

Abstract
Background and Purpose
Opioids remain the most efficient medications against severe pain; they act on receptors that couple to heterotrimeric G‐proteins in the Gαi/o family. Opioids exert many of their acute effects through modulating ion channels via Gβγ subunits. Many of their side effects are attributed to β‐arrestin recruitment. Several biased agonists that do not recruit β‐arrestins, but activate G‐protein‐dependent pathways, have recently been developed. While these compounds have been proposed to be full agonists of G‐protein signalling in several high throughput pharmacological assays, their effects were not studied on ion channel targets.
Experimental Approach
Here, we used patch‐clamp electrophysiology and Ca2+ imaging to test the effects of TRV130, PZM21, and herkinorin, three G‐protein‐biased agonists of μ‐opioid receptors, on ion channel targets of Gαi/o/Gβγ signalling. We also studied G‐protein dissociation using a FRET‐based assay.
Key Results
All three biased agonists induced smaller activation of G‐protein‐coupled inwardly rectifying K+ channels (Kir3.2) and smaller inhibition of transient receptor potential melastatin (TRPM3) channels than the full μ receptor agonist DAMGO. Co‐application of TRV130 or PZM21, but not herkinorin, alleviated the effects of DAMGO on both channels. PZM21 and TRV130 also decreased the effect of morphine on Kir3.2 channels. The CaV2.2 channel was also inhibited less by PZM21 and TRV130 than by DAMGO. We also found that TRV130, PZM21, and herkinorin were less effective than DAMGO at inducing dissociation of the Gαi/Gβγ complex.
Conclusion and Implications
TRV130, PZM21, and potentially herkinorin are partial agonists of μ receptors.
Abbreviations
- DAMGO
l‐tyrosyl‐d‐alanyl‐glycyl‐N‐methyl‐l‐phenylalanyl‐glycinol
- DRG
dorsal root ganglion
- Kir3.2
G‐protein‐coupled inwardly rectifying K+ channel
- TRPM3
transient receptor potential melastatin 3
What is already known
PZM21, TRV130, and herkinorin were described as high efficacy G‐protein‐biased agonists of μ receptors.
What this study adds
We studied the effects of PZM21, TRV130, and herkinorin on ion channel targets of μ receptors.
We found that PZM21, TRV130, and potentially herkinorin are partial agonists of G‐protein signalling.
What is the clinical significance
Our data provide a new perspective on how these novel analgesics exert their effects.
1. INTRODUCTION
Chronic pain is an unsolved medical problem, causing immense suffering to millions of people worldwide (Basbaum, Bautista, Scherrer, & Julius, 2009). Opioids remain the main therapy against severe chronic pain, but these medications cause multiple side effects limiting their application, and they are also highly addictive. The lack of better pain control is believed to be a significant driving force behind the opioid addiction crisis (Skolnick & Volkow, 2016).
Opioids exert their effects via binding to μ, δ, or κ receptors (Stein, 2016). All these receptors act via inhibitory G‐proteins in the Gαi/o family. Upon receptor activation, the Gα subunits dissociate from Gβγ, and both subunits activate multiple downstream effectors. Opioid receptor activation also induces recruitment of β‐arrestins, leading to receptor desensitization and internalization; β‐arrestins, however, are not only important for desensitization but they are also emerging as independent signalling mediators regulating various effectors (DeWire, Ahn, Lefkowitz, & Shenoy, 2007), including ion channels (Liu et al., 2017). For several GPCRs, including opioid receptors, ligands were developed that preferentially activate either the G‐protein or the arrestin pathway, a phenomenon called biased signalling. Most clinically useful analgesic effects of opioids are mediated by μ receptors. Some side effects of opioids, such as tolerance, have been attributed to μ receptor signalling via β‐arrestins (Raehal, Schmid, Groer, & Bohn, 2011).
On the cellular level, opioid receptor activation leads to decreased cAMP production, an effect mediated by inhibition of the adenylate cyclase enzyme by Gαi. Many acute effects of opioids are mediated by a reduction in neuronal activity via modulation of ion channels by Gβγ subunits. The two classical actions of Gβγ signalling are activation of G‐protein‐coupled inwardly rectifying K+ channels (GIRK also known as Kir3) and inhibition of N‐ and P/Q‐type voltage‐gated Ca2+ channels (Cav2.2 and Cav2.1; Clapham & Neer, 1997), both effects leading to reduced neuronal activity.
We and others recently identified transient receptor potential melastatin 3 (TRPM3) as a novel ion channel target of Gβγ subunits; these channels are robustly inhibited by activation of a variety of Gαi‐coupled receptors, including μ receptors in dorsal root ganglion (DRG) neurons, as well as in expression systems (Badheka et al., 2017; Dembla et al., 2017; Quallo, Alkhatib, Gentry, Andersson, & Bevan, 2017). TRPM3 is a heat‐activated Ca2+ permeable non‐selective cation channel; its genetic deletion in mice results in reduced heat sensitivity (Vandewauw et al., 2018; Vriens et al., 2011). TRPM3 also has chemical activators such as the endogenous neurosteroid pregnenolone sulfate (PregS) (Wagner et al., 2008) as well as synthetic chemical activators including CIM0216 and nifedipine (Held et al., 2015). This channel is also involved in regulating glucose homeostasis and is expressed in many other tissues including pancreatic beta cells, retina, and brain (Oberwinkler & Philipp, 2014).
Several biased agonists of μ receptors that activate G‐protein signalling but do not lead to β‐arrestin recruitment have recently been described with the goal of developing painkillers with reduced side effects. Herkinorin was the first G‐protein‐biased agonist that was claimed not to lead to β‐arrestin recruitment and receptor internalization (Groer et al., 2007). TRV130 (oliceridine), developed by Trevena pharmaceuticals (DeWire et al., 2007), has shown promising analgesic effects in clinical trials (Singla et al., 2017; Viscusi et al., 2016). The latest G‐protein‐biased μ receptor agonist PZM21 was developed based on structure‐guided computational modelling and showed promising analgesic results in mice (Manglik et al., 2016). The study showed that PZM21 as well as TRV130 did not lead to β‐arrestin recruitment, but it was found that herkinorin evoked β‐arrestin recruitment similar to that induced by full agonists (Manglik et al., 2016). The effects of PZM21 and TRV130 have not yet been tested on ion channel targets of Gβγ signalling.
Here, we tested the effects of the G‐protein‐biased μ receptor agonists TRV130, PZM21, and herkinorin on three ion channel targets of Gβγ. Surprisingly, we found that all three biased agonists induced significantly smaller inhibition of TRPM3, and CaV2.2 voltage‐gated Ca2+ channels and smaller activation of Kir3.2 (GIRK2) channels than the full agonist DAMGO. Furthermore, PZM21 and TRV130, but not herkinorin, attenuated DAMGO‐induced inhibition of TRPM3 and reduced DAMGO‐induced activation of Kir3.2 channels. PZM21 and TRV130 also reduced morphine‐induced activation of Kir3.2 channels. All three G‐protein‐biased agonists were less efficient in inducing G‐protein dissociation than DAMGO. Overall, our data indicate that PZM21 and TRV130 and potentially herkinorin act as partial μ receptor agonists.
2. METHODS
2.1. Isolation and culture of DRG neurons
Animal procedures were approved by the Institutional Animal Care and Use Committee at Rutgers New Jersey Medical School. DRG neurons were isolated from adult C57Bl6 mice of either sex (2–4 months old) purchased from the Jackson laboratories (RRID:IMSR_JAX:000664). Sex was not treated as a biological variable in either the experimental design or data analysis and that it was assumed that there are no sex differences in the measure of interest in this study. The mice were housed in a 12‐hr light/dark cycle in a controlled temperature room, up to four mice per cage, and were allowed free access to water and food. For the DRG neuron preparation, we used the slightly modified protocol based on that of Malin, Davis, and Molliver (2007), as described previously (Lukacs et al., 2007; Yudin, Lutz, Tao, & Rohacs, 2016). Briefly, animals were anaesthetized with an i.p. injection of ketamine (100 mg·kg−1) and xylazine (12 mg·kg−1). The anaesthetized animals were then perfused via the left ventricle with ice‐cold HBSS (Invitrogen). This procedure was shown to reduce protease activity and improve cell survival and quality (Malin et al., 2007). DRGs were collected from all spinal segments after laminectomy and maintained in ice‐cold HBSS during the isolation procedure. The anaesthetized animal was killed by removing its heart during the surgery. After isolation and trimming of dorsal and ventral roots, ganglia were incubated in an HBSS‐based enzyme solution containing 2‐mg·ml−1 Type I collagenase (Worthington) and 5‐mg·ml−1 Dispase (Sigma) at 37°C for 25–30 min, followed by repetitive trituration for dissociation. After centrifugation at 80×g for 10 min, cells were resuspended and plated on round coverslips pre‐coated with poly‐l‐lysine (Invitrogen) and laminin (Sigma), allowed to adhere for 1 hr, maintained in culture in DMEM/F12 supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific), 100‐IU·ml−1 penicillin, and 100‐μg·ml−1 streptomycin, and were kept in a humidity‐controlled tissue‐culture incubator with 5% CO2 at 37°C for 12–36 hr before experiments. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology.
2.2. Maintenance of HEK293 cells
HEK293 cells were purchased from American Type Culture Collection, Manassas, VA, (catalogue # CRL‐1573), RRID:CVCL_0045. Passage number of the cells was monitored, and cells were used up to passage number 25–30, when a new batch of cells was thawed with low passage number. The cells were maintained in minimal essential medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% (v/v) FBS, 100‐IU·ml−1 penicillin, and 100‐μg·ml−1 streptomycin in an incubator (37°C in 5% CO2).
2.3. Ca2+ imaging
Ca2+ imaging measurements were performed with an Olympus IX‐51 inverted microscope equipped with a DeltaRAM excitation light source (Photon Technology International [PTI], Horiba), as described previously (Lukacs et al., 2013). Briefly, DRG neurons or HEK293 cells were loaded with 1‐μM fura‐2 AM (Invitrogen) at 37°C for 40–50 min before the measurement, and dual‐excitation images at 340 and 380 nm excitation wavelengths were detected at 510 nm with a Roper Cool‐Snap digital CCD camera. Measurements were conducted in the same bath solution that we used for the whole‐cell patch‐clamp recording of TRPM3 channels. Raw data analysis was performed using the Image Master software (PTI). We calculated the normalized change in the relative 340/380 ratio, according to the following formula: ΔR2/ΔR1 = 100(R2 − R0)/(R1 − R0), where R1 and R2 are the maximum ratios during the first and second applications of PregS and R0 is the basal fluorescence ratio before stimulation.
2.4. TRPM3 channel electrophysiology
Whole‐cell patch‐clamp measurements were performed as described previously (Badheka, Borbiro, & Rohacs, 2015). The cells were transiently transfected with cDNA encoding the mouse TRPMα2 splice variant of TRPM3, in the bicistronic pCAGGS/IRES‐GFP vector (Oberwinkler, Lis, Giehl, Flockerzi, & Philipp, 2005; Vriens et al., 2011), and human μ receptor (cDNA Resource Center, catalogue # OPRM10FN0) in ratio 1:1 using the Effectene reagent (Qiagen) according to the manufacturer's protocol, and used in experiments 48–72 hr later. Measurements were carried out on GFP positive cells, in an extracellular solution containing (in mM) 137 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 10 glucose, pH 7.4. The intracellular solution contained (in mM) 140 potassium gluconate, 5 EGTA, 1 MgCl2, 10 HEPES, and 2 NaATP, pH 7.3. Patch‐clamp pipettes were prepared from borosilicate glass capillaries (Sutter Instruments) using a P‐97 pipette puller (Sutter Instrument) and had a resistance of 4–6 MΩ. In all experiments, after the formation of gigaohm‐resistance seals, the whole‐cell configuration was established and currents were recorded using a ramp protocol from −100 to +100 mV over 500 ms preceded by a −100 mV step for 200 ms; the holding potential was −60 mV, and this protocol was applied once every 2 s. The currents were measured with an Axopatch 200B amplifier, filtered at 5 kHz, and digitized through a Digidata 1440A interface. In all experiments, cells that had a passive leak current of more than 100 pA were discarded. Leak was defined as currents at the −60 mV holding potential before the application of PregS. Data were collected and analysed with the pClamp 10.6 acquisition software (RRID:SCR_011323; Molecular Devices, Sunnyvale, CA) and further analysed and plotted with the Origin 8.0 software (Microcal Software Inc., Northampton, MA, USA; RRID:SCR_002815).
2.5. Kir3.2 channel (GIRK2) electrophysiology
All recordings were performed in HEK293 cells after transient transfection with plasmids Kir3.2 (gift of Dr. Tooraj Mirshahi), μ receptors, and pEYFP using the Effectene reagent (Qiagen) according to the manufacturer's protocol with the ratio of cDNA of 1:1:0.1. Patch‐clamp recordings were performed 48–72 hr after transfection. Measurements were carried out on YFP positive cells, in an extracellular solution containing (in mM) 137 KCl, 5 NaCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 10 glucose, pH 7.4. Electrodes (resistance, 4–6 megaohms) were filled with pipette solution containing (in mM) 130 KCl, 2 MgCl2, 10 HEPES, 5 Na‐EGTA, 3 NaATP, and 0.5 NaGTP (pH 7.25). After the whole‐cell configuration was established, the currents were recorded using a voltage protocol starting with a step to −100 mV for 50 ms, followed by a 200‐ms ramp to 40 mV. This protocol was applied once every 2 s; the holding potential was −40 mV. Cells that had a passive leak current of more than 100 pA were discarded. Leak was defined as currents at the −40 mV holding potential before the application of any stimuli. The currents were measured with an Axopatch 200B amplifier, filtered at 5 kHz, and digitized through a Digidata 1440A interface.
2.6. CaV2.2 electrophysiology
All plasmids with CaV2.2 channels and auxiliary subunits in pMT2 vector were purchased from Addgene (Raghib et al., 2001). All recordings were performed in HEK293 cells after transient transfection with plasmids CaV2.2‐GFP (catalogue # 58737), β1b‐mCherry (catalogue # 89892), α2δ1 subunits (catalogue # 58726), and μ receptors using the Effectene reagent (Qiagen) according to the manufacturer's protocol with the ratio of calcium channel subunits and μ receptors in a ratio of 1:1:1:1. Patch‐clamp recordings were performed 48–72 hr after transfection. Transfected cells were visually identified using fluorescence of GFP and mCherry. The whole‐cell Ca2+ currents, carried by barium (I Ba), were recorded using an extracellular recording solution consisting of (in mM) 30 tetraethylammonium chloride, 100 NaCl, 5 CsCl, 1 MgCl2, 10 BaCl2, 10 glucose, and 10 HEPES (pH 7.4). Electrodes (resistance, 4–6 megaohms) were filled with pipette solution containing (in mM) 120 CsCl, 2 MgCl2, 10 HEPES, 10 EGTA, 4 NaATP, and 0.35 NaGTP (pH 7.25). After cells were voltage‐clamped in the conventional whole‐cell configuration, the currents were recorded using a voltage step protocol from the −80 mV holding potential to +10 mV (50 ms), which was applied once every second. The currents were measured with an Axopatch 200B amplifier, filtered at 5 kHz, and digitized through a Digidata 1440A interface. Series resistance was partially compensated using the Axopatch circuitry (about 75–80% prediction and correction; 10‐µs lag). Offline leak and capacitive current subtraction used P/4 protocol with the leak pulses applied following the test pulses was carried out using the Clampfit 10.6 software (Molecular Devices).
2.7. FRET‐based monitoring of Gαi1 activation
FRET measurements were performed as described previously (Borbiro, Badheka, & Rohacs, 2015). Briefly, HEK293 cells were co‐transfected with the mTurqoise2‐tagged Gαi1 (FRET donor), a Venus‐tagged Gγ2 (FRET acceptor), and Gβ1 expressed from a tricistronic vector (Gβ‐2A‐cpV‐Gγ2‐IRES‐Gαi1‐mTq2; Addgene, catalogue # 69623; van Unen et al., 2016) and μ receptors. Fluorescence was detected using a photomultiplier‐based dual‐emission system mounted on an inverted Olympus IX‐71 microscope equipped with 40× oil objective (numerical aperture 1.3). Excitation light (430 nm) was provided by a DeltaRAM light source (PTI). Emission was measured at 480 and 535 nm using two interference filters and a dichroic mirror to separate the two wavelengths. Raw data were analysed with the Felix3.2 programme (PTI).
2.8. Application of solutions and reagents
Coverslips with attached cells were placed in a recording bath (volume ~200 μl), which was continually perfused with fresh extracellular solution at a flow rate of ~3–4 ml·min−1 from gravity‐fed reservoirs and viewed using an Olympus IX‐51 inverted microscope. PregS and naloxone were purchased from Sigma, DAMGO and herkinorin were purchased from Abcam, PZM21 from MedChem Express, morphine sulfate, stock 10 mg·ml−1 from West‐Ward, and TRV130 from Adooq Bioscience. DAMGO (10 mM) and naloxone (10 mM) were dissolved in H2O and morphine stock was diluted up to 5‐mM final concentration in H2O. PregS (250 mM), herkinorin (10 mM), PZM21 (10 mM), and TRV130 (20 mM) were dissolved in DMSO and further diluted in the experimental buffers on the day of the experiment. For most experiments, the final concentration of DMSO is very low (up to 0.01%). For the Kir3.2 concentration‐ dependence measurements in Figure 5, the highest PZM21 and herkinorin concentrations (50 μM) introduced 0.5% DMSO, which when tested alone did not induce any substantial channel activity, and it did not have an effect on DAMGO‐induced currents.
Figure 5.
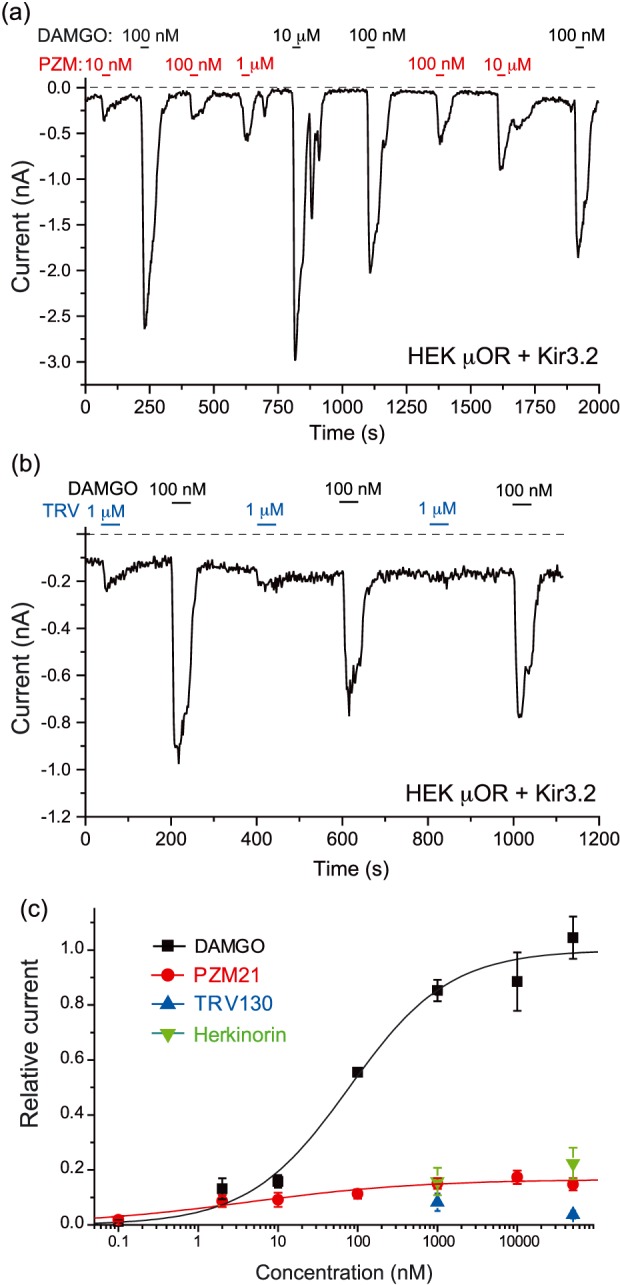
Differential activation of Kir3.2 channels by μ receptor (μOR) agonists in HEK293 cells. Whole‐cell patch‐clamp measurements in HEK293 cells transfected with μ receptor and Kir3.2 channels were performed as described in Section 2. (a–b) Representative traces for inward (−100 mV) currents induced by the various μ receptor agonists. (c) Summary of the concentration–response relationship for DAMGO (n = 6–21 cells), PZM21 (n = 7–16 cells), TRV130 (n = 7–8 cells), and herkinorin (n = 5–7 cells) for individual data points. Kir3.2 currents were measured as a difference in inward currents before the application of the agonist and the peak current induced by each agonist. All data points were first normalized to responses induced by 100‐nM DAMGO in the same experiment. The individual data points were fitted with the Hill equation using the Sigmoid fit function of Microcal Origin 8.0. For visualization, all data points were renormalized to the maximum effect of DAMGO calculated from the Hill fit
2.9. Data and statistical analysis
Data analysis was performed in Microsoft Excel (RRID:SCR_016137) and Microcal Origin 8.0. The normality of the data was verified with the Kolmogorov–Smirnov test. The homogeneity of sample variance was confirmed with Levine test for all data subjected to ANOVA. Data were analysed using Student's t test, or ANOVA with Bonferroni post hoc test. Significance levels are indicated as *P < .05 for all figures. Data were collected in a randomized fashion; the number of experiments was not determined prospectively. Neither data collection nor data analysis was blinded, as electrophysiology and Ca2+ measurements are impractical to perform in a blinded fashion. Data are displayed as box whisker and scatter plots for most figures; mean values are described in Section 3 for most measurements. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology.
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos, et al., 2017; Alexander, Striessnig, et al., 2017).
3. RESULTS
3.1. G‐protein‐biased μ receptor agonists inhibit TRPM3 channels less than the full agonist DAMGO
We tested the effects of the three G‐protein‐biased μ receptor agonists PZM21, TRV130, and herkinorin on Ca2+ signals induced by the TRPM3 agonist PregS (12.5 μM) in DRG neurons. TRPM3 channels are mainly expressed in small nociceptive neurons, 20–25% of all neurons respond to this concentration of PregS (Badheka et al., 2017). For this study, we analysed all PregS responsive neuron and did not make further distinctions within this group. We have found that the full μ receptor agonist DAMGO (1 μM) induced strong inhibition in the vast majority of the PregS responsive neurons (61 of 64; 95%; Figure 1a); while application of the other three agonists at the same concentration affected a much smaller fraction of the PregS responsive neurons (Figure 1b–c). For 1‐μM herkinorin, we observed inhibition in 38 out of the 69 (55%) neurons tested (Figure 1b), for 1‐μM PZM21 in 14 out of 22 (63%; Figure 1c), and for 1‐μM TRV130 in 12 out of 22 neurons (54%; Figure 1d). See Figure S1 for individual traces and scatter plots. We also observed that the inhibitory effects of these three biased agonists were longer lasting compared to the quickly reversible effects of DAMGO.
Figure 1.

Differential inhibition of PregS‐induced Ca2+ signals by μ receptor agonists in DRG neurons. Ca2+ imaging experiments in Fura‐2 loaded mouse DRG neurons were performed as described in Section 2. (a) Black trace shows mean ± SEM of the effect of three consecutive applications of 12.5‐μM PregS (PS) from neurons responsive to this compound, n = 23 cells; 30‐mM KCl was applied at the end of the experiment. In red colour, 1‐μM DAMGO was applied before the second application of PregS, out of 64 neurons, inhibition was observed in 61, the non‐responding cells were not included in the average trace from four independent neuron isolations. (b) Similar measurements with the application of 1‐μM herkinorin; the two traces show the average ratios ± SEM in cells in which PregS‐induced Ca2+ signals were inhibited by herkinorin (red, n = 38 cells) and in cells where inhibition was not observed (blue, n = 31 cells; from four independent neuron isolations). (c) Similar measurements with 1‐μM PZM21; the two traces show the average ratios ± SEM in cells where PregS‐induced Ca2+ signal was inhibited by PZM21 (red, n = 14 cells) and in cells where it was not inhibited (blue, n = 8 cells) from three independent neuron isolations. (d) Similar measurements with 1‐μM TRV130; the two traces show the average ratios ± SEM in cells where PregS‐induced Ca2+ signals were inhibited by TRV130 (red, n = 12 cells) and in cells where they were not inhibited (blue, n = 10 cells) from four independent neuron isolations
DRG neurons express all three types of opioid receptors—μ, δ, and κ, which may potentially confound interpretation of these data, as some of the μ receptor agonists may cross‐react with other opioid receptors. Therefore, we co‐expressed μ receptors and TRPM3 channels in HEK293 cells and performed Ca2+ imaging experiments similar to those in DRG neurons. We found that similar to DRG neurons, PZM21, TRV130, and herkinorin evoked significantly smaller inhibition of PregS‐induced Ca2+ signals than DAMGO (Figure 2). DAMGO (0.5 μM) evoked, on average, an 89.3% inhibition of the PregS‐induced Ca2+ signal, for 1‐μM PZM21, average inhibition was 29.3%, for 1‐μM TRV130, it was 19.7%, and for 1‐μM herkinorin, it was 37.1%. Also, the number of cells where inhibition was more than 20% of the PregS‐induced Ca2+ signal was much higher for DAMGO (207 out of 208 cells, 99.5%) than for herkinorin (96 out of 173; 55.5%) or for TRV130 (76 out of 227; 33.5%) or for PZM21 (98 out of 187; 52.4%).
Figure 2.
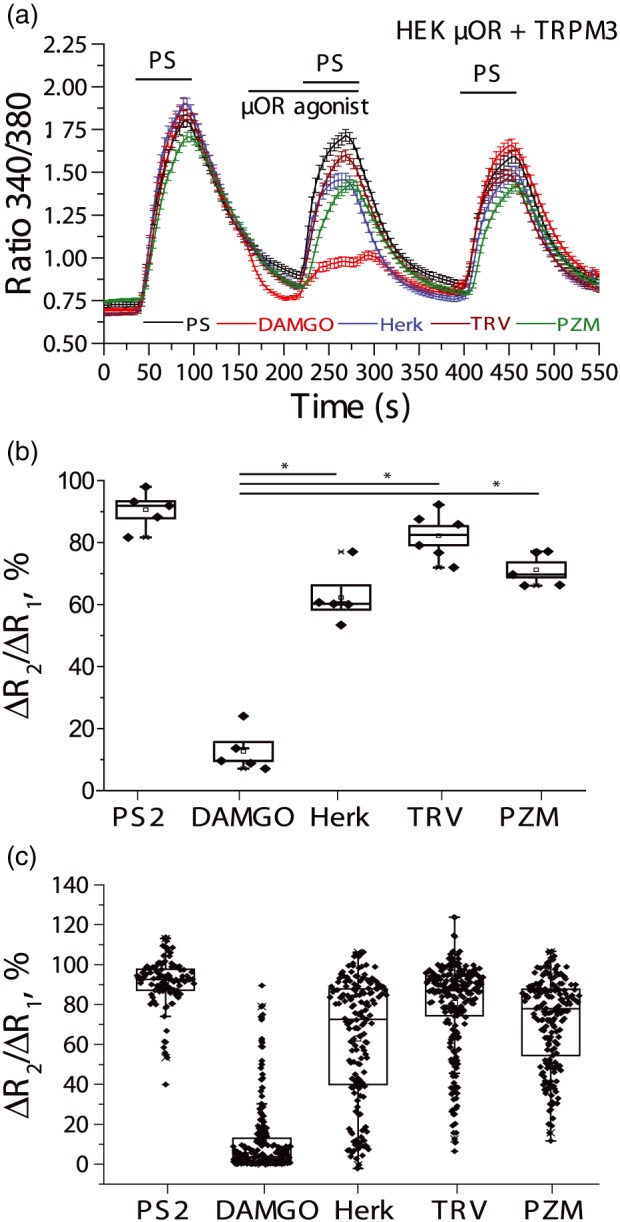
Differential inhibition of PregS‐induced Ca2+ signals by μ receptor (μOR) agonists in HEK293 cells. Ca2+ imaging experiments in HEK293 cells transfected with mTRPM3 and μ receptors were performed as described in Section 2. (a) Traces show mean ± SEM of Ca2+ signals induced by three consecutive applications of 12.5‐μM PregS and the effects of four μ receptor agonists, 0.5‐μM DAMGO (red line), 1‐μM herkinorin (blue), 1‐μM TRV130 (brown), and 1‐μM PZM21 (green). Black trace denotes control experiments with three consecutive applications of 12.5‐μM PregS. (b) Changes in the relative fluorescence ratio according to the following formula: ΔR2/ΔR1 = 100(R2 − R0)/(R1 − R0) normalized to the first pulse of PregS. Each data point represents the average response of all PregS responsive cells on one individual cover glass (n = 5–6). Statistical significance was determined with ANOVA, *P < .05. Statistical significance is shown for the difference between DAMGO and PZM21, TRV130, and herkinorin. All four agonists induced statistically significant inhibition compared to control, asterisks not shown for simplicity. (c) Box whisker and scatter plots of the same data with each data point corresponding to one cell, n = 108 cells for PregS alone, n = 208 cells for DAMGO, n = 173 cells for herkinorin, n = 227 cells for TRV130, and n = 187 cells for PZM21
Ca2+ signals are a convenient way to monitor the activity of Ca2+ permeable ion channels, but they are not linear readouts of channel activity, thus it is difficult to draw quantitative conclusions on the extent of channel inhibition. Therefore, we also performed patch‐clamp experiments to investigate how these biased μ receptor agonists affect PregS‐induced currents in HEK293 cells transfected with μ receptors and TRPM3 (Figure 3). In HEK293 cells transfected with μ receptors only, 25‐μM PregS did not induce any currents (data not shown). In cells transfected with μ receptors and TRPM3, 25‐μM PregS evoked robust outwardly rectifying currents. In each measurement, we applied two pulses of 25‐μM PregS and compared the effect of the test agonist and to the effect of DAMGO. For each agonist, we tested two concentrations, 100 nM and 1 μM. In all cases, the application of DAMGO at both 100 nM and 1 μM induced almost full inhibition of TRPM3 currents. For the three substances tested, the inhibitory effects were smaller compared to DAMGO. Both 100‐nM and 1‐μM PZM21 induced a smaller inhibition of TRPM3 currents than 100‐nM DAMGO; representative traces are shown in Figure 3a,b, summary data are provided in Figure 3c. Similarly, herkinorin both at 100 nM and at 1 μM induced a smaller current inhibition than 100‐nM or 1‐μM DAMGO (Figure 3d–f). TRV130 (100 nM and 1 μM) also induced smaller inhibition of TRPM3 currents than 100‐nM DAMGO (Figure 3g–i). A more detailed analysis of these data is shown in Table S1.
Figure 3.
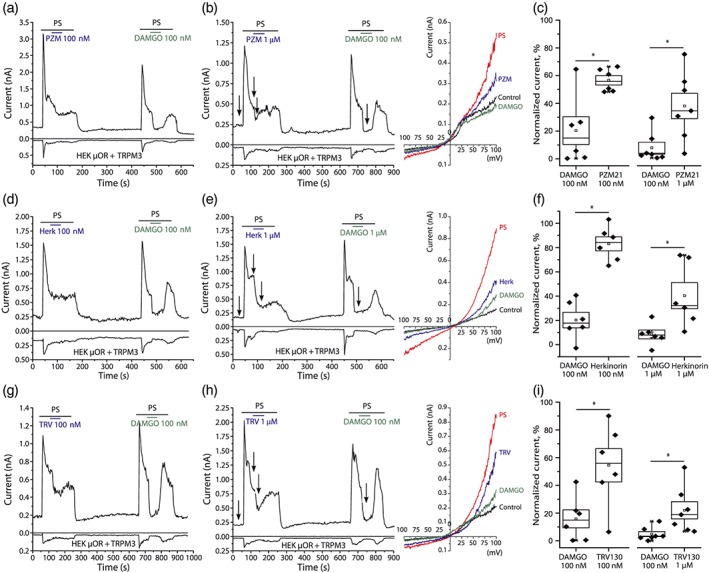
Differential inhibition of PregS‐induced TRPM3 currents by μ receptor agonists in HEK293 cells. Whole‐cell patch‐clamp measurements in HEK293 cells transfected with mTRPM3 channels and μ receptors were performed as described in Section 2. (a, b) Representative traces for 100‐nM PZM21 (n = 6 cells), 1‐μM PZM21 (n = 7 cells), and 100‐nM DAMGO on PregS‐induced current; upper trace shows currents at 100 mV, lower trace at −100 mV. The applications of 25‐μM PregS, DAMGO, and PZM21 are indicated by the horizontal lines. Right panel shows representative I–V curves for time points marked by the arrows. (c) Summary of the data at 100 mV. Current values were normalized to steady‐state PregS (PS)‐induced currents 2 s prior to μ receptor agonist application. Statistical analysis was performed with paired sample t test *P < .05, comparing the effect of DAMGO to the test compound in the same experiment. (d, e) Representative traces for 100‐nM herkinorin and 1‐μM DAMGO (n = 6 cells), 1‐μM herkinorin, and 1‐μM DAMGO (n = 6 cells) on PregS‐induced currents. The applications of 25‐μM PregS, DAMGO, and herkinorin are indicated by the lines. Right panel shows representative I–V curves for time points marked by the arrows. (f) Summary of the data at 100 mV, analysed the same way as in panel c. (g, h) Representative traces for 100‐nM TRV130 (n = 6 cells), 1‐μM TRV130 (n = 6 cells), and 100‐nM DAMGO on PregS‐induced current. The applications of 25‐μM PregS, DAMGO, and TRV130 are indicated by the horizontal lines. Right panel shows representative I–V curves for time points marked by the arrows. (i) Summary of the data at 100 mV analysed the same way as in panel c. All representative traces show test compound followed by DAMGO, but for each condition, at least two measurements were performed in the reverse order
3.2. PZM21 and TRV130 attenuate DAMGO‐induced inhibition of TRPM3 currents
The smaller effects of PZM21, TRV130, and herkinorin compared to DAMGO suggest that these compounds may act as partial agonists. Partial agonists exert a smaller maximal effect than full agonists, and therefore, they reduce the effect of the full agonist when the two are co‐applied. To test if these new reagents behave as partial agonists, we performed patch‐clamp recordings where TRPM3 was first inhibited by 100‐nM DAMGO, then PZM21 or TRV130, was co‐applied with DAMGO (Figure 4). We found that 100‐nM DAMGO inhibited PregS‐induced currents by 97.5 ± 3.2% for the outward (+100 mV) and 94.9 ± 2.8% for the inward (−100 mV) currents. When 1‐μM PZM21 was co‐applied with DAMGO, TRPM3 currents recovered substantially up to 32.7 ± 7.5% inhibition (P < .05 vs. DAMGO inhibition) for the outward (+100 mV) and 33.3 ± 8.9% (P < .05 vs. DAMGO inhibition) for the inward (−100 mV) currents, n = 9 (Figure 4a,b). When PZM21 was washed out in the continuous presence of DAMGO, currents returned to the level of the initial inhibition by DAMGO (Figure 4a,b).
Figure 4.
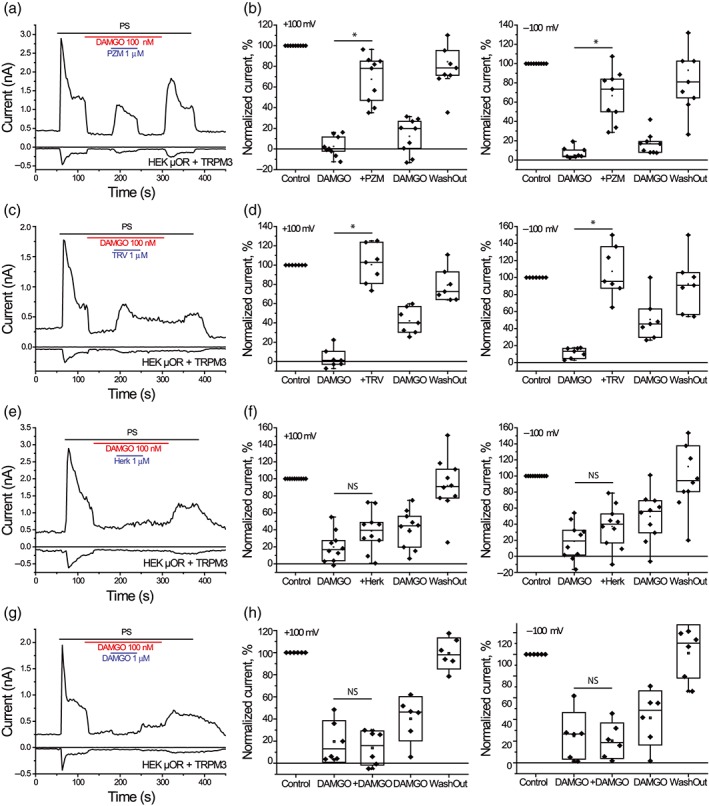
PZM21 and TRV130 oppose the inhibitory effect of DAMGO on PregS‐induced TRPM3 currents in HEK293 cells. Whole‐cell patch‐clamp measurements in HEK293 cells transfected with mTRPM3 channels and μ receptors were performed as described in Section 2. (a) Representative trace for n = 9 cells; the applications of 25‐μM PregS, 100‐nM DAMGO, and 1‐μM PZM21 are indicated by the horizontal lines. (b) Summary of normalized outward (+100 mV, left) and inward currents (−100 mV, right panel). Currents were normalized to steady‐state PregS (PS)‐induced currents 2 s prior to DAMGO application. (c) Representative trace for n = 7 cells; the applications of 25‐μM PregS (PS), 100‐nM DAMGO, and 1‐μM TRV130 are indicated by the horizontal lines. (d) Summary of normalized outward (left) and inward currents (right panel) for the effect of TRV130. (e) Representative trace for n = 10 cells; the applications of 25‐μM PregS, 100‐nM DAMGO, and 1‐μM herkinorin are indicated by the horizontal lines. (f) Summary of normalized outward (left) and inward currents (right panel) for the effect of DAMGO. (g) Representative traces n = 6 cells; the applications of 25‐μM PregS, 100‐nM DAMGO, and 1‐μM DAMGO are indicated by the horizontal lines. (h) Summary of normalized outward (left) and inward current (right panel) for the effect of DAMGO. Statistical analysis was performed with paired t test *P < .05
Similar effects were also observed for 1‐μM TRV130 (Figure 4c,d). In these experiments, DAMGO inhibited PregS‐induced currents by 96.8 ± 3.7% for +100 mV and 88.4 ± 2.2% for −100 mV, and TRV130 reversed this inhibition to 3.9 ± 7.5% potentiation (P < .05 vs. DAMGO inhibition) at +100 mV and 7.2 ± 11.4% potentiation (P < .05 vs. DAMGO inhibition) at −100 mV, n = 7. As opposed to the effect of PZM21, the effect of TRV130 was not quickly reversible (Figure 4c).
As opposed to PZM21 and TRV130, application of 1‐μM herkinorin did not induce a robust recovery from DAMGO‐induced inhibition (Figure 4e,f). In this set of experiments, we observed that 100‐nM DAMGO inhibited PregS‐induced currents by 81.2 ± 5.6% at +100 mV; after co‐application of herkinorin, inhibition decreased to 61.5 ± 7.4%, but this effect was not statistically significant (P > .05 vs. DAMGO inhibition). At −100‐mV, DAMGO induced an 81.4 ± 7.2% inhibition, which decreased to 62.8 ± 8.4% after the co‐application of herkinorin (P > .05 vs. DAMGO inhibition, n = 10).
As a control, we co‐applied 1‐μM DAMGO in the presence of 100‐nM DAMGO, which slightly but not statistically significantly increased the initial inhibitory level (Figure 4g,h). After the initial 100‐nM DAMGO application outward currents were inhibited by 80.3 ± 7.6%, and 1‐μM DAMGO further increased inhibition to 86.3 ± 6.2%, P > .05. Similarly, inward currents changed from 75.1 ± 8.7% to 79.6 ± 6.6% (P > .05) inhibition when 1‐μM DAMGO was co‐applied with 100‐nM DAMGO, n = 6.
Altogether, our data show that PZM21, TRV130, and herkinorin induced significantly smaller inhibition of TRPM3 currents than the full μ receptor agonist DAMGO. We also showed that PZM21 and TRV130 behave as partial agonists, as they induce recovery from DAMGO‐induced inhibition of TRPM3 currents. Next, we tested the effects of activating μ receptors with these compounds on two well‐established Gβγ regulated ion channels Kir3.2 and CaV2.2.
3.3. PZM21 and TRV130 behave as partial μ receptor agonists in activating Kir3.2 channels
Gβγ activation of Kir3 channels is one of the classical downstream effects of Gαi signalling. We co‐expressed Kir3.2 (GIRK2) channels with μ receptors in HEK293 cells and measured inward currents in high extracellular K+ in response to μ receptor agonists in whole‐cell patch‐clamp experiments at −100 mV (Figure 5). DAMGO (1 μM) did not induce any currents in HEK293 cells transfected with only μ receptors using the same protocol (data not shown). Application of the DAMGO induced clear inward currents in HEK293 cells transfected with Kir3.2 channels and μ receptors in a concentration‐dependent manner (Figure 5). In each experiment, we applied 100‐nM DAMGO as a normalizing pulse repetitively throughout the experiment, to correct for different expression levels of channels and receptors between different cells, and for any time‐dependent change in responsiveness. The EC50 for DAMGO was 78 ± 18 nM. We also attempted to generate a full dose‐response curve for PZM21; the responses to this compound showed marked desensitization in some cells, despite the finding that they maintained responsiveness to DAMGO, which made the dose‐response curve difficult to fit reliably. Overall, PZM21 at concentrations up to 50 μM induced responses that were less than 20% of the maximal effect of DAMGO. Both TRV130 (1 or 50 μM) and herkinorin (1 or 50 μM) induced responses less than 25% of the maximal effect of DAMGO (Figure 5 ).
The μ receptor antagonist naloxone (1 μM) almost completely blocked the effects of both 1‐μM PZM 21 (95.8 ± 2.7% inhibition, n = 5) and 1‐μM TRV130 (90.5 ± 2.9% inhibition, n = 6) on GIRK channel activation (Figure S2).
To test if PZM21, TRV130, and herkinorin act as partial agonists, we tested if they reverse the effect of DAMGO on Kir3.2 channels when they are co‐applied with this full agonist. After activation by DAMGO, co‐application of PZM21 or TRV130 quickly induced significant and prominent reduction of the potassium current amplitude, while herkinorin did not have a clear inhibitory effect (Figure 6). Kir3.2 currents induced by 100‐nM DAMGO were inhibited upon the application 100‐nM PZM21 by 52.6 ± 7.9% (P < .05, n = 5) and for 1‐μM PZM21, Kir3.2 currents were inhibited by 73.9 ± 5.3% (P < .05, n = 8). TRV130 (100 nM) inhibited Kir3.2 currents by 51.4 ± 4.6% (P < .05, n = 6) and 1‐μM TRV130 by 73.4 ± 4.8% (P < .05, n = 8).
Figure 6.
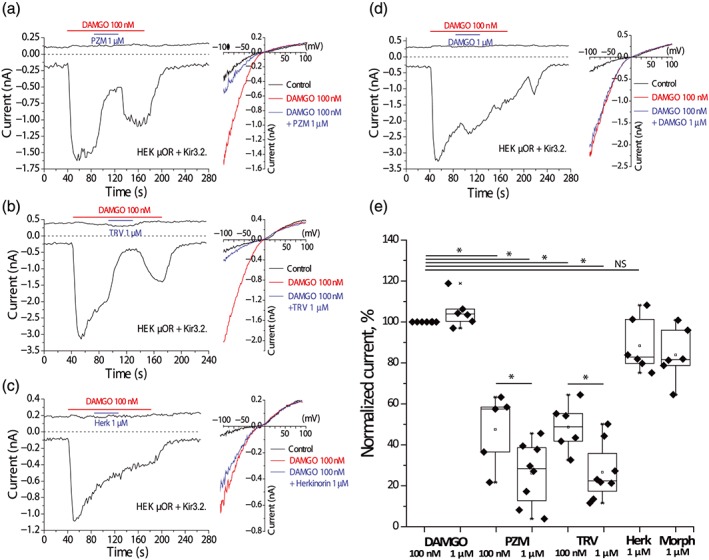
Inhibition of DAMGO‐induced Kir3.2 currents by PZM21 and TRV130 in HEK293 cells. Whole‐cell patch‐clamp measurements in HEK293 cells transfected with μ receptors (μOR) and Kir3.2 channels were performed as described in Section 2. (a) Representative trace for the effects of 1‐μM PZM21 on DAMGO‐activated Kir3.2 currents, at −100 mV (bottom trace) and 40 mV (top trace) n = 8 cells. The applications of 100‐nM DAMGO and 1‐μM PZM21 are indicated by the horizontal lines. Right panel shows I–V curves representative for the corresponding time point measurements. (b) Representative trace for the effect of 1‐μM TRV130 on DAMGO‐induced Kir3.2 currents, n = 8 cells. The applications of 100‐nM DAMGO and 1‐μM TRV130 are indicated by the horizontal lines. Right panel shows I–V curves representative for the corresponding time point measurements. (c) Representative traces for the effect of 1‐μM herkinorin on DAMGO‐induced Kir3.2 currents, n = 6 cells. The applications of 100‐nM DAMGO and 1‐μM herkinorin are indicated by the horizontal lines. Right panel shows representative I–V curve traces. (d) Representative traces for the effect of 1‐μM DAMGO on Kir3.2 currents activated by 100‐nM DAMGO, n = 6 cells. The applications of 100‐nM DAMGO and 1‐μM DAMGO are indicated by the horizontal lines. Right panel shows representative I–V curves. (e) Summary of normalized Kir3.2 inward currents (−100 mV). Currents induced 100‐nM DAMGO were measured before the application, and after wash out of the test compounds; the mean of these two current amplitudes was used as the normalizing point to assess the effects of all test compounds. Statistical analysis was performed with paired t test *P < .05
Herkinorin at 1 μM resulted in a current decrease corresponding to 11.7 ± 5.4% inhibition (P > .05), n = 6, but the currents showed substantial desensitization; therefore, this small decrease may not be due to herkinorin (Figure 6c). To account for the spontaneous current decrease, inhibition levels were calculated by taking the average of the Kir3.2 currents before and after application of the test compounds as the control level. In a control experiment, application of 1‐μM DAMGO in the presence of 100‐nM DAMGO leads to a slight potentiation 5.0 ± 3.1% (P > .05), n = 6 (Figure 6d).
While clinically a strong agonist, morphine is also considered as a partial agonist in some in vitro assays (Livingston, Mahoney, Manglik, Sunahara, & Traynor, 2018). Morphine (1 μM) also induced only a small statistically non‐significant inhibition of DAMGO‐induced Kir3.2 currents 17.9 ± 7.4% (P > .05, n = 6; Figure 6e). When Kir3.2 channels were activated by 1‐μM morphine, co‐application of either PZM21 or TRV130 reversed this activation (Figure 7). For 100‐nM PZM21, inhibition of morphine‐induced Kir3.2 currents reached 59.6 ± 2.3% of the initial level (n = 8, P < .05) and 100‐nM TRV130 induced 76.6 ± 7.4% inhibition (n = 8, P < .05). In the same recordings, DAMGO (100 nM) significantly increased morphine‐induced Kir3.2 currents by 54.2 ± 11.1% (n = 9).
Figure 7.

The effects of PZM21, TRV130, and DAMGO on morphine‐induced Kir3.2 currents. Whole‐cell patch‐clamp measurements in HEK293 cells transfected with μ receptors (μOR) and Kir3.2 channels were performed as described in Section 2. (a) Representative trace for the effects of 100‐nM PZM21 (n = 8 cells); 100‐nM TRV130 (n = 8 cells); and 100‐nM DAMGO (n = 9 cells) on morphine‐activated Kir3.2 currents. The applications of morphine, PZM21, TRV130, and DAMGO are indicated by the horizontal lines. (b) Summary of normalized Kir3.2 inward current (−100 mV). Statistical analysis was performed with t test *P < .05
3.4. G‐protein‐biased μ receptor agonists inhibit N‐type voltage‐gated channels less efficiently than the full agonist DAMGO
N‐type voltage‐gated Ca2+ channels (CaV2.2) are also classical targets for Gβγ subunits. In cells co‐transfected with μ receptors and CaV2.2 channel subunits, 1‐μM DAMGO inhibited the Ba2+ currents by 34.4 ± 4.7%, n = 12. For the two other tested reagents, the level of inhibition was less prominent, 17.5 ± 8% for 1‐µM PZM21 (n = 5) and 13.8 ± 2.3% for 1‐µM TRV130 (n = 7; Figure 8). Both the effects of PZM21 (Figure 8c) and TRV130 (Figure 8f) were significantly smaller than that of DAMGO. The same voltage protocol did not induce any currents in HEK293 cells transfected only with μ receptors (n = 5, data not shown). Note that our protocol does not differentiate between Gβγ‐mediated voltage‐dependent and non‐Gβγ‐mediated voltage‐independent inhibition.
Figure 8.
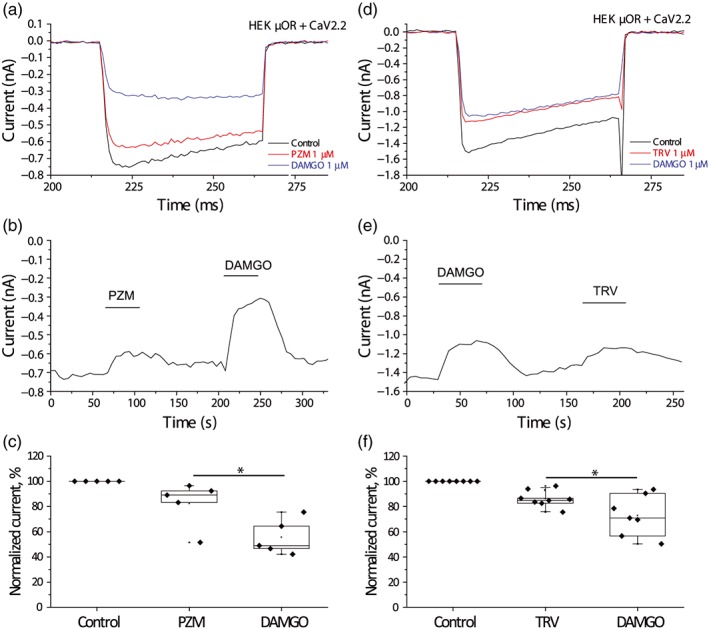
The effects of μ receptor (μOR) agonists on CaV2.2 channels in HEK293 cells. Whole‐cell patch‐clamp measurements in HEK293 cells transfected with μ receptors and CaV2.2 channel subunits were performed as described in Section 2. (a) Representative traces of CaV2.2 barium currents activated by voltage steps from V h − 80 mV to +10 mV and the inhibitory effect of 1‐μM PZM21 and 1‐μM DAMGO. (b) Time course of the IBa in the presence of PZM21 and DAMGO. (c) Summary of normalized currents for 1‐μM PZM21 and 1‐μM DAMGO (n = 5 cells). (d) Representative traces of CaV2.2 barium currents activated by voltage steps from V h − 80 mV to +10 mV and the inhibitory effect of 1‐μM TRV130 and 1‐μM DAMGO. (e) Time course of the IBa in the presence of TRV130 and DAMGO. (f) Summary of normalized currents for 1‐μM TRV130 and 1‐μM DAMGO (n = 8 cells). Statistical significance was calculated with t test, *P < .05
Our data show that G‐protein‐biased μ receptor agonists were less efficient than the full agonist DAMGO in modulating three different ion channel targets to Gαi–Gβγ signalling. We next tested if the G‐protein‐biased agonists were also less efficient than DAMGO on G‐protein dissociation, using a FRET‐based assay.
3.5. G‐protein μ receptor‐biased agonists are less efficient than DAMGO at inducing G‐protein dissociation
To assess the dissociation level of Gα and Gβγ during μ receptor activation, we used a FRET‐based assay in cells transfected with the μ receptor and a tricistronic construct expressing the mTurqoise2‐tagged Gαi1 (FRET donor), the Venus‐tagged Gγ2 (FRET acceptor), and Gβ1 (van Unen et al., 2016). A reduction in the FRET signal in these experiments indicates dissociation of the Gα–Gβγ heterotrimer complex, or a change in relative conformation between the FRET donor and acceptor. In this experiment, cells treated with different DAMGO concentrations demonstrated clear antiparallel signals, an increase at 480 nm and a decrease at 530‐nm emission wavelengths indicating a decrease in FRET (Figure 9a). For 10‐nM DAMGO, the FRET signal showed a slight 0.48 ± 0.12% decrease; 100 nM evoked 2.49 ± 0.36%; and 1‐μM DAMGO resulted in 3.92 ± 0.27% decrease in the FRET signal (Figure 9b). We observed significantly weaker responses for 1‐μM PZM21 (1.58 ± 0.41% decrease), 1‐μM TRV130 (1.03 ± 0.17% decrease), and 1‐μM herkinorin (1.52 ± 0.32% decrease; Figure 9c,d). These data indicate a smaller level of dissociation of Gαi1 and Gβγ compared to the effect of the full μ receptor agonist DAMGO by these agonists. As 1‐μM herkinorin did not evoke a clear reversal of DAMGO‐induced inhibition of TRPM3 and DAMGO‐induced activation of Kir3.2, we also tested higher concentrations of this drug in the FRET assay. For 10‐μM herkinorin, we observed 0.88 ± 0.14% of FRET signal decrease, an effect very similar to that induced by 1‐μM herkinorin. The effects of PZM21, TRV130, and herkinorin were significantly smaller than that induced by 1‐μM DAMGO (Figure 9d).
Figure 9.
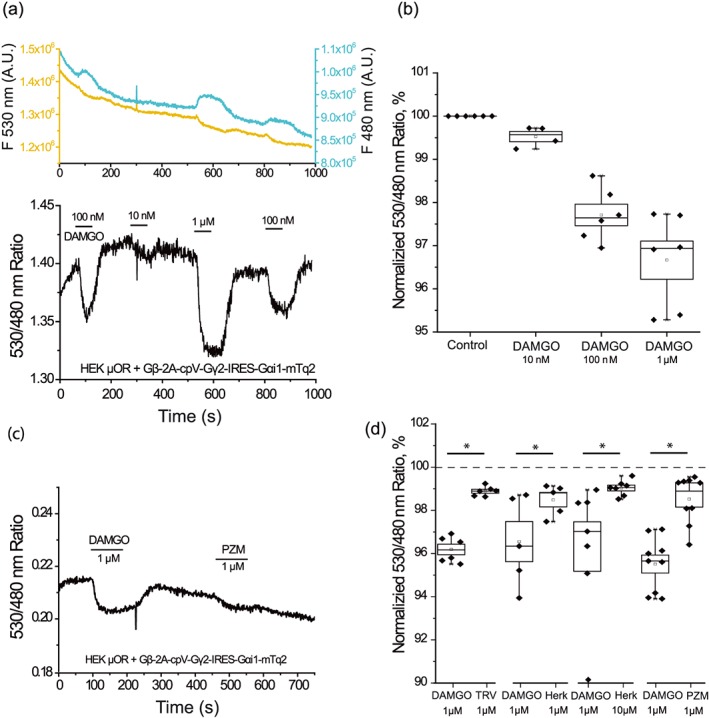
Dissociation of Gα and Gβγ evoked by μ receptor (μOR) agonists in FRET experiments. Fluorescence measurements were performed in HEK293 cells transfected with mTurqoise2‐tagged Gαi1 (FRET donor), a Venus‐tagged Gγ2 (FRET acceptor), and Gβ1 expressed from a tricistronic vector (Gβ‐2A‐cpV‐Gγ2‐IRES‐Gαi1‐mTq2) and μ receptor as described in Section 2. (a) Upper panel, fluorescence traces detected at 480 for FRET donor (blue line) and 535 nm for FRET acceptor (yellow line); excitation wavelength was 430 nm. Lower panel shows the ratio of the individual traces (FRET signal). The applications of 10‐nM, 100‐nM, and 1‐µM DAMGO are indicated by the horizontal lines. (b) Summary of the data. (c) Representative measurement showing the effect of 1‐μM DAMGO and 1‐μM PZM21. (d) Summary of the normalized responses of FRET signals evoked by the applications of 1‐μM TRV130 (n = 6 cells), 1‐μM herkinorin (n = 5 cells), 10‐μM herkinorin (n = 7 cells), and 1‐μM PZM21 (n = 9 cells), compared to the effect of 1‐μM DAMGO in the same experiment for each compound. All fluorescence ratio traces were normalized to the ratio level before the application of the stimulus. Statistical significance was calculated using t test comparing the effect of DAMGO to the tested agonist in each experiment *P < .05
In conclusion, we demonstrated that the recently described Gαi‐biased agonists PZM21 and TRV130 behave as weak partial agonists using three ion channel targets for the Gβγ subunits as a functional readout.
4. DISCUSSION
Most clinically used opioid medications activate the Gαi‐coupled μ receptors. Ligand binding to these receptors leads to the dissociation of Gαi and Gβγ and also induces β‐arrestin recruitment. Both G‐protein subunits have their own effectors; Gαi inhibits adenylate cyclase, and thus decreases cAMP levels. Gβγ subunits have multiple targets including G‐protein‐coupled inwardly rectifying potassium channels (Kir3; Logothetis, Kurachi, Galper, Neer, & Clapham, 1987) and voltage‐gated Ca2+ channels (N‐type and P/Q‐type; Currie, 2010). A recently described target of Gi signalling is the TRPM3 ion channel, which is robustly inhibited by Gβγ subunits (Badheka et al., 2017; Dembla et al., 2017; Quallo et al., 2017). Non‐ion channel targets of Gβγ include phosphoinositide 3‐kinase‐γ (PI3Kγ) and MAPKs (Clapham & Neer, 1997; Khan et al., 2013).
β‐arrestin 1 (arrestin 2) and β‐arrestin 2 (arrestin 3) are important regulators of G‐protein signalling. They were originally described as mediators of agonist‐induced desensitization and internalization (DeWire et al., 2007), and they were presumed to be responsible for many side effects of opioids, such as tolerance and respiratory depression. Recently, β‐arrestins have emerged as independent signalling mediators regulating various effectors (DeWire et al., 2007), including ion channels of the TRP family (Liu et al., 2017). We initiated this study to determine the possible role of the β‐arrestin pathway in regulating TRPM3 channels, the newly described targets of Gβγ signalling.
We found that all three G‐protein‐biased μ receptor agonists, PZM21, TRV130, and herkinorin induced significantly smaller inhibition of TRPM3 channels compared to the full agonist DAMGO both in Ca2+ imaging and patch‐clamp experiments. PZM21 and TRV130 also reversed the inhibitory effect of DAMGO when co‐applied with this full agonist. Similar to their effects on TRPM3 channels, PZM21 and TRV130, but not herkinorin robustly opposed the effect on DAMGO on Kir3.2 channels when co‐applied with this full agonist. Voltage‐gated CaV2.2 channels were also inhibited less by PZM21 and TRV130 compared to DAMGO. These results could indicate two possibilities; either the β‐arrestin pathway is required for the activation of all three ion‐channel targets of Gβγ or PZM21, TRV130, and herkinorin act as partial agonists in G‐protein signalling. To differentiate between these two possibilities, we re‐examined the effects of these compounds on G‐protein dissociation. We performed FRET experiments and found that all the test reagents induced weaker Gα–Gβγ dissociation than DAMGO, indicating that PZM21 and TRV130 behave as weak partial agonists when regulating ion channel targets for Gβγ subunits.
Morphine, while clinically a highly efficacious medication, is also considered a partial agonist in some in vitro assays (Livingston et al., 2018). In our hands, morphine did not induce a clear reversal of DAMGO‐induced Kir3.2 currents. When Kir3.2 channels were activated by morphine first, both PZM21 and TRV130 evoked a clear inhibition when co‐applied with morphine. These data show that PZM21 and TRV130 are lower efficacy agonists than the clinically‐used morphine.
All three compounds tested here were developed as G‐protein‐biased μ receptor agonists to avoid recruitment of β‐arrestin with the promise of novel pain medications with fewer side effects. Herkinorin demonstrated strong antinociceptive effects, similar to that of morphine, in several behavioural assays without inducing tolerance (Lamb, Tidgewell, Simpson, Bohn, & Prisinzano, 2012). At the cellular level, it was demonstrated that herkinorin did not recruit β‐arrestin 2 and did not promote internalization but had Gα agonist activity similar, or stronger than that of DAMGO, detected by a MAPK phosphorylation assay (Groer et al., 2007).
TRV130 was also shown to be effective in a variety of behavioural tests with an efficacy and potency similar to that of morphine, while inducing less constipation and respiratory suppression compared to similar doses of morphine (DeWire et al., 2013). As a biased agonist, it strongly activated Gαi (cAMP assay) without recruitment of β‐arrestin 2 (enzyme complementation assay; DeWire et al., 2013). These findings were confirmed by another independent study on TRV130 (Mori et al., 2017).
The newest Gαi‐biased μ receptor agonist PZM21 was identified based on computational docking of over 3 million compounds to the structure of the μ receptor, and subsequent structure based optimization (Manglik et al., 2016). PZM21 is structurally unrelated to known opioids; it was shown to induce G‐protein activation using various high throughput optical assays, but it induced no recruitment of β‐arrestin 2, with very low levels of receptor internalization. In behavioural experiments, PZM21 had antinociceptive properties in multiple animal tests with significantly less adverse side effects, such as respiratory depression, than known opioids (Manglik et al., 2016). Manglik et al. also compared herkinorin and TRV130 to the effects of PZM21, DAMGO as well as morphine is several high throughput cellular assays. They concluded that PZM21 is a G‐protein biased, potent, selective, and efficacious μ receptor agonist; TRV130 behaved similarly to PZM21, but they found that herkinorin induced substantial β‐arrestin recruitment.
What is the explanation for the difference between our data and those of Manglik et al? The most likely explanation is that the different assays used to assess G‐protein activity have different sensitivities and dynamic ranges. The high throughput optical assays, such as glosensor, are not linear readouts of cAMP levels, and while the assay is highly sensitive at low levels of activity, it may saturate at high activities such as that induced by DAMGO, which could lead to overestimating the maximal effect of a weaker agonist. Indeed, while PZM21 was designated as efficacious in G‐protein stimulation, it induced between 70% and ~100% of the effect of DAMGO depending on the assay (Manglik et al., 2016). Ionic currents measured in patch‐clamp experiments on the other hand are linear readouts of channel activity and currents do not saturate. Channel activity may theoretically saturate if the available channels are maximally activated, but this is unlikely to happen if G‐proteins are not overexpressed. Using channel activity as a readout is therefore more sensitive at high activity levels. Consistent with this, the dose‐response relationship for DAMGO was shifted considerably to the right in our Kir3.2 channel experiments compared to the experiments reported by Manglik et al. In their publication, 1‐μM DAMGO exerted a maximal effect in all assays, and 100‐nM DAMGO exerted a close to maximal effect in most assays. In our Kir3.2 experiments, 1‐μM DAMGO induced ~85% of the maximal effect and 100 nM induced ~60% of the maximal effect.
In our FRET‐based G‐protein dissociation experiments, all three agonists induced well below 50% of the signal induced by DAMGO. In our ion channel experiments, the difference between DAMGO and the biased agonists was variable; depending on the channel, and the concentrations used, but it was again, generally well below the effect induced by DAMGO. We performed the most detailed concentration‐dependence studies with Kir3.2 channels, and none of the compounds exerted more than 25% of the maximal effect of DAMGO even at very high concentrations (50 μM; Figure 5). The difference between DAMGO and the biased agonists was less pronounced for TRPM3 channels (Figure 3). Our most definitive data showing that these compounds act as partial agonists are the clear reversal of the effect of DAMGO on TRPM3 and Kir3.2 channels by PZM21 and TRV130 (Figures 4 and 6), as well as the reversal of the effect of morphine on Kir3.2 channels by PZM21 and TRV130 (Figure 7).
A recent report showed that PZM21 induced respiratory depression similar to that evoked by morphine (Hill et al., 2018), which is in contrast to the earlier report describing this biased agonist (Manglik et al., 2016). They also showed that PZM21 had substantially lower efficacy than DAMGO, using a BRET‐based assay to detect the dissociation of Gαi1 and Gγ2 (Hill et al., 2018). Our data are also consistent with another recent publication, which using conformational biosensor binding to the purified μ receptor, showed that PZM21 had lower efficacy than morphine, or fentanyl, two opioids used clinically (Livingston et al., 2018).
Our data with herkinorin are less conclusive. Herkinorin clearly induced smaller inhibition of TRPM3 and smaller activation Kir3.2 currents than DAMGO even at very high concentrations, which suggests that it is a partial agonist. It also induced smaller G‐protein dissociation than DAMGO even at 10 μM in our FRET‐based assay, again suggesting that it acts as a partial agonist. In contrast, herkinorin, unlike PZM21 and TRV130, did not induce a robust reversal of the effect of DAMGO on TRPM3 currents (Figure 4) and on Kir3.2 channels (Figure 6), even though the marked desensitization of both currents makes a small effect difficult to assess. It is not clear why herkinorin behaves differently from PZM21 and TRV130 in reversing the effects of DAMGO, and we have not further investigated this difference. Also, our data with herkinorin on PregS‐induced Ca2+ signals in DRG neurons are at odds with those of Dembla et al., who showed that herkinorin evoked an inhibitory effect similar to that induced by DAMGO in the same assay (Dembla et al., 2017). It was also shown that in DRG neurons, 10‐μM herkinorin induced an inhibitory effect on N‐type voltage‐gated Ca2+ channels similar to that induced by 1‐μM DAMGO (Rowan et al., 2014).
In summary, our data conclusively demonstrate that the G‐protein‐biased PZM21 and TRV130 act as weak partial μ receptor agonists, when signalling to ion channel targets through G‐proteins.
AUTHOR CONTRIBUTIONS
Y.Y. and T.R. designed the experiments. Y.Y. performed the experiments and analyzed the data. Y.Y. and T.R. wrote the manuscript. T.R. obtained the funding and supervised the overall project.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Table S1. Pair‐wise comparisons of the effects of DAMGO with those of PZM21, TRV130 or herkinorin on inhibition of outward and inward TRPM3 currents from Figure 3. Lower case p values correspond to the significance level of inhibition by the tested agonist and upper case P values correspond to the comparison between the tested agonist and DAMGO in the same experiment.
Figure S1. Differential inhibition of PregS‐induced Ca2+ signals by μ receptor agonists in DRG neurons; individual traces and scatter plots (A‐E) Individual Ca2+ imaging traces from Figure 1, the applications of the various compounds are shown with the horizontal lines. (F) Scatter plots of the responses of individual DRG neurons. Changes in the relative fluorescence ratio were calculated using the following formula: ΔR2/ΔR1 = 100(R2 − R0)/ (R1 − R0) normalized to the first pulse of PregS.
Figure S2. Naloxone inhibits Kir3.2 (GIRK2) currents induced by TRV130 and PZM21 Whole‐cell patch clamp measurements in HEK cells transfected with μ receptor and Kir3.2 channels were performed as described in Methods. (A) Representative traces for the effects of 1 μM naloxone on Kir3.2 currents activated by PZM21, (n = 5) and TRV130 (n = 6). The applications of 1 μM naloxone, PZM21 and TRV130 are indicated by the horizontal lines. (B) Summary of normalized Kir3.2 inward current (−100 mV). Statistical analysis was performed with ANOVA *p < 0.05.
ACKNOWLEDGEMENTS
We thank Drs. Veit Flockerzi and Stephan Philipp for the mouse TRPM3a2 clone; the Kir3.2 clone was a kind gift from Dr Tooraj Mirshahi. This work was supported from National Institute of Neurological Disorders and Stroke Grant NS 055159 and National Institute of General Medical Sciences Grant GM 093290. A previous version of this work was posted on BioRxiv: https://www.biorxiv.org/content/10.1101/445536v1.
Yudin Y, Rohacs T. The G‐protein‐biased agents PZM21 and TRV130 are partial agonists of μ‐opioid receptor‐mediated signalling to ion channels. Br J Pharmacol. 2019;176:3110–3125. 10.1111/bph.14702
REFERENCES
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174(S1), S160–S194. 10.1111/bph.13884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badheka, D. , Borbiro, I. , & Rohacs, T. (2015). Transient receptor potential melastatin 3 is a phosphoinositide dependent ion channel. Journal of General Physiology, 146, 65–77. 10.1085/jgp.201411336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badheka, D. , Yudin, Y. , Borbiro, I. , Hartle, C. M. , Yazici, A. , Mirshahi, T. , & Rohacs, T. (2017). Inhibition of transient receptor potential melastatin 3 ion channels by G‐protein βγ subunits. eLife, 6, e26147 10.7554/eLife.26147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum, A. I. , Bautista, D. M. , Scherrer, G. , & Julius, D. (2009). Cellular and molecular mechanisms of pain. Cell, 139, 267–284. 10.1016/j.cell.2009.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbiro, I. , Badheka, D. , & Rohacs, T. (2015). Activation of TRPV1 channels inhibit mechanosensitive Piezo channel activity by depleting membrane phosphoinositides. Science Signaling, 8, ra15 10.1126/scisignal.2005667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham, D. E. , & Neer, E. J. (1997). G protein βγ subunits. Annual Review of Pharmacology and Toxicology, 37, 167–203. 10.1146/annurev.pharmtox.37.1.167 [DOI] [PubMed] [Google Scholar]
- Currie, K. P. (2010). G protein modulation of CaV2 voltage‐gated calcium channels. Channels (Austin, Tex.), 4, 497–509. 10.4161/chan.4.6.12871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembla, S. , Behrendt, M. , Mohr, F. , Goecke, C. , Sondermann, J. , Schneider, F. M. , … Oberwinkler, J. (2017). Anti‐nociceptive action of peripheral mu‐opioid receptors by Gβγ protein‐mediated inhibition of TRPM3 channels. eLife, 6, e26280 10.7554/eLife.26280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire, S. M. , Ahn, S. , Lefkowitz, R. J. , & Shenoy, S. K. (2007). β‐arrestins and cell signaling. Annual Review of Physiology, 69, 483–510. 10.1146/annurev.physiol.69.022405.154749 [DOI] [PubMed] [Google Scholar]
- DeWire, S. M. , Yamashita, D. S. , Rominger, D. H. , Liu, G. , Cowan, C. L. , Graczyk, T. M. , … Violin, J. D. (2013). A G protein‐biased ligand at the mu‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. The Journal of Pharmacology and Experimental Therapeutics, 344, 708–717. 10.1124/jpet.112.201616 [DOI] [PubMed] [Google Scholar]
- Groer, C. E. , Tidgewell, K. , Moyer, R. A. , Harding, W. W. , Rothman, R. B. , Prisinzano, T. E. , & Bohn, L. M. (2007). An opioid agonist that does not induce μ‐opioid receptor—Arrestin interactions or receptor internalization. Molecular Pharmacology, 71, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held, K. , Kichko, T. , De Clercq, K. , Klaassen, H. , Van Bree, R. , Vanherck, J. C. , … Vriens, J. (2015). Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proceedings of the National Academy of Sciences of the United States of America, 112, E1363–E1372. 10.1073/pnas.1419845112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, R. , Disney, A. , Conibear, A. , Sutcliffe, K. , Dewey, W. , Husbands, S. , … Henderson, G. (2018). The novel mu‐opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. British Journal of Pharmacology, 175, 2653–2661. 10.1111/bph.14224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. M. , Sleno, R. , Gora, S. , Zylbergold, P. , Laverdure, J. P. , Labbe, J. C. , … Hebert, T. E. (2013). The expanding roles of Gβγ subunits in G protein‐coupled receptor signaling and drug action. Pharmacological Reviews, 65, 545–577. 10.1124/pr.111.005603 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, K. , Tidgewell, K. , Simpson, D. S. , Bohn, L. M. , & Prisinzano, T. E. (2012). Antinociceptive effects of herkinorin, a MOP receptor agonist derived from salvinorin A in the formalin test in rats: New concepts in mu opioid receptor pharmacology: From a symposium on new concepts in μ‐opioid pharmacology. Drug and Alcohol Dependence, 121, 181–188. 10.1016/j.drugalcdep.2011.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. H. , Gong, Z. , Liang, Z. L. , Liu, Z. X. , Yang, F. , Sun, Y. J. , … Sun, J. P. (2017). Arrestin‐biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nature Communications, 8, 14335 10.1038/ncomms14335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston, K. E. , Mahoney, J. P. , Manglik, A. , Sunahara, R. K. , & Traynor, J. R. (2018). Measuring ligand efficacy at the mu‐opioid receptor using a conformational biosensor. eLife, 7, e32499 10.7554/eLife.32499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis, D. E. , Kurachi, Y. , Galper, J. , Neer, E. J. , & Clapham, D. E. (1987). The βγ subunits of GTP‐binding proteins activate the muscarinic K+ channel in heart. Nature, 325, 321–326. 10.1038/325321a0 [DOI] [PubMed] [Google Scholar]
- Lukacs, V. , Thyagarajan, B. , Varnai, P. , Balla, A. , Balla, T. , & Rohacs, T. (2007). Dual regulation of TRPV1 by phosphoinositides. The Journal of Neuroscience, 27, 7070–7080. 10.1523/JNEUROSCI.1866-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs, V. , Yudin, Y. , Hammond, G. R. , Sharma, E. , Fukami, K. , & Rohacs, T. (2013). Distinctive changes in plasma membrane phosphoinositides underlie differential regulation of TRPV1 in nociceptive neurons. Journal of Neuroscience, 33, 11451–11463. 10.1523/JNEUROSCI.5637-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin, S. A. , Davis, B. M. , & Molliver, D. C. (2007). Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nature Protocols, 2, 152–160. 10.1038/nprot.2006.461 [DOI] [PubMed] [Google Scholar]
- Manglik, A. , Lin, H. , Aryal, D. K. , McCorvy, J. D. , Dengler, D. , Corder, G. , … Shoichet, B. K. (2016). Structure‐based discovery of opioid analgesics with reduced side effects. Nature, 537, 185–190. 10.1038/nature19112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, T. , Kuzumaki, N. , Arima, T. , Narita, M. , Tateishi, R. , Kondo, T. , … Narita, M. (2017). Usefulness for the combination of G‐protein‐ and β‐arrestin‐biased ligands of mu‐opioid receptors: Prevention of antinociceptive tolerance. Molecular Pain, 13, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberwinkler, J. , Lis, A. , Giehl, K. M. , Flockerzi, V. , & Philipp, S. E. (2005). Alternative splicing switches the divalent cation selectivity of TRPM3 channels. The Journal of Biological Chemistry, 280, 22540–22548. 10.1074/jbc.M503092200 [DOI] [PubMed] [Google Scholar]
- Oberwinkler, J. , & Philipp, S. E. (2014). TRPM3. Handbook of Experimental Pharmacology, 222, 427–459. 10.1007/978-3-642-54215-2_17 [DOI] [PubMed] [Google Scholar]
- Quallo, T. , Alkhatib, O. , Gentry, C. , Andersson, D. A. , & Bevan, S. (2017). G protein βγ subunits inhibit TRPM3 ion channels in sensory neurons. eLife, 6, e26138 10.7554/eLife.26138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal, K. M. , Schmid, C. L. , Groer, C. E. , & Bohn, L. M. (2011). Functional selectivity at the mu‐opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacological Reviews, 63, 1001–1019. 10.1124/pr.111.004598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghib, A. , Bertaso, F. , Davies, A. , Page, K. M. , Meir, A. , Bogdanov, Y. , & Dolphin, A. C. (2001). Dominant‐negative synthesis suppression of voltage‐gated calcium channel Cav2.2 induced by truncated constructs. The Journal of Neuroscience, 21, 8495–8504. 10.1523/JNEUROSCI.21-21-08495.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan, M. P. , Bierbower, S. M. , Eskander, M. A. , Szteyn, K. , Por, E. D. , Gomez, R. , … Jeske, N. A. (2014). Activation of mu opioid receptors sensitizes transient receptor potential vanilloid type 1 (TRPV1) via β‐arrestin‐2‐mediated cross‐talk. PLoS ONE, 9, e93688 10.1371/journal.pone.0093688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla, N. , Minkowitz, H. S. , Soergel, D. G. , Burt, D. A. , Subach, R. A. , Salamea, M. Y. , … Skobieranda, F. (2017). A randomized, Phase IIb study investigating oliceridine (TRV130), a novel μ‐receptor G‐protein pathway selective (μ‐GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. Journal of Pain Research, 10, 2413–2424. 10.2147/JPR.S137952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick, P. , & Volkow, N. D. (2016). Re‐energizing the development of pain therapeutics in light of the opioid epidemic. Neuron, 92, 294–297. 10.1016/j.neuron.2016.09.051 [DOI] [PubMed] [Google Scholar]
- Stein, C. (2016). Opioid receptors. Annual Review of Medicine, 67, 433–451. 10.1146/annurev-med-062613-093100 [DOI] [PubMed] [Google Scholar]
- van Unen, J. , Stumpf, A. D. , Schmid, B. , Reinhard, N. R. , Hordijk, P. L. , Hoffmann, C. , … Goedhart, J. (2016). A new generation of FRET sensors for robust measurement of Gαi1, Gαi2 and Gαi3 activation kinetics in single cells. PLoS ONE, 11, e0146789 10.1371/journal.pone.0146789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewauw, I. , De Clercq, K. , Mulier, M. , Held, K. , Pinto, S. , Van Ranst, N. , … Voets, T. (2018). A TRP channel trio mediates acute noxious heat sensing. Nature, 555, 662–666. 10.1038/nature26137 [DOI] [PubMed] [Google Scholar]
- Viscusi, E. R. , Webster, L. , Kuss, M. , Daniels, S. , Bolognese, J. A. , Zuckerman, S. , … Skobieranda, F. (2016). A randomized, phase 2 study investigating TRV130, a biased ligand of the μ‐opioid receptor, for the intravenous treatment of acute pain. Pain, 157, 264–272. 10.1097/j.pain.0000000000000363 [DOI] [PubMed] [Google Scholar]
- Vriens, J. , Owsianik, G. , Hofmann, T. , Philipp, S. E. , Stab, J. , Chen, X. , … Voets, T. (2011). TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron, 70, 482–494. 10.1016/j.neuron.2011.02.051 [DOI] [PubMed] [Google Scholar]
- Wagner, T. F. , Loch, S. , Lambert, S. , Straub, I. , Mannebach, S. , Mathar, I. , … Oberwinkler, J. (2008). Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nature Cell Biology, 10, 1421–1430. 10.1038/ncb1801 [DOI] [PubMed] [Google Scholar]
- Yudin, Y. , Lutz, B. , Tao, Y. X. , & Rohacs, T. (2016). Phospholipase C δ4 regulates cold sensitivity in mice. The Journal of Physiology, 594, 3609–3628. 10.1113/JP272321 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Pair‐wise comparisons of the effects of DAMGO with those of PZM21, TRV130 or herkinorin on inhibition of outward and inward TRPM3 currents from Figure 3. Lower case p values correspond to the significance level of inhibition by the tested agonist and upper case P values correspond to the comparison between the tested agonist and DAMGO in the same experiment.
Figure S1. Differential inhibition of PregS‐induced Ca2+ signals by μ receptor agonists in DRG neurons; individual traces and scatter plots (A‐E) Individual Ca2+ imaging traces from Figure 1, the applications of the various compounds are shown with the horizontal lines. (F) Scatter plots of the responses of individual DRG neurons. Changes in the relative fluorescence ratio were calculated using the following formula: ΔR2/ΔR1 = 100(R2 − R0)/ (R1 − R0) normalized to the first pulse of PregS.
Figure S2. Naloxone inhibits Kir3.2 (GIRK2) currents induced by TRV130 and PZM21 Whole‐cell patch clamp measurements in HEK cells transfected with μ receptor and Kir3.2 channels were performed as described in Methods. (A) Representative traces for the effects of 1 μM naloxone on Kir3.2 currents activated by PZM21, (n = 5) and TRV130 (n = 6). The applications of 1 μM naloxone, PZM21 and TRV130 are indicated by the horizontal lines. (B) Summary of normalized Kir3.2 inward current (−100 mV). Statistical analysis was performed with ANOVA *p < 0.05.