

Abstract
Background and Purpose
Quinic acid (QA) is an abundant natural compound from plant sources which may improve metabolic health. However, little attention has been paid to its effects on pancreatic beta‐cell functions, which contribute to the control of metabolic health by lowering blood glucose. Strategies targeting beta‐cell signal transduction are a new approach for diabetes treatment. This study investigated the efficacy of QA to stimulate beta‐cell function by targeting the basic molecular machinery of metabolism–secretion coupling.
Experimental Approach
We measured bioenergetic parameters and insulin exocytosis in a model of insulin‐secreting beta‐cells (INS‐1E), together with Ca2+ homeostasis, using genetically encoded sensors, targeted to different subcellular compartments. Islets from mice chronically infused with QA were also assessed.
Key Results
QA triggered transient cytosolic Ca2+ increases in insulin‐secreting cells by mobilizing Ca2+ from intracellular stores, such as endoplasmic reticulum. Following glucose stimulation, QA increased glucose‐induced mitochondrial Ca2+ transients. We also observed a QA‐induced rise of the NAD(P)H/NAD(P)+ ratio, augmented ATP synthase‐dependent respiration, and enhanced glucose‐stimulated insulin secretion. QA promoted beta‐cell function in vivo as islets from mice infused with QA displayed improved glucose‐induced insulin secretion. A diet containing QA improved glucose tolerance in mice.
Conclusions and Implications
QA modulated intracellular Ca2+ homeostasis, enhancing glucose‐stimulated insulin secretion in both INS‐1E cells and mouse islets. By increasing mitochondrial Ca2+, QA activated the coordinated stimulation of oxidative metabolism, mitochondrial ATP synthase‐dependent respiration, and therefore insulin secretion. Bioactive agents raising mitochondrial Ca2+ in pancreatic beta‐cells could be used to treat diabetes.
Abbreviations
- ER
endoplasmic reticulum
- QA
quinic acid
What is already known
Pancreatic beta‐cells modulate metabolic health by lowering blood glucose.
Strategies targeting beta‐cell signal transduction are a new approach for diabetes treatment.
What this study adds
A natural compound quinic acid enhanced glucose‐induced insulin secretion in beta‐cells.
Quinic acid remodelled intracellular Ca2+ and activates mitochondria in beta‐cells.
What is the clinical significance
Bioactive agents modulating mitochondrial Ca2+ in beta‐cells may be used to treat diabetes.
1. INTRODUCTION
Type 2 diabetes is a metabolic disorder characterized by impaired function or reduced mass of pancreatic beta‐cells, which secrete insulin, the only blood glucose‐lowering hormone (Steffes, Sibley, Jackson, & Thomas, 2003). Strategies targeting the beta‐cells are therefore a promising approach for the treatment of Type 2 diabetes (Vetere, Choudhary, Burns, & Wagner, 2014). Pancreatic beta‐cell function is based on metabolism–secretion coupling. By sensing the blood glucose level, these endocrine cells secrete the appropriate amount of insulin, to maintain circulating nutrient levels, according to the metabolic requirements (Rutter, Pullen, Hodson, & Martinez‐Sanchez, 2015).
In the beta‐cell, this process is mediated by glycolysis‐driven production of pyruvate, which is transported in the mitochondrial matrix, where it activates the tricarboxylic acid cycle, enhancing NADH production, which is the fuel for mitochondrial respiratory chain complexes. Activation of mitochondrial respiration promotes the generation of ATP, which inhibits the plasma membrane ATP‐dependent K+ (Kir6.x) channel, resulting in plasma membrane depolarization and the consequent opening of voltage‐dependent Ca2+ channels. The intracellular Ca2+ rise is the final event, which promotes insulin secretion (Rorsman & Ashcroft, 2018). Importantly, mitochondria from pancreatic beta‐cells take up Ca2+ during glucose stimulation (Wiederkehr et al., 2011; Wiederkehr & Wollheim, 2008), and two matrix Ca2+‐dependent processes are then coordinately stimulated (oxidative metabolism and ATP synthase‐dependent respiration) to promote sustained insulin secretion (De Marchi, Thevenet, Hermant, Dioum, & Wiederkehr, 2014).
Given the relevance of pancreatic beta‐cells in the development of diabetes (Butler et al., 2003; Ferrannini, 2010; Steffes et al., 2003; Weir & Bonner‐Weir, 2004), intense investigations have been performed, in the attempt to find antidiabetic compounds, which enhance insulin secretion by promoting beta‐cell metabolism–secretion coupling (Patel, Prasad, Kumar, & Hemalatha, 2012). Several plant‐derived compounds have been demonstrated to modulate insulin secretion (Gray & Flatt, 1999; Norberg et al., 2004). Phenolic compounds from plant origin and particularly caffeic acid and chlorogenic acid have been investigated for their absorption in human and antihyperglycaemic properties (Bhattacharya, Oksbjerg, Young, & Jeppesen, 2014; Jung, Lee, Park, Jeon, & Choi, 2006; Meng, Cao, Feng, Peng, & Hu, 2013; Olthof, Hollman, & Katan, 2001).
Chlorogenic acid, a major phenolic compound in coffee (Olthof et al., 2001), is an ester of caffeic acid and quinic acid (QA). QA is an abundant natural compound found not only in coffee but also in several other plant products like bilberry, prunes, cranberries, sea buckthorns, and kiwifruit (Beveridge, Harrison, & Drover, 2002; Coppola, Conrad, & Cotter, 1978; Heatherbell, Struebi, Eschenbruch, & Withy, 1980; Ryan & Dupont, 1973; Uleberg et al., 2012). Compared with chlorogenic acid and caffeic acid, the study of QA has been rather neglected as is thought to have no biological activity. However, recent studies highlight its antioxidant properties (Pero, Lund, & Leanderson, 2009) and antidiabetic activity (Arya et al., 2014).
The molecular mechanisms underlying the potential health benefit of QA are poorly understood. Some results have been obtained with a closely related analogue of QA, named KZ‐41 (He et al., 2017). A possible interaction between IGF‐1 receptor kinase domain and KZ‐41 was investigated in that study, suggesting that IGF‐1 receptors are possible targets for QA and its analogues. Moreover, the ability of IGF‐1 activation to mobilize calcium from endoplasmic reticulum (ER; Poiraudeau, Lieberherr, Kergosie, & Corvol, 1997) could potentially link the physiological effects of QA with the release of calcium from the ER.
QA is a metabolite of the shikimate pathway (Herrmann & Weaver, 1999) and a normal constituent of our diet, and it can be converted to tryptophan and nicotinamide (Pero et al., 2009), which are precursor in the biosynthesis of nicotinamide adenine dinucleotide (NAD+). The mitochondrial and cytosolic redox balance of NAD+/NADH is essential for cellular metabolism, and in pancreatic beta‐cells, it assures tight coupling between glucose stimulation and insulin secretion (De Marchi et al., 2014; Patterson, Knobel, Arkhammar, Thastrup, & Piston, 2000; Wiederkehr & Wollheim, 2012).
In the light of the abundance of QA in our diet and the link between QA and NADH metabolism, we hypothesized that these compounds could influence metabolism–secretion coupling in pancreatic beta‐cells . In order to evaluate the role of QA in beta‐cell function and to shed light on its mechanism of action, we measured its effects on beta‐cell signal transduction. We combined Ca2+ measurements in different subcellular compartments with the study of bioenergetic (mitochondrial) parameters and insulin secretion. Finally, to validate the activity of QA on islet beta‐cell function in vivo, mice were implanted with mini‐osmotic pumps continuously delivering QA, followed by measuring insulin secretion capacity of islets.
2. METHODS
2.1. Cell culture
INS‐1E cells (Maechler, Antinozzi, & Wollheim, 2000; [RRID]:CVCL_0351) were cultured at 37°C in a humidified atmosphere (5% CO2) in RPMI 1640 medium (Invitrogen) containing 11‐mM glucose, supplemented with 10‐mM HEPES (pH 7.3), 10% (v/v) heat‐inactivated fetal calf serum (Brunschwig AG, Switzerland), 1‐mM sodium pyruvate, 50‐μM β‐mercaptoethanol, 50 μg·ml−1 penicillin, and 100 μg·ml−1 streptomycin (INS‐1 medium). Differentiated human‐induced pluripotent stem cells neurons (iCell neurons) and astrocytes (iCell astrocytes) were obtained from Cellular Dynamics International (CDI, Madison, WI, USA). The cells were thawed according to the manufacturer's instructions. iCell astrocytes were cultured in DMEM supplemented with 10% fetal calf serum and N2 complement. iCell neurons were maintained in a medium supplied by CDI on poly‐l‐ornithine/laminin‐coated dishes for 4–18 days. The medium was changed daily for 5 days followed by growth in maintenance RPMI medium containing B27, dexamethasone (0.1 mM), gentamicin (25 mg·ml−1), and CDI‐specific supplements. Cell cultures were kept in a humidified atmosphere (5% CO2) at 37°C (Thevenet et al., 2016).
2.2. NAD(P)H measurements
Cells were allowed to adhere to glass‐bottom dishes (MatTek, Ashland, MA, USA) for 2 days, and experiments were performed in KRBH. A laser‐scanning confocal microscope (Leica TCS SP5 II MP, Mannheim, Germany) with an HCX IRAPO L 25×/0.95 water objective was utilized to monitor NAD(P)H autofluorescence at 37°C (Life Imaging Services). Laser scans at 727 nm (IR Laser Chameleon Ultra, Coherent) were used for two‐photon NAD(P)H excitation. Each 512 × 512 pixel image represents an average of 16 scans taken with a resonant scanner at 8000 Hz. NAD(P)H emission was collected at 445‐ to 495‐nm wavelength every 20 s. Data were processed with LAS AF software (Leica) and analysed in Excel (Microsoft) and GraphPad Prism 5 (GraphPad). In order to normalize the NAD(P)H responses, each experiment was terminated by adding 1% hydrogen peroxide, which results in an almost complete loss of autofluorescence (minimal value). Fluorescence intensity data were normalized to both basal autofluorescence (= 1) and minimal value (hydrogen peroxide = 0). The change in fluorescence (ΔNAD(P)H) was calculated as the difference between the baseline value and the effect of the added compound.
2.3. Cytosolic, mitochondrial, and ER Ca2+ measurements
Cytosolic, mitochondrial, and ER Ca2+ were measured with the genetically encoded cameleon sensors YC3.6cyto, 4mtD3cpv, and D1ER, respectively (Boss, Bouche, & De Marchi, 2018). For cytosolic Ca2+ measurements, a clonal INS‐1E cell line stably expressing the YC3.6cyto calcium sensor has been recently generated and described in Boss et al. (2017). INS‐1E cells were plated on polyornithin‐treated 35‐mm‐diameter glass‐bottom dishes (MatTek, MA, USA) and, for mitochondrial and ER Ca2+ measurements, transfected with the appropriate sensor, using JetPRIME transfection reagent (Polyplus). Two days after, transfection cells were washed four times, and experiments were performed at 37°C in KRBH. Glass coverslips were inserted in a thermostatic chamber (Life Imaging Services). Cells were imaged on a DMI6000 B inverted fluorescence microscope, using an HCX PL APO 63×/1.40–0.60 numerical aperture oil immersion objective (Leica Microsystems) and an Evolve 512 back illuminated CCD with 16 × 16 pixel camera (Photometrics, Tucson, AZ, USA). Cells were excited at 430 nm through a BP436/20 filter. The two emission images were acquired with BP480/40 and BP535/30 emission filters. Fluorescence ratios were calculated in MetaFluor 7.0 (Meta Imaging Series) and analysed in Excel (Microsoft) and GraphPad Prism 5 (GraphPad). Images were taken every 2 s. Ratio fluorescence intensity data were normalized to the basal fluorescence set to 1.
2.4. Respirometry studies
Oxygen consumption was measured using an XF96 instrument (Seahorse Biosciences, MA, USA). INS‐1E cells were seeded into polyornithine‐coated Seahorse tissue plates at a density of 20,000 cells per well. After 2 days, the cells were washed twice in KRBH 2.5‐mM glucose. Respiration rates were determined every 6 min at 37°C. Respiratory chain inhibitors were added as indicated in the figures. ATP synthase‐dependent respiration was calculated as the difference in respiration rate before and after the addition of oligomycin. Respiration rates were determined every 3 min. All experiments were performed at 37°C.
2.5. Static insulin secretion in INS‐1E cells
For acute treatment of QA, 0.45 x 106 INS‐1E cells were plated on 24‐well plates 48 hr before insulin secretion assay. For chronic treatment of QA, 0.3 x 106 cells were plated, and the following day, QA treatment was started for 72 hr, and new media with QA supplementation were added daily. For secretion, cells were washed and pre‐incubated in KRBH buffer containing 2‐mM glucose with or without of QA for 1 hr. The pre‐incubation buffer was then changed to KRBH solution containing 2‐ or 20‐mM glucose with or without QA for 1 hr to measure basal and glucose‐stimulated insulin release. At the end of the incubation, supernatants were collected to measure insulin release by elisa (Alpco, Salem, NH, USA). Cells were lysed in PBS supplemented with 1% Triton X‐100, and protease inhibitors and protein concentration were measured using DC protein assay (Bio‐Rad, CA, USA). Insulin secretion was normalized to cellular protein content.
2.6. Animal phenotyping
All animal care and experimental procedures were carried according to national Swiss and EU ethical guidelines and approved by the Local Animal Experimentation Committee under Licence VD 3146. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. To investigate the effect of QA on insulin secretion, 12‐week‐old C57BL/6N male mice (Charles River Laboratories, Italy, RRID:MGI:5825016) were used. Upon arrival, mice were 10 weeks old and were randomized according to their body weight. Mice were housed at a maximum of five mice per cage, and cages were enriched with paper tunnels and paper. Mice were acclimatized 2 weeks to the animal facility and fed with normal chow diet (3234, Kliba Provimi Kliba AG, Kaiseraugst, Switzerland). QA and control treatments were conducted in a blinded fashion, and mice were housed individually on a 12‐hr light/12‐hr dark cycle at 21°C and were fed with standard chow diet (AIN‐93G, Research Diet Inc, USA). For osmotic pump experiments, saline or QA was delivered to mice via surgically implanting mini‐osmotic pumps subcutaneously (Alzet, Cupertino, CA, USA), in the interscapular area. Mini‐osmotic pumps had mean pumping rate of 22 μl·hr−1, resulting in QA delivery of 75 mg·kg−1·day−1 for a mouse. During 12 weeks, treatment pumps were changed twice, and mice were given free access to food and water. After 12‐week QA infusion with osmotic pumps, pancreatic islets were isolated from mouse pancreas using collagenase type 4 (Worthington, NJ, USA; 422 U·ml−1) and handpicked. Islets were used in the experiments after overnight culture at 37°C in 5% CO2 in RPMI 1640 supplemented in 10% fetal calf serum, 2‐mM glutamax, 100 U·ml−1 penicillin, and 100 μg·ml−1 streptomycin. For static insulin secretion, 20 islets in triplicate were first incubated with KRBH with 2‐mM glucose for 1 hr to stabilize the insulin secretion. The pre‐incubation buffer was then changed to KRBH solution containing 2‐mM glucose for 1 hr to measure basal glucose‐stimulated insulin secretion rate. Then islets were changed to KRBH with 20‐mM glucose for 1 hr to measure glucose‐stimulated insulin release. Insulin from the incubation buffer and insulin content of the islets lysed with ethanol acid were measured by elisa (Alpco, Salem, NH, USA). For diet supplementation, QA was given in 25 mg·kg−1·day−1 for 2 months with standard chow diet. Glucose excursion curves were determined after an intraperitoneal injection of 2 g·kg−1 glucose on 12‐hr‐fasted mice or 0.75 U·kg−1 insulin (human insulin actrapid, Lilly) on 6‐hr‐fasted C57BL/6N, 22‐week‐old mice, chow diet fed, and supplemented with either water (vehicle) or QA (25 mg·kg−1·day−1). No animal was excluded from statistical analysis.
2.7. Data analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. The exact group size (n) for each experimental group/condition is provided, and “n” refers to independent values, not replicates. Data subjected to statistical analysis have n of at least five per group. Statistical analyses were performed with Prism software (GraphPad Software Inc., San Diego, CA, USA, RRID:SCR_002798). Data are expressed as means ± SEM. Scatter points of single experiments/single cells are included in graphs and bar charts. Statistically significant differences between means were determined using Student's t test for comparison between two means with n ≥ 5. ANOVA test was used when comparing mean values from multiple groups. P values <.05 were accepted as significant.
2.8. Materials
Chemicals were from Sigma, Invitrogen, or VWR, unless otherwise indicated. QA was dissolved in Krebs–Ringer bicarbonate HEPES buffer (KRBH), containing (in mM): 140 NaCl, 3.6 KCl, 0.5 NaH2PO4, 0.5 MgSO4, 1.5 CaCl2, 10 HEPES, and 5 NaHCO3, pH 7.4. Glucose was 2.5 mM unless otherwise specified. A concentration range of 10‐ to 100‐μM QA was used for cell biology experiments. The YC3.6cyto pcDNA3 (Nagai, Yamada, Tominaga, Ichikawa, & Miyawaki, 2004), 4mtD3cpv (Palmer et al., 2006), and D1ER (Palmer, Jin, Reed, & Tsien, 2004) constructs to measure cytosolic, mitochondrial, and endoplasmic [Ca2+], respectively, were kindly provided by Dr A. Miyawaki (Riken Brain Science Institute, Wako, Japan) and Drs R. Y. Tsien and A. Palmer (University of California, San Diego).
2.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017; Alexander, Striessnig et al., 2017).
3. RESULTS
3.1. QA promotes oxidative metabolism in insulin‐secreting cells and astrocytes but not in neurons
The effects of QA (Figure 1a) on cell metabolism have been poorly studied. To investigate the effects of this natural product on metabolism, we measured NAD(P)H autofluorescence in INS‐1E cells (Figure 1b), a model of pancreatic β cells. NAD(P)H is formed from NAD(P)+ as a result of mitochondrial oxidative metabolism and is exclusively fluorescent in its reduced form. In control experiments, INS‐1E cells were stimulated with glucose (Figure 1c), which immediately caused a strong rise of the fluorescence. This NADH production reflects glucose oxidation by matrix dehydrogenases of the TCA cycle (De Marchi et al., 2014; De Marchi et al., 2017). In parallel experiments (Figure 1d), the dose–response effect of QA was tested in insulin‐secreting cells, demonstrating the ability of this natural compound to enhance NAD(P)H formation. Quantification of the fluorescence signal on normalized data demonstrated a significant effect of QA at a concentration of 10 μM and a peak of NAD(P)H generation in the range of 30–100 μM (Figure 1e).
Figure 1.
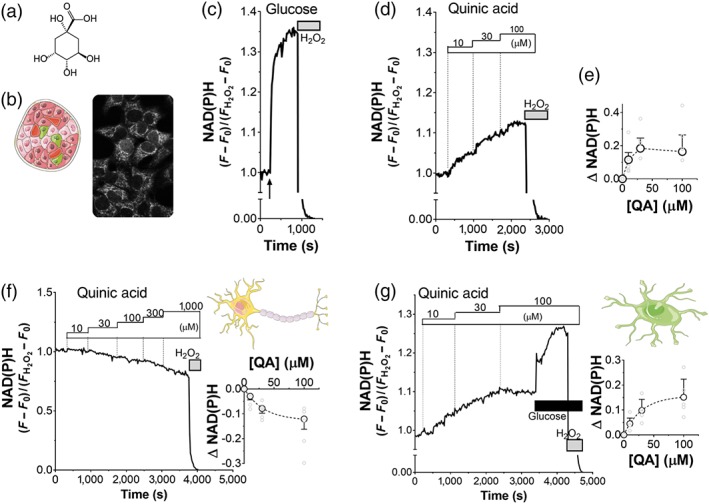
Quinic acid (QA) enhances oxidative metabolism in INS‐1E cells and iCell astrocytes but not in iCell neurons. (a) QA (structure) enhances oxidative metabolism in INS‐1E cells by stimulating Ca2+‐dependent mitochondrial dehydrogenases, which produce NADH. (b) NAD(P)H autofluorescence in INS‐1E cells at 2.5‐mM glucose. Representative traces of NAD(P)H autofluorescence of INS‐1E cells, stimulated with (c) 16.7‐mM glucose and (d) QA (dose–response, as indicated). (c–g) The NAD(P)H signal was normalized to the fluorescence measured in the basal Krebs–Ringer bicarbonate HEPES buffer (set to 1) minus the minimal signal after full oxidation to NAD(P)H using 1% H2O2 (set to 0). Data are representative of (c) n = 5 and (d) n = 5 experiments. (e) Dose–response effect of QA on mitochondrial NAD(P)H production. Values shown are means ± SEM from n = 5 experiments. Effect of QA on NAD(P)H production in (f) iCell neurons and (g) iCell astrocytes. Representative traces (N = 5) of NAD(P)H autofluorescence, stimulated with QA, at the indicated concentrations. Normalization as described in (c). (g) Glucose (2mM) was added, as indicated. Dose–response amplitude of NAD(P)H response, evoked by QA in (f) iCell neurons and (g) iCell astrocytes. Values shown are means ± SEM from n = 5 experiments
To determinate if NAD(P)H generation was a general effect of QA on cell metabolism or if it is specific to pancreatic beta‐cells, we investigated the effects of QA also in neurons and astrocytes, differentiated from induced‐pluripotent stem cells (iCell neurons, Figure 1f; iCell astrocytes, Figure 1g). Following QA stimulation, NAD(P)H autofluorescence was measured in iCell neurons, in glucose‐containing medium: QA caused net oxidation of NAD(P)H (Figure 1f), the opposite of the results in INS‐1E cells (Figure 1d,e). In contrast, QA caused net reduction of NAD(P)H in iCell astrocytes (Figure 1g).
These results demonstrated that QA is a natural bioactive agent, which modulates oxidative metabolism in insulin‐secreting cells. QA promotes a net‐reducing or oxidative effect in different specialized cell types.
3.2. QA enhances cytosolic Ca2+ signalling in insulin‐secreting cells
In pancreatic beta‐cells, the tight coupling between glucose stimulation and insulin secretion is mediated by mitochondrial activation, which consume reducing equivalents (NADH) to generate ATP, promoting KATP channel‐dependent Ca2+ entry. Thus, to investigate the effect of QA on beta‐cell activation, we measured cytosolic Ca2+ rise in INS‐1E insulin‐secreting cells (Figure 2a). Cells expressing the ratiometric cytosolic Ca2+ sensor YC3.6cyto were stimulated with KRBH buffer (control, Figure 2b) or with 100‐μM QA (Figure 2c). The fluorescent signal was unchanged in control experiments, while QA caused an immediate transient cytosolic Ca2+ rise (Figure 2c). Quantification of the total Ca2+ increase during QA stimulation (Figure 2f, left) demonstrated a positive effect of QA on the cytosolic Ca2+ mobilization in basal 2.5‐mM glucose.
Figure 2.
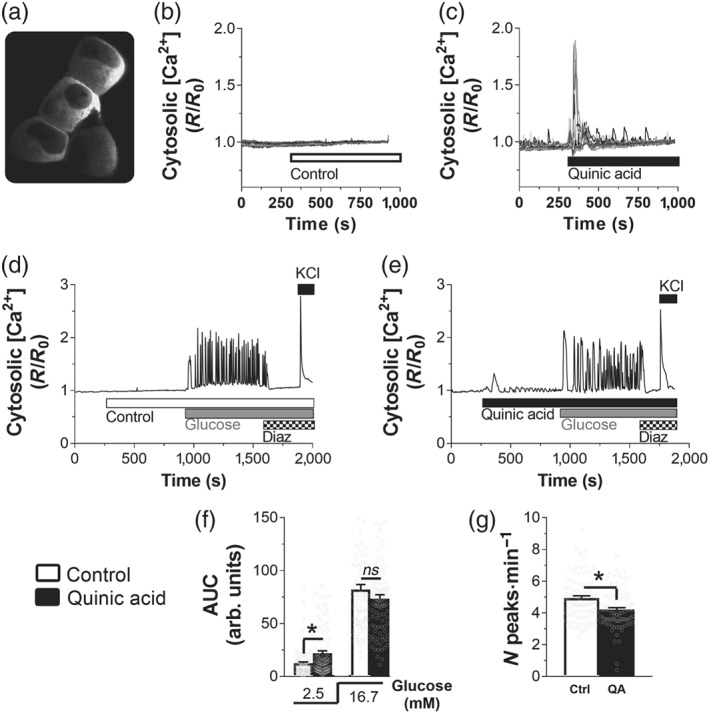
Quinic acid (QA) triggers fast cytosolic Ca2+ rise and modulates the frequency of glucose‐induced cytosolic Ca2+ peaks. (a) Cytosolic YC3.6cyto signal recorded at 535 nm. Example of single‐cell traces of INS‐1E cells, transfected with the cytosolic Ca2+ sensor YC3.6cyto cells at 2.5‐mM glucose and stimulated with (b) Krebs–Ringer bicarbonate HEPES buffer (KRBH; control [Ctrl], n = 5) or (c) 100‐μM QA (n = 5). Representative traces of INS‐1E cells, transfected with YC3.6cyto and stimulated with (d) KRBH (Ctrl, n = 5) or (e) QA (n = 5) and then with 16.7‐mM glucose. KATP channel was activated with 100‐μM diazoxide (Diaz), and then cells were depolarized with addition of 30‐mM KCl (KCl). (f) Statistical evaluation of the total Ca2+ transiting during 5′ stimulation, calculated by integrating the AUC, during KRBH or 100‐μM QA stimulation. (g) Statistical evaluation of the frequency of peaks during glucose stimulation (16.7 mM). (f, g) Values shown are means ± SEM from 97 cells (n = 5; Ctrl) and 93 cells (n = 5; QA); *P <.05, significant effect of QA; Student's t test
The shape of the QA‐dependent cytosolic Ca2+ rise was clearly different compared with glucose‐induced continuous calcium responses (Figure 2d; De Marchi et al., 2014; De Marchi et al., 2017). To investigate if QA acts synergistically with glucose to increase cytosolic [Ca2+], we compared the glucose‐stimulated cytosolic Ca2+ rise in the presence (Figure 2e) or absence of QA (Figure 2d). Statistical evaluation of the total cytosolic [Ca2+] demonstrated that QA enhanced intracellular [Ca2+] only in basal glucose (2.5 mM) but did not show any effect during glucose stimulation (Figure 2f). QA slightly reduced the frequency of calcium transients by 14.7% (Figure 2g). Given that the total Ca2+ rise was not reduced in QA‐stimulated cells, we concluded that the amount of the [Ca2+] rise per peak increased slightly in the presence of QA. QA did not affect calcium changes in response to the KATP channel opener diazoxide or after depolarization of the plasma membrane with KCl (Figure 2d,e). These data suggest that QA does not affect beta‐cell signal transduction at the level of the KATP channel or voltage‐dependent calcium channels.
We conclude that QA affects Ca2+ homeostasis by promoting a fast and transient cytosolic Ca2+ rise. The slight effect on the cytosolic oscillations during glucose stimulation suggests no major effect of this natural bioactive agent on cytosolic [Ca2+] signalling during beta‐cell activation.
3.3. QA promotes Ca2+ release from the ER
The fast peak of QA‐induced cytosolic Ca2+ rise and the absence of oscillations suggested a mechanism different from glucose‐induced Ca2+ entry. The source of calcium may be extracellular or intracellular calcium stores. To distinguish between these possibilities, we measured cytosolic Ca2+ in INS‐1E cells stimulated with QA, in the absence of external Ca2+. As shown in Figure 3a, QA triggered a fast rise of cytosolic Ca2+, even in Ca2+‐free conditions, indicating a second messenger‐mediated Ca2+ release from an internal store, such as the ER. To validate this possibility, we measured ER [Ca2+] with the ER‐targeted Ca2+ indicator D1ER (Figure 3b). To avoid the contribution of the Ca2+ influx from plasma membrane, diazoxide was included in the bath. As shown in Figure 3c,d, QA depleted ER [Ca2+] in both basal (2.5 mM, Figure 3c) and stimulatory (16.7 mM, Figure 3d) glucose. KCl stimulation after depletion was able to replenish the luminal calcium concentrations in both conditions. To confirm the effect of QA on ER [Ca2+] depletion, EGTA was included in the bath (Figure 3e). As shown in Figure 3e, in Ca2+‐free conditions, ER [Ca2+] decreases more rapidly upon stimulation with QA than in control cells. Statistical evaluation of the [Ca2+] depletion (inset, Figure 3e) confirmed a significant effect of QA on the internal [Ca2+] stores. To definitively demonstrate the effect of QA on ER Ca2+ depletion, we measured the effect of QA on cytosolic Ca2+ when the ER was first depleted of Ca2+, by inhibiting the sarco/ER Ca2+‐ATPase (SERCA) pumps with thapsigargin (Figure 3f). As shown in the inset (Figure 3f), in 66% of the cells, QA was not able to increase cytosolic calcium when ER is first depleted. However, a very small cytosolic Ca2+ rise was recorded in a subpopulation of the cells (33%, inset). These data confirm the effect of QA on ER Ca2+ depletion and also indicate that other intracellular Ca2+ stores could potentially slightly contribute to the QA‐dependent Ca2+ rise (e.g., Golgi apparatus or secretory vesicles).
Figure 3.
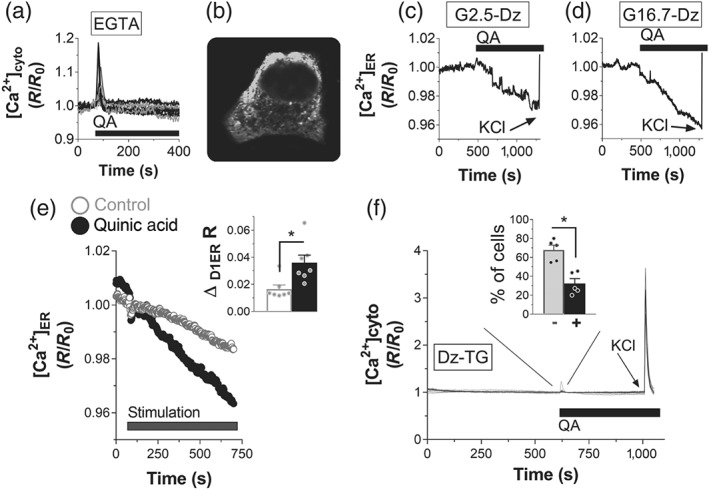
Quinic acid (QA) promotes Ca2+ rise, acting on intracellular Ca2+ stores. (a) Representative traces of cytosolic Ca2+ rise in INS‐1E pancreatic β cells, transfected with the YC3.6cyto (n = 5). The cells were depleted of external Ca2+ by adding 0.5‐mM EGTA in a medium without Ca2+, and then they were stimulated with 100‐μM QA. (b–e) QA promotes Ca2+ release from the endoplasmic reticulum (ER). (b) ER Ca2+ fluorescence was measured with the D1ER sensor (signal recorded at 535 nm). Depletion of ER Ca2+ by 100‐μM QA in INS‐1E cells in the presence of (c) 2.5‐ or (d) 16.7‐mM glucose. The averaged ER signals of the cells in one coverslip are representative of five independent experiments for both (c) and (d). Diazoxide (Dz; 100μM) was included in the bath to avoid the contribution of the plasma membrane Ca2+ influx. KCl (30 μM) was added as indicated. (e) Average of ER Ca2+ signals in INS‐1E cells transfected with the D1ER sensor and then stimulated with Krebs–Ringer bicarbonate HEPES buffer or 100‐μM QA in a medium depleted of external Ca2+ (EGTA 0.5 mM). Statistical evaluation of the amplitude of ER Ca2+ depletion after 10′ stimulation. Data shown are individual values with means ± SEM from n = 7 experiments for both control and QA‐stimulated cells. *P <.05, significant effect of QA; Student's t test. (f) Evaluation of the contribution of intracellular Ca2+ stores to QA‐dependent cytosolic Ca2+ rise. INS‐1E cells, transfected with YC3.6cyto, were incubated in Krebs–Ringer bicarbonate HEPES buffer in the presence of 100‐μM Dz and 1‐μM thapsigargin (TG) to deplete ER Ca2+. Then 100‐μM QA was added to measure the possible contribution of other intracellular Ca2+ stores to the cytosolic Ca2+ elevation. KCl (30 μM) was added as indicated. Single‐cell traces of one representative experiment from n = 5 experiments. The inset shows the number of cells showing (+) or not showing (−) QA‐dependent cytosolic Ca2+ rise. Data shown are individual vales with means ± SEM from n = 5 experiments. *P <.05, significantly different as indicated; Student's t test
Our measurements thus indicate a positive effect of QA on ER Ca2+ release and a possible small effect on other intracellular stores of Ca2+. Taken together, we suggest that QA modulates intracellular Ca2+ homeostasis.
3.4. QA triggers mitochondrial Ca2+ rise during glucose stimulation
Ca2+ homeostasis is a key player for beta‐cell metabolism–secretion coupling (Wiederkehr & Wollheim, 2008). Ca2+ is the main trigger of insulin secretion, but it also serves as the main upstream signal in beta‐cell activation. Calcium also enters mitochondria for the stimulation of energy metabolism (Wiederkehr et al., 2011). Importantly, close contact sites between mitochondria and ER permit the efficient transfer of ER calcium to mitochondria (Giacomello et al., 2010). We followed mitochondrial calcium rises to assess whether QA‐dependent mobilization of calcium was sensed by mitochondria. We measured mitochondrial Ca2+ under both basal and glucose‐stimulated conditions in INS‐1E cells expressing the mitochondrial‐targeted Ca2+ sensor 4mtD3cpv (Figure 4a). In parallel with cytosolic recordings, QA promoted a fast, transient peak of mitochondrial Ca2+ (Figure 4b, stimulation), albeit of small amplitude. We also investigated the effect of QA on mitochondrial Ca2+ in glucose‐stimulated cells (Figure 4b, glucose), and QA showed 48% increase (Figure 4c) over the signal measured in control glucose‐stimulated cells (mock treated 15 min before glucose stimulus).
Figure 4.
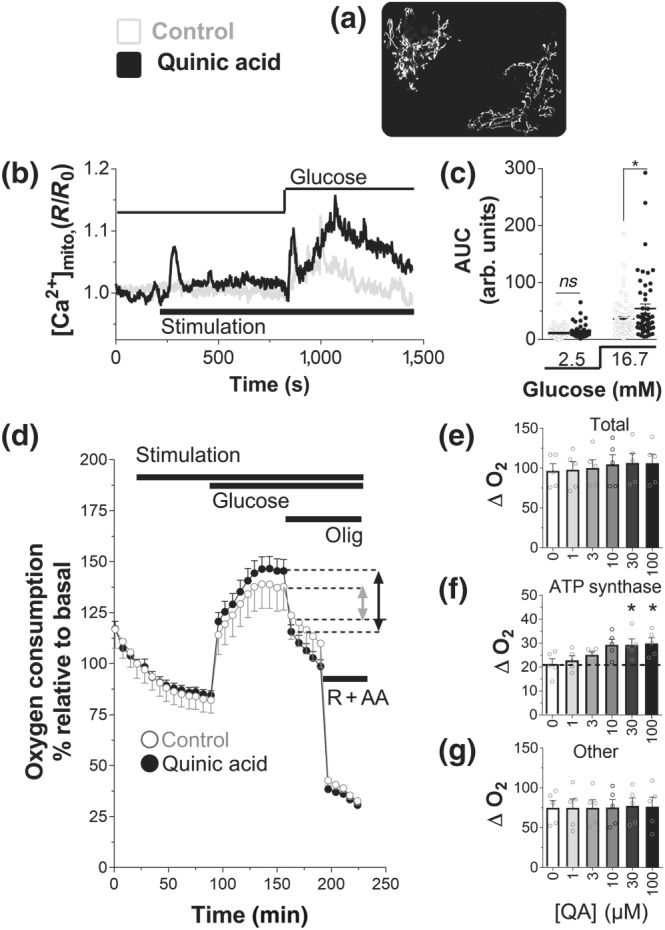
Quinic acid (QA) boosts mitochondrial Ca2+ rise and enhances mitochondrial ATP synthase‐dependent respiration, during glucose stimulation. (a) Mitochondrial fluorescent pattern measured with the mitochondrial Ca2+ sensor 4mtD3cpv, recorded at 535 nm. (b) Representative traces mitochondrial Ca2+ rise in INS‐1E cells, transfected with the 4mtD3cpv. The cells were stimulated with Krebs–Ringer bicarbonate HEPES buffer (KRBH; control [Ctrl]) or 100‐μM QA (in basal 2.5‐mM glucose, and then 16.7‐mM glucose was added (Glucose). (c) Statistical evaluation (scatter plot) of the effect of QA on the amplitude of the mitochondrial [Ca2+] signal in basal (2.5‐mM) glucose or evoked by 16.7‐mM glucose. (b) Traces and (c) scatter points are representative or single points with mean ± SEM, respectively, of 62 cells (n = 5; Ctrl) and 51 cells (n = 5; QA). *P <.05, significant effect of QA; Student's t test. (d–g) Oxygen consumption in INS‐1E cells, assayed in standard KRBH, containing 2.5‐mM glucose. (d) Time course of INS‐1E cells treated with KRBH (Ctrl) or QA (10 μM) as indicated (Stimulation), in basal (2.5‐mM) glucose. Then glucose was added to a final concentration of 16.7 mM (Glucose), and then mitochondrial respiration was inhibited with oligomycin (Olig, 2.5 μg·ml−1) and then rotenone (R, 1 μM) + antimycin (AA, 1 μg·ml−1). Representative trace of five independent experiments. Summary data of the effect of the QA at the indicated concentrations on (e) the total respiration, (f) ATP synthase‐dependent respiration, and (g) other respiration. (e–g) Values shown are means ± SEM from n = 5 experiments. *P <.05, significant effect of QA ; one‐way ANOVA
We concluded that the effect of QA in glucose‐stimulated cells is distinct in different cellular compartments. Therefore, the relatively small level of Ca2+ mobilized by QA diffuses and is diluted in the cytosol and does not influence the total Ca2+ mobilized during glucose stimulation (Figure 2f). On the other hand, the close proximity between ER and mitochondria allows a significant increase in the matrix Ca2+ in QA‐treated cells and could potentially affect mitochondrial function in insulin‐secreting cells.
3.5. QA enhances oligomycin‐sensitive respiration
Mitochondrial matrix Ca2+ signals have been shown to accelerate oxidative metabolism while stimulating ATP synthase‐dependent respiration (De Marchi et al., 2014). Coordinated activation of these two mitochondrial Ca2+‐dependent processes is required to promote robust insulin secretion. Consistent with these findings, we have demonstrated in Figure 1 that QA promotes oxidative metabolism in insulin‐secreting cells. To investigate the effect of QA also on ATP synthase‐dependent respiration, we measured oxygen consumption rate in INS‐1E cells, under basal and glucose‐stimulated conditions (Figure 4d). Incubation of INS‐1E cells with QA did not significantly increase basal respiration, indicating that the small and fast transient increase of mitochondrial Ca2+ did not affect respiration. However, glucose‐dependent respiration was affected by QA treatment. Measurements of total respiration (Figure 4e) showed only a non‐significant increase in QA‐dependent oxygen consumption in dose–response experiments. However, oligomycin‐sensitive respiration reflecting ATP synthase activity was significantly increased by QA (Figure 4f), whereas uncoupled respiration was not affected by QA treatment (Figure 4g). These measurements indicate a positive effect of QA on mitochondrial bioenergetics.
3.6. QA activates metabolism–secretion coupling in insulin‐secreting cells
Coordinated activation of oxidative metabolism and ATP synthase‐dependent respiration by mitochondrial Ca2+ promotes sustained insulin secretion (De Marchi et al., 2014). Given the positive effect of QA on both those processes (Figures 1 and 4), we investigated the ability of QA to promote insulin secretion (Figure 5). In control experiments, stimulation of INS‐1E cells with glucose triggered strong insulin secretion (Figure 5a). Acute treatment of INS‐1E cells with QA significantly increased hormone secretion. Dose–response experiments with QA showed maximal glucose‐induced secretion at a concentration of 50 μM. QA caused a pronounced stimulation of insulin release by 133%, compared with control glucose‐stimulated cells (control, Figure 5a).
Figure 5.
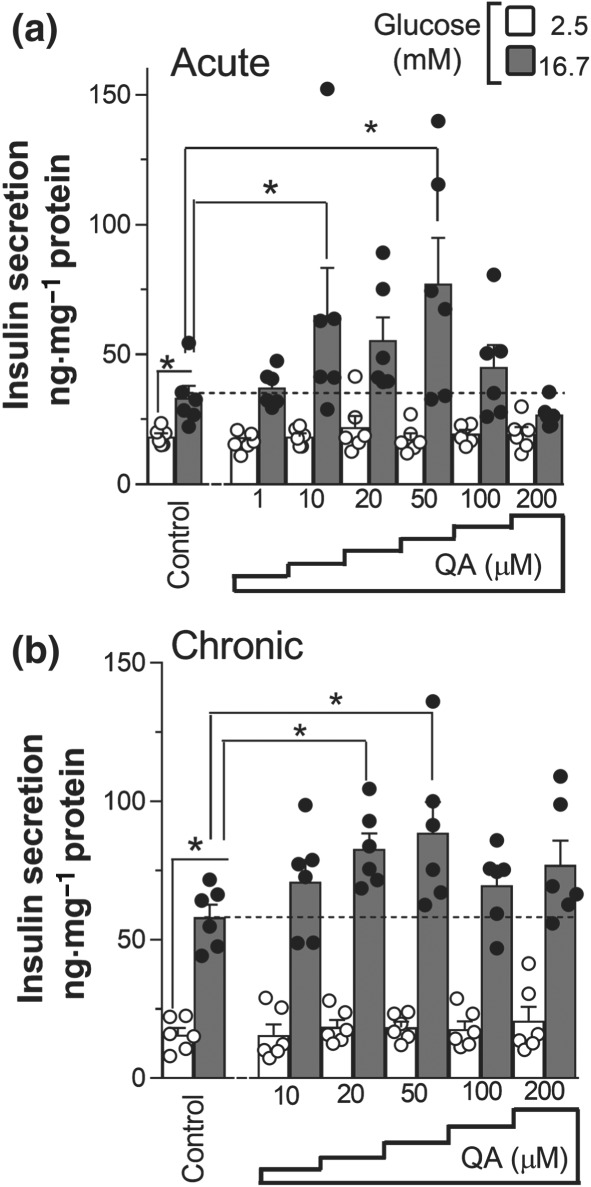
Quinic acid (QA) promotes glucose‐induced insulin secretion. (a) INS‐1E cells were starved for 1 hr in the absence or presence of QA; thereafter, insulin secretion was measured after 1‐hr incubation in 2.5‐ or 16.7‐mM glucose supplemented or not with QA, at the indicated concentration. (b) INS‐1E cells were treated for 3 days with QA, and then insulin secretion was measured with or without QA as above. (a, b) Insulin secretion was normalized to total protein content. Data shown are individual values with means ± SEM from n = 6 experiments. *P <.05, significantly different as indicated; one‐way ANOVA
We also investigated the chronic effects of QA on insulin secretion, by supplementing the cells once per day with fresh QA (Figure 5b). After 3‐days treatment, QA significantly increased the exocytosis of insulin in a dose‐dependent manner. The peak of hormone secretion was recorded from cells treated with a concentration of 20‐μM QA, augmenting insulin exocytosis by 53%, compared with vehicle‐treated cells (control, Figure 5b). These data show that QA promotes insulin secretion after both acute and chronic treatments.
3.7. QA increases glucose‐stimulated insulin release in mice islets, and diet supplementation of QA improves glucose tolerance in mice
As QA induced insulin secretion in INS‐1E cells, we next investigated its effects on islets in vivo. In order to investigate the direct effect of QA and to avoid QA metabolism in the gut, we directly infused QA into the bloodstream for 12 weeks, using an osmotic minipump implanted subcutaneously in mice, containing saline (control) or QA (75 mg·kg−1·day−1; Figure 6a). At the end of the treatment, islets were isolated from mice, and their insulin secretion capacity was measured. Chronic in vivo treatment with QA resulted in significantly increased insulin secretion (Figure 6b) without changing the insulin content of islets (Figure 6c).
Figure 6.
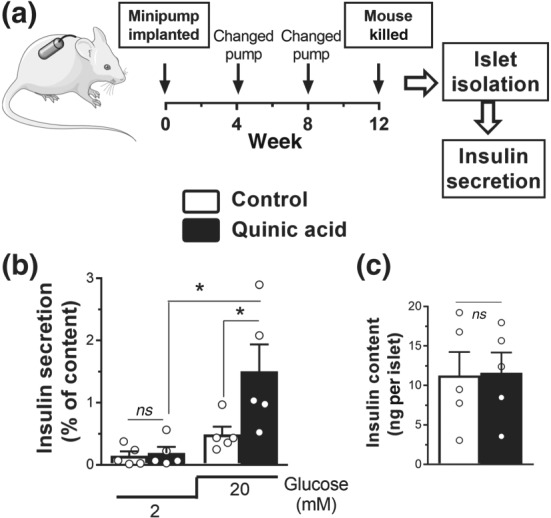
Treatment in vivo with quinic acid (QA) increases glucose‐stimulated insulin release in mouse islets. (a) Experimental schedule. Mice were continuously treated with QA or saline control for 12 weeks using subcutaneously implanted micro‐osmotic pumps. (b) Insulin secretion measured from islets at 2‐ and 20‐mM glucose isolated from mice implanted with QA‐loaded minipump. (c) Insulin content of the islets normalized to islet number. White bars, controls; black bars, QA pump. (b, c) Data shown are individual values with means ± SEM from n = 5 experiments. *P <.05, significantly different as indicated; one‐way ANOVA
We next evaluated whether dietary supplementation with QA could have an effect on glucose metabolism (Figure 7). To that end, QA was added to the diet (25 mg·kg−1) for 2 months to mice followed by intraperitoneal glucose tolerance and insulin tolerance tests (Figure 7). After 16 hr, fasting blood glucose levels were similar between control and QA‐treated mice. However, after glucose infusion, QA‐treated mice showed improved glucose curves having significantly lower glycaemia at 45 min after glucose bolus compared with controls (Figure 7a). Insulin tolerance test performed 2 weeks later showed no significant difference between QA‐treated and control mice (Figure 7b).
Figure 7.
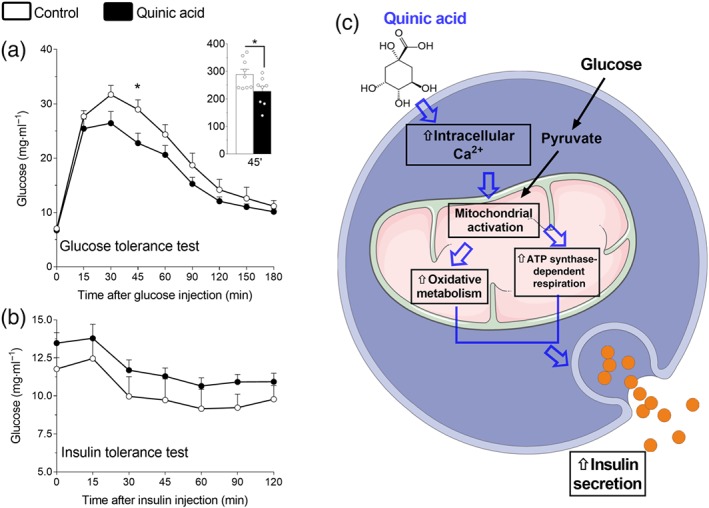
Quinic acid (QA) improves glucose tolerance. Proposed model for QA‐dependent activation of beta‐cells. (a) Glucose tolerance test and (b) insulin tolerance test in mice fed with chow diet supplemented with QA (25 mg·kg−1·day−1) or with chow diet alone for 2 months. Summary data shown at 45 min are individual values with means ± SEM from n = 9–8 mice for each group. *P <.05, significantly different from vehicle group; Student's t test. (c) Proposed model for QA‐dependent activation of beta‐cells. QA boosts intracellular Ca2+ rise in pancreatic beta‐cells, promoting mitochondrial activation. Following glucose stimulation, QA can potentiate, in beta‐cell mitochondria, two matrix Ca2+‐dependent processes (oxidative metabolism and ATP synthase‐dependent respiration), which promotes robust insulin secretion
All together, these results in mice indicate that treatment with QA significantly improved beta‐cell function and dietary QA can improve whole‐body glucose tolerance. Given that insulin tolerance is not changed, the data further suggest that improved glucose tolerance originates from beta‐cells.
4. DISCUSSION
Targeting pancreatic beta‐cells is a promising strategy to treat diabetes, due to the recognition of the pancreatic beta‐cells as central players in the pathogenesis of this multiple organ disorder (Butler et al., 2003; Ferrannini, 2010; Vetere et al., 2014). Current strategies for targeting beta‐cells in the management of diabetes are based on the preservation, expansion, or improved function of this cell type. Approved strategies include the enhancement of insulin secretion, incretin effects, proliferation, protection from cell death, and reprogramming of both acinar and alpha cells to beta‐cells (Vetere et al., 2014). For the next stage of discovery in this field, innovative approaches to modulate biological pathways that regulate beta‐cell function are required (Vetere et al., 2014). Such approaches must be cell specific and regulated. Indeed, compounds that promote insulin secretion should act exclusively under stimulatory glucose concentrations, because uncontrolled insulin secretion can cause lethal hypoglycaemia.
In this context, targeting calcium homeostasis and mitochondria represents an innovative approach to modulate beta‐cell function, promoting beneficial effect for diabetic patients. Therefore, changes in calcium signalling have profound effects on beta‐cell performance and increase risk of developing diabetes (Gilon, Chae, Rutter, & Ravier, 2014; Rorsman, Braun, & Zhang, 2012). In addition, modulation of dynamic cellular calcium homeostasis has been recently demonstrated to prevent cytokine‐mediated beta‐cell loss in diabetes (Clark et al., 2017). Calcium homeostasis and beta‐cell function are substantially linked to mitochondrial function (Wiederkehr & Wollheim, 2012). Indeed mitochondria are versatile intracellular organelles able to take up and release calcium (Santo‐Domingo, Wiederkehr, & De Marchi, 2015). In pancreatic beta‐cells, mitochondrial matrix Ca2+ is an activating signal for insulin secretion (Wiederkehr et al., 2011), and the proposed mechanism of action involves the matrix calcium‐dependent activation of both the mitochondrial oxidative metabolism and the mitochondrial ATP synthase (De Marchi et al., 2014). Moreover, mitochondrial Ca2+ has been proposed to contribute to the improvement of glucose‐stimulated insulin secretion by sulfonylurea compounds in Type 2 diabetic patients (Wiederkehr & Wollheim, 2008). While the effects of Ca2+ homeostasis and mitochondrial function on beta‐cell function is widely accepted, very little therapies based on the modulation intracellular Ca2+ and mitochondrial function have been developed until now. Here, we found a natural compound (QA) that is able to modulate Ca2+ homeostasis, promoting mitochondrial activation and enhancing metabolism–secretion coupling in insulin‐secreting cells. Our dissection of ER, cytosolic, and mitochondrial Ca2+ signals reveals the physiological effects of QA on intracellular Ca2+ homeostasis, indicating the possibility to nutritionally target that process to modulate beta‐cell function. In this context, the ability of QA to promote the release of Ca2+ from the ER appears the key event to activate beta‐cell signal transduction and insulin secretion. Other intracellular Ca2+ stores (Gilon et al., 2014; Lissandron, Podini, Pizzo, & Pozzan, 2010) could potentially contribute to the QA‐dependent Ca2+ remodelling, but the ER appears to be the main target of QA. The direct evidence of the ER Ca2+ release has been previously reported in pancreatic beta‐cells (Graves & Hinkle, 2003; Ravier et al., 2011), and the calcium release channel, ryanodine receptor RyR2, has been recently demonstrated to play a crucial role in the regulation of insulin secretion and glucose homeostasis (Santulli et al., 2015). Consistent with these findings, our results demonstrate that the ability of QA to enhance Ca2+ release from the ER is accompanied by the activation of mitochondrial Ca2+‐dependent processes, which promotes granules exocytosis (De Marchi et al., 2014). Indeed, mitochondria are intracellular organelles located in close proximity to the ER. The physical association between ER and mitochondria, known as mitochondria‐associated ER membrane (Hayashi, Rizzuto, Hajnoczky, & Su, 2009), enables highly efficient transmission of Ca2+ from the ER to mitochondria, stimulating oxidative metabolism. Interestingly, we recorded the major effect of the QA‐dependent increased ER release at mitochondrial level, whereas the cytosolic Ca2+ oscillations were only slightly modulated by that natural bioactive agent. This suggests that the effect of QA on Ca2+ homeostasis occurs mainly at the mitochondrial level, supporting the prominent role of this organelle in metabolism–secretion coupling.
Several natural bioactive compounds have been claimed to exhibit antidiabetic properties, but very little is known about their mechanism of action (Oh, 2015). Natural compounds are indeed very attractive molecule to counteract diabetes, because of their diversity and minimal side effects. Apart from the plant‐derived bioactive agents that target pancreatic beta‐cells , several polyphenols have shown beneficial effects in the treatment of diabetes (Oh, 2015). Plant polyphenols are a large and varied class of natural compounds carrying phenolic hydroxyls, which have attracted great interest in food science (Visioli et al., 2011). As mentioned, the polyphenol caffeic acid is a major compound in coffee, together with chlorigenic acid, which is an ester of caffeic acid and QA (Olthof et al., 2001). While caffeic acid and chlorigenic acid were extensively examined for their effects on cell physiopathology, the abundant natural cyclic polyol QA has received very little attention. By focusing on QA, we have discovered a significant effect of that compound on intracellular calcium homeostasis and a pronounced effect on insulin secretion. Therefore, our results suggest a bioenergetic effect of QA on beta‐cell function, which explains, at least in part, its beneficial effect on granule exocytosis. We cannot exclude, at this point, additional effects of QA on beta‐cell signal transduction. For instance, this compound could directly or indirectly modulate the cytosolic or mitochondrial redox status. Redox reactions are known to contribute to the regulation of insulin secretion (Jezek, Dlaskova, & Plecita‐Hlavata, 2012), and some redox‐active molecules are known to affect beta‐cell function and promote beneficial effect in animal models of diabetes (Kaneto et al., 1999; Tanaka, Gleason, Tran, Harmon, & Robertson, 1999). In the present context, it is possible that QA could integrate calcium and redox signals to promote metabolism–secretion coupling. Consistent with this possibility, we have proposed recently that the combined effects of calcium and redox reactions can trigger granule exocytosis in insulin‐secreting cells (De Marchi et al., 2017). Given that caffeic acid and polyphenols in general are powerful redox‐active compounds (Visioli et al., 2011), synergistic effects would be expected by treating beta‐cells with QA and polyphenols, simultaneously. Further studies should clarify this point and also consider the possibility of specifically targeting the mitochondrial calcium transport. Compounds that increase mitochondrial calcium uptake more efficiently than QA (but transiently, to avoid toxic effects) should promote metabolism–secretion coupling.
Given the effect of QA on pancreatic beta‐cell function, via intracellular Ca2+ rise, additional effects are expected on signal transduction and cell fate. Therefore, it has been recently suggested that activation of GPR 120 (FFA4 receptor) with docosahexaenoic acid increases intracellular Ca2+ levels and thus induces PI3K/Akt signalling‐mediated survival and function of beta‐cells (Zhang et al., 2017). Similarly, QA could potentially modulate that pathway and improve cell protection under stress, which remains to be explored.
Regarding the therapeutic possibilities of an intervention with QA, our results indicate that this naturally occurring polyol can promote glucose‐stimulated insulin release and slightly increase glucose tolerance. In INS‐1E cells, QA increased insulin secretion both acutely and after chronic 3‐day treatment. The positive effects of QA on insulin secretion can be explained by its ability to mobilize calcium and increase the mitochondrial calcium rise. In this manner, QA may prime beta‐cell mitochondria to improve their capacity to synthesize ATP. This explanation is supported by the observed elevated ATP synthase‐dependent respiration after QA treatment. Mitochondrial calcium signals are important for sustained insulin secretion and it is therefore likely that QA stimulates second‐phase insulin secretion. Whether the elevation of the first phase also contributes would need to be tested experimentally.
Chronic 3‐month infusion of QA, systemically, via an osmotic pump increased the glucose‐stimulated insulin secretion in islets, indicating similar mechanisms for both INS‐1E cells and mouse islets. Furthermore, when we gave QA to mice in the diet for 2 months, we were able to improve glucose tolerance, without affecting insulin tolerance in mice. These results support the anti‐diabetic properties of QA. Indeed, chronic 45‐day QA treatment in diet significantly increased blood insulin levels and consequently improved glycaemia on streptozotocin‐induced diabetic rats. This indicates that QA improves beta‐cell function under diabetic stress (Arya et al., 2014). QA has also been shown to have anti‐inflammatory (Jang et al., 2017; Lee, Moon, Kim, & Lee, 2013) and antioxidant properties (Pero et al., 2009), which are both known to be beneficial in diabetic stress. However, one has to keep in mind that QA is metabolized already in the gut, so when QA is given in the diet, we cannot exclude the possibility that the anti‐diabetic effect might be mediated partly by metabolites or conjugates of QA. Taking together our results and those of others, we suggest that QA could be a beneficial natural supplement to alleviate the development of Type 2 diabetes.
In conclusion, we have provided evidence for the ability of QA to modulate pancreatic beta‐cell function, leading to the promotion of insulin secretion and improved glucose tolerance. We have elucidated the mechanism of action of QA (Figure 6d), demonstrating the ability of this bioactive agent to modulate intracellular Ca2+ homeostasis and to activate Ca2+‐dependent mitochondrial function. In this context, the QA‐dependent Ca2+ rise, released from the ER, is taken up by mitochondria and cytosol. Intracellular Ca2+ elevation enhances matrix Ca2+‐dependent function and slightly reshapes cytosolic Ca2+ oscillations. The activation of the mitochondrial Ca2+‐dependent processes, such as oxidative metabolism and ATP synthase‐dependent respiration (De Marchi et al., 2014), enhances insulin exocytosis. Our results indicate that QA and other natural bioactive agents, that modulate Ca2+ homeostasis, enhancing mitochondrial function, could provide a novel approach to activate pancreatic beta‐cells , promoting beneficial effects in the context of Type 2 diabetes.
CONFLICT OF INTEREST
The authors are employees of Nestlé Research—Nestlé Institute of Health Sciences, which is part of the Nestlé Group.
AUTHOR CONTRIBUTIONS
U.D.M. and E.H. conceived the study. U.D.M, E.H., C.C., and A.W. designed the experiments and wrote the manuscript. E.H., A.H., J.T., F.B., S.S.K., J.R., C.C., and U.D.M. performed and analysed the experiments. E.H., J.S‐D., E.H.D., C.C., D.B., and U.D.M. analysed and interpreted the data. All authors wrote and revised the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis and Animal Experimentation and as recommended by funding agencies, publishers, and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
We thank Dr A. Miyawaki (Riken Brain Science Institute, Wako, Japan) and Drs R. Y. Tsien and A. Palmer (University of California, San Diego) for providing the cameleon constructs. We thank the University of Geneva and Professor C. B. Wollheim for the INS‐1E cells. Figures 1, 6, and 7 contain illustrations made by Servier Medical Art, http://www.servier.fr/servier‐medical‐art.
Heikkilä E, Hermant A, Thevenet J, et al. The plant product quinic acid activates Ca2+‐dependent mitochondrial function and promotes insulin secretion from pancreatic beta cells. Br J Pharmacol. 2019;176:3250–3263. 10.1111/bph.14757
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174(Suppl 1), S160–S194. 10.1111/bph.13884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya, A. , Al‐Obaidi, M. M. , Shahid, N. , Bin Noordin, M. I. , Looi, C. Y. , Wong, W. F. , … Mustafa, M. R. (2014). Synergistic effect of quercetin and quinic acid by alleviating structural degeneration in the liver, kidney and pancreas tissues of STZ‐induced diabetic rats: A mechanistic study. Food and Chemical Toxicology, 71, 183–196. 10.1016/j.fct.2014.06.010 [DOI] [PubMed] [Google Scholar]
- Beveridge, T. , Harrison, J. E. , & Drover, J. (2002). Processing effects on the composition of sea buckthorn juice from Hippophae rhamnoides L. Cv. Indian Summer. Journal of Agricultural and Food Chemistry, 50(1), 113–116. 10.1021/jf010369n [DOI] [PubMed] [Google Scholar]
- Bhattacharya, S. , Oksbjerg, N. , Young, J. F. , & Jeppesen, P. B. (2014). Caffeic acid, naringenin and quercetin enhance glucose‐stimulated insulin secretion and glucose sensitivity in INS‐1E cells. Diabetes, Obesity & Metabolism, 16(7), 602–612. 10.1111/dom.12236 [DOI] [PubMed] [Google Scholar]
- Boss, C. , Bouche, N. , & De Marchi, U. (2018). Encapsulated optically responsive cell systems: Toward smart implants in biomedicine. Advanced Healthcare Materials, 7(8), e1701148 10.1002/adhm.201701148 [DOI] [PubMed] [Google Scholar]
- Boss, C. , De Marchi, U. , Hermant, A. , Conrad, M. , Sizzano, F. , Palini, A. , … Bouche, N. (2017). Encapsulation of insulin‐secreting cells expressing a genetically encoded fluorescent calcium indicator for cell‐based sensing in vivo. Advanced Healthcare Materials, 6(4). 10.1002/adhm.201600869 [DOI] [PubMed] [Google Scholar]
- Butler, A. E. , Janson, J. , Bonner‐Weir, S. , Ritzel, R. , Rizza, R. A. , & Butler, P. C. (2003). Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes, 52(1), 102–110. 10.2337/diabetes.52.1.102 [DOI] [PubMed] [Google Scholar]
- Clark, A. L. , Kanekura, K. , Lavagnino, Z. , Spears, L. D. , Abreu, D. , Mahadevan, J. , … Urano, F. (2017). Targeting cellular calcium homeostasis to prevent cytokine‐mediated beta cell death. Scientific Reports, 7(1), 5611 10.1038/s41598-017-05935-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola, E. D. , Conrad, E. C. , & Cotter, R. (1978). High pressure liquid chromatographic determination of major organic acids in cranberry juice. Journal of the Association of Official Analytical Chemists, 61(6), 1490–1492. [PubMed] [Google Scholar]
- De Marchi, U. , Hermant, A. , Thevenet, J. , Ratinaud, Y. , Santo‐Domingo, J. , Barron, D. , & Wiederkehr, A. (2017). A novel ATP‐synthase‐independent mechanism coupling mitochondrial activation to exocytosis in insulin‐secreting cells. Journal of Cell Science, 130(11), 1929–1939. 10.1242/jcs.200741 [DOI] [PubMed] [Google Scholar]
- De Marchi, U. , Thevenet, J. , Hermant, A. , Dioum, E. , & Wiederkehr, A. (2014). Calcium co‐regulates oxidative metabolism and ATP synthase‐dependent respiration in pancreatic beta cells. The Journal of Biological Chemistry, 289(13), 9182–9194. 10.1074/jbc.M113.513184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini, E. (2010). The stunned beta cell: A brief history. Cell Metabolism, 11(5), 349–352. 10.1016/j.cmet.2010.04.009 [DOI] [PubMed] [Google Scholar]
- Giacomello, M. , Drago, I. , Bortolozzi, M. , Scorzeto, M. , Gianelle, A. , Pizzo, P. , & Pozzan, T. (2010). Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store‐operated Ca2+ channels. Molecular Cell, 38(2), 280–290. 10.1016/j.molcel.2010.04.003 [DOI] [PubMed] [Google Scholar]
- Gilon, P. , Chae, H. Y. , Rutter, G. A. , & Ravier, M. A. (2014). Calcium signaling in pancreatic beta‐cells in health and in type 2 diabetes. Cell Calcium, 56(5), 340–361. 10.1016/j.ceca.2014.09.001 [DOI] [PubMed] [Google Scholar]
- Graves, T. K. , & Hinkle, P. M. (2003). Ca2+‐induced Ca2+ release in the pancreatic beta‐cell: Direct evidence of endoplasmic reticulum Ca2+ release. Endocrinology, 144(8), 3565–3574. 10.1210/en.2002-0104 [DOI] [PubMed] [Google Scholar]
- Gray, A. M. , & Flatt, P. R. (1999). Insulin‐secreting activity of the traditional antidiabetic plant Viscum album (mistletoe). The Journal of Endocrinology, 160(3), 409–414. 10.1677/joe.0.1600409 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … Nc, I. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, T. , Rizzuto, R. , Hajnoczky, G. , & Su, T. P. (2009). MAM: More than just a housekeeper. Trends in Cell Biology, 19(2), 81–88. 10.1016/j.tcb.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, H. , Weir, R. L. , Toutounchian, J. J. , Pagadala, J. , Steinle, J. J. , Baudry, J. , … Yates, C. R. (2017). The quinic acid derivative KZ‐41 prevents glucose‐induced caspase‐3 activation in retinal endothelial cells through an IGF‐1 receptor dependent mechanism. PLoS ONE, 12(8), e0180808 10.1371/journal.pone.0180808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatherbell, D. A. , Struebi, P. , Eschenbruch, R. , & Withy, L. M. (1980). A new fruit wine from kiwifruit: A wine of unusual composition and Riesling Sylvaner character. American Journal of Enology and Viticulture, 31(2), 114–121. [Google Scholar]
- Herrmann, K. M. , & Weaver, L. M. (1999). The shikimate pathway. Annual Review of Plant Physiology and Plant Molecular Biology, 50, 473–503. 10.1146/annurev.arplant.50.1.473 [DOI] [PubMed] [Google Scholar]
- Jang, S. A. , Park, D. W. , Kwon, J. E. , Song, H. S. , Park, B. , Jeon, H. , … Kang, S. C. (2017). Quinic acid inhibits vascular inflammation in TNF‐α‐stimulated vascular smooth muscle cells. Biomedicine & Pharmacotherapy, 96, 563–571. 10.1016/j.biopha.2017.10.021 [DOI] [PubMed] [Google Scholar]
- Jezek, P. , Dlaskova, A. , & Plecita‐Hlavata, L. (2012). Redox homeostasis in pancreatic beta cells. Oxidative Medicine and Cellular Longevity, 2012, 932838, 1–16. 10.1155/2012/932838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, U. J. , Lee, M. K. , Park, Y. B. , Jeon, S. M. , & Choi, M. S. (2006). Antihyperglycemic and antioxidant properties of caffeic acid in db/db mice. The Journal of Pharmacology and Experimental Therapeutics, 318(2), 476–483. 10.1124/jpet.106.105163 [DOI] [PubMed] [Google Scholar]
- Kaneto, H. , Kajimoto, Y. , Miyagawa, J. , Matsuoka, T. , Fujitani, Y. , Umayahara, Y. , … Hori, M. (1999). Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta‐cells against glucose toxicity. Diabetes, 48(12), 2398–2406. 10.2337/diabetes.48.12.2398 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & NC3Rs Reporting Guidelines Working Group (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. Y. , Moon, E. , Kim, S. Y. , & Lee, K. R. (2013). Quinic acid derivatives from Pimpinella brachycarpa exert anti‐neuroinflammatory activity in lipopolysaccharide‐induced microglia. Bioorganic & Medicinal Chemistry Letters, 23(7), 2140–2144. 10.1016/j.bmcl.2013.01.115 [DOI] [PubMed] [Google Scholar]
- Lissandron, V. , Podini, P. , Pizzo, P. , & Pozzan, T. (2010). Unique characteristics of Ca2+ homeostasis of the trans‐Golgi compartment. Proceedings of the National Academy of Sciences of the United States of America, 107(20), 9198–9203. 10.1073/pnas.1004702107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler, P. , Antinozzi, P. A. , & Wollheim, C. B. (2000). Modulation of glutamate generation in mitochondria affects hormone secretion in INS‐1E beta cells. IUBMB Life, 50(1), 27–31. 10.1080/15216540050176557 [DOI] [PubMed] [Google Scholar]
- Meng, S. , Cao, J. , Feng, Q. , Peng, J. , & Hu, Y. (2013). Roles of chlorogenic acid on regulating glucose and lipids metabolism: A review. Evidence‐Based Complementary and Alternative Medicine, 2013, 801457, 1–11. 10.1155/2013/801457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, T. , Yamada, S. , Tominaga, T. , Ichikawa, M. , & Miyawaki, A. (2004). Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proceedings of the National Academy of Sciences of the United States of America, 101(29), 10554–10559. 10.1073/pnas.0400417101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg, A. , Hoa, N. K. , Liepinsh, E. , Van Phan, D. , Thuan, N. D. , Jornvall, H. , … Ostenson, C. G. (2004). A novel insulin‐releasing substance, phanoside, from the plant Gynostemma pentaphyllum . The Journal of Biological Chemistry, 279(40), 41361–41367. 10.1074/jbc.M403435200 [DOI] [PubMed] [Google Scholar]
- Oh, Y. S. (2015). Plant‐derived compounds targeting pancreatic beta cells for the treatment of diabetes. Evidence‐Based Complementary and Alternative Medicine, 2015, 629863, 1–12. 10.1155/2015/629863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olthof, M. R. , Hollman, P. C. , & Katan, M. B. (2001). Chlorogenic acid and caffeic acid are absorbed in humans. The Journal of Nutrition, 131(1), 66–71. 10.1093/jn/131.1.66 [DOI] [PubMed] [Google Scholar]
- Palmer, A. E. , Giacomello, M. , Kortemme, T. , Hires, S. A. , Lev‐Ram, V. , Baker, D. , & Tsien, R. Y. (2006). Ca2+ indicators based on computationally redesigned calmodulin‐peptide pairs. Chemistry & Biology, 13(5), 521–530. 10.1016/j.chembiol.2006.03.007 [DOI] [PubMed] [Google Scholar]
- Palmer, A. E. , Jin, C. , Reed, J. C. , & Tsien, R. Y. (2004). Bcl‐2‐mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proceedings of the National Academy of Sciences of the United States of America, 101(50), 17404–17409. 10.1073/pnas.0408030101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, D. K. , Prasad, S. K. , Kumar, R. , & Hemalatha, S. (2012). An overview on antidiabetic medicinal plants having insulin mimetic property. Asian Pacific Journal of Tropical Biomedicine, 2(4), 320–330. 10.1016/S2221-1691(12)60032-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, G. H. , Knobel, S. M. , Arkhammar, P. , Thastrup, O. , & Piston, D. W. (2000). Separation of the glucose‐stimulated cytoplasmic and mitochondrial NAD(P)H responses in pancreatic islet beta cells. Proceedings of the National Academy of Sciences of the United States of America, 97(10), 5203–5207. 10.1073/pnas.090098797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pero, R. W. , Lund, H. , & Leanderson, T. (2009). Antioxidant metabolism induced by quinic acid. Increased urinary excretion of tryptophan and nicotinamide. Phytotherapy Research, 23(3), 335–346. 10.1002/ptr.2628 [DOI] [PubMed] [Google Scholar]
- Poiraudeau, S. , Lieberherr, M. , Kergosie, N. , & Corvol, M. T. (1997). Different mechanisms are involved in intracellular calcium increase by insulin‐like growth factors 1 and 2 in articular chondrocytes: Voltage‐gated calcium channels, and/or phospholipase C coupled to a pertussis‐sensitive G‐protein. Journal of Cellular Biochemistry, 64(3), 414–422. [DOI] [PubMed] [Google Scholar]
- Ravier, M. A. , Daro, D. , Roma, L. P. , Jonas, J. C. , Cheng‐Xue, R. , Schuit, F. C. , & Gilon, P. (2011). Mechanisms of control of the free Ca2+ concentration in the endoplasmic reticulum of mouse pancreatic beta‐cells: Interplay with cell metabolism and [Ca2+]c and role of SERCA2b and SERCA3. Diabetes, 60(10), 2533–2545. 10.2337/db10-1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman, P. , & Ashcroft, F. M. (2018). Pancreatic beta‐cell electrical activity and insulin secretion: Of mice and men. Physiological Reviews, 98(1), 117–214. 10.1152/physrev.00008.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman, P. , Braun, M. , & Zhang, Q. (2012). Regulation of calcium in pancreatic alpha‐ and beta‐cells in health and disease. Cell Calcium, 51(3–4), 300–308. 10.1016/j.ceca.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter, G. A. , Pullen, T. J. , Hodson, D. J. , & Martinez‐Sanchez, A. (2015). Pancreatic beta‐cell identity, glucose sensing and the control of insulin secretion. The Biochemical Journal, 466(2), 203–218. 10.1042/BJ20141384 [DOI] [PubMed] [Google Scholar]
- Ryan, J. J. , & Dupont, J. A. (1973). Identification and analysis of the major acids from fruit juices and wines. Journal of Agricultural and Food Chemistry, 21(1), 45–49. 10.1021/jf60185a018 [DOI] [Google Scholar]
- Santo‐Domingo, J. , Wiederkehr, A. , & De Marchi, U. (2015). Modulation of the matrix redox signaling by mitochondrial Ca2+ . World Journal of Biological Chemistry, 6(4), 310–323. 10.4331/wjbc.v6.i4.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli, G. , Pagano, G. , Sardu, C. , Xie, W. , Reiken, S. , D'Ascia, S. L. , … Marks, A. R. (2015). Calcium release channel RyR2 regulates insulin release and glucose homeostasis. The Journal of Clinical Investigation, 125(5), 1968–1978. 10.1172/JCI79273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffes, M. W. , Sibley, S. , Jackson, M. , & Thomas, W. (2003). Beta‐cell function and the development of diabetes‐related complications in the diabetes control and complications trial. Diabetes Care, 26(3), 832–836. 10.2337/diacare.26.3.832 [DOI] [PubMed] [Google Scholar]
- Tanaka, Y. , Gleason, C. E. , Tran, P. O. , Harmon, J. S. , & Robertson, R. P. (1999). Prevention of glucose toxicity in HIT‐T15 cells and Zucker diabetic fatty rats by antioxidants. Proceedings of the National Academy of Sciences of the United States of America, 96(19), 10857–10862. 10.1073/pnas.96.19.10857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenet, J. , De Marchi, U. , Domingo, J. S. , Christinat, N. , Bultot, L. , Lefebvre, G. , … Wiederkehr, A. (2016). Medium‐chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte–neuron lactate and ketone body shuttle systems. The FASEB Journal, 30(5), 1913–1926. 10.1096/fj.201500182 [DOI] [PubMed] [Google Scholar]
- Uleberg, E. , Rohloff, J. , Jaakola, L. , Trost, K. , Junttila, O. , Haggman, H. , & Martinussen, I. (2012). Effects of temperature and photoperiod on yield and chemical composition of northern and southern clones of bilberry (Vaccinium myrtillus L.). Journal of Agricultural and Food Chemistry, 60(42), 10406–10414. 10.1021/jf302924m [DOI] [PubMed] [Google Scholar]
- Vetere, A. , Choudhary, A. , Burns, S. M. , & Wagner, B. K. (2014). Targeting the pancreatic beta‐cell to treat diabetes. Nature Reviews. Drug Discovery, 13(4), 278–289. 10.1038/nrd4231 [DOI] [PubMed] [Google Scholar]
- Visioli, F. , De La Lastra, C. A. , Andres‐Lacueva, C. , Aviram, M. , Calhau, C. , Cassano, A. , … Edeas, M. (2011). Polyphenols and human health: A prospectus. Critical Reviews in Food Science and Nutrition, 51(6), 524–546. 10.1080/10408391003698677 [DOI] [PubMed] [Google Scholar]
- Weir, G. C. , & Bonner‐Weir, S. (2004). Five stages of evolving beta‐cell dysfunction during progression to diabetes. Diabetes, 53(Suppl 3), S16–S21. 10.2337/diabetes.53.suppl_3.S16 [DOI] [PubMed] [Google Scholar]
- Wiederkehr, A. , Szanda, G. , Akhmedov, D. , Mataki, C. , Heizmann, C. W. , Schoonjans, K. , … Wollheim, C. B. (2011). Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metabolism, 13(5), 601–611. 10.1016/j.cmet.2011.03.015 [DOI] [PubMed] [Google Scholar]
- Wiederkehr, A. , & Wollheim, C. B. (2008). Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta‐cell. Cell Calcium, 44(1), 64–76. 10.1016/j.ceca.2007.11.004 [DOI] [PubMed] [Google Scholar]
- Wiederkehr, A. , & Wollheim, C. B. (2012). Mitochondrial signals drive insulin secretion in the pancreatic beta‐cell. Molecular and Cellular Endocrinology, 353(1–2), 128–137. 10.1016/j.mce.2011.07.016 [DOI] [PubMed] [Google Scholar]
- Zhang, D. , So, W. Y. , Wang, Y. , Wu, S. Y. , Cheng, Q. , & Leung, P. S. (2017). Insulinotropic effects of GPR120 agonists are altered in obese diabetic and obese non‐diabetic states. Clinical Science (London, England), 131(3), 247–260. 10.1042/CS20160545 [DOI] [PubMed] [Google Scholar]