

Abstract
Background and Purpose
Cerebral vasospasm and neuronal apoptosis after subarachnoid haemorrhage (SAH) is the major cause of morbidity and mortality in SAH patients. So far, single‐target agents have not prevented its occurrence. Memantine, a non‐competitive NMDA re3ceptor antagonist, is known to alleviate brain injury and vasospasm in experimental models of SAH. Impairment of NO availability also contributes to vasospasm. Recently, we designed and synthesized a memantine nitrate MN‐08, which has potent dual functions: neuroprotection and vasodilation. Here, we have tested the therapeutic effects of MN‐08 in animal models of SAH.
Experimental Approach
Binding to NMDA receptors (expressed in HEK293 cells), NO release and vasodilator effects of MN‐08 were assessed in vitro. Therapeutic effects of MN‐08 were investigated in vivo, using rat and rabbit SAH models.
Key Results
MN‐08 bound to the NMDA receptor, slowly releasing NO in vitro and in vivo. Consequently, MN‐08 relaxed the pre‐contracted middle cerebral artery ex vivo and increased blood flow velocity in small vessels of the mouse cerebral cortex. It did not, however, lower systemic blood pressure. In an endovascular perforation rat model of SAH, MN‐08 improved the neurological scores and ameliorated cerebral vasospasm. Moreover, MN‐08 also alleviated cerebral vasospasm in a cisterna magna single‐injection model in rabbits. MN‐08 attenuated neural cell apoptosis in both rat and rabbit models of SAH. Importantly, the therapeutic benefit of MN‐08 was greater than that of memantine.
Conclusion and Implications
MN‐08 has neuroprotective potential and can ameliorate vasospasm in experimental SAH models.
Abbreviations
- EBI
early brain injury
- ISDN
isosorbide dinitrate
- MCA
middle cerebral artery
- MDA
malondialdehyde
- RBCs
red blood cells
- SAH
subarachnoid haemorrhage
What is already known
Memantine alleviates brain injury and attenuates vasospasm in experimental models of SAH.
Impairment of NO availability has been reported to contribute to vasospasm.
What this study adds
A memantine nitrate MN‐08 was designed with dual functions: NMDA receptor binding and NO releasing.
MN‐08 was effective in rat and rabbit SAH models and was more effective than memantine.
What is the clinical significance
MN‐08 may serve as a promising new clinical candidate for the treatment of SAH.
1. INTRODUCTION
Subarachnoid haemorrhage (SAH), a severe condition with high mortality and disability, is the extravasation of blood into the subarachnoid space, which is normally filled with CSF. Rupture of an intracranial aneurysm is the leading cause of SAH, accounting for about 80% of cases (Lawton & Vates, 2017). Cerebral vasospasm is an important pathological feature of SAH and can lead to delayed cerebral ischaemia and infarction. These delayed effects start 3–12 days after SAH, in 70% of patients, and remain a major cause of poor outcome and death (Connolly et al., 2012). The pathogenesis of vasospasm is complex and still not fully elucidated. Nimodipine, a calcium channel antagonist, is thought to dilate brain blood vessels and it is the only drug that is effective in preventing delayed ischaemic neurological complications and reducing mortality after SAH (Macdonald, 2014). Researchers are still looking for new therapeutic options for prevention and treatment of SAH.
Several studies in rats and patients have shown that, after SAH, NMDA receptors were over‐stimulated by excessive release of glutamate (Hillered, Vespa, & Hovda, 2005; Westermaier, Jauss, Eriskat, Kunze, & Roosen, 2011). Over‐activation of NMDA receptors can induce massive Ca2+ influx into neurons and initiate signalling pathways leading to cell death (Villmann & Becker, 2007) and glutamate concentrations correlate with the neurological status of SAH patients (Sehba, Plata, & Zhang, 2011). NMDA receptor antagonists have been proposed to provide neuroprotection to attenuate early and delayed neuronal damage after SAH (Fujii et al., 2013). Memantine, a low‐affinity NMDA receptor antagonist, reduced early brain injury (EBI) and cerebral vasospasm after SAH (Huang et al., 2015; Huang, Wang, Shan, Pan, & Tsai, 2015).
NO is a primary vasodilator and regulator of cerebral blood flow and the NO signalling pathway was disrupted after SAH (Pluta, 2008; Sabri, Ai, Lass, D'Abbondanza, & Macdonald, 2013). Levels of NO and its metabolites were significantly decreased in brain tissue after SAH (Khaldi, Zauner, Reinert, Woodward, & Bullock, 2001; Sehba, Schwartz, Chereshnev, & Bederson, 2000). Additionally, the haemoglobin released from haemolysis of red blood cells (RBCs) in cisterns is an important cause of vasospasm after SAH (Macdonald & Weir, 1991). The affinity of NO to haemoglobin is about 1,000 times higher than that of oxygen (Suhardja, 2004) and haemoglobin thus serves as a potent NO scavenger. Our recent understanding of NO‐related pathophysiological changes that ultimately lead to vasospasm in cerebral arteries opens up new possibilities for NO‐based therapy for vasospasm. Several animal and clinical studies have suggested that intravenous injection of sodium nitroprusside or nitroglycerin alleviated cerebral vasospasm (Fathi et al., 2011; Jeffrey & McGinnis, 2002). However, because of the systemic effects of NO in peripheral (non‐cerebral) vessels, there is a risk of undesirable systemic hypotension in patients, which limits the clinical use of these drugs for treatment of SAH (Allen, 1976). NO donor compounds that specifically release NO in brain tissue but not in the rest of the body, however, would have great potential for treatment of cerebrovascular disease.
Recently, a series of memantine nitrate derivatives with dual effects of neuroprotection and vasodilation have been designed and synthesized in our lab (Liu et al., 2017). These compounds were designed by combining the memantine skeleton and a nitrate moiety with the hope that they would simultaneously inhibit NMDA receptors and release NO in the CNS. Several memantine nitrates are effective in protecting neurons against glutamate‐induced injury and can dilate aortic rings, pre‐contracted with phenylephrine. Among them, MN‐08 (as shown in Table 1) is the most effective. In the present study, we further investigated the NMDA receptor binding, NO releasing, and vasodilatory effects of MN‐08 in vitro and explored the therapeutic effect of MN‐08 on rat and rabbit SAH models in vivo.
Table 1.
Chemical structures of MN‐08 and memantine, and the IC50 values of binding to NMDA receptors
| Compound | Chemical structure | NMDA receptor binding assay IC50 (μM) |
|---|---|---|
| MN‐08 |
|
12.8 ± 2.8 |
| Memantine |
|
6.0 ± 1.8 |
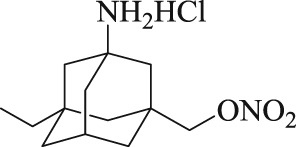
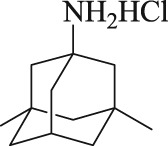
Data are expressed as mean ± SD (n = 8).
2. METHODS
2.1. Animals
All animal care and experimental protocols were performed in accordance with the National Institutes of Health guidelines (USA) and approved by the Ethics Committee of the Institute of Laboratory Animal Science at Jinan University, Guangzhou, China. Animal studies are reported in compliance with the ARRIVE guidelines and with the recommendations made by the British Journal of Pharmacology (Kilkenny et al., 2010; McGrath & Lilley, 2015). The experimental designs were based on the rule of the replacement, refinement, and reduction (the 3Rs) to reduce suffering of the animals and use the minimum number of animals.
2.2. Sourcing and care of animals
Adult male mice (C57BL/6J, 12 weeks old, 25–28 g, RRID:IMSR_JAX:000664), adult male rats (Sprague–Dawley, 8 weeks old, 250–280 g, RRID:MGI:5651135), and adult male rabbits (New Zealand white rabbit, 12 weeks old, 2.5–3 kg) were purchased from Guangdong Medical Laboratory Animal Center (Guangzhou, China). Mice (cage size: 325 × 210 × 180 mm) and rats (cage size: 475 × 350 × 200 cm) were housed in specific pathogen free conditions with free access to food and water (five mice or rats per cages). The room was kept at a controlled temperature (23–25°C) and humidity (45–65%) and on a 12:12 light/dark cycle. The rabbits were single housed in cages and the room temperature was controlled in 23–25°C, and the light was kept in a 12:12 light/dark cycle. The rabbits were free access to water and food.
The sample size calculation in the rat SAH experiment was based on detecting a 100‐μm difference in the perimeter of basilar artery between the MN‐08 and vehicle groups with a type I error (α) of .05 and power of 0.9. We estimated that we would need a minimum sample size of six rats per group given the SD of 90 μm found in our pilot study. Based on this power calculation, for the proposed experiments, we used a sample size of 10 rats (including 7–8 rats for analysis) per SAH operation group to control for possible loss of animals. Using the same approach to calculate the necessary sample size of the rabbit experiments, we assumed a detectable difference of 110 μm in the perimeter of the basilar artery, a type I error (α) of .05, a power of 0.9, and an SD of 90 μm. A sample size of seven rabbits (included 5–6 rabbits for analysis) per SAH operation group was calculated.
2.3. Patch clamp recordings
Patch assay was performed in the IonWorksTM Barracuda system (Molecular Devices, California, US) using HEK293 cells (ATCC, Manassas, USA, RRID:CVCL_0045), which were transfected with the appropriate ion channel or receptor cDNA(s) encoding NR1 (GluN1) and NR2A (GluN2A). Stock solutions of MN‐08 (100 μM) and memantine (150 μM) were prepared and diluted into working solutions: MN‐08 (0.8, 1.6, 3.2, 6.5, 12.5, 25, 50, and 100 μM) and memantine (1.17, 2.34, 4.69, 9.38, 8.8, 37.5, 75, and 150 μM). The patch procedure has been described in previous reports (Jambrina et al., 2016; Maki, Cummings, Paganelli, Murthy, & Popescu, 2014). In short, electrodes were filled with an intracellular solution of 50‐mM CsCl, 90‐mM CsF, 2‐mM MgCl2, 5‐mm EGTA, and 10‐mM HEPES, which was then adjusted to pH 7.2 with CsOH. In preparation for a recording session, the intracellular solution was loaded into the intracellular compartment of the PPC planar electrode. Extracellular solution (HBPS) was prepared as follows: NaCl, 137 mM; KCl, 1.0 mM; CaCl2, 5 mM; HEPES, 10 mM; and glucose, 10 mM. The pH was adjusted to 7.4 with NaOH and the solution was refrigerated until use. The holding potential was set at −70 mV; the potential during agonist/PAM application was set at −40 mV. For recording the cell current, the extracellular buffer was loaded into the PPC plate wells at 11 μl per well. The cell suspension was pipetted into the wells (9 μl per well) of the PPC planar electrode. The whole‐cell recording configuration was established via patch perforation with membrane currents recorded by on‐board patch clamp amplifiers. Two recordings (scans) were performed: first, during pre‐application of MN‐08, and the second, during co‐application of MN‐08 with agonist (EC50 L‐glutamate) to detect inhibition of NR1/NR2A receptors by MN‐08. The application consisted of two additions of the 20‐μl test solution containing memantine at 10 μl·s−1 (2‐s total application time). The addition of either 2× concentrated MN‐08 or memantine was first pre‐applied for 5 min before the second addition of the 1× stock solution.
The inhibitory effects of MN‐08 and memantine on the channel was calculated as % inhibition = (ITA/IEC50)·100%, where ITA was the NR1/NR2A EC50‐elicited current in the presence of various concentrations of MN‐08; IEC50 was the mean current elicited with L‐glutamate EC50.
2.4. Measurement of NO in vitro and in vivo
To measure NO levels in vitro, the Griess reagent was applied to detect the concentration of NO in PBS (pH 7.4). First, stock solutions of MN‐08, isosorbide dinitrate (ISDN; 40 mM in dd H2O), PBS (0.1 M), and PBS containing 25‐mM L‐cysteine were prepared. The reaction protocol began by adding stock solution (200 μl) and PBS with 25‐mM L‐cysteine (1,800 μl) to a 4‐ml tube. The reaction mixture was incubated at 37°C, and Griess reagent (500 μl) was added at appropriate time points (0.5, 1, 2, 4, 6, 8, 12, 16, 24, and 32 hr). The nitrate was allowed to react with the Griess reagent for 30 min. And then, the absorbance of each sample was determined by microplate reader (model ELX800, BioTek Instruments) at 540 nm.
To measure NO levels in vivo, a total NO assay kit was used. Plasma levels of NO were assessed by detecting the amounts of nitrates and nitrites. Rat blood (0.4 ml) was drawn from the jugular vein after intravenous injection with MN‐08 (25 mg·kg−1) or ISDN (3 mg·kg−1) at 0, 0.25, 0.5, 1, 2, and 4 hr. The blood was collected in heparin tubes and centrifuged in 4°C at 1,301× g for 10 min (Eppendorf Biotech Company, Hamburg, Germany). The levels of NO in plasma was measured by detecting absorbance at 540 nm by microplate reader after reaction with Griess reagent. Results were expressed as mean ± SD from six independent experiments.
2.5. Artery ring preparation and isometric force measurement
Adult male Sprague–Dawley rats (250–280 g) were killed by cervical dislocation during anaesthesia with sodium pentobarbital (100 mg·kg−1). The endothelium‐intact middle cerebral artery (MCA) was dissected and isolated from adhering connective tissues in ice‐cold oxygenated Krebs solution (composition in mM: 119 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 1.5 CaCl2, 24.9 NaHCO3, 0.01 EDTA, 11 glucose) The pH was adjusted to 7.4. The MCA rings (approx. 2 mm in length) were mounted in a 10‐ml organ bath of Multi Myograph System (620 m, Danish Myo Technology A/S, Aarhus, Denmark) in Krebs solution maintained at 37°C and continuously aerated with a mixture of gas (95% O2/5% CO2). Arterial rings were set to an optimal tension (4 mN) and stabilized for 60 min. The data were collected by PowerLab 8/35 (ADinstruments, Australia, RRID:SCR_001620) and analysed by LabChart software (version 8.0, ADinstruments, Australia).
KCl (60 mM), 5‐HT (3 μM), or U46619 (1 μM) were used to induce a steady, contracted tone in the MCA rings. Concentration–response curves were subsequently generated by the cumulative addition of MN‐08 at concentrations 1–100 μM. A time‐matched vehicle (ddH2O) control protocol was also performed.
To investigate if inhibition of extracellular Ca2+ influx was involved in the vasorelaxant effect of MN‐08, CaCl2 (3 mM) was added in Ca2+‐free, KCl (60 mM) solution after pre‐incubation with MN‐08 (10, 30, or 100 μM), memantine (10 or 30 μM), or nimodipine (an L‐type calcium channel blocker, 30 nM) for 20 min.
To further investigate other possible mechanisms involved in the vasorelaxation effect of MN‐08, MCA rings were exposed to ODQ (a guanylyl cyclase inhibitor, 10 μM), propranolol (a non‐selective β‐adrenoreceptor antagonist, 1 μM), atropine (an inhibitor of muscarinic acetylcholine receptors (100 nM), glibenclamide (a K+‐ATP channel blocker, 10 μM), indomethacin (a cyclooxygenase inhibitor, 10 μM), BaCl2 (an inward rectifier K+ channel inhibitor, 10 μM), and tetraethylammonium chloride (a non‐selective K+ channel inhibitor, 10 mM). Arteries were constricted using U46619 (1 μM) after a 20‐min incubation period with each inhibitor. MN‐08 (1–100 μM) was then added.
2.6. Two‐photon imaging
A thinned‐skull window was created for brain imaging. We followed the technique reported by Shih et al. (2012). Adult male C57BL/6 mice (25–28 g) were anaesthetized with isoflurane (2.5%, v/v), and the depth of anaesthesia was checked by toe pinch. The body temperature was maintained with a warm pad (RWD Life Science Co., Shenzhen, China) during the procedure. The head area of mice was shaved and sterilized with 70% alcohol followed by iodopovidone. The skull surface was exposed and a custom‐made frame was mounted to the cranium with dental cement. A 2 × 2 mm2 region was selected to thin a circular window by micro‐drill (RWD Life Science Co., Shenzhen, China). The window region was polished with micro‐bled when it was 20–30 μm thick, and the pial vessels were clearly visible through the bone. Mice blood serum was labelled by injecting 200 ul (2.5%, w/v) FITC‐dextran (MW 20,000).
The imaging procedure was described in a previous study (Shih et al., 2012). In short, an Olympus two‐photon microscope (FV1000MPE, Olympus Corporation, Tokyo, Japan) equipment with a 40× water‐immersion objective (NA = 0.80) was used to acquire fluorescence images. In this imaging procedure, the near infrared laser was tuned to 900 nm. For measurement of RBCs velocity, blood vessels with a diameter of 3–6 μm were selected. We chose a scanning rate of 4 μs per pixel and 1,000 lines in total to image a line path in the centre of blood vessels. The average of RBCs velocity speed was calculated from the space–time image. These images were processed by an algorithm using MATLAB® R2015b (MathWorks Inc., Massachusetts, USA; Chhatbar & Kara, 2013).
2.7. Measurement of blood pressure
Rats were anaesthetized with pentobarbital sodium (50 mg·kg−1, supplemented with 10 mg·kg−1 per hour). The anaesthetic depth was evaluated with reflex response to pain induced by toe pinch. During the experiment, the body temperature of rats was monitored and kept in 36.5–37°C with a heating pad (RWD Life Science Co., Shenzhen, China). The femoral artery catheterization was performed to determine haemodynamic parameters. Briefly, a PE 50 catheter, filled with heparinized saline (200 U·ml−1), was inserted into the femoral artery. The artery catheter was connected to the physiological pressure transducer (MLT844, ADinstruments, Australia). The blood pressure was continuously recorded using PowerLab 8/35. The data were collected and analysed by LabChart software.
2.8. Rat endovascular perforation SAH model
Adult male Sprague–Dawley rats (250–280 g) were randomly divided into seven groups: the sham group, the SAH + vehicle group, the SAH + MN‐08 (3 mg·kg−1) group, the SAH + MN‐08 (6 mg·kg−1) group, the SAH + MN‐08 (12 mg·kg−1) group, the SAH + mematine (10 mg·kg−1) group, and the SAH + nimodipine (0.1 mg·kg−1) group. MN‐08 and memantine were dissolved in saline, and a stock solution of nimodipine was prepared in Solutol HS15. Rats were treated with compounds or vehicle (saline) control via intravenous injection at 3 and 6 hr after SAH surgery. On Days 2–7, drugs were administered at 9:00 a.m. and 3:00 p.m. The rat SAH model was performed by endovascular perforation as reported in previous studies (Chen et al., 2015). Briefly, rats were anaesthetized with 2.5% (v/v) isoflurane via animal anaesthesia ventilator system (RWD Life Science Co., Shenzhen, China). The body temperature of the rats was monitored and kept in 37°C with a heating pad (YS‐TCS, RWD Life Science Co., Shenzhen, China). The neck area was sterilized by iodopovidone and alcohol (70%). After exposing the right common carotid artery, external carotid artery (ECA), and internal carotid artery (ICA), the ECA was ligated, cut, and shaped into a 3‐mm stump. A sharpened 4–0 monofilament nylon suture was advanced rostrally into the ICA from the ECA stump until resistance was felt (18–20 mm from the common carotid artery bifurcation) and then pushed 3 mm further to perforate the bifurcation of the anterior and MCA. The suture was withdrawn after approx. 15 s. Rats in the sham operation group underwent the same procedure, but without the perforation. Venous blood gas was monitored by a blood gas analyser (ABL80basic, Radiometer‐Copenhagen, Denmark), and blood glucose level was measured by a portable glucometers (Roche Diagnostics, CA, USA) before and 5 min after the SAH surgery. After surgery, animals were transferred into a temperature and humidity‐controlled chamber. Body weights were monitored before SAH surgery and during the experimental period. If the body weights decreased 20% in 2 days, the rats were killed by pentobarbital sodium (100 mg·kg−1, i.p.).
2.9. Rabbit cisterna magna single‐injection SAH model
Adult male rabbits (2.5–3 kg) were randomly assigned to five groups: the sham group, the SAH + vehicle group, the SAH + MN‐08 (6 mg·kg−1) group, the SAH + memantine (5 mg·kg−1) group, and the SAH + nimodipine (0.05 mg·kg−1) group. The sample size, the number of deaths, body weight, and body temperature and blood glucose are provided in Table S2. Animals were injected with the compounds or vehicle (saline) through a marginal ear vein under conscious state (twice daily for 3 days). The time of treatment was at 3 and 6 hr after SAH surgery and at 9:00 a.m. and 3:00 p.m. on Days 2–3.
Experimental rabbit SAH was produced according to the previous study (Munakata, Naraoka, Katagai, Shimamura, & Ohkuma, 2016). Rabbits were anaesthetized with sodium pentobarbital (35 mg·kg−1) after intravenous injection through the auricular vein. The back neck area was sterilized by iodopovidone and alcohol (70%). During spontaneous breathing, a 23‐gauge butterfly needle was inserted percutaneously into the cisterna magna. After withdrawal of 1.5‐ml CSF, the same amount of non‐heparinized fresh autologous auricular arterial blood was slowly injected into the cisterna magna for 1 min under sterile conditions. Animals were then kept in a 30° head down position for 30 min, facilitating blood flow from the cisterna magna into the posterior communicating cistern more easily and quickly. In sham group animals, the same procedure was applied, but with injection of sterile saline instead of blood. The body temperature and blood glucose level was measured before and 5 min after blood injection into cisterna magna. After recovery from anaesthesia, they were returned to the feeding room. Body weights were measured before SAH surgery and at the end of the experiment. If the body weights decreased 20% in 2 days, the animals were killed by pentobarbital sodium (100 mg·kg−1, i.p.).
2.10. Neurological score and SAH severity evaluation
The neurological scores were blindly evaluated at Day 4 (rabbits) or Day 8 (rats) after SAH using the previously described scoring system of Garcia, Wagner, Liu, and Hu (1995). The evaluation consists of six tests that can be scored 0–3 or 1–3. These six tests include spontaneous activity, symmetry in the movement of the four limbs, forepaw outstretching, climbing, body proprioception, and the response to vibrissa touch.
The severity of SAH was blindly quantified according to a previously published grading scale (Sugawara, Ayer, Jadhav, & Zhang, 2008). The rats or rabbits were killed under deep anaesthesia with pentobarbital sodium (100 mg·kg−1, i.p.) on Day 8 and the brains removed rapidly. The basal cistern was divided into six segments; each segment was scored from 0 to 3 depending on the amount of subarachnoid blood clot. The grading was as follows: Grade 0, no subarachnoid blood; Grade 1, minimal subarachnoid blood; Grade 2, moderate blood clot with recognizable arteries; and Grade 3, blood clot obliterating all arteries within the segment.
2.11. Haematoxylin and eosin staining and vasospasm measurement
Animals were deeply anaesthetized with pentobarbital sodium (100 mg·kg−1), and the chest was opened, a needle (18 G) was inserted into the aorta through left ventricle. The intracardiac perfusion for tissue fixation started with PBS (0.1 M) and was followed by 4% paraformaldehyde. The perfusion solutions were placed in 1.77 m high from the animals, and the perfusion pressure was maintained at 130 mmHg (Gage, Kipke, & Shain, 2012). The brain and brainstem were collected and fixed overnight in 4% paraformaldehyde solution with 30% sucrose. The tissue was dehydrated through a series of graded ethanol and embedded in wax blocks. The brainstem region was divided into three segments: The middle segment was selected to be examined via coronal section (5 μm thick). The sections were stained with haematoxylin and eosin. The degree of cerebral vasospasm was blindly evaluated using measurements of the internal perimeter of lumen, the cross‐sectional area of internal lumen, and wall thickness of the basilar artery.
2.12. TUNEL staining
Neural cell apoptosis in the cortex and hippocampus CA1 of rabbit brain tissue was detected by TUNEL staining according to the manufacturer's protocol. Briefly, brain sections were incubated in 0.5% Triton X‐100 (5 min) and TUNEL reaction mixture (60 min) at 37°C. Cell nuclei were stained with DAPI (1:1,000). Images were acquired with a fluorescence microscope (Olympus IX71, Tokyo, Japan), and apoptotic cells were blindly analysed by counting the average number of cells (red fluorescence) per 100 nuclei (blue fluorescence) in three different visual fields.
2.13. Immunofluorescence staining
The antigen was retrieved by citric acid buffer (pH 6.0) microwave antigen retrieval. After that, the sections were incubated with primary antibodies anti‐NeuN (Cell Signaling Technology, Cat#24307, 1:400, RRID:AB_2651140) or rabbit anti‐caspase‐3 (Cell Signaling Technology, Cat#9661, RRID:AB_2341188) overnight at 4°C and then were subjected to TUNEL staining with an in situ cell death detection kit (Beyotime Biotechnology, Shanghai, China) and corresponding secondary antibodies goat anti‐Rabbit FITC (Thermo Fisher Scientific, Cat#65‐6111, 1:100, RRID:AB_2533966) and goat anti‐rabbit TRITC (Thermo Fisher Scientific, Cat#A16101, 1:500, RRID:AB_2534775) for 60 min at room temperature. Finally, cell nuclei were stained with DAPI (1:1,000, Beyotime Biotechnology, Shanghai, China). Images were acquired with a fluorescence microscope (Olympus IX71, Tokyo, Japan). Immunofluorescent positive cells were counted in a blinded manner in six sections per animal.
2.14. Determination of malondialdehyde level and SOD activity in the brain stem of SAH rabbits
The concentration of malondialdehyde (MDA) and the activities of SOD were measured by corresponding detection kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the instructions as previously described (Zhang et al., 2012).
2.15. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All values from in vitro experiments were expressed as mean ± SD, while the data from animal studies were expressed as mean ± SEM. All statistical analyses were performed with Graphpad Prism software version 7.00 (GraphPad Software, Inc., California, USA, RRID:SCR_002798). The animal neurological score and bleeding grade data were analysed by a non‐parametric Kruskal–Wallis test followed by Dunn's multiple comparisons. The other data were assessed by the Shapiro–Wilk normality test for normal distribution and Levene's test for equal variance. When comparing with two groups, we use paired or unpaired Student's t test. For comparisons of more than two groups, one‐way ANOVA was used, followed by Dunnett's multiple comparisons test. Statistical significance was set at P < .05.
2.16. Materials
Memantine (purity: 98%) was synthesized at the Institute of New Drug Research, Jinan University College of Pharmacy (Guangzhou, China). Griess reagent (Cat#S0021), total NO assay kit (Cat#S0024), TUNEL staining kit (Cat#C1089), Triton X‐100 (Cat# ST795), and DAPI (Cat C1002) were supplied by Beyotime Biotechnology (Shanghai, China). Haematoxylin and Eosin Stain Kit was supplied by Solarbio life sciences (Cat#G1120, Beijing, China). Unless otherwise stated, all other reagents were supplied by Sigma‐Aldrich (St. Louis, MO, USA).
2.17. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017; Alexander, Peters, et al., 2017).
3. RESULTS
3.1. MN‐08 binds to NMDA receptors
MN‐08 is a derivative of memantine, which is an open channel, non‐competitive NMDA receptor antagonist. We first tested the NMDA receptor binding activity of MN‐08 by patch clamp recording with NR1/NR2A NMDA receptors expressed in HEK293 cells. As shown in Table 1, MN‐08 had a potent antagonistic effect on the NR1/NR2A glutamate receptor with an IC50 value of 12.8 ± 2.8 μM. Memantine, the positive control antagonist, produced potent inhibition of the receptor with an IC50 value of 6.0 ± 1.8 μM.
3.2. MN‐08 releases NO in vitro and in vivo
MN‐08 contains a nitrate group, which is hypothesized to release NO in vitro or in vivo by an enzymic or non‐enzymic mechanism. We therefore evaluated the NO release ability of MN‐08 in vitro (in PBS with L‐systeine). ISDN was used as the positive control. MN‐08 released NO in a time‐dependent manner during the tested time period (0.5–32 hr). The NO released by MN‐08 was much less than that released from ISDN. After 24‐hr incubation, the NO level in the MN‐08 sample was 24.5 and 82.4 μM for ISDN (Figure 1a). We then examined release of NO from MN‐08, in rat plasma in vivo. Rat blood samples were collected from the jugular vein via a PE50 catheter at various times (0, 0.25, 0.5, 1, 2, and 4 hr) after treatment with either MN‐08 (25 mg·kg−1) or ISDN (3 mg·kg−1). As shown in Figure 1b, MN‐08 released NO time‐dependently in vivo, although the amount of NO released from MN‐08 was obviously lower than that from ISDN. Four hours after drug administration, the concentration of NO in rat' serum of the MN‐08 group was 32.4 μM, compared with 47.9 μM in the ISDN group.
Figure 1.
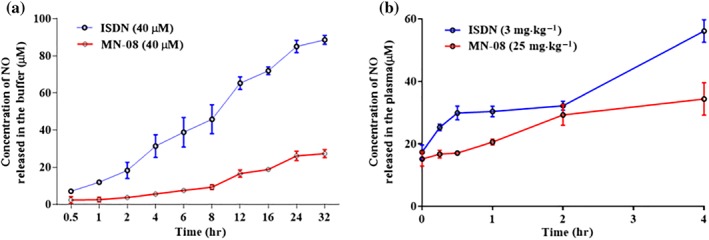
Levels of NO released by MN‐08 in vitro and in vivo. (a) Formation of total NO by MN‐08 (40 μM) and ISDN (40 μM) incubated in PBS buffer with 25‐mM L‐cysteine. (b) NO levels released in rat plasma. After MN‐08 (25 mg·kg−1) or ISDN (3 mg·kg−1) was given intravenously through the tail vein, blood samples were drawn from the jugular vein at 0, 0.25, 0.5, 1, 2, and 4 hr after drug administration. The NO released from MN‐08 or ISDN was assayed with Griess reagent. The absorbance was measured at 540 nm by microplate reader. Data shown are means ± SD (n = 6)
3.3. Vasodilatory effect of MN‐08 on pre‐contracted MCA ex vivo
Next, we examined the vasodilatory effects of MN‐08 on the isolated MCA (Figure 2a) as MN‐08 released NO both in vitro and in vivo. The MCA was pre‐contracted using KCl, 5‐HT, or U46619 (a stable synthetic analogue of the endoperoxide PGH2). After a stable contraction was achieved, increasing concentrations of MN‐08 (1–100 μM) were applied to the incubation system. As shown in Figure 2b–d, MN‐08 induced relaxation in the pre‐contracted blood vessels in a concentration‐dependent manner, no matter which of the three vasoconstrictors had been used. We then investigated the mechanism underlying MN‐08's vasodilatory effects by using inhibitors of the different pathways involved in vasoconstriction and vasodilation events. ODQ, a selective inhibitor of guanylate cyclase, partly attenuated MN‐08‐induced vasodilation (Figure 2e). Other inhibitors, however, including atropine (an inhibitor of ACh M receptors), propranolol (an inhibitor of β‐adrenoceptors), glibenclamide (an inhibitor of ATP sensitive K+ channels), tetraethylammonium (a non‐selective blocker of K+ channels), BaCl2 (an inhibitor of inward rectifier K+ channel), or indomethacin (an inhibitor of COX enzymes), all failed to inhibit MN‐08‐induced vasodilation (Figure S1). Moreover, while ODQ partly inhibited MN‐08‐induced vasodilation, that effect was not abolished by ODQ. We further examined the effect of MN‐08 on extracellular Ca2+‐induced vasocontraction. As shown in Figure 2f, MN‐08, memantine, and nimodipine all significantly attenuated extracellular Ca2+‐induced vasocontraction.
Figure 2.
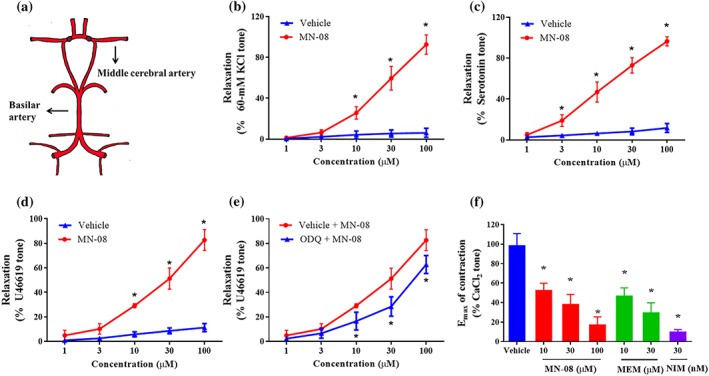
Vasodilator effects of MN‐08 on pre‐contracted MCA. (a) Diagram showing the Circle of Willis. (b–d) Effect of MN‐08 (1–100 μM) on MCA rings, pre‐contracted by (b) KCl (60 mM), (c) 5‐HT (3 μM), or (d) U49916 (1 μM). (e) A selective inhibitor of the guanylate cyclase enzyme (ODQ, 10 μM) inhibited the vasodilator effects of MN‐08 on U49916‐induced vasocontraction. The artery rings were pre‐incubated for 20 min with ODQ before pre‐constriction by U46619. MN‐08 (1–100 μM) was then added cumulatively. (f) Calcium channels were involved in MN‐08‐induced vasodilation. MAC rings were pre‐incubated with different concentrations of MN‐08 (10, 30, and 100 μM), memantine (MEM; 10 and 30 μM), or nimodipine (NIM; 30 nM) for 10 min before being stimulated with KCl (60 mM) in calcium‐free Krebs solution. Subsequently, CaCl2 (3 mM) was added. Calcium‐induced contractile tone was significantly inhibited by treatment with MN‐08 and memantine. Nimodipine served as positive control. The relaxation responses of arterial rings were expressed as the percentage of maximal contraction induced by various vasoconstrictors. Data shown are means ± SD (n = 6). *P < .05, significantly different from vehicle; unpaired Student's t test
3.4. MN‐08 increased RBCs velocity in small vessels of the mouse cerebral cortex
Based on the vasodilatory effect of MN‐08 in vitro, we hypothesized that MN‐08 could improve flow in the cerebral microcirculation. Two‐photon spectroscopy was used to measure the microcirculation in small arterial vessels of mouse located on the coordinates of +3.0 mm from the bregma, 2.5‐mm left of the midline. The study protocol was shown in Figure 3a. The vasculature below the surface of the cerebral cortex was visible after FITC‐dextran labelling (Figure 3b). The method for calculation of blood cell velocity was shown in Figure 3c. Continuous intravenous injection of MN‐08 significantly increased the blood flow velocity when compared with saline treatment (Figure 3d).
Figure 3.
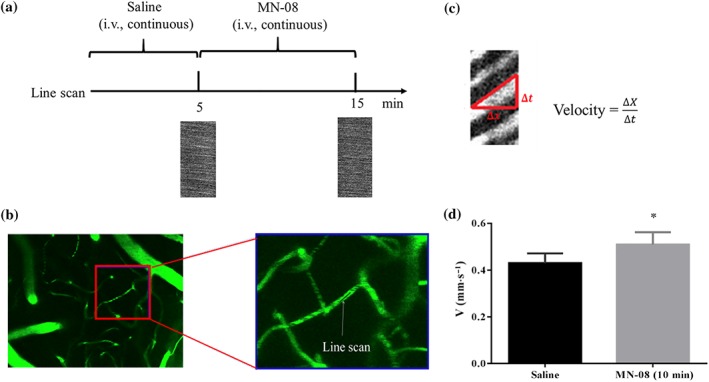
MN‐08 increased RBCs velocity in the cerebral cortex of mice. (a) Diagram of the experimental procedure. Mice were injected with saline (0.02 ml·min−1) for 5 min and then treated with MN‐08 (12 mg·kg−1) for 10 min via tail injection. (b) Fluorescence images of isothiocyanate dextran labelled vessels in the mouse cerebral cortex with a 40× objective (left image) and a magnified image (right). The RBCs velocity was measured using line scan model of two‐photon microscopy. (c) Diagram showing the calculation of RBCs velocity. (d) The RBCs velocity before and after MN‐08 treatment. Data shown are means ± SEM (n = 6). *P < .05, significantly different from saline; paired, two‐tailed Student's t test
3.5. MN‐08 did not lower blood pressure in normal rats
The use of nitrate drugs, such as nitroglycerin and sodium nitroprusside, in animals and patients with SAH is limited by its strong hypotensive effect (Egemen et al., 1993). MN‐08 has a nitrate group, and it can release NO slowly in vitro and in vivo. To determine whether MN‐08 decreases systemic blood pressure, we measured the blood pressure in normal rats during continuous intravenous injection of MN‐08, memantine, and nimodipine (Figure 4a). Unexpectedly, we found that the mean arterial pressure of the MN‐08 group actually increased slightly, about 7.8 mmHg from the baseline (Figure 4b). However, memantine and nimodipine caused the mean arterial pressure to decrease by 5.8 and 7.3 mmHg respectively (Figure 4c,d).
Figure 4.
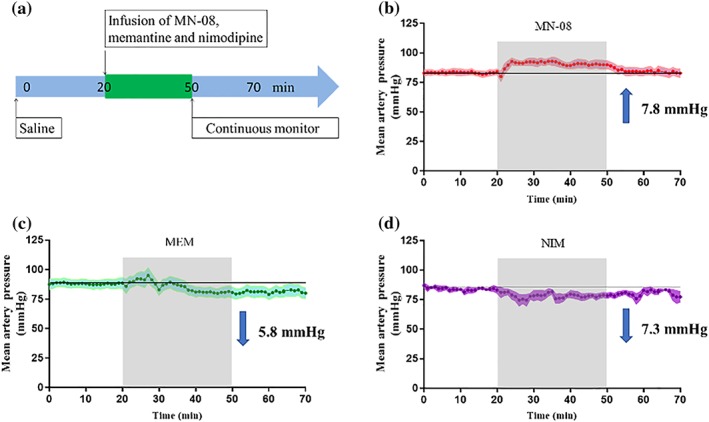
Mean arterial pressure changes after infusion of MN‐08, nimodipine, and memantine in normal rats. (a) Diagram of the experimental procedure. (b–d) The mean blood pressure changes from baseline after (b) MN‐08 (12 mg·kg−1), (c) memantine (MEM; 10 mg·kg−1), and (d) nimodipine (NIM; 0.1 mg·kg−1) administration via continuous tail vein infusion for 30 min. Data shown are means ± SEM (n = 5)
3.6. MN‐08 improved neurological scores and ameliorated SAH bleeding severity grading after SAH in rats
Three animal models have been widely used in studies of SAH ‐ endovascular puncture, cisterna magna blood injection, and cross forebay (Titova, Ostrowski, Zhang, & Tang, 2009). The endovascular puncture model can effectively simulate clinical conditions such as cerebral haemorrhage, decreased cerebral blood flow, and vasospasm (Kooijman et al., 2014). We used this model to test the therapeutic effect of MN‐08. The number of deaths and data of body weight and physiological parameters (pH, P CO2, P O2, and blood glucose) during rat SAH and drug treatment are provided in Tables S1 and S2. SAH surgery resulted in death of some rats and a decrease in body weight. However, there was no significant difference in the number of deaths and body weight among the different treatment groups (Table S1). There were also no significant differences among groups of blood gas parameters before and after SAH surgery (Table S2). Figure 5a,b shows a diagram of the endovascular puncture model of rat SAH and study protocol. Cerebral vasospasm usually occurs between 3–12 days following SAH. The severity of cerebral vasospasm's pathology is dependent on the amount of blood lost and the location of that blood in the basal cisterns. This classification of severity of a SAH patient's condition has been widely utilized in clinical studies. We used a rating system previously described (Sugawara et al., 2008) to evaluate the severity of bleeding in rat model. As shown in Figure 5c,d, the sham group exhibited no bleeding. The basal cisterns of the SAH model group, however, were full of blood clots and in the MN‐08 treatment groups, this score was significantly lower. The memantine and nimodipine groups also exhibited lower SAH bleeding scores. Neurological tests were conducted to evaluate nerve damage on Day 8 after SAH surgery. As shown in Figure 5e, the neurological scores in SAH model rats significantly decreased, compared with those of the sham group. However, MN‐08 at concentrations of 6 and 12 mg·kg−1 significantly improved the neurological scores of SAH rats. Although the neurological scores showed slight improvements in the memantine and nimodipine treatment groups, there was no significant difference in these scores when compared with SAH model groups.
Figure 5.
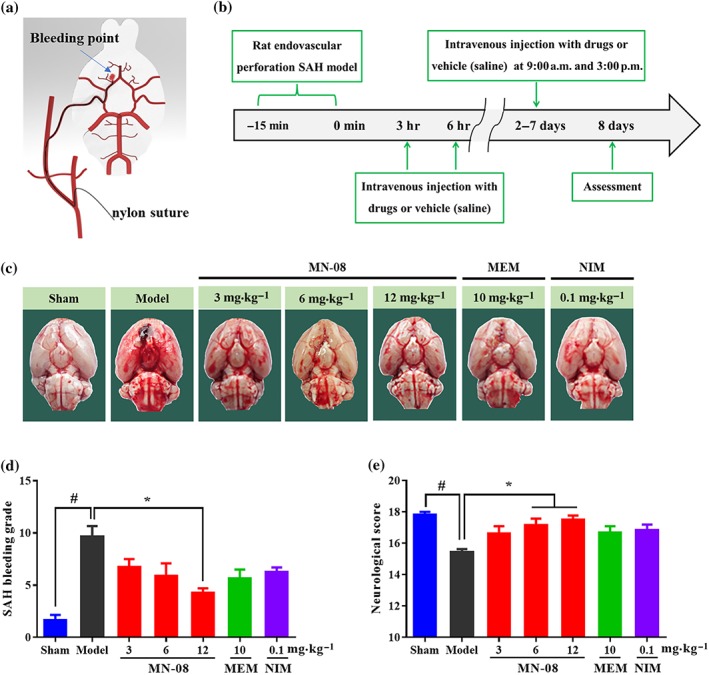
MN‐08 improved neurological scores and lowered bleeding grades after SAH in a rat model. (a) Diagram of endovascular puncture model of SAH. (b) The timeline of this study, rats were treated with MN‐08 (3, 6, and 12 mg·kg−1), memantine (MEM; 10 mg·kg−1), nimodipine (NIM; 0.1 mg·kg−1), or vehicle control (saline) via intravenous injection at 3 and 6 hr after SAH surgery. On Days 2–7, drugs were administered at 9:00 a.m. and 3:00 p.m. (c) Representative photography after endovascular perforation and sham surgery. (d) Effect of MN‐08 on the SAH bleeding grade. (e) Effect of MN‐08 on the neurological score of SAH rats. Data shown are means ± SEM (n = 7 for sham, model, MN‐08 [6 mg·kg−1], memantine and nimodipine groups; n = 8 for MN‐08 [3 mg·kg−1] and MN‐08 [12 mg·kg−1] groups). # P < .05, significantly different from sham group; *P < .05, significantly different from model group; non‐parametric Kruskal–Wallis test followed by Dunn's multiple comparisons
3.7. MN‐08 reduced cerebral vasospasm after SAH in rats
As shown in Figure 6a, the histological images of haematoxylin and eosin staining for the rat basilar arteries indicated severe cerebral vasospasm in the SAH model group compared with the sham operation group. The morphology of cerebral vasoconstriction is characterized by a corrugated internal elastic lamina, a thickened vessel wall, and contracted smooth muscle cells. These morphological changes were attenuated by MN‐08 treatment. Next, sections of basilar artery were semi‐quantitatively analysed (Figure 6b–d); 6 mg·kg−1 MN‐08 treatment clearly increased the perimeter and the cross‐sectional area of the MCA, while 3, 6, and 12 mg·kg−1 concentrations of MN‐08 all significantly attenuated the wall thickness of the basilar artery compared with the SAH rats. By comparison, the memantime and nimodipine treatment groups showed only non‐significant changes in these parameters.
Figure 6.
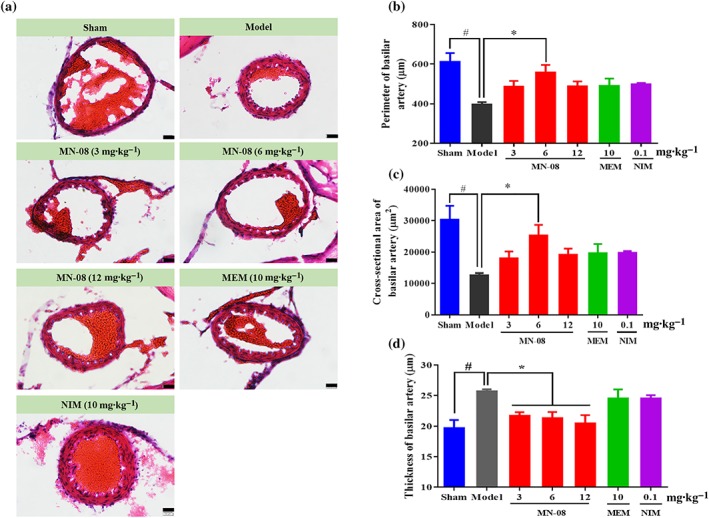
MN‐08 attenuated cerebral vasospasm after SAH in rats. (a) Representative H&E‐stained cross sections of rat basilar arteries after SAH. Scale bars: 20 μm; magnification, ×400. (b–d) Semi‐quantification of the (b) internal perimeter of lumen, (c) cross‐sectional area of internal lumen, and (d) wall thickness of basilar arteries. Data shown are means ± SEM (n = 7 for sham, model, MN‐08 [6 mg·kg−1], memantine (MEM) and nimodipine (NIM) groups; n = 8 for MN‐08 [3 mg·kg−1] and MN‐08 [12 mg·kg−1] groups). # P < .05, significantly different from sham group; *P < .05, significantly different from model group; one‐way ANOVA and Dunnett's multiple comparisons test
3.8. MN‐08 attenuated neuronal cell apoptosis in the cortex after rat SAH
Neural cell apoptosis contributes to EBI after SAH (Zhang et al., 2017). To assess apoptosis in our model, we used NeuN/TUNEL and caspase‐3 staining of neurons in the cortex of the right hemisphere (the endovascular perforated side) after SAH in rats (Figure 7a). As shown in Figure 7b,c, NeuN positive cells in the SAH model group were significantly reduced compared with the sham group, while MN‐08 treatment prevented the neuronal damage. Accordingly, many fewer TUNEL‐positive neurons (Figure 7d) and caspase‐3 stained (Figure 7e,f) cells were observed in the MN‐08‐treated group than in the model group.
Figure 7.
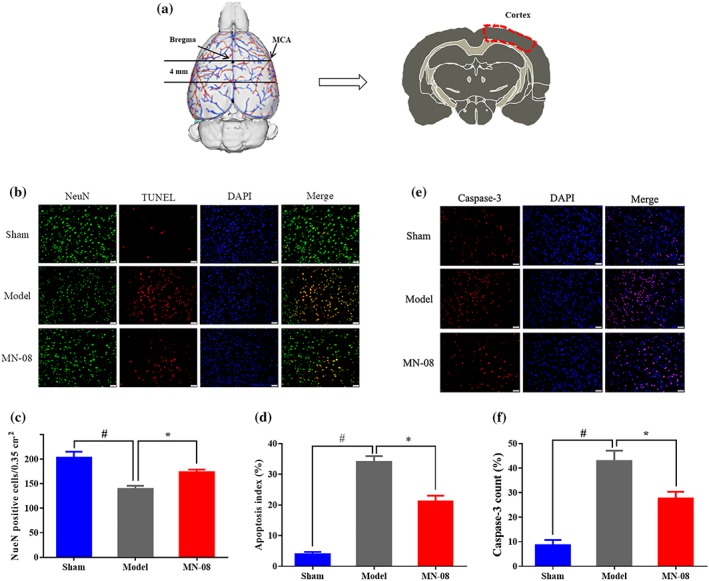
MN‐08 attenuated neuronal cell apoptosis in the cortex after rat SAH. (a) The tested region, the outlines in red area were manual annotations of the cortex (the endovascular perforated side). (b) Images from cortical sections showing NeuN (green)/TUNEL (red) double staining. (c) Quantification of NeuN positive cells. (d) NeuN/TUNEL positive. (e) Caspase‐3 expressions in cortex neurons were detected by immunofluorescence staining (red). (f) Quantification of caspase‐3 positive cells. Scale bars = 50 μm; magnification, ×200. Data shown are means ± SEM (n = 5). # P < .05, significantly different from sham group; *P < .05, significantly different from model; one‐way ANOVA and Dunnett's multiple comparisons test
3.9. MN‐08 attenuated cerebral vasospasm after SAH in rabbits
We further assessed the effects of MN‐08 in a rabbit SAH model, which involves a single injection of blood into the cisterna magna (Figure 8a). This model is suitable for investigating the post‐haemorrhage pathology and the mechanisms of cerebral vasospasm. The experimental protocol is presented in Figure 8b. Blood injection into the cisterna magna resulted in death of several rabbits and a decrease in body weight. However, there were no significant differences in the number of deaths, body weight, and body temperature among different treatment groups (Table S3). The basilar artery was visibly constricted and the arterial wall thickened after the SAH event (Figure 8c). MN‐08 and nimodipine resulted in an clear relaxation of the basilar artery spasm, while memantine treatment showed no significant effect. Moreover, semi‐quantitative analysis of the basilar artery indicated that treatment with MN‐08 and nimodipine, but not memantine, attenuated arterial wall thickness and significantly increased the perimeter and cross‐sectional area of the basilar artery when compared with the untreated SAH model group (Figure 8d–f). We used the same rating system as described for the rat SAH model, to evaluate the severity of bleeding in our rabbit model. As shown in Figure S2, the sham group exhibited no bleeding. The SAH model group and drug treatment groups were all full of blood clots in basal cisterns. There were no statistical significant differences between model group and drug treatment groups.
Figure 8.
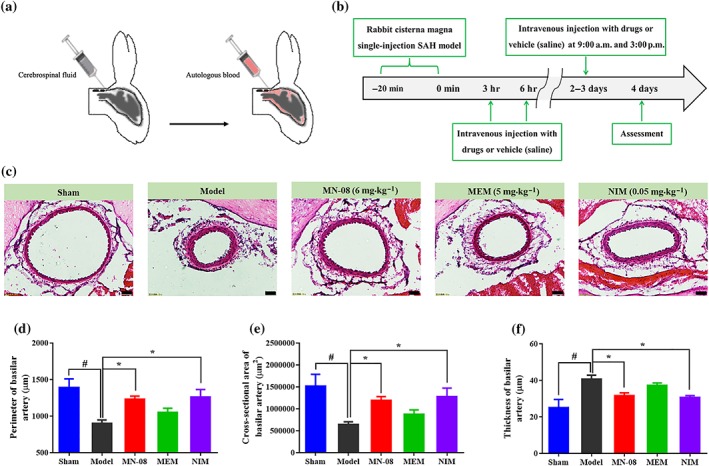
MN‐08 attenuated cerebral vasospasm in rabbits after SAH. (a) Schematic diagram of single blood injection SAH model. (b) The timeline of this study, rabbits were treated with MN‐08 (6 mg·kg−1), memantine (MEM; 5 mg·kg−1), nimodipine (NIM; 0.05 mg·kg−1), or vehicle control (saline) via intravenous injection at 3 and 6 hr after SAH surgery. On Days 2–3, drugs were administered at 9:00 a.m. and 3:00 p.m. (c) Representative H&E‐stained cross sections of the basal artery in rabbits after SAH. Scale bars: 50 μm; magnification, ×200. (d) Semi‐quantification of the internal perimeter of lumen, (e) cross‐sectional area of internal lumen, and (f) wall thickness of the basilar artery. Data shown are means ± SEM (n = 6 for sham, model, MN‐08, and memantine groups; n = 5 for nimodipine group). # P < .05, significantly different from sham group; *P < .05, significantly different from model; one‐way ANOVA and Dunnett's multiple comparisons test
3.10. MN‐08 reduced neural cell apoptosis in the rabbit cerebral cortex and hippocampus after SAH
We also conducted TUNEL staining to detect neuronal cell apoptosis in the cortex and hippocampus of SAH rabbits (Figure 9a). As presented in Figure 9b,c, there were few TUNEL‐positive, apoptotic cells observed in either the cortex or the hippocampus of the sham group. However, there was a clear increase in the number of TUNEL‐positive cells in the SAH model group. MN‐08 treatment, however, significantly reduced the number of apoptotic neurons when compared with the SAH model group (Figure 9d,e).
Figure 9.
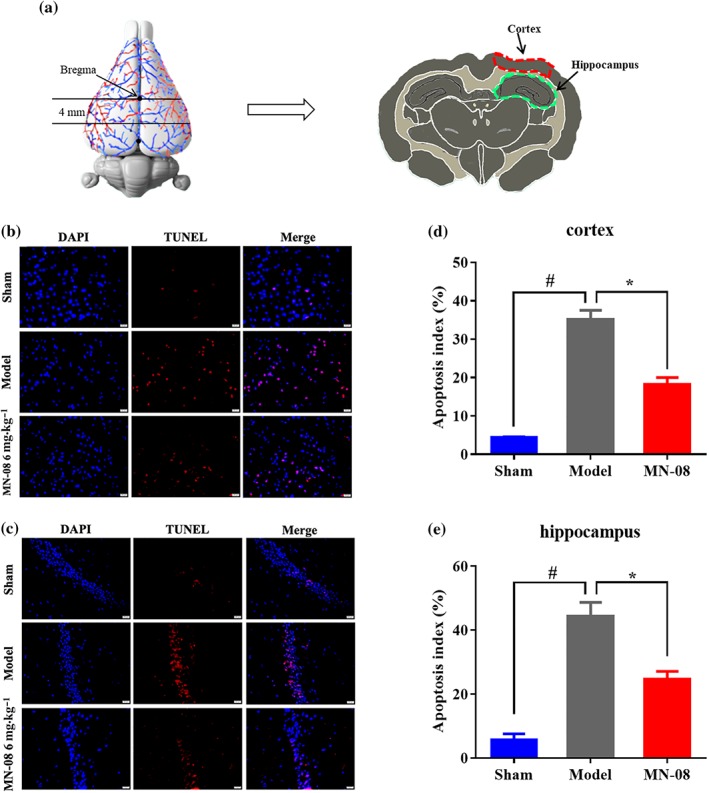
MN‐08 reduced neuronal cell apoptosis in the rabbit cerebral cortex and hippocampus after SAH. (a) The tested region, the outlines in red and green area were manual annotations of the cortex and hippocampus respectively. (b, c) Representative photographs of TUNEL/DAPI double staining of the rabbit cortex and hippocampus after SAH. Scale bars: 20 μm; magnification, ×400. (d, e) Quantification of TUNEL‐positive cells in cortex and hippocampus. Data shown are means ± SEM (n = 6). # P < .05, significantly different from sham group; *P < .05, significantly different from model group; one‐way ANOVA and Dunnett's multiple comparisons test
3.11. MN‐08 attenuated oxidative stress in the brain of rabbit SAH model
To evaluate the effect of MN‐08 on oxidative stress after SAH, we measured the activities of SOD (Figure 10a) and the levels of MDA (Figure 10b) in the brain stem of rabbits after SAH. The activity of SOD decreased and MDA level increased in the untreated model group compared with the sham group; whereas MN‐08 treatment preserved the activities of SOD and markedly suppressed the production of MDA, compared with the model group. This suggests that MN‐08 can attenuate oxidative injury after SAH.
Figure 10.
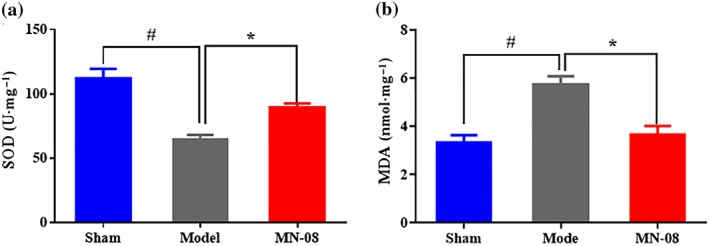
MN‐08 alleviated oxidative stress in SAH rabbits. (a) The activity of SOD and (b) the level of MDA in the brain stem of rabbits. Data shown are means ± SEM (n = 6). # P < .05, significantly different from sham group; *P < .05, significantly different from model group; one‐way ANOVA and Dunnett's multiple comparisons test
4. DISCUSSION
The effects of SAH,‐including cerebral ischaemic injuries, excitotoxicity induction of calcium influx, oxidative stress and vasospasm, have all been previously and comprehensively investigated (Pluta et al., 2009). In this study, we have studied a novel memantine nitrate MN‐08, which possesses the dual functions of binding to NMDA receptors and releasing NO, thereby exerting both neuroprotective and vasodilator effects to confer significant therapeutic benefits in both rat and rabbit SAH models.
The pathogenesis of cerebral vasospasm after SAH is complex and has not yet been fully elucidated. It is believed that levels of powerful vasoconstrictors such as thromboxane A2 endothelin‐1, 5‐HT, PAF, and 20‐hydoxyeicosatetraenoid acid are elevated in the CSF after SAH (Ostergaard et al., 2013). In addition, the depletion of the crucial endogenous vasodilator, NO, is known to cause cerebral vasoconstriction (Schwartz, Sehba, & Bederson, 2000). The cascade of events following SAH is hypothesized as follows: First, SAH triggers erythrocyte haemolysis, resulting in a release of haemoglobin into the subarachnoid space. The oxidation of haemoglobin Fe2+ to methemoglobin Fe3+ causes the release of Fe3+ and ROS formation. ROS can cause severe vasoconstriction either directly or by depleting NO (Rey, Li, Carretero, Garvin, & Pagano, 2002). ROS aside, haemoglobin itself also has 1,000 times greater affinity for NO than for oxygen and can thus act as a scavenger forNO. Finally, SAH affects the expression of NOS, further depleting NO levels (Sehba, Chereshnev, Maayani, Friedrich, & Bederson, 2004). Considerable research has shown that raised levels of NO in the perivascular space can prevent or even reverse vasospasm (Terpolilli et al., 2016). NO donors, such as sodium nitroprusside and nitroglycerin, have been studied in animals and humans. Notably, NO donors reduce vasospasm and improve patient prognosis but they have a major side effect of causing severe systemic hypotension, thereby limiting their clinical use (Siuta, Zuckerman, & Mocco, 2013).
MN‐08 contains a nitrate group. In this study, we demonstrated that MN‐08 slowly released NO in vitro and in vivo. The vasodilation effect of MN‐08 was then confirmed through testing on MCA in vitro, pre‐contracted with a variety of vasoconstrictors. In addition, the vasodilator effects of MN‐08 was, in part, dependent upon the NO‐cGMP pathway and inhibition of extracellular Ca2+ influx, but independent from the β‐adrenergic receptors, mAChRs, COX enzymes, and K+ channels. SAH causes increased intracranial pressure and decreased cerebral blood flow (Macdonald & Schweizer, 2017). We found that MN‐08 increased RBCs velocity in small vessels of mouse cerebral cortices using two‐photon microscopy, suggesting that MN‐08 increases cerebral blood flow.
Although MN‐08 increased NO levels, dilated blood vessels, and improved cerebral microcirculation, unexpectedly, it did not lower systemic blood pressure. We speculate that this result could be due to the following reasons. First, the amount of NO released by MN‐08 is much less than that released by the known NO donor ISDN. Second, MN‐08 has a memantine skeleton, which can bind to NMDA receptors. Apart from being widely distributed in the CNS, NMDA receptors are also present in a variety of non‐neuronal cells and tissues (Bozic & Valdivielso, 2015). Our preliminary pharmacokinetic study showed that, after intravenous administration, MN‐08 quickly disperses into tissues, including the brain. The concentration of MN‐08 in tissue was much higher than that in plasma (data not shown). This is similar to the pharmacokinetics of memantine (Ametamey et al., 2002; Samnick, Ametamey, Gold, & Schubiger, 1997). Therefore, we hypothesize that MN‐08 targets NMDA receptors to release NO, thereby avoiding the undesirable decrease in systemic blood pressure. Furthermore, not only did MN‐08 not cause hypotension but it also even increased blood pressure by a small amount. Central application of a NO donor (intracerebroventricular administration of 3‐morpholinosydnonimine) induced a marked elevation of adrenaline levels and a slight elevation of noradrenaline levels in the plasma (Murakami, Yokotani, Okuma, & Osumi, 1998). We propose that the NO released by the brain following MN‐08 treatment could also slightly elevate plasma adrenaline and noradrenaline levels, thus leading to a mild increase in blood pressure. However, further elucidation of the actual mechanisms underlying MN‐08's effect on blood pressure is needed.
MN‐08's ability to slightly increase blood pressure may be uniquely beneficial to SAH treatment. This was supported by the concept of triple “H” therapy (hypervolemia, hypertension, and hemodilution), which has been employed for many years in order to prevent or reverse vasospasm (Lee, Lukovits, & Friedman, 2006). Diringer et al. (2011) concluded that increased systemic blood pressure could promote cerebral blood flow. Our observation of increased RBCs velocity in small vessels of the mouse cerebral cortex supports the suggestion that MN‐08 serves not only as a vasodilator for vasospasm but also as a stimulator for systemic blood pressure. Both of these effects would be beneficial to the treatment of vasospasm after SAH.
The NMDA receptors play a vital part in the development of the CNS, including synaptic plasticity, memory formation, learning, and formation of neural networks. Over‐activation of the receptors can cause excessive influx of Ca2+ and lead to the early stage of excitotoxicity. The Ca2+ overload will trigger the formation of ROS, such as •OH, O2•−, and ONOO−, which can initiate neuronal injury and apoptosis, including lipid peroxidation, direct DNA damage, and protein oxidation. Cellular apoptosis is an aspect of EBI after SAH reported by several studies (Aoki, Zubkov, Ross, & Zhang, 2002; Cahill, Calvert, & Zhang, 2006). Our research shows that treatment with MN‐08 significantly decreased MDA levels and increased SOD activity in the brain tissue. We further investigated neuronal damage and apoptosis via NeuN/TUNEL and caspase‐3 staining and found that MN‐08 inhibited neuronal apoptosis after SAH in rat and rabbit model. These results support that the MN‐08 may decrease ROS formation and suppress apoptosis of neurons, thus relieving EBI and improving neurological deficits.
Overactive NMDA receptors are involved with the pathogenesis of neuronal death in stroke, trauma and Alzheimer'sdisease (Kemp & Mckernan, 2002). Given the substantial evidence of increased glutamate levels after SAH, the use of NMDA receptor antagonists as treatment is logical. Memantine alleviates brain injury and attenuates vasospasm in experimental models of SAH (Huang, Wang, Shan, et al., 2015; Huang, Wang, Wang, et al., 2015). In our study, MN‐08 was significantly more effective than memantine both in rat and rabbit SAH models, when given at equimolar doses (in rats, 12 mg·kg−1 MN‐08 is equimolar to 10 mg·kg−1 memantine. In rabbits, 6 mg·kg−1 MN‐08 is equimolar to 5 mg·kg−1 memantine). It has also been speculated that a high binding affinity for NMDA receptors may result in increased clinical side effects, consequent on disrupting the normal physiological function of NMDA receptors. For example, the compound MK‐801 has a high affinity for NMDA receptors and is also associated with a number of side effects, including cognitive disruption and psychotic‐spectrum reactions. MK‐801 also blocks NMDA receptor‐mediated synaptic transmission and LTP in rat hippocampal slices (Coan, Saywood, & Collingridge, 1987). Unlike MK‐801, memantine has a low affinity for NMDA receptors and when memantine blocks NMDA receptors, the normal physiological function of the receptors is not affected. Memantine has relatively good safety profile and high tolerance in humans and has been approved as a treatment for Alzheimer's disease. In this study, MN‐08 displayed a lower activity (about two times lower), compared with memantine, in inhibiting NMDA receptors. The normal physiological function of NMDA re3cptors is thus less likely to be affected by MN‐08. Moreover, our preliminary acute toxicity study revealed that MN‐08 had a similar LD50 value as memantine in mice (data not shown). Thus, MN‐08 may provide therapeutic effects at clinically tolerable doses, with few adverse effects.
The pathological mechanism(s) underlying cerebral vasospasm and neuronal cell apoptosis after SAH is multifactorial. Thus far, single‐target agents have not prevented the occurrence of SAH (Hasegawa, Suzuki, Sozen, Altay, & Zhang, 2011). Combination therapies or multifunctional agents may avoid some of the limitations of single‐target therapy by addressing multiple pathophysiological aspects of this complex disease. Our present study provides a proof of concept for the treatment of SAH with the dual functional MN‐08, which concurrently inhibits NMDA receptors and releases NO. MN‐08 may not only protect cortical and hippocampal neurons from apoptosis but also reduce cerebral vasospasm. Taken together, the advantages of the dual‐functional MN‐08 are likely to provide promising therapeutic strategies for SAH, thus meriting further investigation and development of MN‐08 into a clinical drug.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Y.Q.W. and Z.J.Z. designed and supervised the project. G.X.Z., Z.Y.Z., Z.L., S.P.L., Q.Z., and J.J. performed the SAH rat experiments, collected, and analysed the data. L.M.W., N.L., Y.W.S., Z.X.Z., and X.F.Y. performed the SAH rabbit experiments, collected, and analysed the data. F.C.L., B.J.G., and S. M performed the haemodynamic experiments, collected, and analysed the data. L.M.W., F.C.L., and Y.F.H. performed the vitro experiments, collected, and analysed the data. Z.J.Z. and Y.Q.W. wrote the manuscript. All authors have read and approved the final manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1. The MN‐08‐induced changes of body weight in rats before and after SAH
Table S2. Physiological parameters in rats before and after SAH
Table S3. Physiological parameters in rabbits SAH
Figure S1. The effect of inhibitors on MN‐08‐induced vasodilatation
Figure S2. The bleeding grades after SAH in a rabbit model
ACKNOWLEDGEMENTS
We thank Ms. Linda Wang for editing this manuscript. This work was supported by grants from the Natural National Science Foundation of China (Grants NSFC 81603106, 81703339, and U1801287), the National Science and Technology Major Project of China (Innovative Drug Project Grant 2018ZX09301031‐002), the Scientific Projects of Guangdong Province (Grant 2017A030313742, GD‐HK Cooperative Project 2016A050503030), the Scientific Projects of Guangzhou (Grant 201804010495), ITSP‐Guangdong‐Hong Kong Technology Cooperation Funding Scheme (Grant GHP/012/16GD), and Shenzhen Basic Research Program (Grant JCYJ20160331141459373).
Luo F, Wu L, Zhang Z, et al. The dual‐functional memantine nitrate MN‐08 alleviates cerebral vasospasm and brain injury in experimental subarachnoid haemorrhage models. Br J Pharmacol. 2019;176:3318–3335. 10.1111/bph.14763
Fangcheng Luo and Liangmiao Wu contributed equally to this work.
Contributor Information
Gaoxiao Zhang, Email: zhanggaoxiao2005@163.com.
Zaijun Zhang, Email: zaijunzhang@163.com.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017) The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, G. S. (1976). Cerebral arterial spasm. Part 8: The treatment of delayed cerebral arterial spasm in human beings. Surgical Neurology, 6(2), 71–80. [PubMed] [Google Scholar]
- Ametamey, S. M. , Bruehlmeier, M. , Kneifel, S. , Kokic, M. , Honer, M. , Arigoni, M. , … Schubiger, P. A. (2002). PET studies of 18F‐memantine in healthy volunteers. Nuclear Medicine & Biology, 29(2), 227–231. 10.1016/S0969-8051(01)00293-1 [DOI] [PubMed] [Google Scholar]
- Aoki, K. , Zubkov, A. Y. , Ross, I. B. , & Zhang, J. H. (2002). Therapeutic effect of caspase inhibitors in the prevention of apoptosis and reversal of chronic cerebral vasospasm. Stroke, 9(6), 672–677. 10.1054/jocn.2002.1088 [DOI] [PubMed] [Google Scholar]
- Bozic, M. , & Valdivielso, J. M. (2015). The potential of targeting NMDA receptors outside the CNS. Expert Opinion on Therapeutic Targets, 19(3), 399–413. 10.1517/14728222.2014.983900 [DOI] [PubMed] [Google Scholar]
- Cahill, J. , Calvert, J. W. , & Zhang, J. H. (2006). Mechanisms of early brain injury after subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism, 26(11), 1341–1353. 10.1038/sj.jcbfm.9600283 [DOI] [PubMed] [Google Scholar]
- Chen, J. , Qian, C. , Duan, H. , Cao, S. , Yu, X. , Li, J. , … Chen, G. (2015). Melatonin attenuates neurogenic pulmonary edema via the regulation of inflammation and apoptosis after subarachnoid hemorrhage in rats. Journal of Pineal Research, 59(4), 469–477. 10.1111/jpi.12278 [DOI] [PubMed] [Google Scholar]
- Chhatbar, P. Y. , & Kara, P. (2013). Improved blood velocity measurements with a hybrid image filtering and iterative Radon transform algorithm. Frontiers in Neuroscience, 7, 106 10.3389/fnins.2013.00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coan, E. J. , Saywood, W. , & Collingridge, G. L. (1987). MK‐801 blocks NMDA receptor‐mediated synaptic transmission and long term potentiation in rat hippocampal slices. Neuroscience Letters, 80(1), 111–114. 10.1016/0304-3940(87)90505-2 [DOI] [PubMed] [Google Scholar]
- Connolly, E. S. Jr. , Rabinstein, A. A. , Carhuapoma, J. R. , Derdeyn, C. P. , Dion, J. , Higashida, R. T. , … Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology (2012). Guidelines for the management of aneurysmal subarachnoid hemorrhage: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke, 43(6), 1711–1737. 10.1161/STR.0b013e3182587839 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diringer, M. N. , Bleck, T. P. , Claude Hemphill, J. C. , Menon, D. , Shutter, L. , Vespa, P. , … Neurocritical Care Society (2011). Critical care management of patients following aneurysmal subarachnoid hemorrhage: Recommendations from the Neurocritical Care Society's Multidisciplinary Consensus Conference. Neurocritical Care, 15(2), 211–240. 10.1007/s12028-011-9605-9 [DOI] [PubMed] [Google Scholar]
- Egemen, N. , Türker, R. K. , Sanlidilek, U. , Zorlutuna, A. , Bilgiç, S. , Baskaya, M. , … McCormick, J. M. (1993). The effect of intrathecal sodium nitroprusside on severe chronic vasospasm. Neurological Research, 15(5), 310–315. 10.1080/01616412.1993.11740153 [DOI] [PubMed] [Google Scholar]
- Fathi, A. R. , Marbacher, S. , Graupner, T. , Wehrli, F. , Jakob, S. M. , Schroth, G. , & Fandino, J. (2011). Continuous intrathecal glyceryl trinitrate prevents delayed cerebral vasospasm in the single‐SAH rabbit model in vivo. Acta Neurochirurgica, 153(8), 1669–1675. 10.1007/s00701-011-1049-7 [DOI] [PubMed] [Google Scholar]
- Fujii, M. , Yan, J. , Rolland, W. B. , Soejima, Y. , Caner, B. , & Zhang, J. (2013). Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Translational Stroke Research, 4(4), 432–446. 10.1007/s12975-013-0257-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage, G. J. , Kipke, D. R. , & Shain, W. (2012). Whole animal perfusion fixation for rodents. Journal of Visualized Experiments, 65, e3564 10.3791/3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, J. H. , Wagner, S. , Liu, K. F. , & Hu, X. J. (1995). Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke, 26(4), 627–634. 10.1161/01.STR.26.4.627 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, Y. , Suzuki, H. , Sozen, T. , Altay, O. , & Zhang, J. H. (2011). Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochirurgica. Supplement, 110(Pt 1), 43–48. 10.1007/978-3-7091-0353-1_8 [DOI] [PubMed] [Google Scholar]
- Hillered, L. , Vespa, P. M. , & Hovda, D. A. (2005). Translational neurochemical research in acute human brain injury: The current status and potential future for cerebral microdialysis. Journal of Neurotrauma, 22(1), 3–41. 10.1089/neu.2005.22.3 [DOI] [PubMed] [Google Scholar]
- Huang, C. Y. , Wang, L. C. , Shan, Y. S. , Pan, C. H. , & Tsai, K. J. (2015). Memantine attenuates delayed vasospasm after experimental subarachnoid hemorrhage via modulating endothelial nitric oxide synthase. International Journal of Molecular Sciences, 16(6), 14171–14180. 10.3390/ijms160614171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. Y. , Wang, L. C. , Wang, H. K. , Pan, C. H. , Cheng, Y. Y. , Shan, Y. S. , … Tsai, K. J. (2015). Memantine alleviates brain injury and neurobehavioral deficits after experimental subarachnoid hemorrhage. Molecular Neurobiology, 51(3), 1038–1052. 10.1007/s12035-014-8767-9 [DOI] [PubMed] [Google Scholar]
- Jambrina, E. , Cerne, R. , Smith, E. , Scampavia, L. , Cuadrado, M. , Findlay, J. , … Ursu, D. (2016). An integrated approach for screening and identification of positive allosteric modulators of N‐methyl‐D‐aspartate receptors. Journal of Biomolecular Screening, 21(5), 468–479. 10.1177/1087057116628437 [DOI] [PubMed] [Google Scholar]
- Jeffrey, E. , & McGinnis, G. (2002). Safety of intraventricular sodium nitroprusside and thiosulfate for the treatment of cerebral vasospasm in the intensive care unit setting. Stroke, 33(2), 486–492. 10.1161/hs0202.103410 [DOI] [PubMed] [Google Scholar]
- Kemp, J. A. , McKernan, R. M. (2002). NMDA receptor pathways as drug targets. Nature Neuroscience, 5(Suppl), 1039–1042. [DOI] [PubMed] [Google Scholar]
- Khaldi, A. , Zauner, A. , Reinert, M. , Woodward, J. J. , & Bullock, M. R. (2001). Measurement of nitric oxide and brain tissue oxygen tension in patients after severe subarachnoid hemorrhage. Neurosurgery, 49(1), 33–40. 10.1097/00006123-200107000-00005 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & NC3Rs Reporting Guidelines Working Group (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijman, E. , Nijboer, C. H. , van Velthoven, C. T. , Kavelaars, A. , Kesecioglu, J. , & Heijnen, C. J. (2014). The rodent endovascular puncture model of subarachnoid hemorrhage: Mechanisms of brain damage and therapeutic strategies. Journal of Neuroinflammation, 11(1), 2 10.1186/1742-2094-11-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton, M. T. , & Vates, G. E. (2017). Subarachnoid hemorrhage. New England Journal of Medicine, 377(3), 257–266. 10.1056/NEJMcp1605827 [DOI] [PubMed] [Google Scholar]
- Lee, K. H. , Lukovits, T. , & Friedman, J. A. (2006). “Triple‐H” therapy for cerebral vasospasm following subarachnoid hemorrhage. Neurocritical Care, 4(1), 68–76. 10.1385/NCC:4:1:068 [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Yang, S. , Jin, X. , Zhang, G. , Guo, B. , Chen, H. , … Wang, Y. (2017). Synthesis and biological evaluation of memantine nitrates as a potential treatment for neurodegenerative diseases. Medchemcomm, 8(1), 135–147. 10.1039/C6MD00509H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, R. L. (2014). Delayed neurological deterioration after subarachnoid haemorrhage. Nature Reviews. Neurology, 10(1), 44–58. 10.1038/nrneurol.2013.246 [DOI] [PubMed] [Google Scholar]
- Macdonald, R. L. , & Schweizer, T. A. (2017). Spontaneous subarachnoid haemorrhage. Lancet, 389(10069), 655–666. 10.1016/S0140-6736(16)30668-7 [DOI] [PubMed] [Google Scholar]
- Macdonald, R. L. , & Weir, B. K. (1991). A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke, 22(8), 971–982. 10.1161/01.STR.22.8.971 [DOI] [PubMed] [Google Scholar]
- Maki, B. A. , Cummings, K. A. , Paganelli, M. A. , Murthy, S. E. , & Popescu, G. K. (2014). One‐channel cell‐attached patch‐clamp recording. Journal of Visualized Experiments, 88, e51629 10.3791/51629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munakata, A. , Naraoka, M. , Katagai, T. , Shimamura, N. , & Ohkuma, H. (2016). Role of cyclooxygenase‐2 in relation to nitric oxide and endothelin‐1 on pathogenesis of cerebral vasospasm after subarachnoid hemorrhage in rabbit. Translational Stroke Research, 7(3), 220–227. 10.1007/s12975-016-0466-6 [DOI] [PubMed] [Google Scholar]
- Murakami, Y. , Yokotani, K. , Okuma, Y. , & Osumi, Y. (1998). Thromboxane A2 is involved in the nitric oxide‐induced central activation of adrenomedullary outflow in rats. Neuroscience, 78(1), 197–205. 10.1016/S0306-4522(98)00133-X [DOI] [PubMed] [Google Scholar]
- Ostergaard, L. , Aamand, R. , Karabegovic, S. , Tietze, A. , Blicher, J. U. , Mikkelsen, I. K. , … Sørensen, J. C. H. (2013). The role of the microcirculation in delayed cerebral ischemia and chronic degenerative changes after subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism, 33(12), 1825–1837. 10.1038/jcbfm.2013.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta, R. M. (2008). Dysfunction of nitric oxide synthases as a cause and therapeutic target in delayed cerebral vasospasm after SAH. Acta Neurochirurgica. Supplement, 104, 139–147. 10.1007/978-3-211-75718-5_28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta, R. M. , Hansen‐Schwartz, J. , Derier, J. , Vajkoczy, P. , Macdonald, R. , Nishizawa, S. , … Zhang, J. H. (2009). Cerebral vasospasm following subarachnoid hemorrhage: Time for a new world of thought. Neurological Research, 31(2), 151–158. 10.1179/174313209X393564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, F. E. , Li, X. C. , Carretero, O. A. , Garvin, J. L. , & Pagano, P. J. (2002). Perivascular superoxide anion contributes to impairment of endothelium‐dependent relaxation role of gp91(phox). Circulation, 106(19), 2497–2502. 10.1161/01.CIR.0000038108.71560.70 [DOI] [PubMed] [Google Scholar]
- Sabri, M. , Ai, J. , Lass, E. , D'Abbondanza, J. , & Macdonald, R. L. (2013). Genetic elimination of eNOS reduces secondary complications of experimental subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism, 33(7), 1008–1014. 10.1038/jcbfm.2013.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samnick, S. , Ametamey, S. , Gold, M. R. , & Schubiger, P. A. (1997). Synthesis and preliminary in vitro evaluation of a new memantine derivative 1‐amino‐3‐[18F]fluoromethyl‐5‐methyl‐adamantane: A potential ligand for mapping the N‐methyl‐D‐aspartate receptor complex. Journal of Labelled Compounds & Radiopharmaceuticals, 39(3), 241–250. [DOI] [Google Scholar]
- Schwartz, A. Y. , Sehba, F. A. , Bederson, J. B. (2000). Decreased nitric oxide availability contributes to acute cerebral ischemia after subarachnoid hemorrhage. Neurosurgery, 47(1), 208–214. 10.1097/00006123-200007000-00042 [DOI] [PubMed] [Google Scholar]
- Sehba, F. A. , Chereshnev, I. , Maayani, S. , Friedrich, V. Jr. , & Bederson, J. B. (2004). Nitric oxide synthase in acute alteration of nitric oxide levels after subarachnoid hemorrhage. Neurosurgery, 55(3), 677–678. 10.1227/01.NEU.0000134557.82423.B2 [DOI] [PubMed] [Google Scholar]
- Sehba, F. A. , Plata, R. M. , & Zhang, J. H. (2011). Metamorphosis of subarachnoid hemorrhage research: From delayed vasospasm to early brain injury. Molecular Neurobiology, 43(1), 27–40. 10.1007/s12035-010-8155-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehba, F. A. , Schwartz, A. Y. , Chereshnev, I. , & Bederson, J. B. (2000). Acute decrease in cerebral nitric oxide levels after subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism, 20(3), 604–611. 10.1097/00004647-200003000-00018 [DOI] [PubMed] [Google Scholar]
- Shih, A. Y. , Driscoll, J. D. , Drew, P. J. , Nishimura, N. , Schaffer, C. B. , & Kleinfeld, D. (2012). Two‐photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. Journal of Cerebral Blood Flow and Metabolism, 32(7), 1277–1309. 10.1038/jcbfm.2011.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuta, M. , Zuckerman, S. L. , & Mocco, J. (2013). Nitric oxide in cerebral vasospasm: Theories, measurement, and treatment. Neurology Research International, 2013, 972417 10.1155/2013/972417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara, T. , Ayer, R. , Jadhav, V. , & Zhang, J. H. (2008). A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. Journal of Neuroscience Methods, 167(2), 327–334. 10.1016/j.jneumeth.2007.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhardja, A. (2004). Mechanisms of disease: Roles of nitric oxide and endothelin‐1 in delayed cerebral vasospasm produced by aneurysmal subarachnoid hemorrhage. Nature Clinical Practice Cardiovascular Medicine, 1(2), 110–116. 10.1038/ncpcardio0046 [DOI] [PubMed] [Google Scholar]
- Terpolilli, N. A. , Feiler, S. , Dienel, A. , Müller, F. , Heumos, N. , Friedrich, B. , … Plesnila, N. (2016). Nitric oxide inhalation reduces brain damage, prevents mortality, and improves neurological outcome after subarachnoid hemorrhage by resolving early pial microvasospasms. Journal of Cerebral Blood Flow and Metabolism, 36(12), 2096–2107. 10.1177/0271678X15605848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titova, E. , Ostrowski, R. P. , Zhang, J. H. , & Tang, J. (2009). Experimental models of subarachnoid hemorrhage for studies of cerebral vasospasm. Neurological Research, 31(6), 568–581. 10.1179/174313209X382412 [DOI] [PubMed] [Google Scholar]
- Villmann, C. , & Becker, C. M. (2007). On the hypes and falls in neuroprotection: Targeting the NMDA receptor. The Neuroscientist, 13(6), 594–615. 10.1177/1073858406296259 [DOI] [PubMed] [Google Scholar]
- Westermaier, T. , Jauss, A. , Eriskat, J. , Kunze, E. , & Roosen, K. (2011). The temporal profile of cerebral blood flow and tissue metabolites indicates sustained metabolic depression after experimental subarachnoid hemorrhage in rats. Neurosurgery, 68(1), 223–230. 10.1227/NEU.0b013e3181fe23c1 [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Wu, Q. , Zhang, Q. , Lu, Y. , Liu, J. , Li, W. , … Hang, C. (2017). Resveratrol attenuates early brain injury after experimental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation. Frontiers in Neuroscience, 11, 611 10.3389/fnins.2017.00611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Cui, W. , Li, G. , Yuan, S. , Xu, D. , Hoi, M. P. , … Lee, S. M. (2012). Baicalein protects against 6‐OHDA‐induced neurotoxicity through activation of Keap1/Nrf2/HO‐1 and involving PKCα and PI3K/AKT signaling pathways. Journal of Agricultural and Food Chemistry, 60(33), 8171–8182. 10.1021/jf301511m [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The MN‐08‐induced changes of body weight in rats before and after SAH
Table S2. Physiological parameters in rats before and after SAH
Table S3. Physiological parameters in rabbits SAH
Figure S1. The effect of inhibitors on MN‐08‐induced vasodilatation
Figure S2. The bleeding grades after SAH in a rabbit model