

Abstract
High expression of VEGF is associated with immature angiogenesis within the urinary bladder wall and bladder afferent nerve sensitization, leading to visceral hyperalgesia and pelvic pain. Research suggests a shift in VEGF alternative splice variant (VEGF-Axxxa and VEGF-Axxxb) expression with several pathologies (e.g., neuropathic pain and inflammation) as well as differing effects on pain. Translational studies have also demonstrated increased total VEGF expression in the bladders of women with interstitial cystitis/bladder pain syndrome. In the present study, we quantified VEGF alternative splice variant expression in lower urinary tract tissues under control conditions and with cyclophosphamide (CYP)-induced cystitis. Using conscious cystometry and intravesical instillation of a potent and selective VEGF receptor 2 (VEGFR2) tyrosine kinase inhibitor (Ki-8751, 1 mg/kg) in Wistar rats (male and female) with acute and chronic CYP-induced cystitis and control (no CYP) rats, we further determined the functional effects of VEGFR2 blockade on bladder function. With VEGFR2 blockade, bladder capacity increased (P ≤ 0.01) in male and female control rats as well as in male and female rats with acute (P ≤ 0.05) or chronic (P ≤ 0.01 or P ≤ 0.05, respectively) CYP-induced cystitis. Void volume also increased in female control rats (P ≤ 0.01) and female rats with acute (P ≤ 0.05) or chronic (P ≤ 0.05) CYP-induced cystitis as well as in male control rats (P ≤ 0.05) and male rats with chronic CYP-induced cystitis (P ≤ 0.01). These data suggest that VEGF may be a biomarker for interstitial cystitis/bladder pain syndrome and that targeting VEGF/VEGFR2 signaling may be an effective treatment.
Keywords: growth factors, inflammation, interstitial cystitis, micturition, pelvic pain, vascular endothelial growth factor
INTRODUCTION
Interstitial cystitis/bladder pain syndrome (IC/BPS) is a chronic pelvic pain condition with no known etiology. IC/BPS is described by patients as a myriad of unpleasant sensations, including pain, pressure, and discomfort throughout the pelvic region. Patients also experience increased urgency and frequency of voiding, without the presence of infection or other secondary causes (20). Risk factors for developing IC/BPS have not been determined, and diagnosis is based on symptoms followed by an exclusion of other pathologies (14). Neither a universal biomarker nor an effective treatment has been identified. A clinically relevant biomarker for IC/BPS could aid in understanding the pathophysiology of the disease, provide a useful diagnostic tool, and offer a potential treatment target.
In IC/BPS, inflammation causes damage to the mucosal as well as the glycosaminoglycan layer. The urothelium is a large component of the mucosa, and, upon inflammation, cytokines and chemokines are recruited to the sites of damage, where mast cell activation occurs (18). Furthermore, the urothelium barrier becomes leaky, allowing excess cations (e.g., K+) to enter the inner layers of the urinary bladder. This influx of K+ causes sprouting and sensitization of afferent C-fibers in the urinary bladder and further increases mast cell recruitment (18, 34). During this process, research suggests that damaged urothelial cells release substances (e.g., ATP, nitric oxide, and nerve growth factor) that further sensitize bladder afferent nerves. In turn, the nerves release neurochemicals locally in the bladder as well as centrally, creating a vicious, positive, proinflammatory feedback loop that contributes to hyperalgesia and allodynia (5, 7, 18, 43, 44).
VEGF-A (hereafter referred to as VEGF) is a central growth factor underlying pain associated with the lower urinary tract (LUT). Alternative splicing of VEGF yields two isoforms, VEGF-Axxxa and VEGF-Axxxb (where xxx represents the number of amino acids in the mature protein) (21), which differ only in six COOH-terminal amino acids in the mature protein. Previous research by Hulse et al. (24) has suggested an opposing role for the isoforms in models of neuropathic pain. Using exogenous protein as well as splicing inhibitors to control endogenous splicing in the environment of peripheral nerve injury, they demonstrated that VEGF-Axxxa increases pain, whereas VEGF-Axxxb decreases pain severity (24).
Both VEGF isoforms primarily signal through two receptors (VEGFR), VEGFR1 and VEGFR2, both of which are receptor tyrosine kinases. However, the downstream effects of VEGFR2 activation vary between the splice variants, since VEGFxxxa is a full agonist for VEGFR2 and VEGFxxxb lacks the ability to cause efficient autophosphorylation of the receptor (21, 26, 40). VEGFR2 is the best-characterized VEGF receptor, while the function of VEGFR1 remains more uncertain. VEGFR1 may regulate and dampen VEGF/VEGFR2 signaling, in that it acts as a “decoy” by binding and trapping VEGF (33). VEGF binding to VEGFR2 typically results in angiogenesis via intracellular phosphorylation and activation of several downstream signaling cascades that, ultimately, result in endothelial cell proliferation and blood vessel formation (11, 28). Under normal conditions, angiogenesis is necessary to ensure that target tissues receive adequate blood supply. If an organ is hypoxic, target cells secrete VEGF to promote angiogenesis. VEGF is then downregulated once the target organ receives an adequate supply of oxygen (8, 9). However, under pathological conditions, secretion of VEGF contributes to the formation of immature blood vessels that, subsequently, become leaky and hemorrhagic (16, 27).
Angiogenic factors, such as VEGF, are associated with, and contribute to, several neurological conditions (31, 39, 41). Although VEGF is a potent angiogenic factor contributing to angiogenesis and lymphangiogenesis, it may have additional roles in the urinary bladder during inflammation. VEGF and its receptors are expressed in urothelial, lamina propria, and intramural ganglia cells within the bladder wall under normal conditions. Additionally, under pathological conditions, such as inflammation, VEGF expression is upregulated in these areas (13, 27, 36). Furthermore, in the inflamed bladder, research suggests that VEGF promotes nerve plasticity. VEGF infusion into the mouse urinary bladder increased the number of sensory fibers expressing transient receptor potential vanilloid subfamily 1, substance P, and calcitonin gene-related peptide to levels similar to those reported in chronically inflamed mouse urinary bladder (37). VEGF infusion also caused an increase in vasodilation, edema, and recruitment of macrophages, all of which are associated with inflammation. Infusion of a VEGF-neutralizing antibody prevented inflammation-induced afferent nerve plasticity (37) and attenuated pelvic sensitivity, as determined by von Frey filament testing (29). The literature shows conflicting roles of VEGF in frequency of micturition; for example, anti-VEGF treatment involving anti-VEGF-neutralizing antibodies decreased pelvic hypersensitivity but failed to affect voiding frequency after cyclophosphamide (CYP)-induced cystitis in mice (29). In contrast, 2 wk of intravesical instillation of VEGF reduced bladder capacity in mice evaluated with conscious cystometry (31). Furthermore, the urinary bladders of patients with IC/BPS exhibit increased VEGF, and the levels of VEGF positively correlated to reported pain severity (27, 30). By blocking VEGF/VEGR2 signaling using Ki-8751, a potent selective VEGFR2 tyrosine kinase inhibitor, we hypothesized that functional improvements in micturition, as shown by increased bladder capacities and void volumes, will occur when this positive feedback loop is interrupted. We further aimed to examine VEGF splice variants in different LUT tissues under control and CYP conditions.
MATERIALS AND METHODS
Experimental animals.
These experiments from the Vizzard laboratory were performed in accordance with institutional and national guidelines and regulations. The University of Vermont Institutional Animal Care and Use Committee approved all experimental protocols (no. 14-060 and no. 13-030) involving animal use. Animal care was under the supervision of University of Vermont’s Office of Animal Care and Management in accordance with the Association for the Assessment and Accreditation of Laboratory Animal Care and National Institutes of Health guidelines. All efforts were made to minimize the potential for animal pain, stress, or distress.
Male and female Wistar rats (n = 6–8 male rats and 6–8 female rats per treatment group) were bred locally at the Larner College of Medicine at the University of Vermont. Rats were maintained under standard laboratory conditions: they were housed two per cage and maintained on a 12:12-h light-dark cycle, with free access to food and water. Estrous cycle status was not determined in female rats before use.
Tissue collection for mRNA analysis.
Rats were anesthetized with isoflurane (5% in oxygen) and euthanized via thoracotomy, and spinal cord segments (L6 and S1), dorsal root ganglia (DRG; L6 and S1), and urinary bladders were harvested from all treatment groups. The urinary bladder was quickly dissected under RNase-free conditions. The bladder was cut open along the midline and pinned to a Sylgard-coated dish, and the urothelium, including the suburothelium, was removed from the detrusor muscle with fine forceps and iris scissors (19); the urothelium and detrusor were evaluated separately by mRNA analysis.
Quantification of VEGF splice variant mRNA.
Stat-60 total RNA/mRNA isolation reagent (Tel-Test B Labs, Friendswood, TX) was used to extract total RNA from samples (n = 6). One microgram (bladder and spinal cord) or 0.1 µg (DRG) of RNA sample was used to synthesize cDNA using a mix of random hexamers and oligo(dT) primers with Moloney murine leukemia virus reverse transcriptase (Promega). Primers were designed using Oligo Primer Analysis Software 7.0. VEGF-Axxxa and VEGF-Axxxb isoform primers were designed against exon 7/8. The forward primer spans the intron/exon boundary: VEGF forward primer 5′-AGCATTTGTTTGTCCAAGATCC-3′. The reverse primers cover the region where the six different amino acids are coded: VEGF-Axxxa reverse primer 5′-TCACCGCCTTGGCTTGTCACAT-3′ and VEGF-Axxxb reverse primer 5′-TTAATCGGTCTTTCCTTTGAGA-3′. The quantitative PCR standard for each transcript was prepared with the amplified cDNA products ligated directly into the pCR2.1 TOPO vector using the TOPO TA cloning kit (Invitrogen). The nucleotide sequences of the inserts were verified by automated fluorescent dideoxy dye terminator sequencing (Vermont Integrative Genomics Resource). cDNA templates were diluted 10-fold and assayed using Luna universal quantitative PCR master mix (New England Biolabs). Real-time quantitative PCR was performed (7500 Fast real-time PCR system, Applied Biosystems, Foster City, CA) under standard conditions as follows: 1) serial heating at 94°C for 2 min and 2) amplification over 45 cycles at 94°C for 15 s and 56–60°C, depending on primer sets, for 30 s. The amplified product was subjected to SYBR green I melting analysis by ramping the temperature of the reaction samples from 60 to 95°C. A single DNA melting profile was observed under these dissociation assay conditions, demonstrating amplification of a single unique product free of primer dimers or other anomalous products. For data analyses, a standard curve was constructed by amplification of serially diluted plasmids containing the target sequence. Data were analyzed at the termination of each assay using sequence detection software (version 1.3.1, Applied Biosystems, Norwalk, CT). In standard assays, default baseline settings were selected. The increase in SYBR green I fluorescence intensity (ΔRn) was plotted as a function of cycle number, and the threshold cycle was determined by the software as the amplification cycle at which ΔRn first intersects the established baseline. Normalized data were expressed first as the relative quantity of the gene of interest normalized to the relative quantity of the housekeeping gene L32 or 18S. To standardize presentation of these results, expression of each gene in the control samples was set at 100, and values for other regions were calculated relative to the control. To evaluate the percent expression of VEGF-Axxxa and VEGF-Axxxb mRNA isoforms, data were expressed as the absolute quantity of the gene of interest generated by the standard curve.
Adult bladder catheter implantation.
Male and female Wistar rats (n = 6–8 male rats and 6–8 female rats per treatment group) were anesthetized with isoflurane in oxygen (3–4%). A lower midline abdominal incision was made, and polyethylene tubing (PE-50, Clay Adams, Parsippany, NJ) with a flared end was inserted into the bladder dome and secured while the opposite end was tunneled subcutaneously and externalized at the dorsal neck, as previously described (17, 38). Animals received both pre- and postoperative analgesics (carprofen, 5 mg/kg) once per day for a total of 2 days after surgery.
CYP dosing and administration.
Rats were anesthetized with isoflurane (2–3%), and acute cystitis was induced with a single injection of CYP (150 mg/kg ip). For cystometry experiments, chronic cystitis was induced with a total of three injections of CYP (75 mg/kg ip), one injection every 72 h, with a final injection 4 h before conscious cystometry on day 7. For mRNA analyses, chronic cystitis was induced with a total of three injections of CYP (75 mg/kg ip), one injection every 72 h with a final injection 24 h before tissue collection. Rodents were used in studies either 4 h after treatment (acute, 4-h CYP) or 7–8 days after initial treatment (chronic CYP). Control rodents received no CYP treatment (17).
Ki-8751 preparation.
Ki-8751 (Tocris, Bristol, UK) was infused intravesically at 1 mg/kg (25). For infusion into the bladder on the day of testing, the stock solution of Ki-8751 was dissolved in 100% DMSO diluted with saline to a final volume such that DMSO volume was <6% of the final solution. The solution was then vortexed, sonicated, and drawn up into a sterile syringe for infusion into the urinary bladder. Vehicle (6% DMSO in sterile saline), which produced no change in bladder function parameters, was also infused into a separate cohort of rats (data not shown).
Conscious, open-outlet cystometry with continuous infusion and effects of Ki-8751, a potent and selective VEGFR2 antagonist.
Conscious cystometry was performed as previously described (17, 38) 3 days after bladder catheter implantation. An unrestrained rat was placed in a Plexiglas cage with a wire bottom, the bladder was emptied, and the catheter was connected via a T tube to a pressure transducer (model PT300, Grass Instruments, West Warwick, RI) and a microinjection pump (model 22, Harvard Apparatus, South Natick, MA). A small animal cystometry laboratory station (MED Associates, St. Albans, VT) was used for urodynamic measurements (17, 38), as previously described. Saline solution was infused at room temperature into the bladder at a rate of 166.7 µl/min for 20–30 min to acclimate the rat to the saline infusion. After the acclimation period, saline was continuously infused to elicit at least six reproducible bladder-voiding contractions (17, 38). Ki-8751 was then infused at 1 mg/kg (25) directly into the bladder over a 30-min period. Immediately after the Ki-8751 infusion, saline solution was infused again as previously described until at least six more reproducible micturition cycles occurred. The following functional cystometric parameters were recorded in each animal during continuous cystometry: minimum pressure (pressure at the beginning of bladder filling), threshold pressure (bladder pressure immediately before micturition), maximum micturition pressure, intermicturition interval (time between micturition events), infused volume, void volume, and presence and amplitude of nonvoiding bladder contractions (NVCs) (17, 38). Bladder capacity was determined functionally (functional bladder capacity) as the volume infused into the bladder beginning when bladder pressure from the preceding voiding event returned to baseline and ending when micturition commenced (increase in bladder pressure with release of urine). Functional bladder capacity will hereafter be referred to as “bladder capacity” (17, 38). At the conclusion of the experiment, rats were euthanized (5% isoflurane in oxygen plus thoracotomy).
Statistical analyses.
Values are means ± SE. Data were compared using one-way ANOVA as well as paired or unpaired two-tailed Student’s t-tests as appropriate with GraphPad Prism statistical software (La Jolla, CA). Differences with P ≤ 0.05 were considered statistically significant.
RESULTS
Relative expression of VEGF splice variants depends on LUT tissue type.
In all tissue types examined, we found greater expression of the splice variant VEGF-Axxxa (≥90%) than VEGF-Axxxb in control (no CYP) animals (Fig. 1). Interestingly, we found differences in the relative expression of VEGF-Axxxa and VEGF-Axxxb depending on the tissue type. In the urothelium, 97.05% of total VEGF was expressed as VEGF-Axxxa and 2.95% as VEGF-Axxxb. Similarly, in the detrusor, 95.54% of VEGF was expressed as VEGF-Axxxa and 4.46% as VEGF-Axxxb. In spinal cord levels L6 and S1, lumbosacral levels that are crucial for micturition, 99.8% of total VEGF was expressed as VEGF-Axxxa and 0.20% as VEGF-Axxxb in L6 and 99.67% was expressed as VEGF-Axxxa and 0.33% as VEGF-Axxxb in S1. In DRG L6 and S1, 93.93% of total VEGF was expressed as VEGF-Axxxa and 6.13% as VEGF-Axxxb in L6 and 91.02% of total VEGF is expressed as VEGF-Axxxa and 8.98% as VEGF-Axxxb in S1.
Fig. 1.
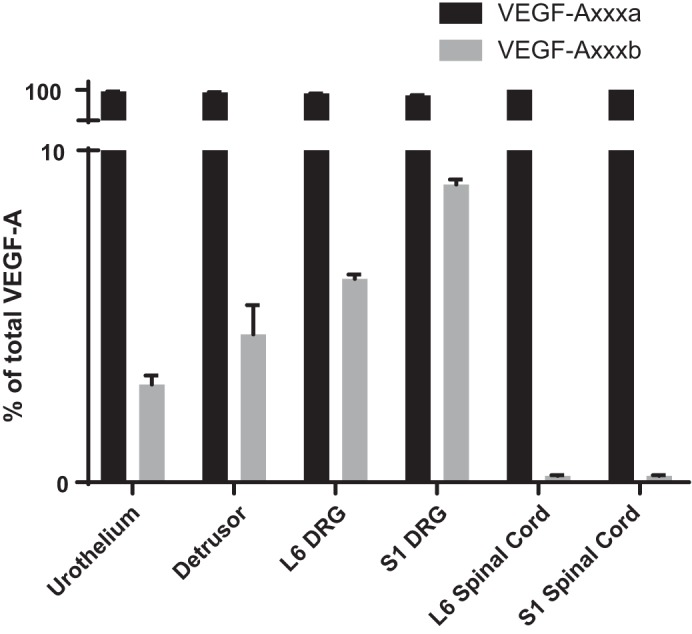
The proportion of total VEGF that is expressed as the VEGF-Axxxa versus VEGF-Axxxb isoform is lower urinary tract tissue specific. Although each tissue expressed significantly more VEGF-Axxxa than VEGF-Axxxb, the highest proportion of VEGF-Axxxa to VEGF-Axxxb occurred in the urinary bladder and the lumbosacral dorsal root ganglion (DRG), whereas the lowest occurred in the lumbosacral spinal cord. Values are means ± SE; n = 6.
Effects of CYP-induced cystitis on VEGF-Axxxa/VEGF-Axxxb isoform expression.
In the urothelium after acute (4 h) CYP-induced cystitis, VEGF-Axxxa and VEGF-Axxxb expression increased (204–243%) significantly relative to control (P ≤ 0.01 and P ≤ 0.05, respectively). In the urothelium after 48 h and 8 days, VEGF-Axxxa significantly decreased, nearing control levels (P ≤ 0.05), whereas VEGF-Axxxb remained elevated. After CYP-induced cystitis, no significant changes in VEGF-Axxxa or VEGF-Axxxb expression occurred in the detrusor at any time (Fig. 2, A and B). In the spinal cord, we did not observe an increase in VEGF-Axxxa in L6. VEGF-Axxxa expression in S1 was increased in the chronic condition compared with acute (4 h) CYP-induced cystitis (P ≤ 0.05). VEGF-Axxxb expression increased significantly 8 days after CYP treatment compared with other time points examined in spinal cord levels L6 and S1 (P ≤ 0.01 for all time points; Fig. 2, C and D). After chronic (8 day) CYP-induced cystitis, expression of VEGF-Axxxa was significantly increased (193%) in DRG L6 compared with both the control (P ≤ 0.05) and 4-h (P ≤ 0.05) treatment. Expression of VEGF-Axxxa also increased (145%) in DRG S1 at 48 h (P ≤ 0.05) of cystitis compared with control. VEGF-Axxxb expression increased significantly in DRG L6 after 48 h of CYP treatment (P ≤ 0.01 for control and P ≤ 0.05 for 4 h) and remained increased at 8 days (P ≤ 0.01) compared with control and 4 h of CYP treatment. VEGF-Axxxb expression was also significantly (P ≤ 0.05) increased in DRG S1 48 h after CYP treatment compared with control (Fig. 2, E and F).
Fig. 2.

A and B: expression of VEGF-Axxxa and VEGF-Axxxb isoforms increased in the urothelium (Uro) of rats with acute (4 h) cyclophosphamide (CYP)-induced cystitis (P ≤ 0.01 and P ≤ 0.05, respectively), returned to control levels after 48 h (P ≤ 0.05), and remained at control levels after 8 days (P ≤ 0.01). Det, detrusor. C and D: expression of VEGF-Axxxa was significantly greater at spinal cord (SC) level S1 after chronic CYP treatment than after 4 and 48 h of CYP treatment (P ≤ 0.05). Expression of VEGF-Axxxb was significantly greater at spinal cord levels L6 and S1 after 8 days of CYP treatment than all other time points (P ≤ 0.01). E and F: in dorsal root ganglion (DRG) L6, VEGF-Axxxa and VEGF-Axxxb were upregulated after 8 days of CYP treatment compared with control levels and 4 h of CYP treatment (P ≤ 0.05 and P ≤ 0.01, respectively). VEGF-Axxxb was significantly increased 48 h after CYP treatment compared with control (P ≤ 0.01) and 4 h after CYP treatment (P ≤ 0.05). In DRG S1, VEGF-Axxxa and VEGF-Axxxb were upregulated significantly 48 h after CYP treatment compared with control levels (P ≤ 0.05). Values are means ± SE; n = 6. Statistical analyses were performed on raw data before transformation. *P < 0.05; **P < 0.01.
Blockade of VEGFR2 increases bladder capacity of male and female rats.
Previous studies have shown that acute (4 h) and chronic (7 day) CYP-induced cystitis decreases bladder capacity and time between voiding events compared with control (no CYP) treatment (2, 15, 42). To evaluate the effect of VEGF signaling through VEGFR2 on bladder dysfunction in animals with CYP-induced cystitis, we infused Ki-8751, a VEGFR2 tyrosine kinase inhibitor, directly into the urinary bladder of male and female rats and observed the effect on various parameters of micturition. With VEGFR2 blockade, bladder capacity increased 1.3-fold in control (no CYP) female rats (P ≤ 0.01), 1.4-fold (P ≤ 0.05) in female rats with acute CYP-induced cystitis, and 1.6-fold in female rats with chronic CYP-induced cystitis (P ≤ 0.05; Fig. 3A). Infusion of Ki-8751 increased bladder capacity 1.4-fold in control male rats (P ≤ 0.01), 1.5-fold in male rats with acute CYP-induced cystitis (P ≤ 0.05), and 2.1-fold in male rats with chronic CYP-induced cystitis (P ≤ 0.01; Fig. 3B). Instillation of vehicle solution did not affect bladder capacity in control or CYP-treated animals (data not shown).
Fig. 3.

Bladder capacity was significantly increased in both male and female control rats (n = 8 female rats and n = 6 male rats), rats with acute (4 h) cyclophosphamide (CYP)-induced cystitis (n = 8 female rats and n = 6 male rats), and rats with chronic CYP-induced cystitis (n = 6 female rats and n = 6 male rats). After VEGF/VEGF receptor 2 (VEGFR2) signaling blockade with Ki-8751, bladder capacities significantly increased in female control rats (P ≤ 0.01) and female rats with acute (P ≤ 0.05) and chronic (P ≤ 0.05) CYP-induced cystitis (A) as well as in male control rats (P ≤ 0.01) and male rats with acute (P ≤ 0.05) and chronic (P ≤ 0.01) CYP-induced cystitis (B). Values are means ± SE. *P < 0.05; **P < 0.01.
Blockade of VEGFR2 increases void volume.
In the same groups of animals, we evaluated void volume by measuring the amount of infused saline excreted by the rats. In control rats after VEGFR2 blockade, void volume increased 1.2-fold in female rats (P ≤ 0.01) and 1.3-fold in male rats (P ≤ 0.05). In female rats with acute CYP-induced cystitis, void volume increased 1.3-fold (P ≤ 0.05) after Ki-8751 infusion; however, no change was observed in male rats. In rats with chronic CYP-induced cystitis after VEGFR2 blockade, void volume increased 1.4-fold in female rats (P ≤ 0.05) and 2.2-fold in male rats (P ≤ 0.01; Fig. 4). Void volume was unchanged after infusion of vehicle solution (data not shown).
Fig. 4.

Void volume was significantly increased in female and male control rats, male and female rats with chronic cyclophosphamide (CYP)-induced cystitis, and female rats with acute (4 h) CYP-induced cystitis (n = 6–8). After VEGF/VEGF receptor 2 (VEGFR2) signaling blockade with Ki-8751, void volume significantly increased in control female rats (P ≤ 0.01) and female rats with acute (P ≤ 0.05) and chronic (P ≤ 0.05) CYP-induced cystitis as well as in control male rats (P ≤ 0.05) and male rats with chronic CYP-induced cystitis (P ≤ 0.01). Values are means ± SE. *P < 0.05, **P < 0.01. ns, not significant.
Blockade of VEGFR2 decreases voiding frequency.
In response to continuous intravesical instillation of saline after Ki-8751 infusion compared with baseline before drug infusion, the intercontraction interval was significantly increased (voiding frequency decreased) by the following amounts in all groups: 1.3-fold (P ≤ 0.01) in control female rats, 1.4-fold (P ≤ 0.01) in control male rats, 1.4-fold (P ≤ 0.05) in female rats with acute CYP-induced cystitis, 1.5-fold (P ≤ 0.01) in male rats with acute CYP-induced cystitis, 1.6-fold (P ≤ 0.05) in female rats with chronic CYP-induced cystitis, and 2.2-fold (P ≤ 0.01) in male rats with chronic CYP-induced cystitis (Fig. 5, male rat data not shown). Blockade of VEGF/VEGFR2 signaling with Ki-8751 in male rats with chronic CYP-induced cystitis decreased threshold (P ≤ 0.01), minimum (P ≤ 0.01), and average (P ≤ 0.05) bladder pressure, although no other changes in bladder pressures were observed in any other groups (Table 1). Infusion of vehicle solution did not affect intercontraction interval or bladder pressure in any of the groups. In addition, NVCs were not consistently observed in either experimental group (control vs. CYP) or sex; therefore, analyses were not performed.
Fig. 5.

Representative cystometry traces from female control rats (A) and female rats with acute (B) and chronic (C) cyclophosphamide (CYP)-induced cystitis before and after infusion of Ki-8751 into the urinary bladder (n = 6–8). Traces show increased intercontraction intervals after infusion of the VEGF receptor 2 (VEGFR2) antagonist Ki-8751 in all groups. Additionally, there was no difference in amplitude or number of nonvoiding bladder contractions in control rats or rats with CYP-induced cystitis.
Table 1.
Mean bladder pressures during open-outlet, conscious cystometry before and after Ki-8751 infusion in no CYP control, acute CYP, and chronic CYP male and female rats
| Threshold Pressure, cmH2O | Minimum Pressure, cmH2O | Average Pressure, cmH2O | Maximum Pressure, cmH2O | |
|---|---|---|---|---|
| Female rats | ||||
| Control | ||||
| Before Ki-8751 | 13.2 ± 0.9 | 10.2 ± 0.8 | 13.1 ± 1.3 | 35.5 ± 3.8 |
| After Ki-8751 | 14.1 ± 0.9 | 8.4 ± 0.3 | 12.0 ± 0.5 | 36.5 ± 3.6 |
| 4-h CYP | ||||
| Before Ki-8751 | 21.0 ± 3.7 | 16.7 ± 3.0 | 19.0 ± 3.2 | 43.9 ± 5.9 |
| After Ki-8751 | 18.6 ± 3.1 | 14.4 ± 2.5 | 16.6 ± 2.7 | 41.0 ± 6.2 |
| Chronic CYP | ||||
| Before Ki-8751 | 37.4 ± 7.2 | 25.6 ± 6.2 | 30.2 ± 6.7 | 71.5 ± 9.7 |
| After Ki-8751 | 36.8 ± 5.0 | 22.5 ± 3.9 | 28.1 ± 4.7 | 73.5 ± 6.7 |
| Male rats | ||||
| Control | ||||
| Before Ki-8751 | 12.8 ± 1.1 | 9.0 ± 0.8 | 11.4 ± 1.1 | 40.9 ± 3.6 |
| After Ki-8751 | 12.6 ± 0.9 | 8.3 ± 1.0 | 10.5 ± 1.0 | 35.3 ± 3.7 |
| 4-h CYP | ||||
| Before Ki-8751 | 16.3 ± 0.3 | 12.6 ± 0.4 | 14.3 ± 0.4 | 41.2 ± 2.1 |
| After Ki-8751 | 16.2 ± 1.0 | 11.3 ± 0.5 | 13.9 ± 0.6 | 40.0 ± 2.3 |
| Chronic CYP | ||||
| Before Ki-8751 | 32.3 ± 2.5 | 23.0 ± 2.2 | 26.6 ± 2.3 | 61.8 ± 3.4 |
| After Ki-8751 | 22.7 ± 1.1† | 13.6 ± 1.1† | 16.3 ± 1.3* | 52.7 ± 4.2 |
Values are means ± SE; n = 6–8 rats/group. CYP, cyclophosphamide. A paired, two-tailed Student’s t-test was used to compare urinary bladder pressure parameters before versus after infusion of Ki-8751. Significant difference between before and after Ki-8751 infusion:
P ≤ 0.05 and
P ≤ 0.01.
DISCUSSION
The present study extends our understanding of VEGF splice variant isoform expression and the functional role of VEGF/VEGFR2 signaling in bladder inflammation in response to acute (4 h) and chronic (7−8 day) CYP-induced cystitis. In the present study, we demonstrated tissue-specific distribution of VEGF-Axxxa/VEGF-Axxxb under control conditions and changes in splice variant expression during various durations of CYP-induced cystitis compared with control. We also showed functional improvements in micturition by blockade of VEGF/VEGFR2 signaling in rats with CYP-induced cystitis and control rats. We demonstrated that blockade of VEGFR2 within the urinary bladder leads to a significant increase in bladder capacity in control and CYP-treated rats of both sexes. Additionally, we found that void volume increases after infusion of the VEGFR2 antagonist in all groups except for male rats with acute CYP-induced cystitis. The magnitude of the change in bladder capacity before versus after Ki-8751 infusion was greater in male rats with chronic CYP-induced cystitis than control rats (P ≤ 0.05). Although there were no significant differences in the magnitude of change between male and female rats before versus after infusion of the VEGFR2 antagonist, for female rats, the drug effect was the same for each experimental group (control vs. acute CYP vs. chronic CYP); for male rats, however, the increase in bladder capacity and void volumes was greatest in the chronic condition followed by the acute condition and, finally, the control condition. To further understand the role of VEGF/VEGFR2 signaling, specifically under inflammatory conditions, future research could include dose-response studies that may highlight differences between the effect of Ki-8751 on no CYP (control) and CYP conditions. The present data suggest that blockade of VEGF/VEGFR2 signaling has a greater effect on male mice with chronic CYP-induced bladder inflammation and highlight the importance of including both sexes in experimental design.
Although previous studies have quantified VEGF protein expression and reported a significant proportion of both VEGF isoforms in various tissues (4, 21, 24), we found that, with respect to mRNA expression, VEGF-Axxxa predominated over VEGF-Axxxb in all LUT tissue types examined. The difference in VEGF-Axxxa and VEGF-Axxxb expression between our findings and those reported in the literature may be due to the differences in the quantification of mRNA versus protein. Specifically, the difference in mRNA expression and protein expression of VEGF splice variants suggests posttranscriptional modification as a possible mechanism for the increased expression of VEGF-Axxxb in different tissue types. Alternatively, differences in isoform expression may be due to variation among different tissue types, since the role of splice variant differential expression remains to be elucidated. Similar to studies that used a model of neuropathic pain, we used the CYP-induced cystitis model of bladder inflammation to demonstrate differences in VEGF splice isoform expression. Our model suggests that both VEGF-Axxxa and VEGF-Axxxb increase in LUT tissues after CYP-induced cystitis. Specifically, we found that levels of VEGF-Axxxa and VEGF-Axxxb increase in the urothelium 4 h after CYP. All changes in VEGF-Axxxa and VEGF-Axxxb in the lumbosacral spinal cord and DRG occurred after longer durations (between 48 h and 8 days) of CYP-induced cystitis. This time course may indicate that the urothelium is the first, local, target of CYP, whereas a shift in mRNA in the lumbosacral spinal cord and DRG, distal to the initial effects of CYP, may require more time.
It has previously been shown in our laboratory that, after acute (2–48 h) CYP-induced cystitis, VEGF protein expression increases in the whole bladder as well as in the urothelium, suburothelial vasculature, and detrusor smooth muscle (12). Furthermore, the Multidisciplinary Approach to the Study of Chronic Pelvic Pain Research Network recently showed that urine levels of VEGF are increased in patients with urological chronic pelvic pain syndrome, which could indicate upregulation of VEGF signaling in the urinary bladder of these patients (14). However, the mechanism by which VEGF becomes upregulated during bladder inflammation is unknown. One hypothesis involves the ischemic environment of the urinary bladder in patients with IC/BPS. Although basal bladder blood flow is the same in patients with and without IC/BPS, research has suggested that blood flow during the filling stage of micturition is significantly different. Specifically, bladder blood perfusion increases in control individuals during bladder filling but decreases in patients with IC/BPS (35). Since ischemia stimulates angiogenesis, VEGF, as a proangiogenic factor, may increase in the urinary bladder during this time in individuals with IC/BPS (14, 32, 35). Another hypothesis involves the role of VEGF in vascular permeability. Research has determined that VEGF increases the permeability of blood vessels in several ways, for example, by disrupting the tight junction barrier of endothelial cells directly by phosphorylating the tight junction regulator protein zonula occludens 1 (ZO-1) (1). In the urothelium, ZO-1 is one of the tight junction proteins responsible for the barrier properties of the urothelium that prevent toxic metabolites and ions in the urine from penetrating into the deeper layers of the walls of the urinary bladder. It is possible that VEGF in the urine acts on ZO-1 in urothelial cells, thereby increasing urothelial permeability and increasing the amount of VEGF and other molecules that can access deeper bladder structures, including bladder vasculature and nerves.
Once VEGF accesses the layers of the urinary bladder, it can have profound effects on the vasculature by increasing angiogenesis and sensitizing afferent nerve endings. Mediators of angiogenesis are actively being considered as potential targets for a variety of chronic inflammatory pathologies. In models of chronic urinary bladder inflammation, there is evidence supporting a marked increase in the innervation of sensory neurons expressing transient receptor potential vanilloid subfamily 1 (10), substance P, and calcitonin gene-related peptide (37). This increase in sensory nerve density has been recapitulated in control mice that receive an instillation of VEGF into their bladder (37). In this model, functional bladder changes mirroring the symptoms of IC/BPS (e.g., reduced micturition pressure and intercontraction interval), along with increased pelvic sensitivity, were seen (37). These results suggest that VEGF is directly linked to the peripheral nervous system and may impact the neural control of the bladder under inflammatory conditions.
Blockade of VEGF/VEGR2 signaling in the urinary bladder leads to increased bladder functional outcomes; however, many questions remain. Future experiments will include examination of the role of VEGF/VEGFR1 signaling in CYP-induced cystitis, since VEGFR1 has also been shown to be involved in angiogenesis and inflammation (3, 22, 23). Furthermore, because both VEGFRs have been shown to be present and functionally active in urothelial and neuronal cells of both human and animal models of bladder inflammation (6, 36), these receptors are potential targets for the regulation of angiogenesis in a model of CYP-induced cystitis. To understand the effect of VEGF on inflammatory mediators, we could examine different proinflammatory molecules in the bladders of animals with CYP-induced cystitis before and after Ki-8751 administration to determine if VEGF signaling plays a direct role in the inflammatory milieu. von Frey filament testing of pelvic hypersensitivity in rats with CYP-induced cystitis after VEGFR2 blockade could provide insight into the VEGF/VEGFR2 contribution to bladder dysfunction as well as pelvic pain. In summary, our study demonstrates that VEGF/VEGFR2 signaling blockade improves bladder function, as demonstrated by the increase in bladder capacity and void volume in control rats and those with acute and chronic urinary bladder inflammation.
GRANTS
This work was funded by National Institute of Diabetes and Digestive and Kidney Diseases Grants RO1-DK-051369 and RO1-DK-060481 (to M. A. Vizzard).
DISCLAIMERS
The National Institutes of Health had no role in the experiments described, including the design, data collection, and analysis of studies performed in the Vizzard laboratory, decision to publish, or preparation of the manuscript. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.T., B.M.G., and M.A.V. conceived and designed research; K.T. and B.M.G. performed experiments; K.T., B.M.G., and M.A.V. analyzed data; K.T., B.M.G., and M.A.V. interpreted results of experiments; K.T., B.M.G., and M.A.V. prepared figures; K.T., B.M.G., and M.A.V. drafted manuscript; K.T., B.M.G., and M.A.V. edited and revised manuscript; K.T., B.M.G., and M.A.V. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Susan Campbell for assistance with rodent surgeries and Jacqueline Ojala for help with data collection.
REFERENCES
- 1.Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem 274: 23463–23467, 1999. doi: 10.1074/jbc.274.33.23463. [DOI] [PubMed] [Google Scholar]
- 2.Arms L, Vizzard MA. Role for pAKT in rat urinary bladder with cyclophosphamide (CYP)-induced cystitis. Am J Physiol Renal Physiol 301: F252–F262, 2011. doi: 10.1152/ajprenal.00556.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Autiero M, Luttun A, Tjwa M, Carmeliet P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J Thromb Haemost 1: 1356–1370, 2003. doi: 10.1046/j.1538-7836.2003.00263.x. [DOI] [PubMed] [Google Scholar]
- 4.Bevan HS, van den Akker NM, Qiu Y, Polman JA, Foster RR, Yem J, Nishikawa A, Satchell SC, Harper SJ, Gittenberger-de Groot AC, Bates DO. The alternatively spliced anti-angiogenic family of VEGF isoforms VEGFxxxb in human kidney development. Nephron, Physiol 110: p57–67, 2008. doi: 10.1159/000177614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birder LA, Apodaca G, De Groat WC, Kanai AJ. Adrenergic- and capsaicin-evoked nitric oxide release from urothelium and afferent nerves in urinary bladder. Am J Physiol Renal Physiol 275: F226–F229, 1998. doi: 10.1152/ajprenal.1998.275.2.F226. [DOI] [PubMed] [Google Scholar]
- 6.Burgu B, Medina Ortiz WE, Pitera JE, Woolf AS, Wilcox DT. Vascular endothelial growth factor mediates hypoxic stimulated embryonic bladder growth in organ culture. J Urol 177: 1552–1557, 2007. doi: 10.1016/j.juro.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Burnstock G. Purine-mediated signalling in pain and visceral perception. Trends Pharmacol Sci 22: 182–188, 2001. doi: 10.1016/S0165-6147(00)01643-6. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P. Angiogenesis in health and disease. Nat Med 9: 653–660, 2003. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature 436: 193–200, 2005. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- 10.Charrua A, Reguenga C, Cordeiro JM, Correiade-Sá P, Paule C, Nagy I, Cruz F, Avelino A. Functional transient receptor potential vanilloid 1 is expressed in human urothelial cells. J Urol 182: 2944–2950, 2009. doi: 10.1016/j.juro.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Yang G, Song JH, Xu H, Li D, Goldsmith J, Zeng H, Parsons-Wingerter PA, Reinecker HC, Kelly CP. Probiotic yeast inhibits VEGFR signaling and angiogenesis in intestinal inflammation. PLoS One 8: e64227, 2013. doi: 10.1371/journal.pone.0064227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheppudira BP, Girard BM, Malley SE, Schutz KC, May V, Vizzard MA. Upregulation of vascular endothelial growth factor isoform VEGF-164 and receptors (VEGFR-2, Npn-1, and Npn-2) in rats with cyclophosphamide-induced cystitis. Am J Physiol Renal Physiol 295: F826–F836, 2008. doi: 10.1152/ajprenal.90305.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chun SY, Lim GJ, Kwon TG, Kwak EK, Kim BW, Atala A, Yoo JJ. Identification and characterization of bioactive factors in bladder submucosa matrix. Biomaterials 28: 4251–4256, 2007. doi: 10.1016/j.biomaterials.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 14.Dagher A, Curatolo A, Sachdev M, Stephens AJ, Mullins C, Landis JR, van Bokhoven A, El-Hayek A, Froehlich JW, Briscoe AC, Roy R, Yang J, Pontari MA, Zurakowski D, Lee RS, Moses MA; MAPP Research Network . Identification of novel non-invasive biomarkers of urinary chronic pelvic pain syndrome: findings from the Multidisciplinary Approach to the Study of Chronic Pelvic Pain (MAPP) Research Network. BJU Int 120: 130–142, 2017. doi: 10.1111/bju.13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dugan C, Malley S, Arms L, May V, Vizzard MA. Role of c-Jun N-terminal kinase (JNK) activation in micturition reflexes in cyclophosphamide (CYP)-induced cystitis in female rats. J Mol Neurosci 54: 360–369, 2014. doi: 10.1007/s12031-014-0308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res 60: 1388–1393, 2000. [PubMed] [Google Scholar]
- 17.Gonzalez EJ, Peterson A, Malley S, Daniel M, Lambert D, Kosofsky M, Vizzard MA. The effects of tempol on cyclophosphamide-induced oxidative stress in rat micturition reflexes. ScientificWorldJournal 2015: 1–13, 2015. doi: 10.1155/2015/545048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grover S, Srivastava A, Lee R, Tewari AK, Te AE. Role of inflammation in bladder function and interstitial cystitis. Ther Adv Urol 3: 19–33, 2011. doi: 10.1177/1756287211398255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo M, Chang P, Hauke E, Girard BM, Tooke K, Ojala J, Malley SM, Hsiang H, Vizzard MA. Expression and function of chemokines CXCL9-11 in micturition pathways in cyclophosphamide (CYP)-induced cystitis and somatic sensitivity in mice. Front Syst Neurosci 12: 9, 2018. doi: 10.3389/fnsys.2018.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanno PM, Burks DA, Clemens JQ, Dmochowski RR, Erickson D, Fitzgerald MP, Forrest JB, Gordon B, Gray M, Mayer RD, Newman D, Nyberg L Jr, Payne CK, Wesselmann U, Faraday MM; Interstitial Cystitis Guidelines Panel of the American Urological Association Education and Research, Inc . AUA guideline for the diagnosis and treatment of interstitial cystitis/bladder pain syndrome. J Urol 185: 2162–2170, 2011. doi: 10.1016/j.juro.2011.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer 8: 880–887, 2008. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He J, Wang H, Liu Y, Li W, Kim D, Huang H. Blockade of vascular endothelial growth factor receptor 1 prevents inflammation and vascular leakage in diabetic retinopathy. J Ophthalmol 2015: 1–11, 2015. doi: 10.1155/2015/605946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang H, Shen J, Vinores SA. Blockade of VEGFR1 and 2 suppresses pathological angiogenesis and vascular leakage in the eye. PLoS One 6: e21411, 2011. doi: 10.1371/journal.pone.0021411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hulse RP, Beazley-Long N, Hua J, Kennedy H, Prager J, Bevan H, Qiu Y, Fernandes ES, Gammons MV, Ballmer-Hofer K, Gittenberger de Groot AC, Churchill AJ, Harper SJ, Brain SD, Bates DO, Donaldson LF. Regulation of alternative VEGF-A mRNA splicing is a therapeutic target for analgesia. Neurobiol Dis 71: 245–259, 2014. doi: 10.1016/j.nbd.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Im E, Rhee SH, Park YS, Fiocchi C, Taché Y, Pothoulakis C. Corticotropin-releasing hormone family of peptides regulates intestinal angiogenesis. Gastroenterology 138: 2457–2467, 2010. doi: 10.1053/j.gastro.2010.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawamura H, Li X, Harper SJ, Bates DO, Claesson-Welsh L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res 68: 4683–4692, 2008. doi: 10.1158/0008-5472.CAN-07-6577. [DOI] [PubMed] [Google Scholar]
- 27.Kiuchi H, Tsujimura A, Takao T, Yamamoto K, Nakayama J, Miyagawa Y, Nonomura N, Takeyama M, Okuyama A. Increased vascular endothelial growth factor expression in patients with bladder pain syndrome/interstitial cystitis: its association with pain severity and glomerulations. BJU Int 104: 826–831, 2009. doi: 10.1111/j.1464-410X.2009.08467.x. [DOI] [PubMed] [Google Scholar]
- 28.Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem 272: 32521–32527, 1997. doi: 10.1074/jbc.272.51.32521. [DOI] [PubMed] [Google Scholar]
- 29.Lai HH, Shen B, Vijairania P, Zhang X, Vogt SK, Gereau RW 4th. Anti-vascular endothelial growth factor treatment decreases bladder pain in cyclophosphamide cystitis: a Multidisciplinary Approach to the Study of Chronic Pelvic Pain (MAPP) Research Network animal model study. BJU Int 120: 576–583, 2017. doi: 10.1111/bju.13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JD, Lee MH. Increased expression of hypoxia-inducible factor-1α and vascular endothelial growth factor associated with glomerulation formation in patients with interstitial cystitis. Urology 78: 971.e11–971.e15, 2011. doi: 10.1016/j.urology.2011.05.050. [DOI] [PubMed] [Google Scholar]
- 31.Malykhina AP, Lei Q, Erickson CS, Epstein ML, Saban MR, Davis CA, Saban R. VEGF induces sensory and motor peripheral plasticity, alters bladder function, and promotes visceral sensitivity. BMC Physiol 12: 15, 2012. doi: 10.1186/1472-6793-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marti HH, Risau W. Angiogenesis in ischemic disease. Thromb Haemost 82, Suppl 1: 44–52, 1999. doi: 10.1055/s-0037-1615552. [DOI] [PubMed] [Google Scholar]
- 33.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signaling—in control of vascular function. Nat Rev Mol Cell Biol 7: 359–371, 2006. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 34.Parsons CL, Greenberger M, Gabal L, Bidair M, Barme G. The role of urinary potassium in the pathogenesis and diagnosis of interstitial cystitis. J Urol 159: 1862–1866, 1998. doi: 10.1016/S0022-5347(01)63178-1. [DOI] [PubMed] [Google Scholar]
- 35.Pontari MA, Hanno PM, Ruggieri MR. Comparison of bladder blood flow in patients with and without interstitial cystitis. J Urol 162: 330–334, 1999. doi: 10.1016/S0022-5347(05)68552-7. [DOI] [PubMed] [Google Scholar]
- 36.Saban MR, Backer JM, Backer MV, Maier J, Fowler B, Davis CA, Simpson C, Wu XR, Birder L, Freeman MR, Soker S, Hurst RE, Saban R. VEGF receptors and neuropilins are expressed in the urothelial and neuronal cells in normal mouse urinary bladder and are upregulated in inflammation. Am J Physiol Renal Physiol 295: F60–F72, 2008. doi: 10.1152/ajprenal.00618.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saban MR, Davis CA, Avelino A, Cruz F, Maier J, Bjorling DE, Sferra TJ, Hurst RE, Saban R. VEGF signaling mediates bladder neuroplasticity and inflammation in response to BCG. BMC Physiol 11: 16, 2011. doi: 10.1186/1472-6793-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnegelsberg B, Sun TT, Cain G, Bhattacharya A, Nunn PA, Ford AP, Vizzard MA, Cockayne DA. Overexpression of NGF in mouse urothelium leads to neuronal hyperinnervation, pelvic sensitivity, and changes in urinary bladder function. Am J Physiol Regul Integr Comp Physiol 298: R534–R547, 2010. doi: 10.1152/ajpregu.00367.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shim JW, Madsen JR. VEGF signaling in neurological disorders. Int J Mol Sci 19: e275, 2018. doi: 10.3390/ijms19010275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens M, Oltean S. Modulation of VEGF-A alternative splicing as a novel treatment in chronic kidney disease. Genes (Basel) 9: 98, 2018. doi: 10.3390/genes9020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallon M, Chang J, Zhang H, Kuo CJ. Developmental and pathological angiogenesis in the central nervous system. Cell Mol Life Sci 71: 3489–3506, 2014. doi: 10.1007/s00018-014-1625-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wróbel A, Doboszewska U, Rechberger E, Rojek K, Serefko A, Poleszak E, Skalicka-Woźniak K, Dudka J, Wlaź P. Rho kinase inhibition ameliorates cyclophosphamide-induced cystitis in rats. Naunyn Schmiedebergs Arch Pharmacol 390: 613–619, 2017. doi: 10.1007/s00210-017-1361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshimura N, Oguchi T, Yokoyama H, Funahashi Y, Yoshikawa S, Sugino Y, Kawamorita N, Kashyap MP, Chancellor MB, Tyagi P, Ogawa T. Bladder afferent hyperexcitability in bladder pain syndrome/interstitial cystitis. Int J Urol 21, Suppl 1: 18–25, 2014. doi: 10.1111/iju.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu Y, de Groat WC. Sensitization of pelvic afferent nerves in the in vitro rat urinary bladder-pelvic nerve preparation by purinergic agonists and cyclophosphamide pretreatment. Am J Physiol Renal Physiol 294: F1146–F1156, 2008. doi: 10.1152/ajprenal.00592.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]