

Abstract
Objective:
Individuals with urea cycle disorders (UCDs) often present with intellectual and developmental disabilities. The major aim of this study was to evaluate the impact of diagnostic and therapeutic interventions on cognitive outcomes in UCDs.
Methods:
This prospective, observational, multicenter study includes data from 503 individuals with UCDs who had comprehensive neurocognitive testing with a cumulative follow-up of 702 patient-years.
Results:
The mean cognitive standard deviation score (cSDS) was lower in symptomatic than in asymptomatic (p<0.001, t-test) individuals with UCDs. Intellectual disability (Intellectual quotient < 70; cSDS < −2.0) was associated with the respective subtype of UCD and early disease onset, while height of the initial peak plasma ammonium concentration was inversely associated with neurocognitive outcomes in mitochondrial (i.e. proximal) rather than cytosolic (i.e. distal) UCDs. In ornithine transcarbamylase and argininosuccinate synthetase 1 deficiencies, we did not find evidence that monoscavenger therapy with sodium or glycerol phenylbutyrate was superior to sodium benzoate in providing cognitive protection. Early liver transplantation appears to be beneficial for UCDs. Noteworthy, individuals with argininosuccinate synthetase 1 and argininosuccinate lyase deficiencies identified by newborn screening had better neurocognitive outcomes than those diagnosed after the manifestation of first symptoms.
Interpretation:
Cognitive function is related to interventional and non-interventional variables. Early detection by newborn screening and early liver transplantation appear to offer greater cognitive protection, but none of the currently used nitrogen scavengers was superior with regard to long-term neurocognitive outcome. Further confirmation could determine these variables as important clinical indicators of neuroprotection for individuals with UCDs.
Keywords: Urea cycle disorders, newborn screening, cognitive function, nitrogen scavenger, liver transplantation
Introduction
Urea cycle disorders (UCDs) are a group of rare inherited metabolic diseases with an estimated cumulative prevalence of 1 in 35,000 to 52,000 newborns1. Individuals with UCDs are often affected by morbidity due to neurocognitive deficits and reduced life expectancy1–4. UCDs consist of five enzymopathies directly affecting the urea cycle, i.e. deficiencies of carbamoyl-phosphate synthetase 1 (CPS1-D; MIM #237300), ornithine transcarbamylase (OTC-D; MIM #311250), argininosuccinate synthetase 1 (ASS1-D; MIM #215700), argininosuccinate lyase (ASL-D; MIM #207900), arginase 1 (ARG1-D; MIM #207800), two transporter defects, i.e. deficiencies of the citrin or aspartate/glutamate carrier (MIM #603471 and #605814) and the mitochondrial ornithine transporter 1 causing hyperornithinemia-hyperammonemia-homocitrullinuria syndrome (HHH-S; MIM #238970), and two associated enzymes producing HCO3- (carbonic anhydrase VA; MIM #615751) and N-acetyl-L-glutamate (N-acetylglutamate synthase; NAGS-D; MIM #237310), which are required as substrate and cofactor of CPS1, respectively5, 6.
Individuals with early onset disease (EO) characterized by hyperammonemia prior to 28 days of age often present with life-threatening symptoms and have a high risk of mortality4, 7. In surviving individuals, poor neurological outcome is often observed. In individuals who present with symptoms after the newborn period, i.e. late onset disease (LO), the phenotype is variable and typically less severe than in individuals with EO3, 8, 9. Growing clinical awareness, development of diagnostic and therapeutic recommendations, and the implementation of national newborn screening (NBS) programs hold out the potential for better outcomes. However, there is still uncertainty about the benefit of NBS for individuals with ASS1-D and ASL-D, diseases that are rarely included in NBS disease panels in Europe but more commonly in the United States10. Importantly, effects of long-term scavenger treatment by either sodium/glycerol phenylbutyrate (PBA) or sodium benzoate (BZA) are unclear and the superiority of one or the other remains uncertain11, especially if clinical endpoints were used. Previous studies have highlighted that disease onset (EO vs. LO), initial peak plasma ammonium concentration (NH4+max), duration of hyperammonemic episodes, and hyperammonemic encephalopathy with coma are among the best predictors of mortality and severity of the neurological impairment2–4, 12, 13.
Large international registry studies from North America – the Urea Cycle Disorders Consortium (UCDC; https://www.rarediseasesnetwork.org/cms/ucdc) activated in February 2006 – and from Europe – the European registry and network for intoxication type metabolic disease (E-IMD; https://www.eimd-registry.org/) activated in January 2011 – have been systematically collecting comprehensive longitudinal clinical, neurological, biochemical, and therapy-related data14–16. While recent studies from both consortia have significantly improved our understanding of the natural history of UCDs, the impact of early diagnosis (by NBS) and long-term scavenger treatment on the overall neurocognitive outcome is still unclear13. Therefore, we compared and combined data from the UCDC and E-IMD registries to increase statistical power and investigated the largest cohort worldwide consisting of 1,095 individuals with UCDs17. This approach enabled us to unravel the effect of early diagnosis (by NBS) and compare various treatment modalities [conservative treatment vs. liver transplantation (LTx)] with regard to individuals’ neurocognitive outcomes.
Materials and Methods
Clinical studies, eligibility criteria, and overview of the E-IMD and UCDC data
The UCDC database is registered in the U.S. National Library of Medicine (https://clinicaltrials.gov), whereas the E-IMD registry is recorded on the German Clinical Trials Register (https://www.drks.de). Requirements set forth by the ICMJE (International Committee of Medical Journal Editors) are met. Written informed consent was obtained from all participants before enrolment into the observational studies. The protocols used for the E-IMD and UCDC databases have received prior approval by the appropriate Institutional Review Boards. All procedures were in accordance with the ethical standards of the Helsinki Declaration of 1975, as revised in 2013.
Data from individuals who died before the start date of the E-IMD registry (1st January 2011) were not recorded. Data were retrieved from the UCDC and E-IMD electronic databases with cut-off dates allowing for comparability with previous publications13, 17. Both registries use remote data entry via electronic case report forms comprising clinical and biochemical data regarding diagnosis and treatment from baseline, scheduled regular follow-ups, and unscheduled (emergency) visits to the hospital or emergency room. Definition of variables (e.g. symptomatic vs. asymptomatic, etc.) collected by both registries as well as a detailed description of the study population, upon which these data analyses have been performed, were published recently17.
Variables used for data analyses
The following independent variables were used for data analyses: type of UCD, mode of diagnosis [NBS, selective metabolic investigation after onset of symptoms (SX), positive family history that triggered diagnostic work-up, prenatal testing], clinical status at diagnosis (presymptomatic vs. symptomatic17), disease onset [EO, LO, asymptomatic (ASx)], initial peak plasma ammonium concentration (NH4+max), diagnostic delay (difference between age at diagnosis and age at first symptoms), therapy escalation level (EL), and peak plasma L-glutamine concentration at initial presentation. Individuals were assigned either to the early diagnosis group (i.e. identification by NBS, prenatal testing or postnatal testing of individuals with positive family history within the neonatal period) or to the late diagnosis group (i.e. SX or positive family history after the neonatal period). Individuals who underwent LTx were also classified by onset type and age at transplantation. We investigated the effects of the independent variables on the neurocognitive outcome [cognitive standard deviation score (cSDS)]. Mortality could not be examined due to a low number of deceased individuals within the observation period17.
Neurocognitive tests
The UCDC and E-IMD both use extensive psychological test batteries to evaluate various cognitive domains, which has been described in detail18, 19. For each Hamburg-Wechsler-Intelligenztest für Kinder (HAWIK), Wechsler Adult Intelligence Scale (WAIS), Wechsler Abbreviated Scale of Intelligence (WASI), Wechsler Intelligence Scale for Children (WISC), and Wechsler Preschool and Primary Scale of Intelligence (WPPSI) full scale IQ was selected, for Bayley Scales of Infant Development (Bayley) mental developmental index and cognitive scale were used19. We used test results from the last regular study visit and intraindividual results from repeated psychological testing, where available. To achieve comparability, z-scores were calculated using the normative data (mean and standard deviation) from the standardization sample of each psychological test and labelled it as cSDS.
Among the 1,095 individuals with UCDs from 18 countries who were enrolled in the UCDC and E-IMD registries between 2006 and 2016, those who were cognitively or physically unable to be tested, and individuals who refused or were too young to be tested were excluded from the analysis. Individuals who withdrew from the study were likewise excluded from the data analysis. 503 individuals were tested with the HAWIK (Symptomatic/ASx: n=21/3), WAIS (Symptomatic/ASx: n=17/10), WASI (n=187/99), WISC (Symptomatic/ASx: n=31/2), WPPSI (Symptomatic/ASx: n=41/14), and Bayley (Symptomatic/ASx: n=60/18). Since the number of individuals for each specific subsequent analysis varies, the respective numbers of UCD individuals and applied statistical test methods will be reported in brief in the corresponding results sections, descriptive characteristics will be provided in detail in Suppl. Table 1.
Data availability
The datasets generated and analyzed during the current study are not publicly available due to existing data protection laws. Furthermore, data ownership is retained by the members of the UCDC and E-IMD consortia making data available for specific research purposes. Data availability is subject to the consent of both consortia upon request.
Statistical analysis
All statistical analyses were performed using R20, a language for statistical computing and graphics (https://www.r-project.org). Multiple regression analysis was used to model a numeric dependent outcome variable with respect to several independent predictor variables, and the results were displayed in analysis of variance (ANOVA) tables with type two F-tests21. Post-hoc comparisons were carried out with estimated marginal means22. Two groups were compared with the Welch’s two sample t-test. To detect interactions between variables, model-based recursive partitioning was applied23, 24. Ordinal response variables were analyzed by cumulative link models25. Count data were analyzed by log-linear models and visualized by mosaic plots with Pearson residual-based shadings26.
Results
Study population
Median age at neuropsychological testing was 5.3 years in EO, 15.1 years in LO, and 30.5 years in ASx individuals. In symptomatic individuals, 41.5% (n=148) were male and 58.5% (n=209) female, whereas in ASx individuals the male-to-female ratio was skewed in favor of females [ASx male: 18.5%, n=27; ASx female: 81.5%, n=119] due to high prevalence of ASx individuals with female OTC-D (fOTC-D). Neurocognitive testing showed a significantly lower cSDS in symptomatic (n=357) compared to ASx UCD individuals (n=146; p<0.001, t-test). Among symptomatic UCDs, EO individuals (n= 132) performed worse than LO individuals (n=225; p<0.001, t-test).
Impact of non-interventional variables on neurocognitive function
To investigate the influence of underlying UCD on cognitive function of symptomatic individuals, we used model-based recursive partitioning23, 24. Compared to the cytosolic (synonym: distal) UCDs, i.e. ASS1-D, ASL-D, and ARG1-D (hereafter referred to as group 1), height of the initial NH4+max was associated with impaired neurocognitive outcome in individuals with mitochondrial (synonym: proximal) UCDs (i.e. NAGS-D, CPS1-D, mOTC-D, fOTC-D), HHH-S, and citrin deficiency (hereafter referred to as group 2). The initial NH4+max inversely correlated with the cSDS of symptomatic individuals from group 2 (β=−0.0012, p=0.006, R2=0.047; n=138) but not from group 1 (β=−0.0003, p=0.19, R2=0.011; n=71; Fig. 1), indicating a decrease of 0.12 cSDS per 100 μmol/l of increased initial NH4+max in group 2, notwithstanding that individuals from group 1 had significantly higher initial NH4+max than individuals of group 2 (p<0.001, t-test). To investigate whether this apparent effect was largely due to symptomatic individuals with fOTC-D, we repeated the analysis by excluding symptomatic fOTC-D and found a robust, analogous association between height of initial NH4+max and neurocognitive outcome (group 2 without fOTC-D: β=−0.0011, p=0.051, R2=0.054; n=54).
Figure 1: Model-based recursive partitioning for 209 symptomatic UCD individuals.

Cognitive SDS from 138 symptomatic individuals in group 2 (NAGS-D: n=2, CPS1-D: n=2, mOTC-D: n=45, fOTC-D: n=84, HHH-S: n=4, Citrin deficiency: n=1) was associated inversely with the height of initial NH4+max (β=−0.0012, p=0.006). The association was unchanged if individuals with fOTC-D were excluded from the analysis. UCD individuals belonging to group 1 have lower cSDS than individuals from group 2 (p<0.001, t-test). Gray lines are fitted cSDS from the linear model; the gray shaded area corresponds to 95% confidence interval (CI).
To this end, we included fOTC-D in all subsequent subgroup analyses and divided symptomatic individuals into three groups with normal cSDS (IQ ≥ 85; cSDS ≥ −1.0), low normal to borderline cSDS (70 ≤ IQ < 85; −2.0 ≤ cSDS < −1.0), and intellectual disabilities (IQ < 70; cSDS < −2.0)27. High initial NH4+max and affiliation with group 1 were associated with intellectual disability (each p≤0.001, cumulative link model). Interestingly, however, interaction between initial NH4+max and group affiliation was significant (p=0.018, cumulative link model), suggesting a higher probability of cognitive impairment in individuals from group 2 than from group 1, when initial NH4+max was high (Fig. 2A–F).
Figure 2: Height of initial NH4+max is associated with group affiliation in 240 symptomatic UCD individuals.

The left column (Fig 2A, 2B, and 2C) includes individuals from group 1; the right column (Fig 2D, 2E, and 2F) includes individuals from group 2. The first line (Fig 2A, and 2D) includes individuals with normal cSDS (IQ ≥ 85; cSDS ≥ −1.0), the second line (Fig 2B, and 2E) individuals with low normal to borderline cSDS (70 ≤ IQ < 85; −2.0 ≤ cSDS < −1.0), and the third line (Fig 2C, and 2F) includes individuals with intellectual disability (IQ < 70; cSDS < −2.0). Individuals from group 1 (ASS1-, ASL-, and ARG1-D) suffer more likely from intellectual disability (p<0.001, cumulative link model). Since interaction between initial NH4+max and group affiliation is significant (p=0.018, cumulative link model) with regard to cognitive outcome, height of initial NH4+max is associated with impaired neurocognition in group 2 (NAGS-D, CPS1-D, mOTC-D, fOTC-D, HHH-S, and citrin-deficiency) rather than group 1, despite higher initial NH4+max in group 1 (p<0.001, t-test). Gray lines are fitted probabilities to be assigned to a specific group (normal cSDS, low normal to borderline cSDS or intellectual disability) predicted by the cumulative link model; the gray shaded area corresponds to 95% CI.
Early diagnosis by NBS suggests an improved neurocognitive outcome
Individuals with ASS1-D (n=15) and ASL-D (n=23) identified by NBS had normal mean cSDS that are higher than in individuals diagnosed after the manifestation of first symptoms (each p<0.001, t-test; Fig. 3). The beneficial effect of NBS was further observed in subgroup analyses for each disease (Table 1). Since individuals identified by NBS were younger than those identified by SX, we next modelled the effect of the diagnostic mode on cognitive function exclusively in preschool children (younger than seven years of age) to avoid overestimation of a potential age effect. Importantly, age-matched preschool ASS1-D (n=11) and ASL-D (n=17) individuals performed better if identified by NBS (each p<0.05, ANOVA post-hoc comparison; Fig.4, Table 1).
Figure 3: Boxplots reflecting the beneficial cognitive effect of NBS for individuals with ASS1-D (n=49) and ASL-D (n=56).
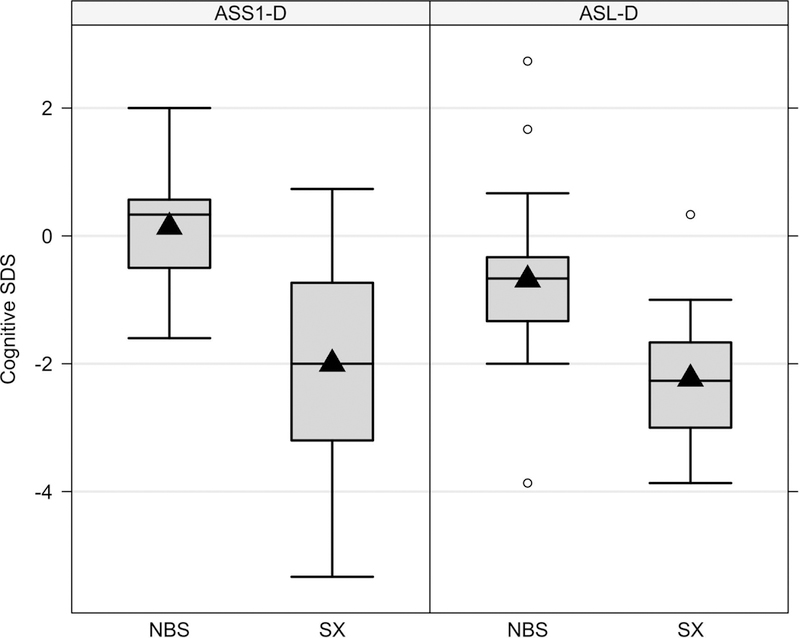
Descriptive characteristics are presented separately in Table 1. Data are shown as median (black line) and mean (triangle), length of the box corresponds to interquartile range (IQR), upper and lower whiskers correspond to max. 1.5 x IQR, each point represents an outlier. NBS, newborn screening; SX, selective metabolic investigation after the onset of symptoms.
Table 1.
Cognitive SDS and age at testing of individuals with ASS1-D or ASL-D
| cSDS – NBS Mean, SD Median [Q1, Q3] Min, Max; n |
cSDS – SX Mean, SD Median [Q1, Q3] Min, Max; n |
Age at testing NBS (years) Mean, SD Median [Q1, Q3] Min, Max; n |
Age at testing SX (years) Mean, SD Median [Q1, Q3] Min, Max; n |
|
|---|---|---|---|---|
| ASS1-D | ||||
| All | 0.14, 0.85 0.33 [−0.50, 0.57] −1.60, 2.00; 15 |
−2.00, 1.48 −2.00 [−3.17, −0.77] −5.33, 0.73; 34 |
4.90, 3.87 4.04 [1.61, 7.52] 0.54, 13.23; 15 |
12.73, 8.25 10.41 [6.01, 18.82] 0.57, 29.30; 34 |
| EO | −0.20, 0.83 −0.33 [−0.53, 0.30] −1.60, 1.00; 7 |
−2.04, 1.56 −2.20 [−3.17, −0.77] −5.33, 0.73; 26 |
4.79, 5.07 3.20 [0.98, 7.29] 0.54, 13.23; 7 |
12.86, 8.50 10.41 [6.81, 18.82] 0.57, 29.30; 26 |
| LO | 0.47, n/a 0.47 [0.47, 0.47] 0.47, 0.47; 1 |
−1.89, 1.25 −1.93 [−3.00, −0.88] −3.47, −0.13; 8 |
8.41, n/a 8.41 [8.41, 8.41] 8.41, 8.41; 1 |
12.31, 7.91 12.56 [5.42, 18.67] 2.87, 22.05; 8 |
| ASx | 0.43, 0.86 0.33 [−0.10, 0.70] −0.53, 2.00; 7 |
n/a |
4.50, 2.65 4.23 [2.73, 5.61] 1.61, 9.00; 7 |
n/a |
|
Preschool (≤7 years) |
0.15, 0.97 0.33 [−0.50, 0.70] −1.60, 2.00; 11 |
−1.97, 1.70 −1.93 [−2.75, −0.65] −5.33, 0.20; 10 |
2.93, 1.92 3.20 [1.32, 4.13] 0.54, 6.64; 11 |
3.51, 1.94 3.55 [2.38, 4.69] 0.57, 6.50; 10 |
| ASL-D | ||||
| All | −0.69, 1.30 −0.67 [−1.33, −0.33] −3.87, 2.73; 23 |
−2.24, 0.89 −2.27 [−3.00, −1.67] −3.87, 0.33; 33 |
3.96, 3.14 2.98 [1.20, 6.63] 0.73, 11.07; 23 |
14.64, 11.55 13.81 [4.31, 21.30] 0.75, 43.35; 33 |
| EO | −0.98, 1.65 −0.67 [−1.40, −0.60] −3.87, 1.67; 7 |
−2.24, 1.06 −2.33 [−3.20, −1.67] −3.87, 0.33; 17 |
5.56, 3.16 4.95 [4.07, 6.70] 1.34, 11.07; 7 |
12.46, 12.39 4.34 [2.54, 22.97] 0.75, 38.69; 17 |
| LO | −1.55, 0.33 −1.50 [−1.65, −1.40] −2.00, −1.20; 4 |
−2.24, 0.70 −2.17 [−2.85, −1.83] −3.47, −1.00; 16 |
5.41, 3.16 6.00 [3.43, 7.98] 1.59, 8.05; 4 |
16.96, 10.47 14.74 [10.11, 19.31] 1.96, 43.35; 16 |
| ASx | −0.23, 1.15 −0.40 [−0.83, −0.08] −1.33, 2.73; 12 |
n/a |
2.54, 2.64 1.40 [0.87, 2.80] 0.73, 8.12; 12 |
n/a |
|
Preschool (≤7 years) |
−0.60, 0.93 −0.67 [−1.33, −0.33] −2.00, 1.67; 17 |
−1.65, 0.93 −1.67 [−2.25, −1.33] −3.00, 0.33; 10 |
2.36, 1.63 1.73 [0.94, 3.22] 0.73, 5.47; 17 |
2.45, 1.32 2.39 [1.37, 3.32] 0.75, 4.34; 10 |
Cognitive SDS and age at testing in ASS1-D and ASL-D individuals identified by NBS and SX. Overall, individuals with ASS1-D or ASL-D perform better if identified by NBS rather than by SX (each p<0.001, t-test). ASx ASS1-D and ASL-D individuals identified by NBS performed normal in cognitive testing and achieve better than ASS1-D and ASL-D individuals identified by SX (each p<0.05, ANOVA). EO ASS1-D individuals performed better (p=0.021, ANOVA) and EO ASL-D individuals tended to perform better (p=0.075, ANOVA) than their counterparts identified by SX. Individuals identified by NBS were younger than their SX counterparts. Age-matched preschool ASS1-D and ASL-D subsamples achieved better in cognitive testing if identified by NBS (each p<0.05, ANOVA post-hoc comparison). ASx, asymptomatic; EO, early onset; LO, late onset; NBS, newborn screening; n/a, not available; SX, selective metabolic investigation after the onset of symptoms.
Figure 4: Modelling the effect of early diagnosis by NBS for age-matched preschool children with ASS1-D (n=21) and ASL-D (n=27).

Descriptive characteristics are presented separately in Table 1. NBS improves cognitive outcome for ASS1-D (p<0.001, ANOVA post-hoc comparison) and ASL-D individuals (p=0.021, ANOVA post-hoc comparison). Gray lines are fitted cSDS values from the linear model; the gray shaded area corresponds to 95% CI. NBS, newborn screening. SX, selective metabolic investigation after the onset of symptoms.
Use of phenylbutyrate is not associated with better global cognitive outcome as compared to benzoate
To evaluate treatment with PBA and/or BZA on cognitive outcome, we studied OTC-D and ASS1-D, the two most frequently reported UCDs. Since therapeutic intensity is thought to reflect the individual disease severity (Fig. 5), we specifically investigated onset-, and age-matched individuals who received exclusively either PBA or BZA during the entire observation period.
Figure 5: Mosaic plot presenting the association of disease severity with therapy intensity from 359 UCD individuals at last psychological testing.

Individuals with information on the variables disease onset, cSDS tests, initial NH4+max, and specific therapy are presented. Individuals belonging to escalation level (EL) 0 (n=50) receive no therapy, whereas individuals on EL 1 (n=96) receive a natural protein intake below 100% WHO safe values (NPI) or L-arginine/-citrulline/-ornithine ± carglumic acid for NAGS-D. EL 2 (n=29) corresponds to NPI and L-arginine/-citrulline/-ornithine ± carglumic acid for NAGS-D. EL 3 (n=147) applies to individuals receiving one scavenger (BZA or PBA) ± NPI ± L-arginine/-citrulline/-ornithine ± carglumic acid for NAGS-D, and EL 4 (n=37) includes individuals receiving two scavengers (BZA and PBA) ± NPI ± L-arginine/-citrulline/-ornithine ± carglumic acid for NAGS-D. The height of the boxes represent the proportion of EL, the widths of the boxes in a row represent the proportions of individuals with a specific disease onset (EO, LO, ASx) within that EL. The intensities of color reflect the discrepancies between observed and expected frequencies. Blue color shows that observed frequencies are higher than expected, red color shows that observed frequencies are lower than expected. Pearson residuals indicate that ASx individuals are overrepresented in EL 0 and 1 and underrepresented in EL 2 to 4. Symptomatic UCD individuals (EO, LO) are similarly distributed within EL 2 to 4. However, (severe) metabolic decompensations increase continuously between EL 2 to 4, as mirrored by the median initial NH4+max (EL 2: median initial NH4+max 198.0 μmol/l, n=14/29; EL 3: median initial NH4+max 270.0 μmol/l, n=111/147; EL 4: median initial NH4+max 335.5 μmol/l, n=26/37).
We investigated 77 individuals with OTC-D and 21 with EO ASS1-D receiving long-term monoscavenger therapy. Individuals with EO OTC-D performed worse than their LO counterparts (p=0.032, main effect ANOVA), however independent from the disease onset, use of neither PBA nor BZA was superior with regard to the cognitive outcome (p=0.94, main effect ANOVA). Interaction between type of monoscavenger therapy and disease onset was neither significant for age at testing (p=0.60, ANOVA) nor for cSDS (p=0.41, ANOVA), demonstrating that for age-and onset-matched OTC-D, long-term monoscavenger therapy with neither PBA nor BZA is superior to one another regarding neurocognitive outcome. Also, age-matched individuals with EO ASS1-D performed equally poor after long-term monoscavenger therapy with either PBA or BZA (p=0.531, t-test; Fig. 6).
Figure 6: Boxplots reflecting the effects of long-term monoscavenger therapy on cognition of individuals with OTC-D (n=77) and EO ASS1-D (n=21).

77 OTC-D individuals (EO-PBA: n=10; EO-BZA: n=3; LO-PBA: n=52; LO-BZA: n=12) and 21 EO ASS1-D individuals receiving all monoscavenger therapy (EO-PBA: n=17; EO-BZA: n=4) were analyzed with regard to their cognitive outcome. Monoscavenger treatment with PBA is not superior to BZA neither for age-matched EO or LO OTC-D (p=0.41, ANOVA) nor for age-matched EO ASS1-D individuals (p=0.531, t-test). For OTC-D and EO ASS1-D individuals below the weight of 20 kg with sufficient scavenger dose information, both median sodium-phenylbutyrate and median sodium benzoate doses were within the recommended ranges5. Data are shown as median (black line) and mean (triangle), length of the box corresponds to interquartile range (IQR), upper and lower whiskers correspond to max. 1.5 x IQR, each point represents an outlier. PBA, sodium/glycerol phenylbutyrate; BZA, sodium benzoate.
Comparison of the cognitive function of OTC-D individuals treated with long-term mono-or combined scavenger therapy also revealed lower cSDS in EO than in LO individuals (p=0.05, ANOVA). Performance of age-matched individuals with EO and LO OTC-D was independent from the utilization of one or two scavengers (p=0.38, ANOVA), suggesting that use of intensified, i.e. combined scavenger therapy is important for the individuals’ metabolic control but is not associated with higher cSDS scores. This is substantiated by age-matched EO ASS1-D individuals who were cognitively more affected when combined long-term scavenger therapy was utilized (p=0.023, t-test). Use of a second scavenger appears not to improve neurocognition but rather reflects individual disease severity and metabolic instability of those individuals.
Timely liver transplantation tends to be associated with higher cSDS
To investigate which variables are associated with the receipt of a liver graft, we investigated 51 liver-transplanted individuals (CPS1-D: n=6; mOTC-D: n=18; fOTC-D: n=5; ASS1-D: n=12; ASL-D: n=10) and 303 untransplanted individuals with conservative treatment. Individuals with former CPS1-D and mOTC-D received more frequently, and fOTC-D received less frequently than expected a liver transplant (p<0.001, log-linear model). Non-interventional variables, such as early disease onset (p<0.001, log-linear model), high initial NH4+max (p<0.001, t-test) as well as high peak plasma L-glutamine concentration at initial presentation (p<0.001, t-test) were more likely associated with the fact that UCD individuals underwent liver transplantation.
Subsequently, we assessed whether the time point of liver transplantation might affect the neurocognitive outcome in UCD individuals. To this end, we investigated 45 liver-transplanted individuals who received cSDS testing exclusively after transplantation (CPS1-D: n=6; mOTC-D: n=18; fOTC-D: n=5; ASS1-D: n=8; ASL-D: n=8). Particularly LO individuals (p=0.026, ANOVA) and those who received a timely transplantation independent from the disease onset (p=0.087, ANOVA) tended to perform better. However, interaction between disease onset and time of transplantation was not significant (p=0.76, ANOVA), suggesting that delayed liver transplantation appears to be associated with cognitive impairment, regardless of the disease onset (Fig. 7).
Figure 7: Cognitive SDS of psychological testing after liver transplantation by age at transplantation (months) and disease onset (EO vs. LO).
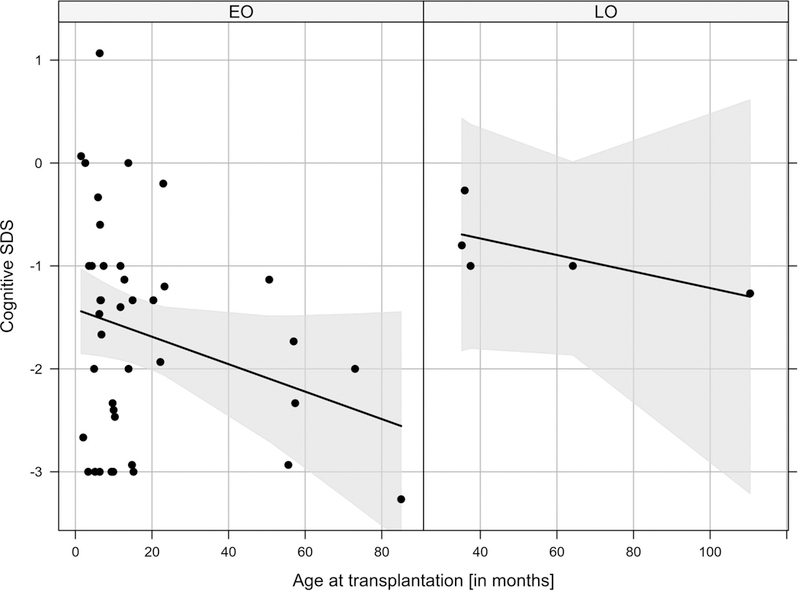
Individuals with EO (n=40) perform worse than those with LO (n=5; p=0.026, ANOVA). If indication for LTx is made, timely liver transplantation tends to be associated with improved neurocognitive outcome (p=0.087, ANOVA). The interaction between disease onset and age at transplantation is not significant (p=0.76, ANOVA). Gray lines are fitted cSDS values from the linear model; the gray shaded area corresponds to 95% CI.
Discussion
The major aim of this prospective, observational, multicenter study of UCDs was to investigate predictors of cognitive function associated with interventional [e.g. (early) diagnosis and therapy] and non-interventional (e.g. initial NH4+max and disease onset) variables. The study revealed four major results: (1) Association between height of initial NH4+max and cognitive impairment is disease-dependent, being more pronounced in individuals with mitochondrial (proximal) UCDs, HHH-S, and citrin deficiency than in those with cytosolic (distal) UCDs. (2) Individuals with ASS1-D and ASL-D had better cognitive outcome when early treatment followed identification by NBS. (3) Long-term monoscavenger therapy with PBA was not associated with higher cSDS as compared to treatment with BZA in individuals with OTC-D and EO ASS1-D. Although combined scavenger therapy is thought to be important for achieving better metabolic control, use of a second scavenger appears not to improve neurocognitive outcome. (4) UCD individuals that underwent LTx appear to benefit from timely intervention.
Vulnerability to initial NH4+max is disease-dependent
While others have described specific deficits of the neurocognitive profile in UCDs, a recent study demonstrated that cognitive function in individuals with OTC-D is associated with global impairment and IQ serves as a valid marker for describing global (neuro)cognitive outcome19, 28, which was studied in this work. Symptomatic individuals with cytosolic UCDs perform below the age norm and worse than those with mitochondrial UCDs, HHH-S, and citrin deficiency. Except for selected UCDs such as ASL-D, showing progressive neurological and hepatic disease18, this is unexpected and not consistent with previous studies29. Since it is known that UCD individuals with neonatal onset disease manifestation have a high risk of mortality4, often those with mitochondrial UCDs such as OTC-D are less likely to be captured by registry studies2, 8, 9. However, ascertainment bias does not seem to fully explain the observed discrepancy in outcomes, and alternative pathomechanisms are also to be considered. This is supported by the finding that height of initial NH4+max was inversely associated with cognitive outcome in mitochondrial UCDs, HHH-S, and citrin deficiency rather than cytosolic UCDs, despite higher initial NH4+max in the latter. The observed discrepancy highlights the need for a careful reevaluation of variables that are commonly used to describe disease severity and to predict survival and neurocognitive outcome1, 3, 4, 12. This is substantiated by a study, demonstrating that cumulative exposures to disease-specific neurotoxic biomarkers – i.e. plasma ammonium, L-glutamine, L-citrulline, and L-arginine – appear to be sensitive predictors of global neuropsychological outcome in ASS1-D, ASL-D, and ARG1-D18.
Recently, ASS1, the deficient enzyme in individuals with ASS1-D, has been identified as an RNA-binding protein and as such could potentially interfere with RNA metabolism30. Individuals suffering from ASL-D often develop impaired motor and cognitive functions and progressive hepatic disease despite metabolic control5, 8, 9, 28, 31. Interestingly, an enzymatic and structural function of ASL, the deficient protein in ASL-D, was proposed32. Moreover, high concentrations of argininosuccinic acid and guanidinosuccinic acid in the brain are anticipated to exert a neurotoxic effect33, 34. In the future, a better understanding of genotype-phenotype correlations for ASS1-D, ASL-D, and ARG1-D are required to provide more insight into phenotypic variability of these putatively neurodegenerative diseases.
Newborn screening – an intervention with favorable impact on neurocognition
NBS for UCDs is carried out heterogeneously worldwide. Due to deficits in national tracking systems, the impact of NBS on neurocognitive outcome is insufficiently reported and only few preliminary analyses demonstrate a putatively beneficial effect13, 35–37. Thus, current data analysis offers the unique possibility to study whether NBS for ASS1-D and ASL-D might positively influence cognitive function. Interestingly, ASS1-D and ASL-D individuals perform better in psychological testing if identified early by NBS. Modelling the effects of early diagnosis via NBS on age-matched preschool children suggested superiority of NBS to SX for both diseases. It is difficult to separate the effect of NBS from differences in the underlying severity of UCDs identified by NBS or SX, and thus, repetition of psychological testing, identification of possible benign mutations and a longer follow-up period should be performed in the future to elucidate whether NBS might unequivocally exert a positive life-long effect on cognition.
PBA is not superior to BZA in preventing global neurocognitive impairment
The impact of pharmacotherapy on lowering plasma NH4+ concentrations has been shown earlier12, 38; however, our understanding of optimal long-term scavenger treatment is hampered by the strong effect of initial NH4+max and by the heterogeneous implementation of treatment recommendations13. Apart from studies that shed light on the efficacy and safety of scavengers39, 40, preliminary data suggesting that PBA might positively affect short-term neurocognitive outcome in UCDs is scarce41. Theoretically, on a mole-per-mole basis, nitrogen-disposing efficacy of PBA should be twice that of BZA, and biochemical superiority of PBA has been demonstrated11.
However, no systematic studies regarding the effects of long-term pharmacotherapy (with either PBA, BZA, or a combination of both), as defined by clinical endpoints, are available so far. Since therapeutic intensity reflects the phenotypic severity of UCD individuals, as demonstrated by the EL pattern, comparison of UCD individuals receiving different treatment intensities is invalid. To this end, we compared age-and onset-matched OTC-D and EO ASS1-D individuals receiving long-term monoscavenger treatment (PBA or BZA) and found no superiority of PBA to BZA with regard to cognitive outcome in any group. Importantly, severities of initial and long-term recurrent brain-damaging (hyperammonemic) events appear to overshadow putatively positive effects of long-term monoscavenger therapy on cognition. Unfortunately, the choice of combined scavenger therapy does not appear to improve neurocognitive abilities in severely affected individuals with OTC-D and EO ASS1-D. These disenchanting pharmacotherapeutic data along with the poor cognitive outcome in symptomatic UCD individuals suggest an urgent need for new therapies to mitigate the cascade of serious neurotoxic long-term sequelae. Intriguingly, a potential new therapeutic approach was proposed by specific and irreversible inhibition of ornithine aminotransferase in zebrafish correcting hyperammonemia-induced biochemical hallmarks and preventing neurotoxicity and mortality in that species42. Mammalian transfer of this principle might be a promising new therapeutic option.
Timely liver transplantation appears to protect neurocognitive function
Liver transplantation is influenced by age at onset and underlying disease severity, and is considered when conservative treatment has failed to prevent individuals from recurrent, life-threatening metabolic decompensations. Current recommendations propose introducing this treatment early in life5. A retrospective study reported that individuals with an initial NH4+max above 300 μmol/L might already benefit from LTx43, 44. We demonstrate that individuals who are suffering from a severe phenotype – i.e. individuals with high initial NH4+max and high initial L-glutamine concentrations as well as early disease onset – are more likely to receive a liver graft. Timely liver transplantation appears to be neuroprotective, since with each year of delayed LTx a cognitive deterioration was evident in this cross-sectional approach. Cognitive dysfunction might even be more pronounced if timely transplantation is not feasible, since individuals included in this study must present in a sufficient physical and cognitive condition to undergo psychological testing at all. Due to an indication/ascertainment bias and the cross-sectional nature, this study faces typical limitations of observational data analysis. More intraindividual and interindividual long-term data as well as pre-and post-transplant investigations are required to evaluate the optimal time point of transplantation, and to study whether the cognitive outcome can indeed be positively influenced by early LTx in UCD individuals.
Limitations
Despite a cumulative follow-up of 702 patient-years, it is not possible to draw unequivocal conclusions on the evolving natural cognitive phenotype of UCD individuals, as reflected by repeated intraindividual psychological testing. This would require multiple follow-up psychological testings with adequate group sizes, stratification to specific observed treatment escalation levels without changes in management, comparable case mixes, and particularly a sufficient observation period.
Correlations between the frequency, severity, area under the curve of repeated hyperammonemic episodes, or duration of hospital stay and long-term cSDS values are beyond the scope of this project but will be the subject to future studies. Furthermore, the impact of other non-interventional variables (ethnic origin, socio-economic factors, sex, difference in disease severity) on cognition requires clarification, since cognitive differences for symptomatic and asymptomatic UCD individuals reported in this work might reflect, at least in part, sex and case mix differences. Future investigations will also need to focus on the impact of genotypic variability and residual enzyme function on clinical outcome in order to understand predictors and modifiers of long-term outcome in individuals with UCDs more profoundly.
Conclusion
We demonstrate prototypically that combining cross-sectional data from the UCDC and E-IMD databases provides important and indispensable information for the identification of important associations that need to be confirmed in a longitudinal manner for providing evidence-based recommendations that are translated into patient care. We propose that any future clinical trial for UCDs would benefit from evaluation against clinical outcomes instead of biochemical surrogate parameters and must be carefully matched against variables that have a known impact on the clinical outcome.
Supplementary Material
Acknowledegements
All UCDC and E-IMD sites contributed to the dataset of the longitudinal study. Principal investigators and personnel with key contributions are listed as UCDC and E-IMD consortia study group members. Furthermore, we gratefully acknowledge subsequent study coordinators – Sondra Bloxam, Linnea Brody, Liora Caspi, Sara Elsbecker, Luca Fierro, Vimal Gunasekaran, Joan Hart, Audrey Lynn, Mary Mullins, Ulrike Mütze, Cassandra Papaleo, Kendall Parks, Irma Payan, Thu Quan, Kara Simpson, Rebecca Singer, and Kimberly Wallis – and study neuropsychologists – Fabienne Dietrich Alber, Talin Babikian, Heidi Bender, Christopher Boys, David Breiger, Corinna Buerger, Magdalena Kowoll, Susan E. Caudle, Mina Nguyen-Driver, Benjamin Goodlett, Elizabeth Kerr, Casey Krueger, Eva Mamak, Jacqueline H. Sanz, and Rachel Tangen. We would also like to acknowledge Rima Izem for the helpful biostatistical work, as well as the contributions of (former) longitudinal study PIs: Stephen Cederbaum, Annette Feigenbaum, Douglas S. Kerr, Uta Lichter-Konecki, Margretta R. Seashore, George A. Diaz, Nicholas Ah Mew, Robert McCarter, Mendel Tuchman, and James Weisfeld-Adams. In particular, we are indebted to all our UCD individuals and their families for their trust, patience, and participation in both longitudinal registry studies for many years.
The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The Urea Cycle Disorders Consortium is also supported by the O’Malley Foundation, the Rotenberg Family Fund, the Dietmar-Hopp Foundation, the Kettering Fund, and the National Urea Cycle Disorders Foundation. In addition, support for neuropsychological testing is provided by an NIH grant for Intellectual and Developmental Disability Research Centers (U54HD090257). The E-IMD patient registry has received funding by the European Union (E-IMD; EAHC no 2010 12 01; coordinator: Stefan Kölker) in the framework of the Health Programme. After the end of the EU funding period, the E-IMD patient registry has been sustained by funding from the Kindness-for-Kids Foundation (Munich, Germany), the Kettering Fund, and Dietmar Hopp Foundation. SK, PB, and GFH (Heidelberg, Germany) receive funding from the Dietmar Hopp Foundation (St. Leon-Rot, Germany) for coordinating the study “Newborn Screening and Metabolic Medicine 2020 (NBS2020)” including individuals with urea cycle disorders. MZ (Heidelberg, Germany) was supported by the Physician-Scientist Program at University of Heidelberg and by a Career Development Fellowship provided by the Heidelberg Research Center for Molecular Medicine (HRCMM) in the framework of Excellence Initiative II of the German Research Foundation. MRB and JH (Zurich, Switzerland) are supported by radiz – Rare Disease Initiative Zurich, a clinical research priority programme of the University of Zurich.
SK receives funding from Horizon Pharma Ireland Limited for the European Post-Authorization Registry for Ravicti® (glycerol phenylbutyrate) oral liquid in partnership with the E-IMD (RRPE) (EU PAS Register no. EUPAS17267; http://www.encepp.eu/). AS, SEMC, JLM 2nd, RCG, and GE receive personal or consultant fees from Horizon Pharma Ireland Limited. JKB received honorarium for an Advisory Board Meeting in 07/2018 on “using Ravicti® for newborns under 2 months of age” from Horizon Pharma Ireland Limited. COH and SAB received payment for the support of clinical trial(s) from Horizon Pharma Ireland Limited, and SAB is a member of the clinical advisory board of Horizon Pharma Ireland Limited. The company manufactures a drug that was studied amongst other scavenger drugs in this analysis regarding the impact of long-term scavenger therapy on cognitive outcome in individuals with urea cycle disorders.
Abbreviations
- ARG1-D
Arginase 1 deficiency
- ASx
Asymptomatic
- ASL-D
Argininosuccinate lyase deficiency
- ASS1-D
Argininosuccinate synthetase 1 deficiency
- Bayley
Bayley Scales of Infant Development
- BZA
Sodium benzoate
- CPS1-D
Carbamoyl-phosphate synthetase 1 deficiency
- cSDS
Cognitive standard deviation score
- E-IMD
European registry and network for intoxication type metabolic diseases (www.eimd-registry.org/)
- EO
Early onset (≤28 days)
- EL
Escalation level
- fOTC-D
Female ornithine transcarbamylase deficiency
- HAWIK
Hamburg-Wechsler-Intelligenztest für Kinder
- HHH-S
Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome
- IQ
Intellectual quotient
- LO
Late onset (>28 days)
- LTx
Liver transplantation
- mOTC-D
Male ornithine transcarbamylase deficiency
- NAGS-D
N-acetylglutamate synthase deficiency
- NBS
Newborn screening
- NH4+max
Peak plasma ammonium concentration
- NPI
Natural protein intake below 100% WHO safe values
- PBA
(Sodium or glycerol) phenylbutyrate
- SX
Selective metabolic investigation after onset of symptoms
- UCDC
Urea Cycle Disorders Consortium (www.rarediseasesnetwork.org/cms/ucdc)
- UCD(s)
Urea cycle disorder(s)
- WAIS
Wechsler Adult Intelligence Scale
- WASI
Wechsler Abbreviated Scale of Intelligence
- WISC
Wechsler Intelligence Scale for Children
- WPPSI
Wechsler Preschool and Primary Scale of Intelligence
Footnotes
Potential Conflicts of Interest
All other authors declare that they have nothing to report.
References
- 1.Nettesheim S, Kolker S, Karall D, et al. Incidence, disease onset and short-term outcome in urea cycle disorders -cross-border surveillance in Germany, Austria and Switzerland. Orphanet J Rare Dis. 2017. June 15;12(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008. October;97(10):1420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kido J, Nakamura K, Mitsubuchi H, et al. Long-term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis. 2012. September;35(5):777–85. [DOI] [PubMed] [Google Scholar]
- 4.Burgard P, Kolker S, Haege G, Lindner M, Hoffmann GF. Neonatal mortality and outcome at the end of the first year of life in early onset urea cycle disorders--review and meta-analysis of observational studies published over more than 35 years. J Inherit Metab Dis. 2016. March;39(2):219–29. [DOI] [PubMed] [Google Scholar]
- 5.Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012. May 29;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Karnebeek CD, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet. 2014. March 6;94(3):453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nassogne MC, Heron B, Touati G, Rabier D, Saudubray JM. Urea cycle defects: management and outcome. J Inherit Metab Dis. 2005;28(3):407–14. [DOI] [PubMed] [Google Scholar]
- 8.Kolker S, Garcia-Cazorla A, Valayannopoulos V, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015. November;38(6):1041–57. [DOI] [PubMed] [Google Scholar]
- 9.Kolker S, Valayannopoulos V, Burlina AB, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J Inherit Metab Dis. 2015. November;38(6):1059–74. [DOI] [PubMed] [Google Scholar]
- 10.Loeber JG, Burgard P, Cornel MC, et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis. 2012. July;35(4):603–11. [DOI] [PubMed] [Google Scholar]
- 11.Nagamani SCS, Agarwal U, Tam A, et al. A randomized trial to study the comparative efficacy of phenylbutyrate and benzoate on nitrogen excretion and ureagenesis in healthy volunteers. Genet Med. 2018. July;20(7):708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. 2007. May 31;356(22):2282–92. [DOI] [PubMed] [Google Scholar]
- 13.Posset R, Garcia-Cazorla A, Valayannopoulos V, et al. Age at disease onset and peak ammonium level rather than interventional variables predict the neurological outcome in urea cycle disorders. J Inherit Metab Dis. 2016. September;39(5):661–72. [DOI] [PubMed] [Google Scholar]
- 14.Seminara J, Tuchman M, Krivitzky L, et al. Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium. Mol Genet Metab. 2010;100 Suppl 1:S97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Summar ML, Endo F, Kolker S. On the Creation, Utility and Sustaining of Rare Diseases Research Networks: Lessons learned from the Urea Cycle Disorders Consortium, the Japanese Urea Cycle Disorders Consortium and the European Registry and Network for Intoxication Type Metabolic Diseases. Mol Genet Metab. 2014. Sep-Oct;113(1–2):105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolker S, Dobbelaere D, Haberle J, et al. Networking Across Borders for Individuals with Organic Acidurias and Urea Cycle Disorders: The E-IMD Consortium. JIMD Rep. 2015;22:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Posset R, Garbade SF, Boy N, et al. Transatlantic combined and comparative data analysis of 1095 patients with urea cycle disorders-a successful strategy for clinical research of rare diseases. J Inherit Metab Dis. 2018. July 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waisbren SE, Cuthbertson D, Burgard P, et al. Biochemical markers and neuropsychological functioning in distal urea cycle disorders. J Inherit Metab Dis. 2018. July;41(4):657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buerger C, Garbade SF, Dietrich Alber F, et al. Impairment of cognitive function in ornithine transcarbamylase deficiency is global rather than domain-specific and is associated with disease onset, sex, maximum ammonium, and number of hyperammonemic events. J Inherit Metab Dis. 2019; 42:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2018. [Google Scholar]
- 21.Fox J, Weisberg S. An R Companion to Applied Regression: Sage Publications; 2018. [Google Scholar]
- 22.Lenth RV. Least-squares means: the R package lsmeans. J Stat Softw 2016;69:33. [Google Scholar]
- 23.Zeileis A, Hothorn T, Hornik K. Model-based recursive partitioning. Journal of Computational and Graphical Statistics. 2008;17(2):492–514. [Google Scholar]
- 24.Seibold H, Zeileis A, Hothorn T. Model-Based Recursive Partitioning for Subgroup Analyses. Int J Biostat. 2016;12(1):45–63. [DOI] [PubMed] [Google Scholar]
- 25.Venables V, Ripley B. Modern applied statistics with S. New York, NY: Springer, 2002. [Google Scholar]
- 26.Hornik K, Zeileis A, Meyer D. The strucplot framework: visualizing multi-way contingency tables with vcd. Journal of Statistical Software. 2006;17(3):1–48. [Google Scholar]
- 27.Lenhard W, Lenhard A. Normwertrechner. Bibergau: Psychometrica DOI 1013140/RG2145925363. https://www.psychometrica.de/normwertrechnerhtml. 2015. [Google Scholar]
- 28.Krivitzky L, Babikian T, Lee HS, Thomas NH, Burk-Paull KL, Batshaw ML. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009. July;66(1):96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ah Mew N, Krivitzky L, McCarter R, Batshaw M, Tuchman M, Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research N. Clinical outcomes of neonatal onset proximal versus distal urea cycle disorders do not differ. J Pediatr. 2013. February;162(2):324–9 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012. June 8;149(6):1393–406. [DOI] [PubMed] [Google Scholar]
- 31.Keskinen P, Siitonen A, Salo M. Hereditary urea cycle diseases in Finland. Acta Paediatr. 2008. October;97(10):1412–9. [DOI] [PubMed] [Google Scholar]
- 32.Erez A, Nagamani SC, Shchelochkov OA, et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat Med. 2011. November 13;17(12):1619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erez A, Nagamani SC, Lee B. Argininosuccinate lyase deficiency-argininosuccinic aciduria and beyond. Am J Med Genet C Semin Med Genet. 2011. February 15;157C(1):45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baruteau J, Jameson E, Morris AA, et al. Expanding the phenotype in argininosuccinic aciduria: need for new therapies. J Inherit Metab Dis. 2017. May;40(3):357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merritt JL 2nd, Brody LL, Pino G, Rinaldo P. Newborn screening for proximal urea cycle disorders: Current evidence supporting recommendations for newborn screening. Mol Genet Metab. 2018. June;124(2):109–13. [DOI] [PubMed] [Google Scholar]
- 36.Mercimek-Mahmutoglu S, Moeslinger D, Haberle J, et al. Long-term outcome of patients with argininosuccinate lyase deficiency diagnosed by newborn screening in Austria. Mol Genet Metab. 2010. May;100(1):24–8. [DOI] [PubMed] [Google Scholar]
- 37.Lindner M, Gramer G, Haege G, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases--report of 10 years from South-West Germany. Orphanet J Rare Dis. 2011. June 20;6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Enns GM. Nitrogen sparing therapy revisited 2009. Mol Genet Metab. 2010;100 Suppl 1:S65–71. [DOI] [PubMed] [Google Scholar]
- 39.Husson MC, Schiff M, Fouilhoux A, et al. Efficacy and safety of i.v. sodium benzoate in urea cycle disorders: a multicentre retrospective study. Orphanet J Rare Dis. 2016. September 23;11(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berry SA, Longo N, Diaz GA, et al. Safety and efficacy of glycerol phenylbutyrate for management of urea cycle disorders in patients aged 2months to 2years. Mol Genet Metab. 2017. November;122(3):46–53. [DOI] [PubMed] [Google Scholar]
- 41.Diaz GA, Krivitzky LS, Mokhtarani M, et al. Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology. 2013. June;57(6):2171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zielonka M, Breuer M, Okun JG, Carl M, Hoffmann GF, Kolker S. Pharmacologic rescue of hyperammonemia-induced toxicity in zebrafish by inhibition of ornithine aminotransferase. PLoS One. 2018;13(9):e0203707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kido J, Matsumoto S, Momosaki K, et al. Liver transplantation may prevent neurodevelopmental deterioration in high-risk patients with urea cycle disorders. Pediatr Transplant. 2017. September;21(6). [DOI] [PubMed] [Google Scholar]
- 44.Kido J, Matsumoto S, Mitsubuchi H, Endo F, Nakamura K. Early liver transplantation in neonatal-onset and moderate urea cycle disorders may lead to normal neurodevelopment. Metab Brain Dis. 2018. June 11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are not publicly available due to existing data protection laws. Furthermore, data ownership is retained by the members of the UCDC and E-IMD consortia making data available for specific research purposes. Data availability is subject to the consent of both consortia upon request.