

Abstract
Background and Purpose
Entecavir (ETV), a first‐line antiviral drug against hepatitis B virus (HBV), has the possibility to be used to prevent mother‐to‐child transmission. The aim of present study was to clarify the mechanism of ETV uptake into hepatocytes and evaluate the alteration of ETV's hepatic distribution during pregnancy.
Experimental Approach
The roles of equilibrative nucleotide transporter (ENT) 1 and organic anion transporter (OAT) 2 in ETV accumulation and anti‐HBV efficacy were studied in human ENT1 or OAT2 overexpressed cell models and HepG2.2.15 cells, respectively; meanwhile, the liver‐to‐plasma ETV concentration ratios in non‐pregnant and pregnant mice were measured to evaluate the effect of pregnancy on ETV hepatic distribution.
Key Results
ETV was shown to be a substrate of ENT1 and OAT2. An ENT1 inhibitor significantly decreased the efficacy of ETV in HepG2.2.15 cells, while overexpression of OAT2 increased susceptibility of HBV to ETV. The liver‐to‐plasma ETV concentration ratios in pregnant mice were sharply reduced; whereas, the absolute concentration of ETV in the liver did not obviously alter in pregnancy. Although oestradiol and progesterone showed a concentration‐dependent inhibition on ETV accumulation both in hepatic cell lines and in primary human hepatocytes, a physiologically relevant concentration of oestradiol and progesterone did not affect antiviral activity of ETV.
Conclusions and Implications
OAT2 and ENT1 are the main transporters involved in the hepatic uptake and anti‐HBV efficacy of ETV. The concentration of ETV in the liver was not obviously altered during pregnancy, which indicates that dosage adjustment in pregnancy is not necessary.
Abbreviations
- CNT
concentrative nucleoside transporter
- D22
decynium‐22
- E2
oestradiol
- ENT
equilibrative nucleoside transporter
- ETV
entecavir
- Indo
indomethacin
- HBV
hepatitis B virus
- HCC
hepatocellular cancer
- MATE
multidrug and toxin extrusion
- MPP+
1‐methyl‐4‐phenylpyridinium iodide
- MTT
3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide
- NBTI
S‐(4‐Nitrobenzyl)‐6‐thioinosine
- NMDG
N‐methyl‐d‐glucamine
- OAT
organic anion transporter
- OCT
organic cation transporter
- OCTN
organic cation/carnitine transporter
- P4
progesterone
- PHH
primary human hepatocytes
- siRNA
small interfering RNAs
What is already known
Transporters play key roles in the hepatic accumulation of entecavir.
What this study adds
OAT2 and ENT1 are the main transporters involved in the hepatic uptake and efficacy of entecavir.
The concentration of entecavir in the liver during pregnancy is not obviously altered.
What is the clinical significance
The results help to predict the possibility of any drug–drug interaction associated with entecavir.
The results provide useful information on entecavir application during pregnancy.
1. INTRODUCTION
Hepatitis B virus (HBV) infection is a risk factor for liver cirrhosis and hepatocellular cancer (HCC). Globally, an estimated 240 million persons suffer from chronic hepatitis B, and chronic infections develop in approximately 90% of patients who were infected with the virus during infancy (Tang, Covert, Wilson, & Kottilil, 2018). Antiviral therapy is recommended in late pregnancy with an HBV DNA level >200,000 IU·ml−1 to reduce the risk of mother‐to‐infant transmission (Terrault et al., 2018). Currently, lamivudine, telbivudine, and tenofovir have been explored for use in pregnant women, but these drugs have been found to have high levels of resistance or to have adverse effects such as myopathy and nephrotoxicity (Hermans et al., 2016; Lim, Liao, & Tsai, 2018; Milián et al., 2017). However, as one of the first‐line antiviral drugs with a high efficacy, entecavir (ETV) also has a significant barrier‐related drug resistance (Park et al., 2017). Additionally, fetal exposure to ETV in humans is probably extremely low based on low therapeutic dose (0.5 mg·day−1) and low placental transfer in pregnant mice (Ma et al., 2017). Furthermore, data from the Antiretroviral Pregnancy Registry released in June 2018 showed that ETV had a low defect rate of 2.56%, which was not different from the unexposed group (Antiretroviral Pregnancy Registry Steering Committee, 2018). These findings suggest that there is a pressing need to explore potential antivirals for treatment of HBV infection during pregnancy and that ETV has the possibility to be used in pregnant women to prevent mother‐to‐child transmission.
Cell membrane transporters play crucial roles in the disposition of hydrophilic xenobiotics in vivo, which further affect drug efficacy (Liu et al., 2015). Our previous study demonstrated that ETV is a substrate of organic anion transporter (OAT) 1, OAT3, multidrug and toxin extrusion (MATE) 1, and MATE2‐K, which mediate renal clearance of ETV (Yang et al., 2016). Recent studies revealed that ABCB1 (P‐glycoprotein), ABCC4 (multidrug resistance‐associated protein 4), and ABCG2 (breast cancer resistance protein) (BCRP) are involved in ETV‐related drug–drug interactions in the liver (Chen et al., 2017). However, scant information was available with respect to the transport mechanism of ETV into hepatocytes, which is essential for predicting and clarifying the efficacy of ETV in specific physiological conditions.
Pregnancy involves substantial physiological changes, including delayed gastric emptying, increased cardiac output, and increased glomerular filtration rate (Pariente et al., 2016). In addition, elevated hormones, especially oestradiol (E2) and progesterone (P4), probably regulate the expression of drug transporters, and thereby affect the distribution of drugs (Isoherranen & Thummel, 2013). E2 and P4 have been reported to impair the activity of transporters by internalization or direct inhibition (Tocchetti et al., 2017). ETV is a hydrophilic compound with a pKa value of 10.5, which implies that transporter‐mediated hepatic uptake of ETV plays an important role in its accumulation in the liver. Therefore, we inferred that the ETV‐related transporters might be affected by the increase in E2 and P4 during pregnancy, which would probably result in an alteration in the hepatic distribution of ETV and its anti‐HBV efficacy.
With this in mind, one aim of the present study was to elucidate the main transporters that mediate the hepatic uptake of ETV and to evaluate their roles in the anti‐HBV activity of ETV in HepG2.2.15 cells. The other aim was to assess whether the hepatic distribution of ETV would be altered in pregnancy and the underlying mechanism.
2. METHODS
2.1. Animal welfare and ethical statements
All the experimental procedures were in accordance with the Animals (Scientific Procedures) Act 1986 Amendment Regulations (SI 2012/3039), UK, and animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. The procedures were approved by Institutional Animal Care and Use Committee of Zhejiang University Medical Center (2015‐0026) and complied with the standards of animal welfare in China. All experimental procedures were conducted as humanely as possible.
Specific pathogen‐free female non‐pregnant and pregnant (gestational day 14.5) ICR mice, age of 8–10 weeks, were obtained from Beijing Vital River Laboratory Animal Technology Company (Beijing, China; SCXK [Jing] 2012‐0001). The animals were housed in rectangular cages with bedding at controlled temperature (22.0 ± 0.1°C) and humidity (50 ± 10 %), with a 12‐hr light/dark cycle and free access to food and water before the start of the experiment. The number of mice per cage was less than five.
2.2. Cell culture
The MDCK II cells and HEK293 (RRID:CVCL_0045) cells were acquired from Peking Union Medical College (Beijing, China) and kindly provided by Professor Feng Han, Zhejiang University, respectively, and grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. The human hepatic cell HepG2, Huh‐7, and HL7702 (RRID:CVCL_6926) cells were from the Chinese Academy of Sciences Cell Cultures Library (Wuhan, China), cultured in DMEM and RPMI 1640, respectively, with addition of 10% FBS, 1% penicillin/streptomycin, and 1% non‐essential amino acids. Stable HBV‐expressing human hepatoma HepG2.2.15 cells (RID:CVCL_L855) were donated by Professor Naishuo Zhu (Fudan University), maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 1% non‐essential amino acids, and 0.5 mg·ml−1 G418. Cells were grown in a humidified air/CO2 incubator (5% v/v) at 37°C.
Cryopreserved primary human hepatocytes (PHH, RILD Research Institute for Liver Disease, Shanghai, China) from three female donors (Table S1) were plated in InVitroGRO™ CP Medium (BioreclamationIVT, Baltimore, MD, USA) at 37°C with 5% CO2 before drug treatment or cellular uptake experiments.
2.3. Cellular uptake experiment
The cellular accumulation of ETV in hepatic cell lines or PHH was performed as described in our previous study (Li et al., 2016). Briefly, the cells were pre‐incubated with HBSS at 37°C for 10 min with or without inhibitors, and then HBSS containing ETV in the absence or presence of inhibitors was added to initiate the uptake. The uptake was terminated by removing the incubation buffer and adding ice‐cold PBS quickly at the designated time. Then the cells were washed three times with ice‐cold PBS and lysed with 100 μl of 0.1% sodium dodecyl sulfate.
The intracellular accumulation of ETV, ribavirin, and cGMP was also determined in OAT2, ENT1 transfected, or mock cells using the method described for hepatic cell lines, except the buffer was replaced by Na+‐free KRH (NMDG, 125 mM; d‐glucose, 5.6 mM; KCl, 4.8 mM; MgSO4·7H2O, 1.2 mM; KH2PO4, 1.2 mM; CaCl2, 1.2 mM; HEPES, 25 mM; pH = 7.4) for hENT1, or HBSS containing 10 μM of NBTI for hOAT2, respectively.
The concentrations of ETV, cGMP, and ribavirin in the cells were quantified with LC–MS/MS and normalized to the total protein content, detected by use of a BCA assay, in the lysates to control for variations in cell numbers. The uptake in the presence of inhibitors was expressed as a percentage of the vehicle group (% of control).
2.4. Retroviral transduction of OAT2‐pLent or ENT1‐pLent into MDCK or HEK293 cells
HEK293T cells were transfected with hOAT2‐pLent, hENT1‐pLent, and vector using Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA). Viral production in the supernatant 48 hr after transfection was used for infection of MDCK or HEK293 cells. OAT2, ENT1 overexpressed cells (MDCK‐hOAT2 and HEK‐hENT1), and mock cells (MDCK‐OV and HEK‐OV) were successfully constructed by puromycin screening for more than 1 week and validated by comparing the accumulation of probe substrates of transporters in overexpressed cells with that in mock cells.
2.5. Small interfering RNA transfection
The small interfering RNAs (siRNA) were transfected to HL7702 cells (50% confluence) with Lipofectamine 3000 (Life Technologies, Waltham, MA, USA) according to the manufacturer's protocol. Cells were harvested at designated times after transfection before mRNA extraction or cellular uptake assay. siRNAs were synthesized in GenePharma (Shanghai, China), and the target sequences are listed in Table S2.
2.6. Plasmid transfection
hOAT2‐pEnter were transfected to HL7702 cells (90% confluence) with Lipofectamine 3000. Cells were harvested at 48 hr after transfection before mRNA extraction or cellular uptake assay. hOAT2‐pEnter were transfected to HepG2.2.15 cells (60% confluence) with Lipofectamine 3000. Cells were grown in complete medium with 2% FBS at 24 hr after transfection and then cultured for 3 days followed by cellular uptake assay and mRNA extraction.
2.7. Cytotoxicity assay
Cytotoxicity was measured by MTT assays according to a previously published method (Tu et al., 2014). HepG2.2.15 cells (80% confluence) were incubated with medium containing 0.1% DMSO with or without the indicated concentrations of ETV, NBTI, E2, or P4, dissolved in complete medium containing 2% FBS, for a designated time. At the end of the incubation, the supernatant was collected. Then 200 μl of MTT reagent at a final concentration of 0.5 mg·ml−1 were added to each well, and the mixture was incubated for another 4 hr at 37°C. Subsequently, the incubation medium was thoroughly removed before 150 μl of DMSO was added to dissolve the formazan. The absorbance of each well was measured at 490 nm with 630 nm as a reference by a microplate reader (Spectra Max M2, Molecular Devices, CA, USA). The cell viability is expressed as a percentage of the vehicle group (% control) to control for unwanted sources of variation.
2.8. In vitro antiviral activity
To determine the role of ENT1 in the antiviral activity of ETV, HepG2.2.15 cells were cultured in ETV‐containing medium (2% FBS) with or without NBTI (1 μM) for 6 days. To clarify the role of OAT2 in antiviral activity of ETV, HepG2.2.15 cells after hOAT2‐pEnter or vector transfection for 24 hr were cultured with the indicated concentrations of ETV for 3 days, and 1 μM of NBTI was added to the medium to exclude the effect of ENT1. The level of HBV DNA was detected in supernatants, harvested after 6‐ or 3‐day treatments, with a One Step Hepatitis B viral DNA quantitative fluorescence diagnostic kit (Hunan Sansure Biotech, Hunan, China).
2.9. Hepatic distribution studies in mice
GD = 18.5 of pregnant mice is equivalent to the late pregnancy of humans, which is the period when antiviral therapy is recommended to reduce the risk of mother‐to‐infant transmission. Therefore, pregnant mice of GD = 18.5 were used in hepatic distribution studies. Blood samples (0.25 ml) were collected from the orbital venous sinus into heparin‐containing tubes; liver samples were collected from mice, anaesthetized with ether, killed 0.167, 0.5, 1, and 3 hr after a single oral dose of ETV; the weights of the non‐pregnant mice were 25.1–36.3 g and the pregnant mice 51.0–68.5 g. The dose of ETV was calculated from the human dose (2 mg·day−1) according to body surface area. Pregnant mice with the same gestation period could not be obtained at one time when eight mice were designed in each group; therefore, we had eight non‐pregnant mice in the control group, with only five pregnant mice in the ETV treatment group. The plasma was collected immediately after centrifugation of the blood samples at 8,000× g for 10 min. Tissue samples were weighed accurately, then minced and homogenized thoroughly with 1:8 (w/v) 50% acetonitrile solution. The separated plasma and tissue homogenates were frozen at −40°C until analysis.
2.10. Treatment of hepatic cell lines and primary human hepatocytes with E2 and P4
HepG2, Huh‐7, and HL7702 cells were seeded at a density of 2 × 105 cells·ml−1 in 24‐well plates. After overnight, cells were treated without or with designated concentrations of E2 or/and P4 for 3 days (refreshing the medium per 12 hr) followed with mRNA extraction or cellular uptake of ETV assay (culture supernatant was replaced by HBSS before uptake assay to exclude the direct inhibitory effect of E2 or P4).
PHH were seeded at a density of 2 × 105 cells·ml−1 in collagen‐coated 12‐well plates (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol. After 24 hr, the medium was replaced by DMEM without serum addition, and then the PHH were incubated without or with the indicated concentrations of E2 or/and P4. The medium was changed every 12 hr. After the 24‐hr treatment, the cells were harvested for RNA extraction or cellular uptake of ETV assay.
2.11. LC–MS/MS determination of ETV, ribavirin, and cGMP
The concentrations of ETV, ribavirin, and cGMP in the cellular uptake assay were determined by the modified LC–MS/MS method (Ma et al., 2017), by use of an Agilent 1290‐6460 LC–MS with a triple quadrupole mass spectrometer (Agilent, CA, USA). Three volumes of acetonitrile containing MPP+ (50 nM) as internal standard were added to cell lysates for protein precipitation. After being vortexed for 5 min, the mixture was centrifuged at 16,000× g for 15 min, and 2.0 μl of the supernatant was analysed by LC–MS/MS. The chromatographic separation was performed on a X‐Bridge™ BEH HILIC column (2.5 μm, 2.1 × 50 mm, Waters, USA) with a gradient elution (0–1.6 min, 95% of B; 1.6–2.0 min, 95–60%; 2.0–4.0 min, 60% of B) at 0.2 ml·min−1, where mobile phases A and B were 0.1% formic acid in 10‐mM ammonium formate‐water and 0.1% formic acid in acetonitrile respectively. Mass spectrometric analysis was performed on an ESI source in positive ion mode. Quantification was obtained using multiple reaction monitoring mode, and detailed MS conditions were as listed as Table S3. The method was validated, and satisfactory specificity, precision, and accuracy were demonstrated.
The concentrations of ETV in the lysates of HepG2.2.15 cells in the antiviral activity assay and in the liver or blood samples were detected by a similar method except for the sample preparation. Six volumes of acetonitrile containing phenacetin (10 nM) as internal standard were added for protein precipitation following vortexing and centrifugation. Then 600 μl of the supernatant were transferred to another tube and then evaporated to dryness at 38°C in a vacuum concentrator system (Labconco, Kansas, MO, USA). The residue was reconstituted in 100 μl of acetonitrile/water (9:1, v/v), and the aliquot of 10 μl was injected into the LC–MS/MS system. The values were normalized to the total protein content, measured by BCA assay, in the lysates to control for variations in the number of cells. The accumulation in the presence of E2 or P4 was expressed as a percentage of the vehicle group (% control) to control for unwanted sources of variation.
2.12. Real‐time quantitative PCR analysis
Total RNA was isolated by use of the RNA simple Total RNA Kit (Tiangen, Beijing, China) and then reversely transcribed into cDNA using PrimeScript RT Master Mix (Takara, Tokyo, Japan). The resulting cDNA was amplified by quantitative real‐time PCR with SYBR® Premix EX Taq (Takara, Tokyo, Japan). Relative mRNA levels of target genes were expressed as the ratio of target to GAPDH genes. The specific primers are shown in Table S4.
2.13. Western blot analysis
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. Proteins were extracted with RIPA (P0013K; Beyotime, Nanjing, China), and the protein concentration was determined by the BCA Protein Assay Kit (Beyotime, Nanjing, China). Proteins (100 μg) were mixed with loading buffer (P0015; Beyotime, Nanjing, China) and then boiled at 100°C for 5 min. Aliquots of denatured protein were separated by electrophoresis on SDS‐PAGE (Bio‐Rad, Hercules, CA) and subsequently transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked overnight at 4°C with 5% non‐fat milk in TBST buffer (100‐mM Tris–HCl, pH 7.4, 150‐mM NaCl, and 0.1% Tween 20) and then incubated with specific primary antibodies (anti‐ENT1 at 1:500 and anti‐GAPDH at 1:5,000) in blocking buffer at room temperature for 2 hr. Membranes were then washed with TBST three times (5 min each time) and incubated for 1 hr with HRP‐conjugated secondary antibodies at room temperature. After the samples had been washed with TBST (5 min every time), signals were visualized by chemiluminescent detection using the enhanced chemiluminescence western blotting detection system (LI‐COR Biosciences, Lincoln, NE).
2.14. Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Experimental subjects/preparations were randomized to groups, and group assignments, data recording, and data analysis were blinded to the operator. The Michaelis–Menten constants V max and K M were calculated by fitting the data to the Michaelis–Menten equation V = V max/(1 + (K M/[S])), where V is the initial uptake velocity and [S] is the concentration of substrate. In vitro data, each point represents the mean ± SD for at least five wells, and in vivo data were shown as mean ± SD from at least five animal samples.
Statistical analyses and significance were determined by Student's unpaired two‐tailed t test between two groups. One‐way ANOVA followed by Bonferroni's or Dunnett's post hoc test was applied to the data if more than two groups were analysed, only if F achieved the level of significance P < .05, and no significant variance inhomogeneity was observed. All of the statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA; RRID:SCR_002798). P values <.05 were considered statistically significant.
2.15. Materials
FBS, trypsin, non‐essential amino acid (100×), DMEM, and RPMI 1640 medium were purchased from Gibco Invitrogen Corporation (Carlsbad, CA, USA). Entecavir (ETV) and ribavirin were provided by Dalian Meilun Biotechnology Co., Ltd. SYBR Green was obtained from Takara Bio Inc. (Otsu, Japan). cGMP, indomethacin (Indo), S‐(4‐nitrobenzyl)‐6‐thioinosine (NBTI), and 1‐methyl‐4‐phenylpyridinium (MPP+) were obtained from Sigma‐Aldrich (St. Louis, MO, USA). G418 and 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide (MTT) were obtained from Sangon Biotech Co., Ltd. (Shanghai, China). N‐methyl‐d‐glucamine (NMDG), puromycin, and quinidine were from Aladdin Co., Ltd. (Shanghai, China). Bicinchoninic acid (BCA) protein assay kit was obtained from Beyotime Institute of Biotechnology (Nanjing, China). Acetonitrile was obtained from Tedia (Fairfield, TX, USA). All other chemicals were purchased from commercial sources and were of analytical grade.
Expression plasmid hOAT2‐pEnter (OAT2‐546aa), hOAT2‐pLent‐EF1a‐FH‐CMV‐GFP‐P2A‐puromycin (hOAT2‐pLent), hENT1‐pLent‐EF1a‐FH‐CMV‐GFP‐P2A‐puromycin (hENT1‐pLent), and corresponding vectors were obtained from ViGene Biosciences Inc. (Jinan, China). Antibodies including anti‐equilibrative nucleoside transporter 1 (ENT1; 11337‐1‐AP) and anti‐GAPDH (Mab5465) were purchased from Proteintech (Wuhan, China) and LiankeBio (Hangzhou, China), respectively.
MDCK cells (CVCL_0422) stably transfected with full length hOCT1 cDNA (MDCK‐hOCT1) were established or already present in our laboratory (Tu et al., 2014).
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Kelly, Marrion, Peters, Faccenda, Harding, Pawson, Sharman, Southan, Davies, & CGTP Collaborators, 2017).
3. RESULTS
3.1. ETV uptake by hepatic cell lines was reduced by inhibitors of ENT1 and OAT2
The effects of inhibitors of ENT1/2 (NBTI), OAT2 (Indo), OCT (decynium‐22, D22), and OCTN2 (L‐carnitine) on ETV uptake (20 μM, 3 min) into hepatic cell lines were evaluated. NBTI (1, 100 μM) dramatically reduced the accumulation of ETV in HepG2, Huh‐7, and HL7702 cells. However, no significant inhibitory effect of D22 was observed in any of the above cell lines. OAT2 and OCTN2 inhibitors decreased the ETV accumulation in HepG2 cells to 55.8% and 83.4% of the control, respectively, but displayed no influence in both HL7702 and Huh‐7 cells (Figure 1a). Given that hENT1 is predominantly expressed in the liver compared to hENT2 (Figure S1), we inferred that ENT1 and OAT2 play key roles in the hepatic uptake of ETV.
Figure 1.
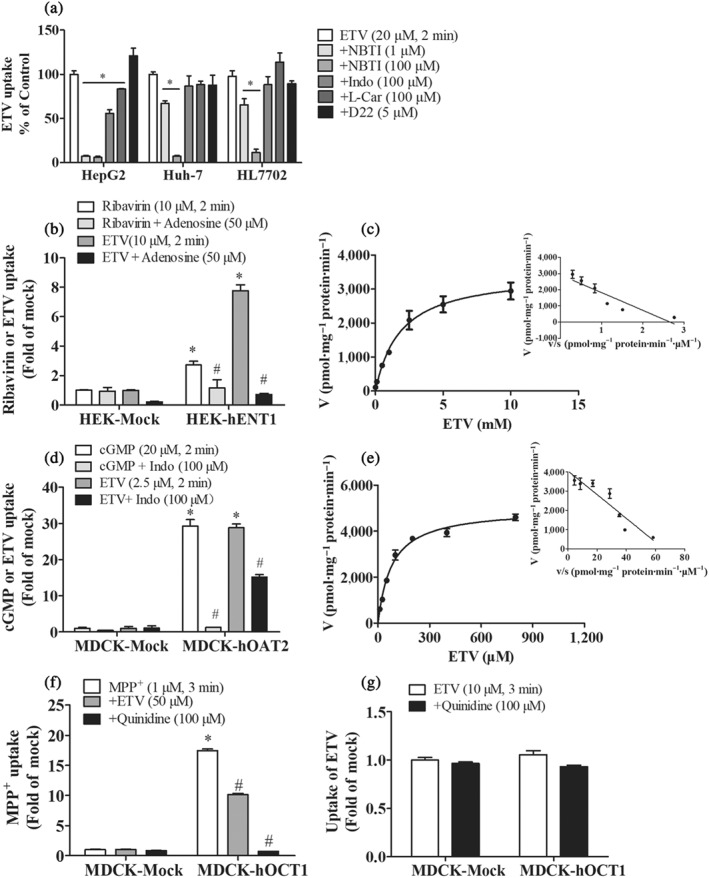
Cellular accumulation of ETV mediated by hENT1 and hOAT2. (a) Inhibitory effect of NBTI (ENT inhibitor), Indo (OAT2 inhibitor), l‐carnitine (OCTN2 inhibitor), and D22 (OCT3 inhibitor) on the intracellular accumulation of ETV in HepG2, Huh‐7, and HL7702 cells. n = 5. *P < .05 in comparison with ETV‐alone group. (b, d) Accumulations of known substrates (ribavirin or cGMP) and ETV in mock and HEK‐hENT1 or MDCK‐hOAT2 cells in the absence or presence of respective inhibitors (adenosine or Indo). (c, e) Concentration‐dependence of ETV accumulation in HEK‐hENT1 or MDCK‐hOAT2 cells. Cells were incubated at 37°C for 2 min with indicated concentration of ETV. (f) The accumulations of MPP+ in mock cells and MDCK‐hOCT1 cells in the absence or presence of ETV or quinidine (OCT1 inhibitor). (g) The accumulations of ETV in mock cells and MDCK‐hOCT1 cells in the absence or presence quinidine. n = 5. *P < .05 in comparison with vector group; # P < .05 in comparison with the accumulation in transporter‐overexpressed cells without inhibitor
3.2. ETV was a substrate of hENT1 and hOAT2 but not of hOCT1
To further determine the cellular uptake of ETV mediated by hENT1 and hOAT2, we investigated whether ETV was a substrate of hENT1 and hOAT2 in transporter‐overexpressed cell models. The functional activity of HEK‐hENT1 and MDCK‐hOAT2 cells was assessed by comparing the accumulations of ribavirin (a known ENT1 substrate) and cGMP (a probe substrate of OAT2) in HEK‐hENT1 and MDCK‐hOAT2 cells, respectively, with that in mock cells (Figure 1b,d). The intracellular accumulations of ETV in HEK‐hENT1 and MDCK‐hOAT2 cells were approximately eightfold and 29‐fold higher than that in mock cells, respectively, which were significantly inhibited by adenosine (50 μM) and Indo (100 μM) (Figure 1b,d), indicating that ETV is a substrate of hENT1 and hOAT2. The kinetic data showed the hENT1‐mediated uptake followed Michaelis–Menten kinetics with the K M (1.8 ± 0.3 mM), V max (3.5 ± 0.6 nmol·mg−1 protein·min−1), and Cl int (V max/K M: 1.9 ± 0.1 ml·g−1 protein·min−1; Figure 1c), indicating that ETV is a low affinity substrate of hENT1. In contrast, ETV was shown to be a high affinity substrate of hOAT2 with the K M (0.08 ± 0.01 mM), V max (5.0 ± 0.2 nmol·mg−1 protein·min−1), and Cl int (V max/K M: 63.0 ± 4.7 ml·g−1 protein·min−1; Figure 1e), respectively.
ETV was reported to be a substrate of OCT2 and OCT3 (Ma et al., 2017; Yang et al., 2016). Considering the overlap of substrates for OCTs and the predominant expression of OCT1 in the liver, we speculated that OCT1 might be involved in the hepatic uptake of ETV. However, our results revealed that the accumulation of ETV in MDCK‐hOCT1 cells was not different from that in the mock cells, indicating that ETV is not a substrate of OCT1, although ETV (50 μM) inhibited the accumulation of MPP+ (1 μM), a typical substrate of OCT1, in MDCK‐hOCT1 cells (Figure 1g,h).
3.3. The roles of ENT1 and OAT2 in hepatic uptake of ETV
To further confirm the role of ENT1 and OAT2 in the hepatic uptake of ETV, the accumulation of ETV in HL7702 cells transfected with siRNAs targeting hENT1 or transiently overexpressing hOAT2 was investigated. The silencing efficiency of siRNAs was determined by RT‐qPCR at the mRNA level (Figure 2a). It was found that siENT1‐s2 reduced ETV uptake into HL7702 cells to 40.2% at 24 hr and to 36.8% at 48 hr of the control, respectively, whereas siENT1‐s1 only attenuated ETV uptake at 48 hr but not at 24 hr (Figure 2b). OAT2‐overexpressed HL7702 cells were evaluated by mRNA expression level and the accumulation of cGMP (a probe substrate of hOAT2; Figure 2c,d). ETV accumulation in OAT2‐overexpressed HL7702 cells was increased to 3.2‐fold of that in the mock cells (Figure 2d), which further confirmed that OAT2 contributes to the hepatic uptake of ETV.
Figure 2.
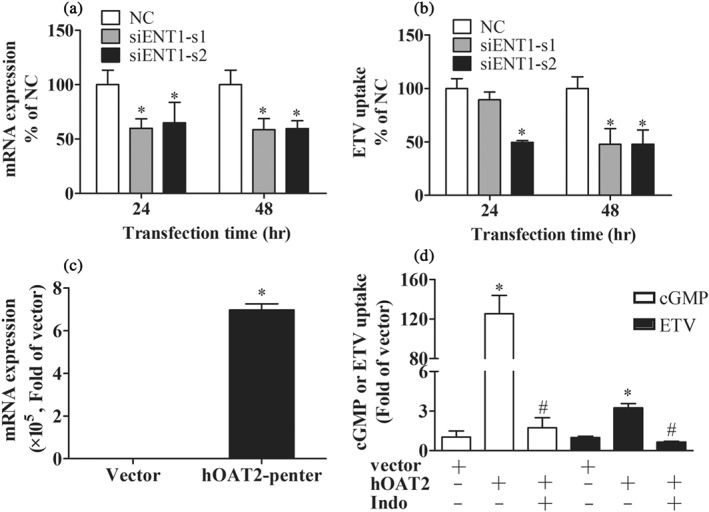
The effect of ENT1 knockdown by siRNA or OAT2 overexpression in HL7702 cells on the accumulation of ETV. hENT1 mRNA expression (a) and ETV uptake (b) in HL7702 cells transfected with hENT1‐siRNA (s1 and s2) and control siRNA for the time indicated. n = 5. *P < .05 in comparison with the vector. (c) The mRNA expression of hOAT2 in HL7702 cells transfected with hOAT2‐pEnter and corresponding vector for 48 hr. (d) Accumulations of cGMP (OAT2 probe substrate) and ETV in HL7702 cells transfected with hOAT2‐pEnter and corresponding vector for 48 hr in the absence or presence of Indo (OAT2 inhibitor). n = 5. *P < .05 in comparison with vector group; # P < .05 in comparison with ETV‐alone group
3.4. Contribution of ENT1 to antiviral activity of ETV on HepG2.2.15 cells
NBTI (0.1–100 μM), an ENT1 inhibitor, significantly reduced ETV uptake into HepG2.2.15 cells (Figure 3a). To investigate the involvement of hENT1 in the antiviral activity of ETV, we examined the alterations in HBV DNA levels in cell culture supernatants after inhibiting the activity of ENT1 with 1 μM NBTI (no cytotoxicity at 1 μM, Figure S2). The level of HBV DNA in the cell supernatants exposed to ETV (3, 6, and 12 nM) for 6 days was markedly reduced (Figure 3b), and NBTI (1 μM) attenuated the susceptibility of HBV to ETV, which was consistent with the effect of NBT1 on ETV uptake (Figure 3c). In addition, treatment of HepG2.2.15 cells with NBTI (1 μM) did not obviously alter the expression of influx and efflux transporters involved in ETV cellular transport (Figure 3d). Taken together, ENT1 contributes to the anti‐HBV efficacy of ETV via mediating its hepatic cellular uptake.
Figure 3.
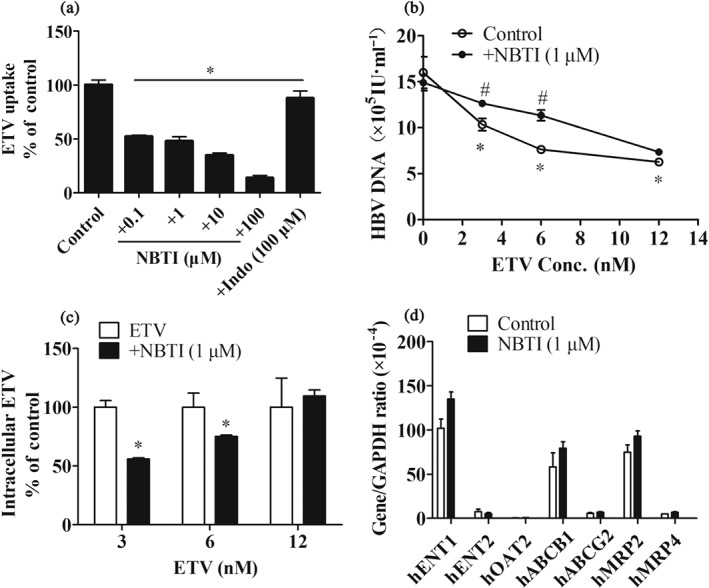
The role of ENT1 in the antiviral activity of ETV. (a) ETV accumulations in HepG2.2.15 cells in the absence or presence of the indicated concentrations of NBTI and Indo. n = 5. *P < .05 in comparison with ETV‐alone group. (b) The effect of NBTI (1 μM) on the level of HBV DNA in HepG2.2.15 cell supernatant. HepG2.2.15 cells were treated with the indicated concentrations of ETV with or without NBTI for 6 days. n = 5. *P < .05 in comparison with vehicle group; # P < .05 in comparison with ETV‐alone group. (c) Intracellular ETV concentrations in HepG2.2.15 cells treated with or without NBTI for 6 days. n = 5. *P < .05 in comparison with ETV‐alone group. (d) Gene expression of influx and efflux transporters in HepG2.2.15 cells after being treated with vehicle or NBTI (1 μM) for 6 days
3.5. OAT2 overexpression increased susceptibility to ETV of HepG2.2.15 cells
Since OAT2 mediated the influx of ETV into cells, we further determined the role of hOAT2 in the antiviral activity of ETV by evaluating the sensitivity of HBV to ETV when hOAT2 was overexpressed. mRNA expression of hOAT2 and accumulation of cGMP and ETV in HepG2.2.5 cells transfected with hOAT2‐pEnter for 96 hr were significantly higher than that of the control (Figure 4a,b), indicating that the cell model could be used in our study. The MTT results showed that 0.1–100 μM of ETV exhibited no cytotoxicity in HepG2.2.5 cells for 3 days (Figure S2). As shown in Figure 4c, compared with the vector cells, HepG2.2.15 cells overexpressing hOAT2 were much more sensitive to ETV with IC50 values of ETV 45.4 ± 5.7 nM versus 13.1 ± 2.0 nM. Concomitantly, the intracellular concentration of ETV was predictably higher in hOAT2‐overexpressed cells (Figure 4d). The above results indicate that hOAT2 also plays an important role in ETV's anti‐HBV efficacy by mediating its hepatic cellular uptake.
Figure 4.
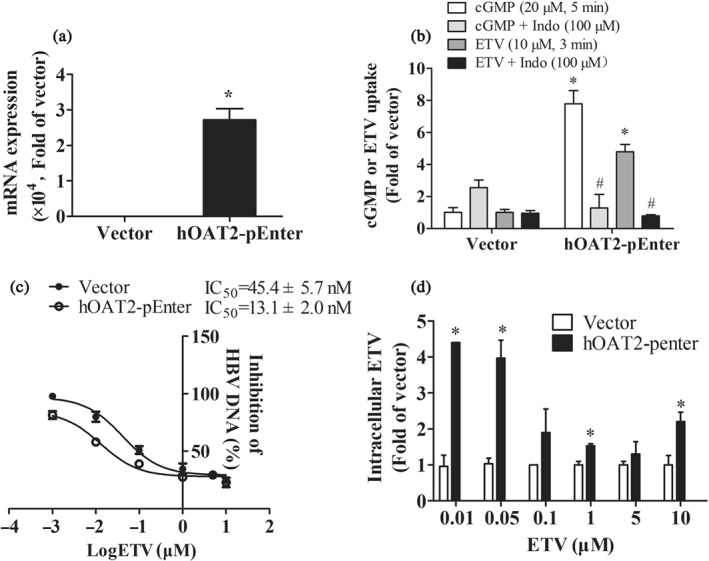
The role of OAT2 in the antiviral activity of ETV. (a) The mRNA expression of hOAT2 in HepG2.2.15 cells transfected with hOAT2‐pEnter and corresponding vector for 96 hr. n = 5. *P < .05 in comparison with vector group. (b) Accumulations of cGMP and ETV in HepG2.2.15 cells transfected with hOAT2‐pEnter and corresponding vector for 96 hr in the absence or presence of Indo. n = 5. *P < .05 in comparison with vector group; # P < .05 in comparison with the accumulation in cells transfected with hOAT2‐pEnter without inhibitor. (c) The antiviral activity and (d) intracellular concentration of ETV in HepG2.2.15 cells transfected with hOAT2‐pEnter or corresponding vector for 3 days. n = 5. *P < .05 in comparison with vector group
3.6. The liver‐to‐plasma ETV concentration ratio was reduced in pregnant mice
To elucidate whether the hepatic distribution of ETV was changed during pregnancy, the concentrations of ETV in the liver and plasma of non‐pregnant and pregnant mice at 0.167, 0.5, 1, and 3 hr after a single oral dose were determined. Our results revealed that the concentrations of ETV in plasma were increased in pregnant mice at 0.167 and 1 hr after dosing (Figure 5a). In non‐pregnant mice, ETV reached the highest concentration (383.3 ng·g−1) in the liver at 0.167 hr but slowly decreased to 134.5 μg·g−1 at 3 hr after a single dose, which were 4.4‐ and 38‐fold of that in plasma, respectively. The concentrations of ETV in the liver of non‐pregnant and pregnant mice were comparable except for at 0.5 hr (Figure 5b). However, the liver‐to‐plasma ETV concentration ratios in pregnant mice were sharply reduced to 41.1%, 67.2%, and 21.1% of those of non‐pregnant mice at 0.167, 0.5, and 1 hr after its administration respectively (Figure 5c). Concomitantly, the P4 concentrations in the plasma of pregnant mice were sevenfold higher than those in non‐pregnant mice, whereas the concentrations of E2 were not obviously altered (Figure 5d). The results above indicate that the relative hepatic distribution (liver‐to‐plasma) of ETV, but not the absolute concentrations, is decreased in late pregnancy.
Figure 5.
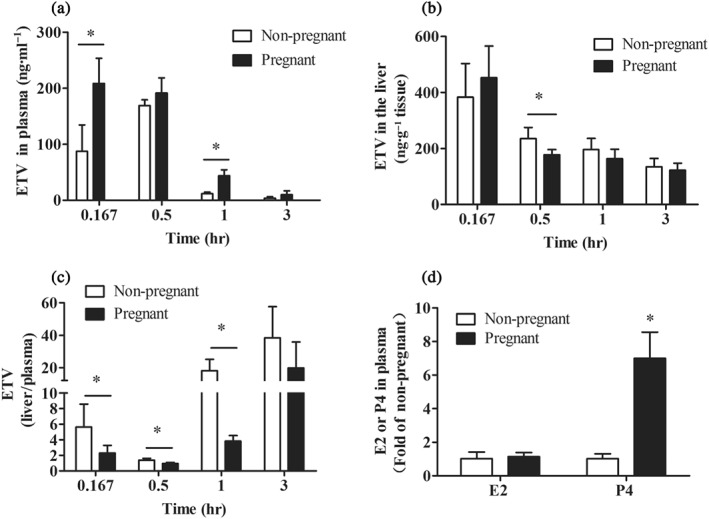
The concentrations of ETV in the plasma (a) and the liver (b) at 0.167, 0.5, 1.0, and 3.0 hr after orally dosing with ETV in non‐pregnant (n = 8) and pregnant mice (GD = 18.5, n = 5). (c) The liver‐to‐plasma ETV concentration ratios. n = 5. (d) The plasma concentrations of E2 and P4 in non‐pregnant and pregnant mice (GD = 18.5), n = 6. *P < .05 in comparison with non‐pregnant mice
3.7. mEnt1 expression was down‐regulated in the mouse liver during pregnancy
Since the hepatic uptake of ETV is mainly mediated by transporters, the lower relative hepatic distribution of ETV in pregnant mice may be related to the decreased expression of a transporter during pregnancy. As expected, the mRNA expression of mEnt1 in the liver of pregnant mice was approximately 50% of that in non‐pregnant mice; however, no significant change was observed in other transporters including mOat2, mOct3, mEnt2, mAbcb1a, mAbcg2, mMrp2, and mMrp4. Our further study revealed that the protein expression of mEnt1 was notably decreased in pregnant mice based on the western blotting result (Figure 6).
Figure 6.
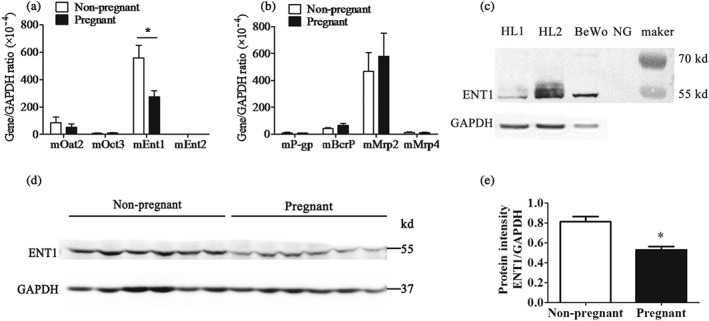
The mRNA expressions of uptake (a) and efflux (b) transporters in the liver of non‐pregnant (n = 6) and pregnant mice (GD = 18.5, n = 6). (c) Human liver (HL) and BeWo cells were used as positive controls in western blot analysis, which verify that the protein band at ~55 kD position was mEnt1. (d) Protein expression of mEnt1 in the liver of non‐pregnant and pregnant mice (GD = 18.5); the band intensity was analysed with ImageJ software (e). n = 6. *P < .05 in comparison with non‐pregnant mice
3.8. The ETV accumulation was reduced by E2 and P4 via inhibiting the activities of hENT1 and hOAT2 but not by modifying their expression
Since mEnt1 expression in the liver was reduced in late pregnancy, we supposed that the hepatic uptake of ETV was affected by the continuous effects of E2 and P4, two important hormones during pregnancy. Unexpectedly, after treatment with the indicated concentration of E2 or/and P4 for 72 hr, ETV accumulations in HepG2, Huh‐7, and HL7702 cells were not obviously altered (culture supernatant was replaced by HBSS before uptake assay to exclude the direct inhibitory effect of E2 or P4; Figure 7), which was consistent with the finding that the mRNA levels of hENT1 and hOAT2 were not changed (data not shown).
Figure 7.
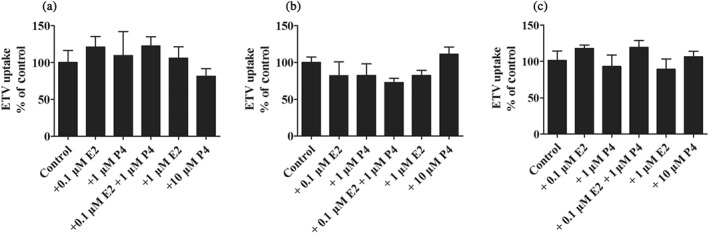
The uptake of ETV (20 μM, 3 min) in HepG2 (a), Huh‐7 (b), and HL7702 (c) cells after being treated with the indicated concentrations of E2 or/and P4 for 72 hr (n = 5)
E2 and P4 are reported to be inhibitors of hOCT3 and hOCTN2 (Hayer‐Zillgen, Bruss, & Bonisch, 2002; Rytting & Audus, 2008). We proposed that E2 and P4 might affect the cellular uptake of ETV via directly inhibiting transporter activity. Co‐incubation with E2 at 0.1–1 μM for 15 min had a negligible effect on the ETV accumulations in HepG2, Huh‐7, and HL7702 cells. In contrast, 2–10 μM of P4 displayed a concentration‐dependent inhibition of the uptake of ETV in HepG2 cells. P4 (1–10 μM) slightly inhibited ETV accumulation in Huh‐7 cells and showed about 50% inhibition in HL7702 cells at 10 μM (Figure 8a–c). To further determine whether E2 or P4 reduce ETV accumulation via inhibition of hENT1 and hOAT2, the ETV accumulation in HEK‐hENT1 and MDCK‐hOAT2 cells in the absence or presence of E2 and P4 was investigated. As shown in Figure 8d–e, 1–100 μM of E2 significantly reduced ETV uptake in both of the cell models, and high concentrations of P4 also reduced the intracellular ETV level, which indicates that E2 and P4 inhibit the activities of the transporters and subsequently reduce the accumulation of ETV.
Figure 8.
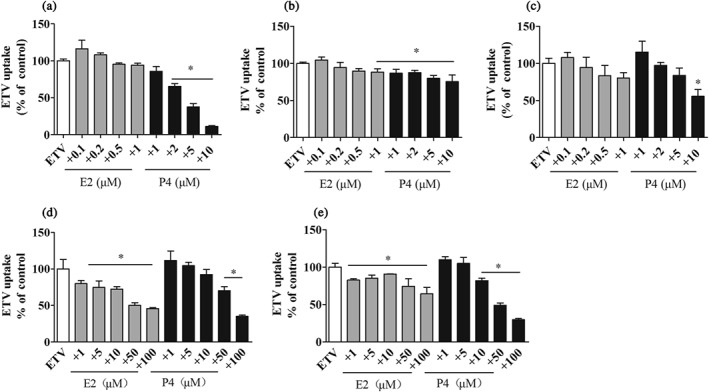
The inhibitory effects of E2 and P4 at the indicated concentrations on the uptake of ETV (20 μM, 3 min) in HepG2 (a), Huh‐7 (b), HL7702 (c), HEK‐hENT1 (d), and MDCK‐hOAT2 (e) cells. n = 5. *P < .05 in comparison with ETV‐alone group
3.9. ETV accumulation in PHH
To investigate whether ENT1 and OAT2 contribute to the uptake of ETV into human hepatocytes, we measured ETV accumulation in PHH from three individual donors in the absence or presence of the transporter inhibitors. Due to the scarcity of human hepatocytes, two tubes of cells from each donor were applied, which were not enough to obtain five wells for each group; thus, each column in Figure 9 represents the mean ± SD for three wells. According to the journal guidelines, these results were not statistically analysed. NBTI (1 μM, ENT1 inhibitor) and Indo (100 μM, OAT2 inhibitor) reduced ETV uptake by PHH to 87.1% and 14.8% of the vehicle control, respectively (Figure 9a). Additionally, P4 (5, 25 μM) also directly reduced the intracellular ETV concentration (Figure 9b) with inhibition of 29.1% and 68.5% respectively. However, neither the mRNA expression of ETV's transporters nor ETV accumulation in PHH seemed to be altered after treatment with physiologically relevant concentrations of E2 (0.1 μM) and P4 (1 μM) for 24 hr (Figure 9c,d).
Figure 9.
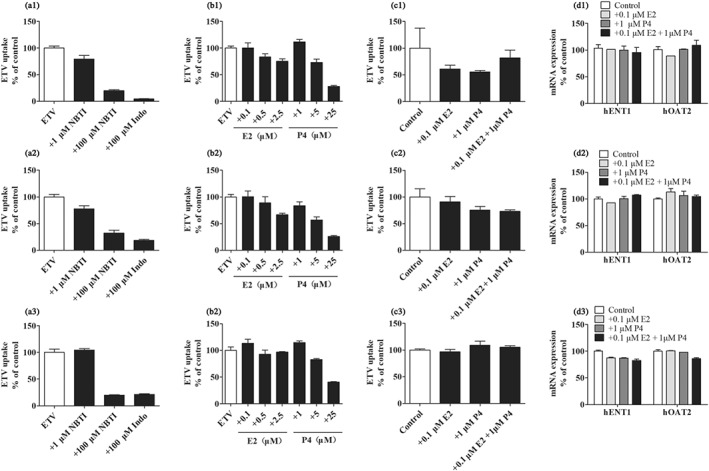
The inhibitory effect of NBTI s(ENT inhibitor), Indo (OAT2 inhibitor), E2, and P4 at the indicated concentrations on the uptake of ETV into PHH of three donors, 1, 2, and 3 (a, b). The effect of treatment with E2 or/and P4 for 72 hr on ETV accumulations and mRNA expressions of ENT1 and OAT2 in PHH of three donors, 1, 2, and 3 (c, d). Each column in this figure represents the mean ± SD for three wells
4. DISCUSSION
Due to high hydrophilicity, ETV requires a “gate” to penetrate the plasma membrane of cells prior to its accumulation, which determines intracellular ETV concentration and further affects ETV anti‐HBV activity. It has been reported that OCT1 and several members of organic anion transporting polypeptide (OATP) family are abundantly expressed in human hepatocytes (Roth, Obaidat, & Hagenbuch, 2012); however, our results indicate that ETV is not a substrate of OCT1 (Figure 1g). Additionally, it was reported that OATP2B1, OATP1B1, and OATP1B3 are not involved in the accumulation of ETV (Furihata et al., 2017).
Our previous study proved that only high concentration of NBTI (100 μM) slightly decreased ETV uptake into BeWo cells expressing high levels of ENT1 (Ma et al., 2017); however, 1 μM of NBTI significantly decreased the ETV uptake into hepatic cell lines, especially in HepG2 cells, with inhibition of 92% (Figure 1a). We speculated that the roles of ENT1 might be eclipsed in BeWo cells because of the co‐expression of concentrative nucleoside transporter (CNT) 3 which predominantly contributes to placental transport of ETV in placenta (Ma et al., 2017). To verify this hypothesis, uptake of ETV by BeWo cells was conducted in Na+ free buffer, in which sodium‐dependent CNT3 is functionally knocked down. Interestingly, in Na+ free buffer, 10 nM of NBTI reduced ETV uptake to 36% of the control (Figure S3). Considering that sodium‐independent ENT1 is predominately expressed in the liver compared with other nucleosides transporters (Iikura et al., 2012), we postulated that ENT1 plays an important role in the hepatic uptake of ETV. Additionally, inhibition of ENT1‐mediated ETV uptake by NBTI significantly attenuated the antiviral activity of the drug as well as its accumulation in HepG2.2.15 cells (Figure 3), which further indicates that ENT1 is a key factor in determining the susceptibility of HBV to ETV.
OAT2, originally called novel liver‐specific transporter, is dominantly expressed in the liver and functionally transports nucleosides with a preference for guanine analogues (Shen, Lai, & Rodrigues, 2017). ETV, a synthetic deoxy‐guanosine analogue, was reported to be a substrate of OAT1/3 (Yang et al., 2016). Thus, we speculated that OAT2 is involved in the hepatic uptake of ETV and its antiviral activity, which was further firmly confirmed by OAT2 overexpression in HL7702 and HepG2.2.15 cells (Figures 2 and 4). It is noteworthy that when OAT2 activity was inhibited by Indo (100 μM) in PHH, ETV accumulations were reduced to 14.8% of the vehicle group, which was lower than that in the presence of the ENT1 inhibitor, 1 μM of NBTI (Figure 9). A high concentration of NBTI (100 μM) was needed to display dramatic decrease in ETV accumulation when abundant OAT2 is co‐expressed in PHH (Figure 9). Moreover, the intrinsic clearance (Cl int) value of hENT1 was 1.9 ml g−1·min−1, much lower than that of hOAT2 (63 ml·g−1·min−1), indicating that hOAT2 contributes more than hENT1 to the uptake of ETV from the circulation into hepatocytes. HBV infection is a risk factor for HCC. To further predict alterations in the hepatic distribution of ETV in HCC patients, we analysed five independent microarray datasets from Oncomine database (Chen et al., 2002; Mas et al., 2009; Roessler et al., 2010; Wurmbach et al., 2007) and found that OAT2 was down‐regulated but ENT1 was not in most HCC tissues compared with adjacent controls (Figure S4). In the present study, we did not evaluate the role of OAT2 and ENT1 inhibitors, such as Indo and NBTI, in the hepatic uptake of ETV in vivo because specific transporter inhibitors are unavailable. However, ENT1 or OAT2 knockout animal models would be good choices to be used in further studies. The roles of the two transporters in the antiviral efficacy of ETV during pregnancy should also be confirmed by data from clinical trials in pregnant women.
A small sample of clinical data from the Antiretroviral Pregnancy Registry showed that the defect rate of ETV was 2.56%, which was no different from the natural birth defects (Antiretroviral Pregnancy Registry Steering Committee, 2018). Additionally, our previous study indicated that fetal exposure to ETV is very low because of low clinical doses and low placental transfer (Ma et al., 2017). Therefore, we speculated that ETV has the potential to be used in pregnant women. Based on various physical changes during pregnancy, we further determined whether the hepatic distribution and antiviral effectiveness of ETV were changed, which would subsequently affect ETV's efficacy during pregnancy. The present study showed that the concentration of ETV in pregnant mice was almost unchanged; however, ETV concentration ratios (liver‐to‐plasma) in pregnant mice were dramatically reduced (Figure 5) with an increase in plasma levels of ETV, which might be attributed to the down‐regulation of mENT1 in pregnancy (Figure 6). An increased plasma concentration of ETV was also observed in pregnant rats (data not shown), which might be attributed to the decreased renal excretion of ETV in pregnancy because E2 and P4 could inhibit the activities of hOAT1/3 and thus attenuate ETV uptake into renal cells (data not shown). The changes in the expression and activity of intestinal transporters probably affect the ETV absorption during pregnancy, which also needs to be clarified in further studies.
It was reported that E2 and P4 could inhibit the activities of hOCT2 and hOCTN2 (Hayer‐Zillgen et al., 2002; Rytting & Audus, 2008). Therefore, we speculated that the substantial increased levels of E2 and P4 during pregnancy may affect the activity of transporters and further contribute to the decreased liver‐to‐plasma ETV concentration ratio. As expected, E2 and P4 displayed a concentration‐dependent inhibition of ETV uptake by hENT1 and hOAT2, which also provided an explanation for the reduction in ETV accumulation in hepatic cell lines and PHH (Figures 8 and 9). However, co‐incubation of ETV with physiologically relevant concentrations of E2 (0.1 μM) and P4 (1 μM) for 6 days neither affected the anti‐HBV activity nor reduced the intracellular concentrations of ETV in HepG2.2.15 cells (Figure S5). Therefore, we deduced that E2 and P4 at levels observed in late pregnancy could not exert an inhibitory effect on the hepatic uptake of ETV via altering the activities of ENT1 and OAT2.
Besides direct inhibition on transporter activities, E2 and P4 treatment also could affect transporter expressions and thereby change the intracellular accumulations of drugs (Morgan et al., 2018; Tasnif, Morado, & Hebert, 2016). Therefore, we further explored the effect of 72 hr co‐treatment of E2 and P4 on ETV accumulation. Our study found that levels of ETV accumulated in three human hepatic cell lines treated with E2 (0.1 and 1.0 μM), P4 (1.0 and 10 μM), or E2 (0.1 μM) plus P4 (1.0 μM) for 72 hr did not show any significant difference from those treated with vehicle (Figure 7). Additionally, physiologically relevant concentrations of E2 or/and P4 did not alter the expressions of the two main transporters (OAT2 and ENT1) for hepatic uptake of ETV, and therefore, it seemed that there was no effect on ETV uptake into hepatocytes (Figure 9c).
5. CONCLUSION
In conclusion, the present study demonstrated that OAT2 and ENT1 are the main transporters involved in the hepatic uptake of ETV and subsequently contribute to its antiviral efficacy. Moreover, ETV's concentration in the liver was not obviously changed during pregnancy, which implies that dosage adjustments in the third trimester of pregnancy are probably not necessary. The results provide helpful information on the application of ETV in pregnant women, as well as for predicting the possibility of drug–drug interactions associated with the use of ETV.
AUTHOR CONTRIBUTIONS
Z.Y.M., S.H.L., and H.D.J. designed the research. Z.Y.M., S.H.L., D.L.S., M.R.B., H.Z., and T.J. performed the research. Z.Y.M., N.M.L., H.Z., and S.Z. analysed data. Z.Y.M., S.H.L., and H.D.J. wrote the paper.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1.
Information of primary hepatocytes
Table S2. siRNA sequence
Table S3. Transition, fragmentor and collision energy of compounds
Table S4.Primer list for real‐time qPCR
Figure S1
The mRNA expression of ENT1 and ENT2 in human liver tissue (n = 5).
Figure S2
The cytotoxicity of NBTI or ETV in HepG2.2.15 cells for 6 or 3 days, respectively (n = 6).
Figure S3
The inhibition of NBTI (ENT inhibitor) at indicated concentration on the uptake of ETV in BeWo cells in KRH buffer or Na+ free buffer. n = 5. *P < 0.05 in comparison with ETV‐alone group.
Figure S4
The mRNA expression of ENT1 and OAT2 in HCC. A meta‐analysis of ENT1 (A) or OAT2 (B) gene expression from three Oncomine databases where colored squares indicate the median rank for OAT2 (vs. Normal tissue) across 5 analyses. Chen Liver (1), Mas Liver (2), Roessler Liver (3, 4), Wurmbach Liver (5). The P value is given for the median‐rank analysis.
Figure S5
The role of E2 and P4 in the antiviral activity of ETV. The cytotoxicity of E2 (A) and P4 (B) in HepG2.2.15 cells for 6 days. (C) The effect of 0.1 μM of E2 and 1 μM of P4 on the level of HBV DNA in HepG2.2.15 cell supernatant. HepG2.2.15 cells were treated with indicated concentrations of ETV with or without E2 and P4 for 6 days (D) Intracellular ETV concentration in HepG2.2.15 cells when were co‐treated with or without 0.1 μM of E2 and 1 μM of P4 for 6 days. n = 5.
Figure S6
Accumulations of 10 μM of cGMP in OAT2‐overexpressed cells in the absence or presence of Indo or NBTI. n = 5. *P < 0.05 in comparison with ETV‐alone group.
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (81573492 and 81803612) and by the Zhejiang Province Natural Science Foundation of China (LY17H310003). We thank Ms. Haihong Hu for managing the instruments and helping in the experiments.
Ma Z, Lu S, Sun D, et al. Roles of organic anion transporter 2 and equilibrative nucleoside transporter 1 in hepatic disposition and antiviral activity of entecavir during non‐pregnancy and pregnancy. Br J Pharmacol. 2019;176:3236–3249. 10.1111/bph.14756
Shuanghui Lu is co‐first author and contributed equally to this work.
REFERENCES
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The concise guide to pharmacology 2017/18: Transporters. British Journal of Pharmacology., 174(S1), S360–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antiretroviral Pregnancy Registry Steering Committee (2018). The antiretroviral pregnancy registry international report for 1 January 1989 through 30 June 2018, Wilmington NC: Registry Coordinating Center. (updated June 2018).
- Chen, Q. , Chen, H. , Wang, W. , Liu, J. , Liu, W. , Ni, P. , … Zhang, J. (2017). Glycyrrhetic acid, but not glycyrrhizic acid, strengthened entecavir activity by promoting its subcellular distribution in the liver via efflux inhibition. European Journal of Pharmaceutical Sciences., 106, 313–327. [DOI] [PubMed] [Google Scholar]
- Chen, X. , Cheung, S. T. , So, S. , Fan, S. T. , Barry, C. , Higgins, J. , … Brown, P. O. (2002). Gene expression patterns in human liver cancers. Molecular Biology of the Cell, 13(6), 1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furihata, T. , Morio, H. , Zhu, M. Y. , Suzuki, Y. , Ide, H. , Tsubota, A. , … Chiba, K. (2017). Human organic anion transporter 2 is an entecavir, but not tenofovir, transporter. Drug Metabolism and Pharmacokinetics, 32(1), 116–119. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer‐Zillgen, M. , Bruss, M. , & Bonisch, H. (2002). Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. British Journal of Pharmacology, 136(6), 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans, L. E. , Svicher, V. , Pas, S. D. , Salpini, R. , Alvarez, M. , Ben Ari, Z. , … Wensing, A. M. (2016). Combined analysis of the prevalence of drug‐resistant hepatitis B virus in antiviral therapy‐experienced patients in Europe (CAPRE). Journal of Infectious Diseases, 213(1), 39–48. [DOI] [PubMed] [Google Scholar]
- Iikura, M. , Furihata, T. , Mizuguchi, M. , Nagai, M. , Ikeda, M. , Kato, N. , … Chiba, K. (2012). ENT1, a ribavirin transporter, plays a pivotal role in antiviral efficacy of ribavirin in a hepatitis C virus replication cell system. Antimicrobial Agents and Chemotherapy, 56(3), 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoherranen, N. , & Thummel, K. (2013). Drug metabolism and transport during pregnancy: How does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metabolism and Disposition, 41(2), 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. P. , Song, F. F. , Weng, Y. Y. , Yang, X. , Wang, K. , Lei, H. M. , … Jiang, H. D. (2016). Role of OCT2 and MATE1 in renal disposition and toxicity of nitidine chloride. British Journal of Pharmacology, 173(16), 2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, S. , Liao, H. , & Tsai, C. (2018). Telbivudine associated mitochondrial myopathy. Liver Internatinal, 38(6), 1139. [DOI] [PubMed] [Google Scholar]
- Liu, Y. P. , Wu, H. Y. , Yang, X. , Xu, H. Q. , Li, Y. C. , Shi, D. C. , … Fu, W. L. (2015). Association between thiopurine S‐methyltransferase polymorphisms and thiopurine‐induced adverse drug reactions in patients with inflammatory bowel disease: A meta‐analysis. PLoS ONE, 10(3), e0121745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Z. Y. , Yang, X. , Jiang, T. , Bai, M. R. , Zheng, C. H. , Zeng, S. , … Jiang, H. D. (2017). Multiple SLC and ABC transporters contribute to the placental transfer of entecavir. Drug Metabolism and Disposition, 45(3), 269–278. [DOI] [PubMed] [Google Scholar]
- Mas, V. R. , Maluf, D. G. , Archer, K. J. , Yanek, K. , Kong, X. , Kulik, L. , … Fisher, R. (2009). Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus‐induced hepatocellular carcinoma. Molecular Medicine, 15(3–4), 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milián, L. , Peris, J. , Gandía, P. , Andújar, I. , Pallardó, L. , Górriz, J. , … Blas‐García, A. (2017). Tenofovir‐induced toxicity in renal proximal tubular epithelial cells: Involvement of mitochondria. Aids, 31(12), 1679–1684. [DOI] [PubMed] [Google Scholar]
- Morgan, E. T. , Dempsey, J. L. , Mimche, S. M. , Lamb, T. J. , Kulkarni, S. , Cui, J. Y. , … Slitt, A. L. (2018). Physiological regulation of drug metabolism and transport: Pregnancy, microbiome, inflammation, infection, and fasting. Drug Metabolism and Disposition, 46(5), 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariente, G. , Leibson, T. , Carls, A. , Adams‐Webber, T. , Ito, S. , & Koren, G. (2016). Pregnancy‐associated changes in pharmacokinetics: A systematic review. Plos Medcine., 13(11), e1002160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. , Kwak, K. , Kim, S. , Jang, M. , Suk, K. , Kim, D. , … Park, C. K. (2017). Comparison of the long‐term efficacy between entecavir and tenofovir in treatment‐naïve chronic hepatitis B patients. BMC Gastroenterology, 17(1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler, S. , Jia, H. L. , Budhu, A. , Forgues, M. , Ye, Q. H. , Lee, J. S. , … Wang, X. W. (2010). A unique metastasis gene signature enables prediction of tumor relapse in early‐stage hepatocellular carcinoma patients. Cancer Research, 70(24), 10202–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, M. , Obaidat, A. , & Hagenbuch, B. (2012). OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. British Journal of Pharmacology, 165(5), 1260–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytting, E. , & Audus, K. L. (2008). Contributions of phosphorylation to regulation of OCTN2 uptake of carnitine are minimal in BeWo cells. Biochemical Pharmacology, 75(3), 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, H. , Lai, Y. R. , & Rodrigues, A. D. (2017). Organic anion transporter 2: An enigmatic human solute carrier. Drug Metabolism and Disposition, 45(2), 228–236. [DOI] [PubMed] [Google Scholar]
- Tang, L. , Covert, E. , Wilson, E. , & Kottilil, S. (2018). Chronic hepatitis B infection: A review. Jama, 319(17), 1802–1813. [DOI] [PubMed] [Google Scholar]
- Tasnif, Y. , Morado, J. , & Hebert, M. F. (2016). Pregnancy‐related pharmacokinetic changes. Clinical Pharmacology and Therapeutics, 100(1), 53–62. [DOI] [PubMed] [Google Scholar]
- Terrault, N. , Lok, A. , McMahon, B. , Chang, K. , Hwang, J. , Jonas, M. , … Wong, J. B. (2018). Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology, 67(4), 1560–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocchetti, G. N. , Arias, A. , Arana, M. R. , Rigalli, J. P. , Domínguez, C. J. , Zecchinati, F. , … Mottina, A. D. (2017). Acute regulation of multidrug resistance‐associated protein 2 localization and activity by cAMP and estradiol‐17β‐d‐glucuronide in rat intestine and Caco‐2 cells. Archives of Toxicology, 92(2), 777–788. [DOI] [PubMed] [Google Scholar]
- Tu, M. J. , Li, L. P. , Lei, H. M. , Ma, Z. Y. , Chen, Z. J. , Sun, S. Y. , … Jiang, H. D. (2014). Involvement of organic cation transporter 1 and CYP3A4 in retrorsine‐induced toxicity. Toxicology, 322, 34–42. [DOI] [PubMed] [Google Scholar]
- Wurmbach, E. , Chen, Y. B. , Khitrov, G. , Zhang, W. , Roayaie, S. , Schwartz, M. , … Llovet, J. M. (2007). Genome‐wide molecular profiles of HCV‐induced dysplasia and hepatocellular carcinoma. Hepatology, 45(4), 938–947. [DOI] [PubMed] [Google Scholar]
- Yang, X. , Ma, Z. Y. , Zhou, S. S. , Weng, Y. Y. , Lei, H. M. , Zeng, S. , … Jiang, H. D. (2016). Multiple drug transporters are involved in renal secretion of entecavir. Antimicrobial Agents and Chemotherapy, 60(10), 6260–6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Information of primary hepatocytes
Table S2. siRNA sequence
Table S3. Transition, fragmentor and collision energy of compounds
Table S4.Primer list for real‐time qPCR
Figure S1
The mRNA expression of ENT1 and ENT2 in human liver tissue (n = 5).
Figure S2
The cytotoxicity of NBTI or ETV in HepG2.2.15 cells for 6 or 3 days, respectively (n = 6).
Figure S3
The inhibition of NBTI (ENT inhibitor) at indicated concentration on the uptake of ETV in BeWo cells in KRH buffer or Na+ free buffer. n = 5. *P < 0.05 in comparison with ETV‐alone group.
Figure S4
The mRNA expression of ENT1 and OAT2 in HCC. A meta‐analysis of ENT1 (A) or OAT2 (B) gene expression from three Oncomine databases where colored squares indicate the median rank for OAT2 (vs. Normal tissue) across 5 analyses. Chen Liver (1), Mas Liver (2), Roessler Liver (3, 4), Wurmbach Liver (5). The P value is given for the median‐rank analysis.
Figure S5
The role of E2 and P4 in the antiviral activity of ETV. The cytotoxicity of E2 (A) and P4 (B) in HepG2.2.15 cells for 6 days. (C) The effect of 0.1 μM of E2 and 1 μM of P4 on the level of HBV DNA in HepG2.2.15 cell supernatant. HepG2.2.15 cells were treated with indicated concentrations of ETV with or without E2 and P4 for 6 days (D) Intracellular ETV concentration in HepG2.2.15 cells when were co‐treated with or without 0.1 μM of E2 and 1 μM of P4 for 6 days. n = 5.
Figure S6
Accumulations of 10 μM of cGMP in OAT2‐overexpressed cells in the absence or presence of Indo or NBTI. n = 5. *P < 0.05 in comparison with ETV‐alone group.