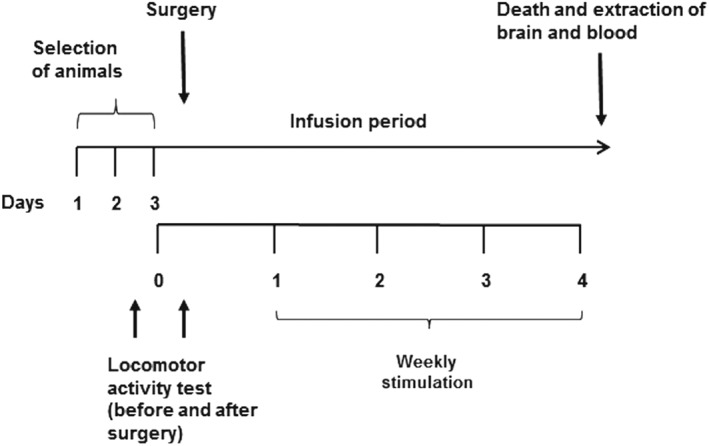
Timeline of the experimental design. Animals were subjected to the active avoidance shuttle box for 3 days. Those that achieved a response between 30% and 70% of conditioned avoidance responses were selected to be subjected to surgery for infusion of amyloid‐β (Aβ), methyllycaconitine (MLA), or both. Before and after the surgery, a locomotion test was performed in order to verify if the surgery influenced this parameter. No difference in locomotion was observed among groups after the surgery (control: 377.4 ± 50.10 units, n = 14; Aβ: 340.7 ± 34.27 units, n = 14; Aβ + MLA: 365.9 ± 44.85, n = 12; and MLA: 415.3 ± 53.13, n = 11). During the infusion period, animals were subjected to weekly stimulations in the same active avoidance shuttle box, as a strategy of attention training. After the behavioural observations, animals were anaesthetized and killed. Blood and brain were collected and frozen for posterior analysis
2.2.2. Surgery for mini‐pump implantation
The selected animals (total of 57) were randomly divided into four groups and subjected to surgery for the implantation of mini‐osmotic pumps (Model 1004, Alzet, Cupertino, CA, USA). The animals were injected with atropine (0.04 mg·kg−1) 15 min before the injection of the anaesthetics (8.3 mg·kg−1 ketamine and 0.30 mg·kg−1 xylazine; anaesthetics and atropine were administered intraperitoneally) in order to avoid bradycardia induced by these drugs. After anaesthesia, a stainless steel cannula (Brain Kit 1, Alzet, Cupertino, CA, USA) was implanted in the animal's lateral ventricle using a stereotaxic instrument at coordinates: −0.8 mm anteroposterior, −1.4 mm mediolateral to the bregma, and −3.5 mm dorsoventral to the cranium (Franklin & Paxinos, 2007). The other extremity of the cannula was attached to a PVC catheter (medical grade, OD = 1.14 mm, ID = 0.69 mm) connected to the mini‐osmotic pump that was implanted s.c. in the dorsum of the animal's neck. The contents of the mini‐osmotic pumps were delivered at a flow of 0.11 μl·hr−1, having a total volume of 100 μl, according to the manufacturer's guidelines (Amaral et al., 2010). As described earlier (Amaral et al., 2010; Frautschy et al., 1998; Viel et al., 2008), E‐64 was infused together with the Aβ peptide to induce neurodegeneration. E‐64 is a cysteine protease inhibitor known for increasing the neurodegeneration caused by Aβ infusion.
The groups were formed as follows: control group (n = 14): mini‐pumps filled with the vehicle (4‐mM HEPES, pH 8.0—plus 0.22‐nmol E‐64); Aβ group (n = 14): mini‐pumps filled with human (1–42) Aβ peptide (0.46 nmol) plus 0.22‐nmol E‐64; Aβ ± MLA group (n = 12): mini‐pumps filled with Aβ (0.46 nmol) plus the α7 nicotinic antagonist MLA (256‐nmol MLA and 0.22‐nmol E‐64); and MLA group (n = 11): mini‐pumps filled only with MLA (256 nmol), following a method previously described (Frautschy et al., 1998; Viel et al., 2008).
After the surgery, all animals received indomethacin (1 mg·kg−1, s.c.) to avoid inflammation and pain and were placed individually in a transparent cage to prevent them from fighting and displacing the cannula.
Six animals out of 57 died during the surgery or during the recuperation period, following surgery, on account of cardiorespiratory arrest probably due to sensitivity to the drugs used during the procedure.
After the behavioural observations, the animals were anaesthetized with isoflurane, and the blood was collected by cardiac puncture. After 30 min in room temperature, blood samples were centrifuged (14,031× g, 15 min, 4°C), and the serum was separated. Animals were, then, killed by decapitation, and the brains were removed and immediately frozen in dimethylbutane at −50°C and stored at −80°C until used. For each animal, one hemisphere was used for immunohistochemical analysis, and the other hemisphere was submitted to lysis in order to be used in elisas.
2.2.3. Quantification of senile plaques
Quantification was performed as described previously (Nunes et al., 2015). From one of the hemispheres, tissue samples (20 μm) were obtained in a cryostat (−20°C to −22°C, Microm HM505N, Francheville, France), and sections were mounted on gelatin‐coated slides, desiccated for 5 min at room temperature, and kept at −80°C until use. For quantification of senile plaques, the slides were warmed to room temperature (22°C) and air dried (5–10 min). Slides were washed five times in PBS and incubated in thioflavin S solution 0.1% in PBS, containing 0.1% Triton‐100, for 5 min. The sections were then washed twice with PBS, incubated with ethanol 70% for 5 min, and washed, again, three times with PBS. At the end, the sections were covered with coverslips using Fluoroshield with DAPI (Sigma, USA). The complete process was performed in a dark room. Analysis of senile plaques of whole brain, labelled with thioflavin S, was performed using an optic microscope (Nikon Eclipse E‐600), with a suitable filter for fluorescence.
A total of three pictures of the hippocampus from each animal was taken to identify the senile plaques just after labelling. According to previous experience of the research team, at the end of Aβ chronic infusion, senile plaques can be observed at the anteroposterior levels approximately close to −1.70 mm with reference to bregma (Franklin & Paxinos, 2007). At this point, CA1, CA3, and dentate gyrus areas can be observed and analysed. Thioflavin S fluorescence was identified using ImageJ software (1.51j8—National Institutes of Health, USA; RRID:SCR_003070) and counted manually. The quantity of plaques was analysed in 2–3 brain slices from each animal, dividing the number of plaques by the number of slices analysed. The operator was blinded to the groups, and only the supervisor knew the groups.
2.2.4. Quantification of brain‐derived neurotrophic factor protein levels
The cortex and the hippocampus were identified according to the description of Franklin and Paxinos (2007). The areas were isolated and immediately homogenized in lysis buffer containing 20‐mM Tris–HCl (pH 8.0), 137‐mM NaCl, 10% glycerol, 1% Triton X‐100, and a tablet of protease inhibitors (Roche Diagnostics). Homogenates were centrifuged at 14,031× g, for 15 min, at 4°C, and the supernatant was separated and frozen at −80°C until use. The concentration of protein in the brain homogenates and serum was evaluated according to the Bradford method (Bradford, 1976).
For the detection of total free brain‐derived neurotrophic factor (BDNF), a sandwich elisa kit was used (Emax ImmunoAssay System, PROMEGA, Cat # G7611), following the protocol of the manufacturer. For the colorimetric reading, Biotek Eon equipment was used, with Gen5 2.0 software, at 450 nm.
2.2.5. Evaluation of AChE activity
Evaluation of AChE activity was done as previously described (Morzelle et al., 2016). The cortex, hippocampus, and serum samples were obtained as described above. The enzyme activity was measured using an elisa kit (Abcam—ab138871) according to the manufacturer's protocol. The colorimetric reading was made using Biotek Eon equipment, with Gen5 2.0 software, at 410 nm.
2.3. Statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. Results are expressed as means ± SEM and analysed with the GraphPad Prism programme (GraphPad Software, San Diego, CA, version 6; RRID:SCR_002798). For animal selection, CARs of the first and third days were compared using Student's paired t test. Data from the sustained attention test (performed weekly) were analysed using two‐way ANOVA (repeated measures) followed by Bonferroni's multiple comparison test. All other data were also analysed using two‐way ANOVA followed by Bonferroni's multiple comparison test. In each dataset, the homogeneity of sample variance was tested using both Brown–Forsythe's and Bartlett's tests. As long as one of the tests indicated no variance among groups, ANOVA was performed. Only probability values (P) less than .05 were considered statistically significant.
2.4. Drugs
Human (1–42) Aβ peptide and MLA were purchased from Sigma‐Aldrich. All other drugs used were of analytical grade.
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Peters, et al., 2017; Alexander, Fabbro, et al., 2017).
3. RESULTS
3.1. Selection of animals and effects of α7 blockade on memory recovery
During the first contact with the active avoidance equipment (acquisition session), the animals showed a learning behaviour corresponding to 5.3 ± 0.5% (n = 51) from a total of 50 tasks, with a pause between Trials 25 and 26. The tasks were repeated over 3 days, and by the third session (retention session), the same animals presented a 14.5‐fold average increase in CAR, which represented long‐term memory retention of 49.5 ± 2.3% (Figure 2a). Only animals presenting a difference of 30–70% (n = 51) between acquisition and retention sessions were selected and then divided randomly into four experimental groups and submitted to surgery for the implantation of the mini‐osmotic pumps. The third day of observation (retention session) was considered as Week 0 of stimulation in Figure 2b, as it represents the animals' behaviours observed before the surgery (Figure 2a).
Figure 2.
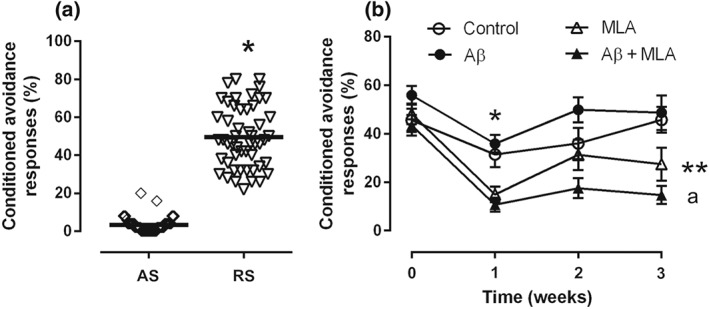
(a) Percentage of conditioned avoidance responses of mice at time of first contact with the equipment (acquisition session [AS]) and 3 days after the AS (retention session [RS]). Symbols are the individual percentages registered. *P < .05. (b) Percentage of conditioned avoidance responses (CARs) of mice with different treatments. (a) Mean values in Week 0 correspond to individual values shown in RS. Infusion of amyloid‐β (Aβ) and/or the α7 nicotinic ACh receptor antagonist methyllycaconitine (MLA) produced a reduction in CAR (*P < .05) in the first test after the beginning of the infusion, when compared with Week 0. Animals infused only with Aβ presented memory recovery after each weekly session of stimulation in the active avoidance equipment. However, animals infused with MLA with or without Aβ did not present memory recovery after any session of weekly stimulation. **P < .05, when compared with Week 0. a P < .05, when compared with the Week 0. On Week 3, there was a difference between control and Aβ + MLA groups. Sample sizes: (a) 51 animals and (b) control group: n = 14; Aβ group: n = 14; Aβ ± MLA group: n = 12; and MLA group: n = 11
One week after surgery, and for the following 3 weeks, all groups were submitted to a weekly sustained attention test, following the method described previously (Viel et al., 2012). Overall, that was difference in time and in behaviour among groups. Also, an interaction among groups was verified, indicating that treatments and time influenced CAR. One week post‐surgery, control animals did not show a reduction in CAR when compared with Week 0 (45.6 ± 4.8%), suggesting that the surgery did not influence the animals' memory (Figure 2b). In addition, the percentage of CAR of these animals increased each week over the 3 weeks and was maintained (45.7 ± 5.4%; Figure 2b). This showed that re‐testing improved memory consolidation, as observed in previous works from our lab (Viel et al., 2012).
In contrast, 1 week after surgery, the animals from the Aβ, Aβ + MLA, and MLA groups presented reduction in CAR of 35.8% (35.9 ± 3.6%), 75.4% (10.7 ± 2.9%), and 69.1% (14.9 ± 3.2), respectively, when compared with the CAR obtained at Week 0 (55.9 ± 3.9%, 43.5 ± 4.2%, and 48.2 ± 4.2%, respectively), although Aβ values were not different from control values, as observed before (Amaral et al., 2010). After the third week, the Aβ group showed a reversion of the memory loss, as CARs (48.7 ± 7.1%) were similar to those obtained before the surgery (Figure 2b). However, animals from the Aβ + MLA group did not present memory recovery during the weekly sessions, as the reductions in CAR after the surgery were maintained until the end (14.7 ± 3.8%), when compared with Week 0. The same phenomenon was observed with animals infused with MLA only, that is, they did not recover the memory of the task, as CAR remained as low as the first week (27.4 ± 6.7%), when compared with Week 0. The responses of the animals infused with Aβ + MLA were 67.9% lower than the control responses and 64.7% lower than the Aβ group responses (Figure 2b).
In order to better understand the weekly stimulation effect of the sessions and the effects of the blockage of the α7nAChR, we analysed and compared the percentage of CAR for each of the 10 trials of the 50 total trials observed each week, as described previously (Viel et al., 2008).
At Week 0 (before the surgery), all selected and randomly assigned animals presented an increase in CAR throughout the 50 trials, showing that although they initially did not know the task, they learned during the 50 trials (Figure 3). No difference was observed in Trials 41–50 among the groups.
Figure 3.
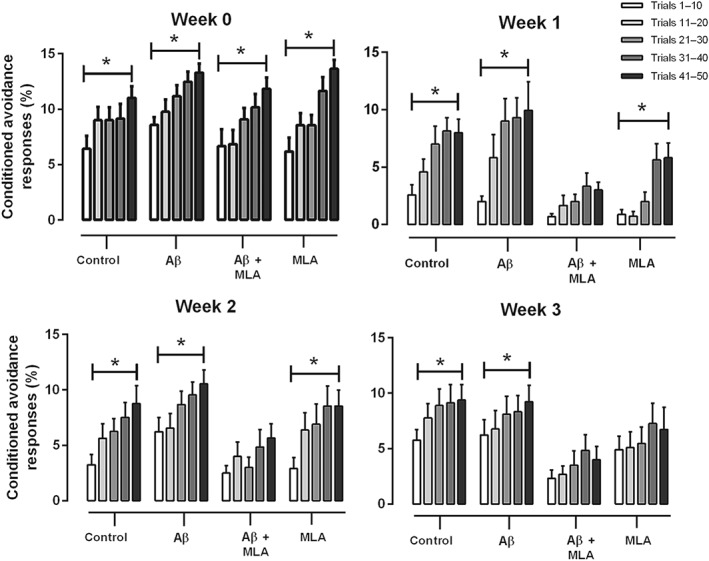
Conditioned avoidance responses (%) for each week of control and amyloid‐β (Aβ) animals infused or not infused with methyllycaconitine (MLA) and re‐subjected to the active avoidance apparatus. Week 0 refers to the test before surgery, where all animals showed increases in conditioned avoidance responses within the 50 trials. Weeks 1, 2, and 3 refer to tests after the surgery where it is possible to note the effects of infused drugs on behaviour. Each set of 50 trials was divided into five groups of 10 trials. Data are presented as means ± SEM. *P < .05. Sample sizes: control group: n = 14; Aβ group: n = 14; Aβ ± MLA group: n = 12; and MLA group: n = 11
For the control and the Aβ groups, following submission to the equipment, a similar behaviour was observed: Each week, during the first 10 trials, the animals did not recognize the task, but they could re‐learn during the other 40 trials, showing an increase in CAR percentage. In contrast, soon after the surgery (Week 1), the Aβ + MLA group showed no increase in CAR during the trials, which indicates a difficulty in context recognition or a deficit in memory recovery. A reduction in CAR in Trials 41–50 of the Aβ + MLA group was also observed, when compared with the Aβ group. In the two last sessions (Weeks 2 and 3), the Aβ + MLA group did not show any increase in CAR percentage, suggesting that the absence of the α7nAChR prevented the behavioural recovery promoted by the weekly stimulation (Figure 3).
Finally, the MLA group continued to remember the task in Weeks 1 and 2 but failed to perform as well as the control and Aβ groups with the weekly stimulation (Figure 3). This observation reinforces the hypothesis that α7nAChRs are necessary for the memory recovery promoted by the weekly sustained attention stimulation sessions.
3.2. Quantification of senile plaques
Formation of senile plaques was observed in animals infused with the Aβ peptides 1–42 (0.41 ± 0.03 plaques per slice) but not in the vehicle (control) or MLA‐infused groups. The association of the α7 antagonist MLA with Aβ produced a 2.82‐fold increase in the number of senile plaques (1.15 ± 0.26 plaques per slice, Figure 4).
Figure 4.
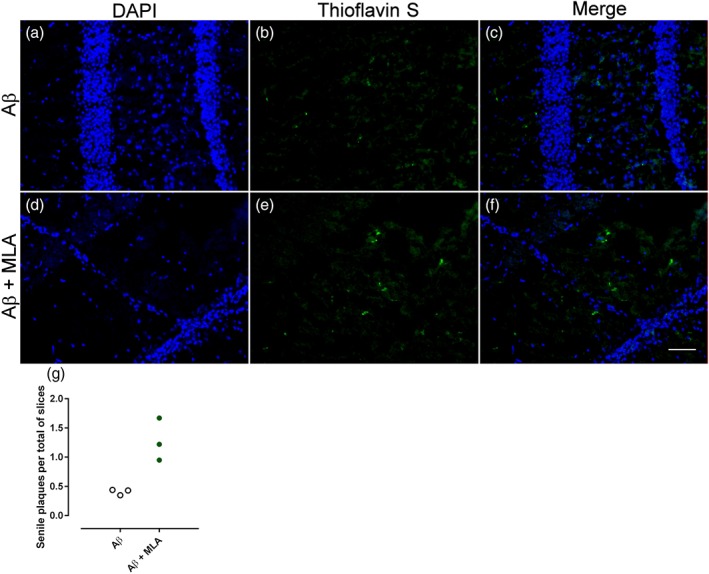
Representative fluorescent images of coronal brain sections depicting hippocampus in C57Bl/6 mice infused with (a–c) amyloid‐β (Aβ) peptide or (d–f) Aβ + methyllycaconitine (MLA). (e) An increased number of Aβ plaques was found in Aβ + MLA animals' hippocampi. (a, d) Cell nuclei stained with DAPI in blue, (b, e) senile plaques stained with thioflavin S in green, and (c, f) merged images. Scale bar: 50 μm. (g) Exploratory quantification of senile plaques found in hippocampus of animals infused with Aβ peptide. Results are presented as pooled data of three animals per group. Although the sample size is low, an increase in number of senile plaques per slice was verified. Vehicle‐infused (control) or MLA‐infused animals did not present any plaques
3.3. Quantification of BDNF protein levels
In order to assess a relationship between the function of α7 AChR and synaptic plasticity, the levels of the neurotrophin BDNF were evaluated in brain areas related to memory, once BDNF is well known to influence structural and functional aspects of synaptic transmission (Kowianski et al., 2018). The alteration of this neurotrophin was also evaluated in the serum. No differences were observed in the cortex of all animals, despite the presence of the Aβ peptide and/or the presence of the MLA (Figure 5a). In the hippocampus, an interaction was observed, indicating that both the induction of neurodegeneration and the presence of MLA altered the protein levels of BDNF. There was an increase of 2.8‐fold in the neurotrophin levels in the group treated with MLA (7.65 ± 1.84 pg·μg−1 protein) when compared with the control group (2.10 ± 0.79 pg·μg−1 protein). Nevertheless, the presence of neurodegeneration significantly affected this increase, as the infusion of both MLA and Aβ caused a reduction of 78.8% in the BDNF levels (1.62 ± 0.89 pg·μg−1 protein), when compared with animals infused with MLA but without Aβ (Figure 5b).
Figure 5.
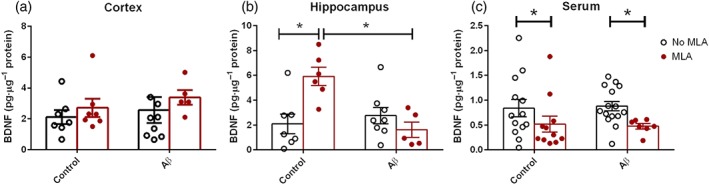
Quantification of the brain‐derived neurotrophic factor (BDNF) levels in the (a) cortex, (b) hippocampus, and (c) serum of mice subjected chronically to the different infusions. Data are presented as means ± SEM. *P < .05. Sample sizes: cortex: control: n = 7; methyllycaconitine (MLA): n = 7; amyloid‐β (Aβ): n = 9; and Aβ + MLA: n = 5. Hippocampus: control: n = 7; MLA: n = 6; Aβ: n = 8; and Aβ + MLA: n = 5. Serum: control: n = 13; MLA: n = 11; Aβ: n = 15; and Aβ + MLA: n = 7
In the serum, the presence of MLA caused reductions of 45.9% and 37.6% in the neurotrophin levels in groups with (0.47 ± 0.05 pg·μg−1 protein) or without (0.52 ± 0.16 pg·μg−1 protein) neurodegeneration, respectively, when compared with control animals (0.84 ± 0.17 pg·μg−1) or only with the Aβ peptide (0.88 ± 0.10 pg·μg−1; Figure 5c).
3.4. Evaluation of AChE activity
Activity of brain and serum AChE was analysed by the elisa method. In the cortex, both the blockade of the α7nAChR and the neurodegeneration promoted by the chronic infusion of Aβ altered the enzyme activity. There was an increase of 1.8‐fold in the AChE activity in animals infused with MLA (10.53 ± 0.77 U·ml−1), compared with that of control animals (5.67 ± 2.03 U·ml−1). Chronic infusion with Aβ promoted an increase in enzyme activity (9.83 ± 0.52 U·ml−1), when compared with control animals (Figure 6a).
Figure 6.
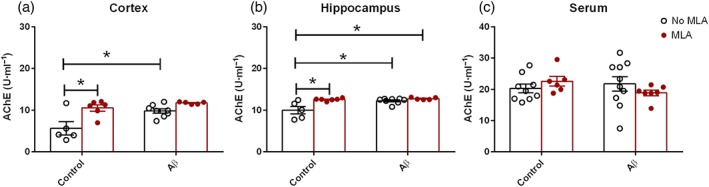
Activity of the AChE enzyme measured in the cortex (a), hippocampus (b), and serum of animals from the different groups. Data are presented as means ± SEM. *P < .05. Sample sizes: cortex: control: n = 5; methyllycaconitine (MLA): n = 6; amyloid‐β (Aβ): n = 8; and Aβ + MLA: n = 5. Hippocampus: control: n = 5; MLA: n = 6; Aβ: n = 8; and Aβ + MLA: n = 5. Serum: control: n = 9; MLA: n = 6; Aβ: n = 10; and Aβ + MLA: n = 7
In the hippocampus samples, the presence of neurodegeneration and the blockade of the α7nAChR altered the enzyme activity in all groups. There was an increase of 1.2‐fold in the AChE activity in the MLA group (12.57 ± 0.12 U·ml−1), compared with control group (10.00 ± 0.91 U·ml−1). Chronic infusion of Aβ also increased enzyme activity by 1.2‐fold (12.27 ± 0.23 U·ml−1). In addition, infusion of both Aβ and MLA also increased the enzyme activity by 1.2‐fold (12.77 ± 0.17 U·ml−1), compared with the control animals, showing interaction of these treatments in respect of AChE activity (Figure 6b).
In the serum, there were no differences among the groups (Figure 6c).
4. DISCUSSION
In previous studies, our research team showed that the weekly submission of rats chronically infused with the Aβ peptide to the active avoidance apparatus prevented the loss of aversive memory evocation (Viel et al., 2012). This stabilization of the behavioural responses was related to an increase in the α7nAChR in the frontal cortex, hippocampus, and amygdalae and was associated with an increase in the attention of the animals to the task. The strategy was, therefore, described as attention training.
In order to confirm the role of the α7nAChR in the memory recovery observed earlier, in the present study, we sought to assess whether the absence of the α7nAChR would influence the memory recovery promoted by the attention training. Therefore, in addition to the group infused with the Aβ peptide, another group of animals was infused with the Aβ peptide together with the α7 antagonist MLA. Moreover, the effect of MLA alone was also observed in a group infused only with this antagonist.
One week after the surgery, animals infused with Aβ did not present memory loss when compared with the control group, as observed in previous works from the group with both mice and rats (Amaral et al., 2010; Viel et al., 2008). Nevertheless, the addition of MLA to Aβ or only the administration of MLA promoted reduction of memory confirming the already known prejudice of memory with the blockade of α7nAChR (Andriambeloson et al., 2014). On the other way, animals that were infused only with Aβ and submitted to the weekly attention training presented evident and significant improvements in memory, which were not observed with animals infused with Aβ + MLA or only MLA. As we have shown before, the chronic infusion of Aβ during 4–5 weeks promote memory loss in mice and rats with deposition of Aβ senile plaques (Amaral et al., 2010; Morzelle et al., 2016; Viel et al., 2012).
This improvement in memory promoted by the attention training is reported as being related to improvements in α7 cholinergic neurotransmission in areas related to learning and memory formation such as the prefrontal cortex, the amygdalae, and hippocampus (Cheng & Yakel, 2015; Hoyle et al., 2006; Viel et al., 2012; Wallace & Porter, 2011) and to the neuroplasticity of dendritic spines, which reinforce memory consolidation (Hlushchenko et al., 2016; Miermans, Kusters, Hoogenraad, & Storm, 2017).
On the other hand, in the group infused with Aβ and the antagonist MLA, the memory reduction observed 1 week after the surgery was maintained, despite the weekly stimulation in the equipment, showing that the animals did not succeed in maintaining memory. This result differs from that observed with the Aβ group and confirmed our hypothesis, as it is possible to suggest that the joint infusion of Aβ and MLA impaired memory recovery. This was also confirmed in the analysis made week by week (Figure 3), where it was possible to observe that Aβ + MLA animals could not recover or re‐learn the memory trial. Many studies have shown that the activation of nicotinic receptors reduces Aβ toxicity and improves neuroprotection. This protection is proportional to the number of expressed nicotinic receptors (Dineley et al., 2015; Jonnala & Buccafusco, 2001). It was observed that the α7nAChRs play an important role in memory pathways, and, when blocked by MLA, there was no improvement in memory in response to the weekly stimulation. In order to better understand the α7nAChR neuroprotective role in this model, we tested the hypothesis that the isolated infusion of the α7 antagonist would also cause a reduction in the cognitive function of mice. Memory deficits effects were observed immediately after the infusion with MLA, with the MLA mice having worse behaviour performance compared with the control animals. Following the attention training, there was only partial memory recovery. The infusion of the antagonist, therefore, caused similar behaviour impairment but a failure to recover. With these observations, it is possible to link the α7 function to the formation of cognitive reserves, since animals infused with MLA did not present memory recovery. In the presence of Aβ, brain function could be maintained due to structural reserves (integral neuronal density and synaptic connectivity) or functional reserves (efficacy of neuronal circuitry) as happens with animals submitted to an enriched environment (Balthazar et al., 2018; Baraldi et al., 2013; Eckert & Abraham, 2013; Milgram, Siwak‐Tapp, Araujo, & Head, 2006; Petrosini et al., 2009). The evidence suggests that there is a relationship between the α7nAChR and the formation of the brain resilience that would permit the brain to be resistant to injuries, shock, or neuropathological disturbances.
Currently, there are several biomarkers of brain resilience. In the present study, BDNF levels were evaluated as this neurotrophin is a vital component of synaptic plasticity and memory formation. BDNF is also involved in the processes of neuronal growth and regeneration, as well as cell survival, and its epigenetic reprogramming can have a key role in these processes (Karpova, 2014; Kowianski et al., 2018).
In the present study, no differences in BDNF levels were observed in the cortex. However, in the hippocampus, the levels were significantly changed. It is well known that the most acceptable biological process to explain the formation of long‐term memory is the LTP that happens in the hippocampus and is maintained by BDNF action in the tropomyosin receptor kinase B, promoting the synthesis of proteins responsible for structural changes and increased synaptic responses (Choi et al., 2009; Mayford, Siegelbaum, & Kandel, 2012). In addition, the cholinergic system is involved with the amplification of LTP induction (Fujii et al., 1999; Matsuyama & Matsumoto, 2003). Consequently, the cholinergic system alters protein synthesis and is involved with consolidation of memory (Martí Barros et al., 2004; Navakkode & Korte, 2012; Lana et al., 2013; Nees, 2015; Richter et al., 2017). In the present study, the blockade of α7nAChR significantly increased the neurotrophin levels in hippocampus. Once the method used detects total BDNF, this effect could be related to an increase in proBDNF that is known to reduce spine density and NMDA receptor and postsynaptic density protein 95 (Qiao, An, Xu, & Ma, 2017). So, this observation deserves further investigation.
The infusion of Aβ, per se, did not alter the levels of BDNF in the hippocampus, cortex, or serum of the animals. Several studies highlight the reduction of BDNF in serum or plasma as a biomarker for schizophrenia, depression, and AD (Lu et al., 2014; Pláteník et al., 2014; Polyakova et al., 2015; Toyooka et al., 2002). The absence of any change in BDNF content in the brain tissues or in the serum of animals infused with Aβ in the present study may be due to the short infusion period and/or to the weekly stimulation in the active avoidance shuttle box, resulting in, as demonstrated in the behavioural experiments, memory recovery in the animals. In agreement with our findings, it has already being reported that the expression of BDNF is dependent on neuronal activity (Palomer, Carretero, Benvegnù, Dotti, & Martin, 2016). Nevertheless, the blockade of α7nAChR caused a reduction in serum BDNF in all groups, which reinforces the link between α7nAChR and BDNF function and suggests that the absence of the receptor function, even in the short period of these experiments, was a determinant for the memory deficit.
According to the most widely accepted hypothesis for AD, there is an increase in both cholinergic and non‐cholinergic activities of the enzyme AChE in AD patients (Campanari et al., 2016; Rao, Sridhar, & Das, 2007). In fact, some research suggest that the evaluation of changes in activity or levels of this enzyme in the blood or in tissues could be taken as a biomarker to follow the progression of the disease and its co‐morbidities (García‐Ayllón et al., 2010; Rao et al., 2007). In agreement with the literature, in the present study, a significant increase in AChE activity was found in the cortex and in the hippocampus of animals infused with Aβ peptide. It is also well demonstrated that lower concentrations of Aβ peptide may interact with α7nAChR and contribute to neuronal homeostasis, whereas increased concentrations of Aβ can cause toxicity of cholinergic neurons (Puzzo et al., 2008; Lombardo & Maskos, 2015). With the progression of the disease, Aβ peptide accumulates in neurons that express α7nAChR. Internalization of the Aβ peptide seems to be facilitated by its interaction with the α7 receptor in the surface of neuronal cells, which causes damage to the neurons. It appears that the absence of the cholinergic terminals promotes a compensatory increase in the levels and the activity of the AChE (García‐Ayllón et al., 2010), which is in accordance with our findings. Therefore, the observed increase in the activity of AChE can be due to at least two causes: (a) the reduction of α7 function could lead to a compensatory increase in enzyme activity and (b) the reduction of α7 function in glial cells could lead to a reduction in the clearance of Aβ peptide, with an increase in senile plaque number (as observed in the present study) and destruction of cholinergic neurons.
In support of the idea that the blockade of α7nAChR is as harmful to memory as the infusion of Aβ peptide, it was shown that the infusion of both Aβ and MLA, alone or together, also increased AChE activity.
The enzyme activity did not change in the serum of all groups, different from the results observed in the literature (García‐Ayllón et al., 2010; Rao et al., 2007), probably because of the short period of the experimental model, which was not sufficient to promote a peripheral alteration of the enzyme.
In conclusion, it is suggested that α7nAChRs have a determinant role in memory recovery during attention training stimulus in the face of a neurodegenerative process. The absence of the receptor is as harmful for memory maintenance as the presence of Aβ peptide and leads to an increase in AChE activity in brain tissues and affects the recruitment mechanisms of tissue resilience against injuries. In this way, attention training may induce memory recovery in the presence of Aβ in animals with functioning α7nAChR pathways. However, the absence of α7nAChR (with their associated mechanisms of neuroprotection) reduces the resilience of the brain tissue (leading to an increase in AChE activity and a decrease in BDNF levels) and preventing memory recovery. Taken together, these data reinforce that the use of attention strategies can help pharmacological therapies that are currently in use to delay the progression of dementia related to AD. Moreover, these data support the importance of α7nAChRs in the maintenance of memory and neuroplasticity.
AUTHOR CONTRIBUTIONS
M.T.L. and T.A.V. conceived and designed the experiments; M.T.L., D.M., N.M.S., G.C.C., H.N.M., H.S.B., and T.A.V. performed the experiments, data acquisition, and analysis; M.T.L. and T.A.V. wrote the manuscript; and M.T.L., H.S.B., and T.A.V. contributed intellectually to the paper. All authors agreed to the final version of the manuscript and confirmed that they have contributed sufficiently to this work.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis and Animal Experimentation and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
ACKNOWLEDGEMENTS
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, São Paulo, Brazil; Grants 2012/12917‐3 and 2016/07115‐6). N.M.S. received a studentship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil). M.T.‐L., G.C.C., and H.N.M. received studentships from FAPESP (Grants 2011/07365‐9, 2013/06935‐1, and 2016/20574‐0, respectively).
Telles‐Longui M, Mourelle D, Schöwe NM, et al. α7 nicotinic ACh receptors are necessary for memory recovery and neuroprotection promoted by attention training in amyloid‐β‐infused mice. Br J Pharmacol. 2019;176:3193–3205. 10.1111/bph.14744
REFERENCES
- Addy, N. , Nakajama, A. , & Levin, E. (2003). Nicotinic mechanisms of memory: Effects of acute local DHbE and MLA infusions in the basolateral amygdala. Cognitive Brain Research, 16, 51–57. 10.1016/S0926-6410(02)00209-4 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon, M. , Pereira, E. F. , Wonnacott, S. , & Albuquerque, E. X. (1992). Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Molecular Pharmacology, 41(4), 802–808. [PubMed] [Google Scholar]
- Amaral, F. A. , Lemos, M. T. , Dong, K. E. , Bittencourt, M. F. , Caetano, A. L. , Pesquero, J. B. , … Buck, H. S. (2010). Participation of kinin receptors on memory impairment after chronic infusion of human amyloid‐β 1‐40 peptide in mice. Neuropeptides, 44(2), 93–97. 10.1016/j.npep.2009.10.006 [DOI] [PubMed] [Google Scholar]
- Andriambeloson, E. , Huyard, B. , Poiraud, E. , & Wagner, S. (2014). Methyllycaconitine‐ and scopolamine‐induced cognitive dysfunction: Differential reversal effect by cognition‐enhancing drugs. Pharmacology Research & Perspectives, 2(4), e00048 10.1002/prp2.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard, C. , Gauthier, S. , Corbett, A. , Brayne, C. , Aarsland, D. , & Jones, E. (2011). Alzheimer's disease. Lancet, 377(9770), 1019–1031. 10.1016/S0140-6736(10)61349-9 [DOI] [PubMed] [Google Scholar]
- Balthazar, J. , Schöwe, N. M. , Cipolli, G. B. , Buck, H. S. , & Viel, T. A. (2018). Enriched environment significantly reduced senile plaques in a transgenic mice model of Alzheimer's disease, improving memory. Frontiers in Aging Neuroscience, 10, 288 10.3389/fnagi.2018.00288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi, T. , Schöwe, N. M. , Balthazar, J. , Monteiro‐Silva, K. C. , Albuquerque, M. S. , Buck, H. S. , & Viel, T. A. (2013). Cognitive stimulation during lifetime and in the aged phase improved spatial memory, and altered neuroplasticity and cholinergic markers of mice. Experimental Gerontology, 48(8), 831–838. 10.1016/j.exger.2013.05.055 [DOI] [PubMed] [Google Scholar]
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Analytical Biochemistry, 72, 248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- Campanari, M. L. , Navarrete, F. , Ginsberg, S. D. , Manzanares, J. , Sáez‐Valero, J. , & García‐Ayllón, M. S. (2016). Increased expression of read through acetylcholinesterase variants in the brains of Alzheimer's disease patients. Journal of Alzheimer's Disease, 53(3), 831–841. 10.3233/JAD-160220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Q. , & Yakel, J. L. (2015). The effect of α7 nicotinic receptor activation on glutamatergic transmission in the hippocampus. Biochemical Pharmacology, 97(4), 439–444. 10.1016/j.bcp.2015.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. , Li, Y. , Parada, L. F. , & Sisodia, S. S. (2009). Regulation of hippocampal progenitor cell survival, proliferation and dendritic development by BDNF. Molecular Neurodegeneration, 4, 52 10.1186/1750-1326-4-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi, J. , & Bouzat, C. (2016). Understanding the bases of function and modulation of α7 nicotinic receptors: Implications for drug discovery. Molecular Pharmacology, 90(3), 288–299. 10.1124/mol.116.104240 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. A. , Giembycz, M. A. , … McGrath, J. C. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP . British Journal of Pharmacology, 172, 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, A. R. , Hardick, D. J. , Blagbrough, I. S. , Potter, B. V. , Wolstenholme, A. J. , & Wonnacott, S. (1999). Characterisation of the binding of [3H]methyllycaconitine: A new radioligand for labelling α7‐type neuronal nicotinic acetylcholine receptors. Neuropharmacology, 38, 679–690. 10.1016/S0028-3908(98)00221-4 [DOI] [PubMed] [Google Scholar]
- Dineley, K. T. , Pandya, A. A. , & Yakel, J. L. (2015). Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends in Pharmacological Sciences, 36(2), 96–108. 10.1016/j.tips.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorostkar, M. M. , Zou, C. , Blazquez‐Llorca, L. , & Herms, J. (2015). Analyzing dendritic spine pathology in Alzheimer's disease: Problems and opportunities. Acta Neuropathologica, 130(1), 1–19. 10.1007/s00401-015-1449-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, L. , Bhattacharyya, B. J. , Belmadani, A. , Pan, L. , Miller, R. J. , & Kessler, J. A. (2014). Stem cell derived basal forebrain cholinergic neurons from Alzheimer's disease patients are more susceptible to cell death. Molecular Neurodegeneration, 9, 3 10.1186/1750-1326-9-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, M. J. , & Abraham, W. C. (2013). Effects of environmental enrichment exposure on synaptic transmission and plasticity in the hippocampus. Current Topics in Behavioral Neurosciences, 15, 165–187. 10.1007/7854_2012_215 [DOI] [PubMed] [Google Scholar]
- Foucault‐Fruchard, L. , Doméné, A. , Page, G. , Windsor, M. , Emond, P. , Rodrigues, N. , … Antier, D. (2017). Neuroprotective effect of the alpha 7 nicotinic receptor agonist PHA 543613 in an in vivo excitotoxic adult rat model. Neuroscience, 356, 52–63. 10.1016/j.neuroscience.2017.05.019 [DOI] [PubMed] [Google Scholar]
- Franklin, K. B. J. , & Paxinos, G. (2007). The mouse brain in stereotaxic coordinates (third ed.). San Diego: Academic Press. [Google Scholar]
- Frautschy, S. A. , Horn, D. L. , Sigel, J. J. , Harris‐White, M. E. , Mendoza, J. J. , Yang, F. , … Cole, G. M. (1998). Protease inhibitor coinfusion with amyloid β‐protein results in enhanced deposition and toxicity in rat brain. The Journal of Neuroscience, 18(20), 8311–8321. 10.1523/JNEUROSCI.18-20-08311.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii, S. , Ji, Z. , Morita, N. , & Sumikawa, K. (1999). Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Research, 846(1), 137–143. 10.1016/S0006-8993(99)01982-4 [DOI] [PubMed] [Google Scholar]
- García‐Ayllón, M. S. , Riba‐Llena, I. , Serra‐Basante, C. , Alom, J. , Boopathy, R. , & Sáez‐Valero, J. (2010). Altered levels of acetylcholinesterase in Alzheimer plasma. PLoS ONE, 5(1), e8701 10.1371/journal.pone.0008701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Ayllón, M. S. , Silveyra, M. X. , & Sáez‐Valero, J. (2008). Association between acetylcholinesterase and β‐amyloid peptide in Alzheimer's cerebrospinal fluid. Chemico‐Biological Interactions, 175(1–3), 209–215. 10.1016/j.cbi.2008.04.047 [DOI] [PubMed] [Google Scholar]
- Grant, A. , Dennis, N. A. , & Li, P. (2014). Cognitive control, cognitive reserve, and memory in the aging bilingual brain. Frontiers in Psychology, 5, 1401 10.3389/fpsyg.2014.01401 eCollection 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, C. M. , & Dineley, K. T. (2012). Nicotinic acetylcholine receptors in Alzheimer's disease: Neuroprotective, neurotrophic or both? Current Drug Targets, 13, 613–622. [DOI] [PubMed] [Google Scholar]
- Hlushchenko, I. , Koskinen, M. , & Hotulainen, P. (2016). Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton (Hoboken), 73(9), 435–441. 10.1002/cm.21280 [DOI] [PubMed] [Google Scholar]
- Hoyle, E. , Genn, R. F. , Fernandes, C. , & Stolerman, I. P. (2006). Impaired performance of alpha7 nicotinic receptor knockout mice in the five‐choice serial reaction time task. Psychopharmacology, 189(2), 211–223. 10.1007/s00213-006-0549-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonnala, R. R. , & Buccafusco, J. J. (2001). Relationship between the increased cell surface α7 nicotinic receptor expression and neuroprotection induced by several nicotinic receptor agonists. Journal of Neuroscience Research, 66(4), 565–572. 10.1002/jnr.10022 [DOI] [PubMed] [Google Scholar]
- Karpova, N. N. (2014). Role of BDNF epigenetics in activity‐dependent neuronal plasticity. Neuropharmacology, 76(Pt C), 709–718. 10.1016/j.neuropharm.2013.04.002 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowianski, P. , Lietzau, G. , Czuba, E. , Was'kow, M. , Steliga, A. , & Morys, J. (2018). BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cellular and Molecular Neurobiology, 38, 579–593. 10.1007/s10571-017-0510-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulshreshtha, A. , & Piplani, P. (2016). Current pharmacotherapy and putative disease‐modifying therapy for Alzheimer's disease. Neurological Sciences, 37(9), 1403–1435. 10.1007/s10072-016-2625-7 [DOI] [PubMed] [Google Scholar]
- Lana, D. , Cerbai, F. , Di Russo, J. , Boscaro, F. , Giannetti, A. , Petkova‐Kirova, P. , … Giovannini, M. G. (2013). Hippocampal long term memory: Effect of the cholinergic system on local protein synthesis. Neurobiology of Learning and Memory, 106, 246–257. 10.1016/j.nlm.2013.09.013 [DOI] [PubMed] [Google Scholar]
- Lane, C. A. , Hardy, J. , & Schott, J. M. (2018). Alzheimer's disease. European Journal of Neurology, 25(1), 59–70. 10.1111/ene.13439 [DOI] [PubMed] [Google Scholar]
- Lavrencic, L. M. , Richardson, C. , Harrison, S. L. , Muniz‐Terrera, G. , Keage, H. A. D. , Brittain, K. , … Stephan, B. C. M. (2018). Is there a link between cognitive reserve and cognitive function in the oldest‐old? The Journals of Gerontology. Series a, Biological Sciences and Medical Sciences, 73(4), 499–505. 10.1093/gerona/glx140 [DOI] [PubMed] [Google Scholar]
- Lemos, M. T. , Amaral, F. A. , Dong, K. E. , Bittencourt, M. F. , Caetano, A. L. , Pesquero, J. B. , … Buck, H. S. (2010). Role of kinin B1 and B2 receptors in memory consolidation during the aging process of mice. Neuropeptides, 44(2), 163–168. 10.1016/j.npep.2009.12.006 [DOI] [PubMed] [Google Scholar]
- Lu, B. , Nagappan, G. , & Lu, Y. (2014). BDNF and synaptic plasticity, cognitive function, and dysfunction. Handbook of Experimental Pharmacology, 220, 223–250. 10.1007/978-3-642-45106-5_9 [DOI] [PubMed] [Google Scholar]
- Lombardo, S. , & Maskos, U. (2015). Role of the nicotinic acetylcholine receptor inAlzheimer's disease pathology and treatment. Neuropharmacology, 6(PtB), 255–262. 10.1016/j.neuropharm.2014.11.018 [DOI] [PubMed] [Google Scholar]
- Martí Barros, D. , Ramirez, M. R. , Dos Reis, E. A. , & Izquierdo, I. (2004). Participation of hippocampal nicotinic receptors in acquisition, consolidation and retrieval of memory for one trial inhibitory avoidance in rats. Neuroscience, 126(3), 651–656. 10.1016/j.neuroscience.2004.03.010 [DOI] [PubMed] [Google Scholar]
- Masters, C. L. , Bateman, R. , Blennow, K. , Rowe, C. C. , Sperling, R. A. , & Cummings, J. L. (2015). Alzheimer's disease. Nature Reviews. Disease Primers, 1, 15056 10.1038/nrdp.2015.56 [DOI] [PubMed] [Google Scholar]
- Matsuyama, S. , & Matsumoto, A. (2003). Epibatidine induces long‐term potentiation (LTP) via activation of α4β2 nicotinic acetylcholine receptors (nAChRs) in vivo in the intact mouse dentate gyrus: Both α7 and α4β2 nAChRs essential to nicotinic LTP. Journal of Pharmacological Sciences, 93(2), 180–187. 10.1254/jphs.93.180 [DOI] [PubMed] [Google Scholar]
- Mayford, M. , Siegelbaum, S. A. , & Kandel, E. R. (2012). Synapses and memory storage. Cold Spring Harbor Perspectives in Biology, 4(6). pii: a005751. 10.1101/cshperspect.a005751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miermans, C. A. , Kusters, R. P. , Hoogenraad, C. C. , & Storm, C. (2017). Biophysical model of the role of actin remodeling on dendritic spine morphology. PLoS ONE, 12(2), e0170113 10.1371/journal.pone.0170113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgram, N. W. , Siwak‐Tapp, C. T. , Araujo, J. , & Head, E. (2006). Neuroprotective effects of cognitive enrichment. Ageing Research Reviews, 5(3), 354–369. 10.1016/j.arr.2006.04.004 [DOI] [PubMed] [Google Scholar]
- Morzelle, M. C. , Salgado, J. M. , Telles, M. , Mourelle, D. , Bachiega, P. , Buck, H. S. , & Viel, T. A. (2016). Neuroprotective effects of pomegranate peel extract after chronic infusion with amyloid‐β peptide in mice. PLoS ONE, 11(11), e0166123 10.1371/journal.pone.0166123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navakkode, S. , & Korte, M. (2012). Cooperation between cholinergic and glutamatergic receptors are essential to induce BDNF‐dependent long‐lasting memory storage. Hippocampus, 22(2), 335–346. 10.1002/hipo.20902 [DOI] [PubMed] [Google Scholar]
- Nees, F. (2015). The nicotinic cholinergic system function in the human brain. Neuropharmacology, 96(Pt B), 289–301. 10.1016/j.neuropharm.2014.10.021 [DOI] [PubMed] [Google Scholar]
- Nunes, M. A. , Schöwe, N. M. , Monteiro‐Silva, K. C. , Baraldi‐Tornisielo, T. , Souza, S. I. , Balthazar, J. , … Buck, H. S. (2015). Chronic microdose lithium treatment prevented memory loss and neurohistopathological changes in a transgenic mouse model of Alzheimer's disease. PLoS ONE, 10(11), e0142267 10.1371/journal.pone.0142267. Erratum in: PLoS One 2015;10(12):e0145695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomer, E. , Carretero, J. , Benvegnù, S. , Dotti, C. G. , & Martin, M. G. (2016). Neuronal activity controls Bdnf expression via Polycomb de‐repression and CREB/CBP/JMJD3 activation in mature neurons. Nature Communications, 7, 11081 10.1038/ncomms11081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosini, L. , De Bartolo, P. , Foti, F. , Gelfo, F. , Cutuli, D. , Leggio, M. G. , & Mandolesi, L. (2009). On whether the environmental enrichment may provide cognitive and brain reserves. Brain Research Reviews, 61(2), 221–239. 10.1016/j.brainresrev.2009.07.002 [DOI] [PubMed] [Google Scholar]
- Picciotto, M. R. , Higley, M. J. , & Mineur, Y. S. (2012). Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron, 76(1), 116–129. 10.1016/j.neuron.2012.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pláteník, J. , Fišar, Z. , Buchal, R. , Jirák, R. , Kitzlerová, E. , Zvěřová, M. , & Raboch, J. (2014). GSK3β, CREB, and BDNF in peripheral blood of patients with Alzheimer's disease and depression. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 50, 83–93. 10.1016/j.pnpbp.2013.12.001 [DOI] [PubMed] [Google Scholar]
- Polyakova, M. , Stuke, K. , Schuemberg, K. , Mueller, K. , Schoenknecht, P. , & Schroeter, M. L. (2015). BDNF as a biomarker for successful treatment of mood disorders: A systematic & quantitative meta‐analysis. Journal of Affective Disorders, 174, 432–440. 10.1016/j.jad.2014.11.044 [DOI] [PubMed] [Google Scholar]
- Puzzo, D. , Privitera, L. , Leznik, E. , Fà, M. , Staniszewski, A. , Palmeri, A. , & Arancio O. (2008). Picomolar amyloid‐beta positively modulates synaptic plasticity and memory inhippocampus. Journal of Neuroscience. 28(53), 14537–14545. 10.1523/JNEUROSCI.2692-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, H. , An, S. C. , Xu, C. , & Ma, X. M. (2017). Role of proBDNF and BDNF in dendritic spine plasticity and depressive‐like behaviors induced by an animal model of depression. Brain Research, 1663, 29–37. 10.1016/j.brainres.2017.02.020 [DOI] [PubMed] [Google Scholar]
- Rao, A. A. , Sridhar, G. R. , & Das, U. N. (2007). Elevated butyrylcholinesterase and acetylcholinesterase may predict the development of type 2 diabetes mellitus and Alzheimer's disease. Medical Hypotheses, 69(6), 1272–1276. 10.1016/j.mehy.2007.03.032 [DOI] [PubMed] [Google Scholar]
- Richter, N. , Michel, A. , Onur, O. A. , Kracht, L. , Dietlein, M. , Tittgemeyer, M. , … Kukolja, J. (2017). White matter lesions and the cholinergic deficit in aging and mild cognitive impairment. Neurobiology of Aging, 53, 27–35. 10.1016/j.neurobiolaging.2017.01.012 [DOI] [PubMed] [Google Scholar]
- Scheltens, P. , Blennow, K. , Breteler, M. M. , de Strooper, B. , Frisoni, G. B. , Salloway, S. , & Van der Flier, W. M. (2016). Alzheimer's disease. Lancet, 388(10043), 505–517. 10.1016/S0140-6736(15)01124-1 [DOI] [PubMed] [Google Scholar]
- Sivaprakasam, K. (2006). Towards a unifying hypothesis of Alzheimer's disease: Cholinergic system linked to plaques, tangles and neuroinflammation. Current Medicinal Chemistry, 13(18), 2179–2188. 10.2174/092986706777935203 [DOI] [PubMed] [Google Scholar]
- Stern, Y. (2012). Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurology, 11(11), 1006–1012. 10.1016/S1474-4422(12)70191-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal, D. R. , Walter, J. , Saido, T. C. , & Fändrich, M. (2015). Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer's disease. Acta Neuropathologica, 129(2), 167–182. 10.1007/s00401-014-1375-y [DOI] [PubMed] [Google Scholar]
- Toyooka, K. , Asama, K. , Watanabe, Y. , Muratake, T. , Takahashi, M. , Someya, T. , & Nawa, H. (2002). Decreased levels of brain‐derived neurotrophic factor in serum of chronic schizophrenic patients. Psychiatry Research, 110(3), 249–257. 10.1016/S0165-1781(02)00127-0 [DOI] [PubMed] [Google Scholar]
- Turek, J. W. , Kang, C. H. , Campbell, J. E. , Arneric, S. P. , & Sullivan, J. P. (1995). A sensitive technique for the detection of the α7 neuronal nicotinic acetylcholine receptor antagonist, methyllycaconitine, in rat plasma and brain. Journal of Neuroscience Methods, 61, 113–118. 10.1016/0165-0270(95)00032-P [DOI] [PubMed] [Google Scholar]
- Viel, T. A. , Caetano, A. L. , Albuquerque, M. S. , Araujo, M. S. , & Buck, H. S. (2012). Chronic infusion of amyloid‐β peptide and sustained attention altered α7 nicotinic receptor density in the rat brain. Current Alzheimer Research, 9(10), 1210–1220. 10.2174/156720512804142930 [DOI] [PubMed] [Google Scholar]
- Viel, T. A. , Lima Caetano, A. , Nasello, A. G. , Lancelotti, C. L. , Nunes, V. A. , Araujo, M. S. , & Buck, H. S. (2008). Increases of kinin B1 and B2 receptors binding sites after brain infusion of amyloid‐beta 1‐40 peptide in rats. Neurobiology of Aging, 29(12), 1805–1814. 10.1016/j.neurobiolaging.2007.04.019 [DOI] [PubMed] [Google Scholar]
- Waite, L. M. (2015). Treatment for Alzheimer's disease: Has anything changed? Australian Prescriber, 38(2), 60–63. 10.18773/austprescr.2015.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, T. L. , & Porter, R. H. (2011). Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease. Biochemical Pharmacology, 82(8), 891–903. 10.1016/j.bcp.2011.06.034 [DOI] [PubMed] [Google Scholar]
- Wattmo, C. , & Wallin, A. K. (2017). Early‐ versus late‐onset Alzheimer's disease in clinical practice: Cognitive and global outcomes over 3 years. Alzheimer's Research & Therapy, 9(1), 70–83. 10.1186/s13195-017-0294-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse, P. J. , Price, D. L. , Struble, R. G. , Clark, A. W. , Coyle, J. T. , & Delon, M. R. (1982). Alzheimer's disease and senile dementia: Loss of neurons in the basal forebrain. Science, 215(4537), 1237–1239. 10.1126/science.7058341 [DOI] [PubMed] [Google Scholar]