

Abstract
Cytotoxic chemotherapy is the foundation for the treatment of the wide variety of childhood malignancies; however, these therapies are known to have a variety of deleterious side effects. One common chemotherapy used in children, doxorubicin (DOX), is well known to cause cardiotoxicity and cardiomyopathy. Recent studies have revealed that DOX impairs skeletal and smooth muscle function and contributes to fatigue and abnormal intestinal motility in patients. In this study, we tested the hypothesis that systemic DOX administration also affects detrusor smooth muscle (DSM) function in the urinary bladder, especially when administered at a young age. The effects on the DSM and bladder function were assessed in BALB/cJ mice that received six weekly intravenous injections of DOX (3 mg·kg−1·wk−1) or saline for the control group. Systemic DOX administration resulted in DSM hypertrophy, increased voiding frequency, and a significant attenuation of DSM contractility, followed by a slower relaxation compared with the control group. Gene expression analyses revealed that unlike DOX-induced cardiotoxicity, the bladders from DOX-administered animals showed no changes in oxidative stress markers; instead, downregulation of large-conductance Ca2+-activated K+ channels and altered expression of myosin light-chain kinase coincided with reduced myosin light-chain phosphorylation. These results indicate that in vivo DOX exposure caused DSM dysfunction by dysregulation of molecules involved in the detrusor contractile-relaxation mechanisms. Collectively, our findings suggest that survivors of childhood cancer treated with DOX may be at increased risk of bladder dysfunction and benefit from followup surveillance of bladder function.
Keywords: bladder dysfunction, detrusor, doxorubicin, myosin, smooth muscle
INTRODUCTION
Cancer is the leading cause of death, second only to accidents in children (1–14 yr) in the United States, and the overall pediatric cancer incidence rate has increased by 0.7% per year since 1975 (44). In contrast, with the use of systemic cytotoxic chemotherapy as the central part of multimodal therapy, the overall 5-yr survival rates of pediatric cancer have increased from 58% in the mid-1970s to 83% currently (44). However, chemotherapy exposure in childhood has been attributed to a number of deleterious side effects, including cardiac and pulmonary impairment, delayed growth, infertility, intellectual delay, and increased risk of secondary malignancies (35a). Cardiotoxicity has been attributed to anthracyclines, specifically doxorubicin (DOX), which is one of the most frequently used chemotherapeutics for treating hematological malignancies as well as solid tumors, in both children and adults (32, 43, 45). Age has been observed to be an important predictor for DOX-induced cardiotoxicity, where elderly adults and young children are reported to constitute higher risk groups (17, 23). Despite the long-held belief that DOX toxicity is highly specific to cardiac muscle, accumulating evidence suggests that DOX also impacts skeletal and smooth muscle function, providing a link to symptoms, including fatigue, intestinal dysmotility, and vascular smooth muscle senescence (20, 25, 35, 47). Based on those findings, we hypothesized that DOX might affect detrusor smooth muscle (DSM) function, leading to a risk of lower urinary tract dysfunction (LUTD), especially in survivors of childhood cancer exposed to DOX. Data on effects of systemic chemotherapy received in childhood on voiding function are scarce; however, studies have suggested a general association between childhood chemotherapy exposure and LUTD (8, 21, 41).
In the present study, we aimed to investigate how systemic DOX exposure impacts DSM function using a murine model, with focus on the effects in young age.
MATERIALS AND METHODS
Animals.
All procedures using animals were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Colorado Anschutz Medical Campus. BALB/cJ mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed in a 12:12-h light-dark cycle with ad libitum access to food and water. Both male and female mice (n = 28 mice/sex) were used in this study. Mice were given a weekly dose of 3 mg/kg doxorubicin hydrochloride injection USP (DOX; Teva Pharmaceuticals, North Wales, PA) by intravenous injection via a tail vein for 6 wk (cumulative DOX doses of 18 mg/kg) (11). Two male mice and four female mice that received DOX were excluded from the study after 4–5 wk of DOX administration due to the development of inflammation around injection sites that was not relieved by our Animal Care and Use Committee-approved regimen.
Spontaneous void spot assay.
To evaluate voiding habits, void spot assays (VSAs) were conducted at 6 days after the third and sixth administration, as previously described (n = 9–12 mice for each sex per group) (29). Briefly, mice were individually placed onto a steel wire mesh (0.64-cm2 opening), with the floor elevated from the cage bottom by 1 cm. A Whatman-grade filter paper (ThermoFisher Scientific, Hampton, NH) was placed to cover the entire surface of the bottom of the cage. No water or food was provided during the 3-h test period. All VSA experiments started at ∼9 AM. The urine spots on the paper were imaged using ultraviolet light, and the number of all urine spots > 0.3 cm2 (corresponding to 10 µl) was counted as voids. The volume of each spot was summed to determine the total voided volume.
Histological analysis.
Paraformaldehyde-fixed paraffin sections (5 µm thickness) of the urinary bladders from each group at 1 wk after the last injection were subjected to hematoxylin and eosin staining. The area of the DSM layer was measured by immunostaining for smooth muscle actin (SMA), and the proportion of the DSM layer was expressed as a percentage to the total area of the section measured under bright field (n = 8 per group). Collagen fibers were visualized and imaged with second harmonic generation microscopy (Carl Zeiss Microscopy, Thornwood, NY) (10). Sections from at least four animals (n = 2–3 animals of each sex per group) were analyzed for reproducibility. Areas of whole tissue section, DSM layer, and collagen fibers (pseudocolored in gray) in second harmonic generation images of bladder sections were measured using Adobe Photoshop (Adobe Systems, San Jose, CA). Collagen measurement was performed in the entire tissue section and in the DSM layer separately and normalized to the thickness of the respective area. All measurements were conducted in a blind fashion to avoid biased interpretation. For immunostaining, sections were subjected to heat-induced antigen retrieval (10 mM Tris, 1 mM EDTA, and 0.05% Tween 20, pH 9.0) (28). The antibodies used in this study are shown in Tables 1 and 2. Control experiments performed without primary antibodies showed neither nonspecific labeling nor cross-reactivity between secondary antibodies.
Table 1.
Primary antibodies
| Gene Name | Vendor | Catalog No. | Working Dilution (Use) |
|---|---|---|---|
| β-Actin/Actb | Santa Cruz Biotechnology (Dallas, TX) | sc-47778 | 1:1,000 (WB) |
| BKα/Slo1 | Alomone Laboratories (Jerusalem, Israel) | APC-021 | 1:250 (WB) |
| Antibodies (Davis, CA) | 75-408 | 1:50 (IF) | |
| Desmin | Novus Biologicals (Centennial, CO) | NB120-15200 | 1:200 (IF) |
| Mlck/Mylk | Proteintech (Rosemont, IL) | 21642-1-AP | 1:1,000 (WB) |
| Myl9 | Novus Biologicals | NBP1-55383 | 1:500 (WB) |
| Phospho-Myl9 (Ser19) | ThermoFisher Scientific (Waltham, MA) | MA515163 | 1:1,000 (WB) |
| PGP9.5/UCH-L1 | Santa Cruz Biotechnology | sc-23852 | 1:10 (IF) |
| Tagl/Sm22α | St John’s Laboratory (London, UK) | STJ193096 | 1:1,000 (WB) |
| SMA | Santa Cruz Biotechnology | sc-53142 | 1:50 (IF)/1:1,000 (WB) |
| smMHC | Proteintech | 21404-1-AP | 1:2,000 (WB) |
| Smtn | Proteintech | 23567-1-AP | 1:1,000 (WB) |
| Vimentin | Proteintech | 10366-1-AP | 1:2,000 (WB) |
IF, immunofluorescence; WB, Western blot.
Table 2.
Secondary antibodies
| Anti | Label | Vendor | Catalog No. | Working Dilution (Use) |
|---|---|---|---|---|
| Rabbit IgG | HRP | Novus Biologicals (Centennial, CO) | NB7160 | 1:10,000 (WB) |
| CF543 | Biotium (Fremont, CA) | 20308 | 1:1,000 (IF) | |
| Mouse IgG | HRP | Jackson ImmunoResearch (West Grove, PA) | 715-035-150 | 1:10,000 (WB) |
| CF488A | Biotium | 20014 | 1:1,000 (IF) | |
| Goat IgG | CF555 | Biotium | 20039 | 1:1,000 (IF) |
HRP, horseradish peroxidase; IF, immunofluorescence; WB, Western blot.
In vitro detrusor contractility measurements.
In vitro DSM contractility measurements were performed as previously described (27). Briefly, freshly isolated urinary bladders from mice in each group at 1 wk after the last injection (n = 5 mice for each sex per group) were cut into halves longitudinally. Each strip (~3 × 6 mm) was placed in organ baths (Radnoti, Monrovia, CA) filled with oxygenated Tyrode buffer [containing (in mM) 125 NaCl, 2.5 KCl, 23.8 NaHCO3, 0.5 MgCl2, 0.4 NaH2PO4, 1.8 CaCl2, and 5.5 glucose] at 37°C. Tissues were equilibrated for 30 min and then stretched to their optimum length for muscle contraction in which the maximum force for muscle contraction was produced by electrical field stimulation (EFS; 70 V, 32 Hz). Once the optimum length for muscle contraction was determined, each muscle strip was equilibrated for 30 min in fresh Tyrode buffer and then subjected to several tests, including the contractile response to EFS (70 V, 0.5–32 Hz), the acetylcholine receptor (AChR) agonist carbachol (CCh; 0.1–100 µM), and high KCl (125 mM replaced NaCl in Tyrode buffer). Contractile responses to EFS were also recorded after 15 min of incubation of the following substances: 1) 2,2,6,6-tetramethylpiperidin-4-yl heptanoate [TMPH; antagonist of neuronal nicotinic AChRs containing the α3-subunit (nAChR-α3*); 0.3 µM] and 2) the combination of TMPH and atropine [muscarinic AChR (mAChR) antagonist (1 µM)]. Contractile parameters were measured using PowerLab Laboratory Chart (version 8.1.9, ADInstruments, Colorado Springs, CO). Force measurements were performed and analyzed as previously described (27). The rate of relaxation for each detrusor strip was calculated as the time constant (τ) fit to the relaxation phase upon the end of EFS using pCLAMP software (Molecular Devices, San Jose, CA).
Gene expression analysis.
Total RNA isolated from the urinary bladders from each group of mice (n = 4–5 mice/group) was transcribed into cDNA and used in real-time quantitative PCR as previously described (27). Expression levels of each gene were calculated as fold changes based on threshold cycle values. Data were normalized to the mean of two housekeeping genes: β-actin (Actb) and TATA box-binding protein (Tbp). Western blot analysis was performed to assess the protein expression level as previously described (n = 5 per group) (29). The antibodies are shown in Tables 1 and 2. The signals specific for each antibody were quantified using ImageJ software (National Institutes of Health, Bethesda, MD) and normalized to a housekeeping gene Actb.
Statistical analysis.
All data were analyzed using one-way repeated-measures ANOVA using GraphPad InStat (GraphPad Software, La Jolla, CA). GraphPad outlier calculator (GraphPad Software) was used to detect outliers, which were excluded from the analysis in detrusor contractility tests. A probability value of P < 0.05 was regarded as significant.
RESULTS
DOX induced a bladder enlargement through detrusor hypertrophy.
DOX exposure caused a significant growth retardation in mice after the fourth administration compared with the control group. The bladder weight relative to body weight showed a significant increase in the DOX-treated group (122 ± 5%, with the control taken as 100%, n = 9 animals for each sex per group) at 1 wk after the last injection (Fig. 1). Bladder histology was comparable between DOX-treated and control groups in terms of structures of the urothelia, lamina propria, and DSM layers. No signs of fibrosis or inflammation were observed (Fig. 2A). The distribution and proportional amount of collagen contents were comparable in the bladders and in the DSM layer between the control and DOX-treated groups (Fig. 2B). A significant increase in the thickness of the DSM layer was observed in the DOX-treated group compared with the control group (62.1 ± 1.1% vs. 67.4 ± 1.1% of the total area, P = 0.007; Fig. 2C). Both male and female mice in each group showed a similar pattern of changes in body weight, bladder weight relative to body weight, and bladder histology. These results indicate that systemic DOX exposure induced the bladder enlargement through detrusor hypertrophy, similar to previously reported DOX-induced cardiac hypertrophy (34).
Fig. 1.

Comparison of changes in body weights and bladder weights between doxorubicin (DOX)-administered and control (Ctrl) groups. Body weight was measured at 6 days after the administration and expressed as the change relative to that on the day before the first administration, shown as 0 (left). Bladder weight was measured at 1 wk after the sixth administration of agents and expressed as a percentage of the body weight (right). Values are means ± SE. *P < 0.05 and †P < 0.005 vs. control mice.
Fig. 2.
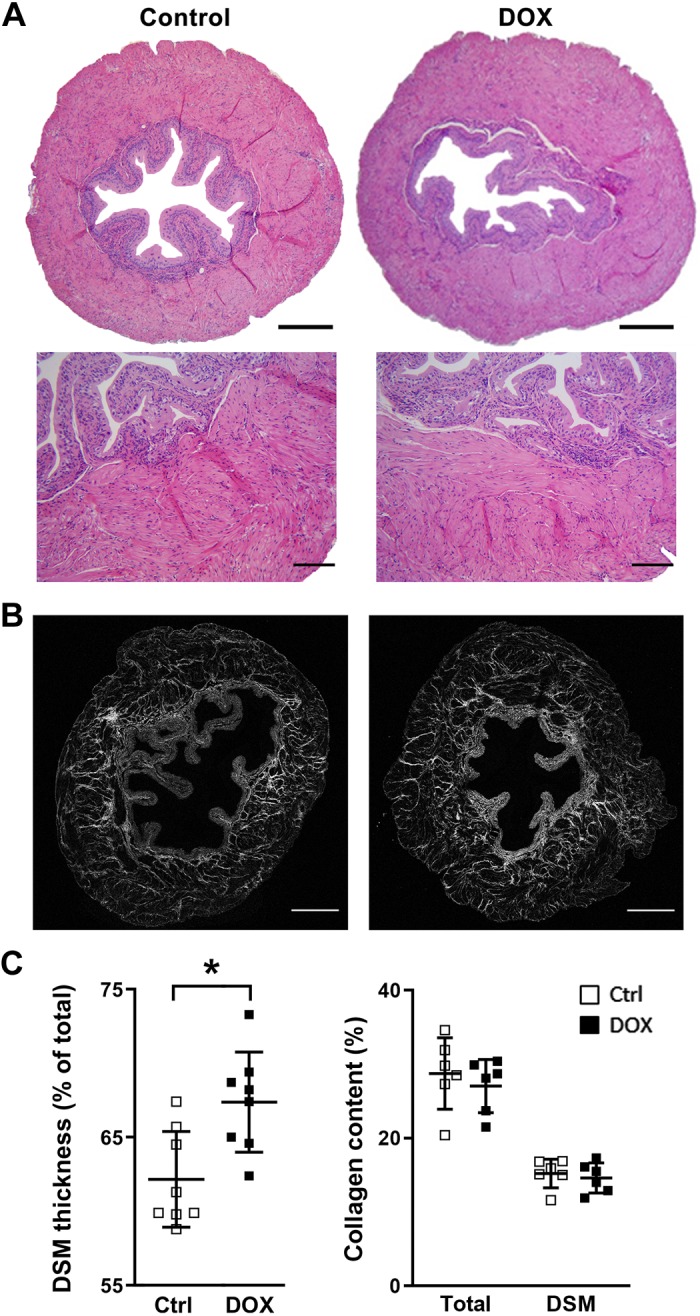
Bladder histology at 1 wk after the sixth administration. A: hematoxylin and eosin staining. Original scale bars = 400 µm (top) and 50 µm (bottom). B: collagen distribution (gray) in the bladder by second harmonic generation imaging. Original scale bars = 500 µm. C: thickness of the detrusor smooth muscle (DSM) layer relative to that of the total bladder wall (left). Values of the ratio of DSM to total tissue section areas represent means ± SE (in %). The total cross-sectional area was taken as 100%. Right, the collagen level in the total area and in the DSM layer. Values represent means ± SE (in %) to the areas of total tissue or DSM layer on each section, respectively; n = 4–5 per group. *P < 0.05 vs. the control (Ctrl) group. DOX, doxorubicin.
An increased voiding frequency in female mice after systemic DOX exposure.
There was no apparent difference in the number of urine spots and total voided volume between two groups in both male and female mice at 6 days after the third and sixth administration (DOX cumulative doses of 9 and 18 mg/kg, respectively). At the later time point, male mice in both groups rarely had countable urine spots. Instead, a large number of tiny urine spots (<10 µl), ranging from 53 to 108, were observed and, therefore, made the analysis uninformative in male mice. This micturition pattern observed in male mice could be associated with the anxiety and territorial nature known for male BALB/cJ mice, which manifested as territorial urine marking behaviors in an unfamiliar environment (5, 6, 30). Female mice showed no apparent differences in the total voided volume and number and mean volume of voids at and over 75 µl between two groups at both time points (Fig. 3A). However, DOX-treated female mice showed a noticeable trend of the frequent voids of <75 µl compared with control mice at both time points (1.6 ± 0.4 vs. 0.4 ± 0.2, P = 0.11, and 2.5 ± 0.5 vs. 0.9 ± 0.3, P = 0.06, at 6 days after the third and sixth administration, respectively; Fig. 3B).
Fig. 3.

Micturition patterns in doxorubicin (DOX)-treated and control (Ctrl) groups at 6 days after the third and sixth administration. A: total number of urine spots (left) and total voided volume (right). B: number of urine spots of <25, 25–<50, 50–<75, 75– <100, and ≥100 µl per void in female mice. Values are means ± SE; n = 9–10 per group.
DOX induced an attenuation of DSM contractility and relaxation response.
Bladder strips from the DOX group demonstrated a significant decrease in detrusor contractility in response to EFS (8, 16, and 32 Hz) and CCh (10 and 100 μM) by 25–32% compared with mice from the control group upon normalization of the response to strip weight (Fig. 4A). Detrusor contractility in response to KCl was also decreased in the DOX-treated group by 22% (P = 0.049). Next, we examined DSM contractility triggered by EFS in the presence of TMPH and atropine, inhibitors of nicotinic and muscarinic AChRs, respectively, to investigate whether neurotransmitter release and/or AChR dysfunction play a role in the decreased DSM contractility in the DOX-treated group. DSM contractility was suppressed with both AChR inhibitors at a comparable level in both control and DOX-treated groups (Fig. 4B), suggesting that the decreased DSM contractility in the DOX-treated group was primarily caused by dysfunction of DSM itself rather than via neural pathways. Analysis of DSM relaxation kinetics at the end of EFS-evoked contraction revealed that the relaxation curve was best fitted by two exponentials, suggesting that the relaxation process includes two components with fast and slow τ (τ1 and τ2, respectively). DOX-exposed mice had a significantly smaller τ1 than control mice (17.4 ± 1.3 vs. 27.1 ± 2.4 mg/s, P < 0.001), whereas no difference was observed for τ2 between the groups (Fig. 4C).
Fig. 4.

Contractility of bladder strips at 1 wk after the sixth administration. A: peak contractile force in response to electric field stimulation (EFS; left), carbachol (CCh; middle), and KCl (right; N = 10 and n = 17 per group, where N is the number of animals and n is the number of tissues used in the specific experiments). Solid lines and open boxes represent the control group; dashed lines and closed boxes represent the doxorubicin (DOX)-treated group. B: peak contractile force in response to EFS at 32 Hz with TMPH and atropine (muscarinic acetylcholine receptor inhibitor). Force was normalized with tissue weight and expressed as relative to the response to EFS in the absence of inhibitors. C: time constant (τ) of bladder strip relaxation upon the end of EFS (32 Hz). Left, representative response curves of contraction evoked by EFS and the following relaxation from each group. Each strip shows two different components of τ of relaxation: τ1 (blue arrows) and τ2 (orange arrows), indicating the fast and slow component of a relaxation curve, respectively. Right, summary of the detrusor smooth muscle relaxation rate (N = 6 and n = 9 per group). Force was normalized with tissue weight. Values are means ± SE. *P < 0.05 and †P < 0.005 vs. the control group.
No changes in the expression levels of oxidative stress and inflammatory markers in the bladders after DOX exposure.
Previous studies have established that the pathophysiology of DOX-induced cardiomyopathy is predominantly determined by oxidative stress, which developed upon overproduction of free radicals: reactive oxygen and nitrogen species (RONS) on one side and a deficiency of intrinsic antioxidant mechanisms on the other side (43). To evaluate if oxidative stress and inflammation pathways were responsible for the DOX-induced impairment of DSM contractility, we examined the expression levels of key components in those pathways: NADPH oxidases [Nox and dual oxidase (Duox)], nitric oxide synthases (NOS), superoxide dismutases (SOD), IL-1β, IL-6, IL-8, and TNF-α (15, 40). There were no apparent changes in expression levels of any tested genes between the two groups (Fig. 5A), suggesting that oxidative stress and inflammation were unlikely the key players for the DOX-induced detrusor dysfunction in our model.
Fig. 5.

Gene expression analyses in the bladders. A: quantitative PCR results for oxidative stress (left) and proinflammatory (right) markers. B: quantitative PCR results for genes known to be involved in detrusor smooth muscle (DSM) contractile and relaxation function. Values are means ± SE of fold differences (with the control group taken as 1). Results were normalized with β-actin (Actb) and TATA box-binding protein (Tbp; N = 4–5 per group). Open and closed bars represent control and doxorubicin (DOX)-treated groups, respectively. *P < 0.05 vs. the control group. C and D: Western blot analysis of genes related to DSM contractility (C) and acto-myosin filaments (D). Nox1 and Nox2, NADPH oxidase 1 and 2; Duox1 and Duox2, dual oxidase 1 and 2; eNos, endothelial nitric oxide synthase; nNos, neuronal nitric oxide synthase; Sod1, Sod2, and Sod3, superoxide dismutase 1, 2, and 3; Cald1, caldesmon 1; smMHC, smooth muscle myosin heavy chain; Mlck, myosin light-chain kinase; Smtn, smoothelin; Tpm1, tropomyosin 1; Cav1.2 and Cav1.3, voltage-dependent Ca2+ channel 1.2 and 1.3; BK, large-conductance Ca2+-activated K+ channel; Adrb2 and Adrb3, β2- and β3-adrenoceptor; Chrm2 and Chrm3, muscarinic receptor 2 and 3; ChAT, choline acetyltransferase; P2X1, purinergic receptor P2X 1; Myl9, myosin light chain 9; P-Myl9, Ser19-phosphorylated Myl9; SMA, smooth muscle actin; Tagl, transgelin.
DOX induced altered myosin light-chain kinase expression paralleled with reduced phosphorylation of myosin light chain in the bladders.
To understand the molecular basis underlying the DOX-induced DSM contractile dysfunction, we examined expression levels of genes that play a role in detrusor contractility in the bladders from each group. Quantitative PCR analysis revealed that no apparent changes in the expression levels of contractile apparatus, including desmin, smooth muscle myosin heavy chain (smMHC), myosin light-chain kinase (Mlck), and smoothelin (Smtn), as well as neuronal markers associated with bladder function (3, 24) between the groups (Fig. 5B). Western blot analysis confirmed the comparable levels of expression for protein related to smooth muscle contractile function, including smMHC, Smtn, vimentin, SMA, and transgelin, from the DOX-treated group compared with the control group (Fig. 5, C and D). However, a 130-kDa isoform of smooth muscle Mlck (smMlck; 108–130 kDa) (39) was strongly expressed in the control group, whereas the DOX-treated group showed significant changes in the proportional expression of two isoforms: a downregulation of a 130-kDa isoform by 69% (P = 0.008) and a 2.7-fold upregulation of the smaller isoform (110 kDa, P = 0.031) compared with the control group (Fig. 5C). Various Mlck protein isoforms derived by alternative splicing or alternative initiation have been suggested to have distinct control of binding and catalytic activity of the enzyme (9). Western blot analysis revealed a significant increase in smooth muscle myosin light chain (Myl)9 expression by 1.9-fold (P = 0.003), besides a marked decrease in Ser19-phosphorylated Myl9 normalized to total Myl9 in the bladders from the DOX-treated group compared with the control group (0.6 ± 0.1 vs. 3 ± 0.7%, P = 0.038; Fig. 5D). These results suggest that systemic DOX exposure caused an altered expression of smMlck isoforms associated with a reduced phosphorylation of myosin light chain in the bladder.
DOX induced a downregulation of the large-conductance Ca2+-activated K+ channel β1-subunit accompanied by reduced large-conductance Ca2+-activated K+channel α-subunit cell surface expression in DSM cells.
Quantitative PCR data showed a significant downregulation of the large-conductance voltage and Ca2+-sensitive K+ [(BK)] channel β1-subunit (BKβ1; 50 ± 6%, with the control group taken as 100%, P = 0.009), along with an ~40% decrease of BK α-subunit (BKα) expression (P > 0.05) in the DOX-treated group (Fig. 5B). Western blot analysis showed a 25% reduction of BKα in the bladders from the DOX-treated group compared with the control group, although the change was statistically insignificant (Fig. 5C). We could not demonstrate the level of BKβ1 protein, as none of three commercially available antibodies tested detected any specific signal with molecular mass close to that predicted (28 and 38 kDa) in Western blot analysis (data not shown). Studies have demonstrated that BKβ1 regulates the activity of the pore-forming α-subunit through modulation of its Ca2+ sensitivity and voltage dependence as well as stimulation of cell surface expression of BKα (2, 7). We analyzed BKα distribution in the bladders from both DOX-treated and control groups. Robust BKα immunoreactivity (IR) was distributed throughout the cytoplasm of interstitial cells/fibroblasts residing within the lamina propria in both DOX-treated and control groups. In contrast, reduced BKα IR was detected in the DSM layer, with a decreasing intensity from the inner layer next to the lamina propria toward the outer layer. No colocalization of BKα IR with a neuronal marker, protein gene product 9.5, was observed in the bladders (Fig. 6A). Double labeling of BKα with desmin revealed that BKα IR localized primarily on the plasma membrane of DSM cells; however, BKα IR on the DSM cell surface was less intense in the DOX-treated group than that in the control group (Fig. 6B). These results suggest that DOX-induced downregulation of BKβ1 in the bladders interfered with cell surface expression of BKα in DSM cells, leading to potentially reduced BK channel activity in DSM cells.
Fig. 6.

Distribution of the large-conductance Ca2+-activated K+channel α-subunit (BKα) in the bladders. A: representative immunostainings for BKα (green; left), protein gene product 9.5 (PGP9.5; red; middle), and merged image (right) with DAPI (blue) for nuclear staining. White, dotted lines outline the boundaries among the urothelial (U), lamina propria (L), and detrusor smooth muscle (D) layers. Original scale bars = 50 µm. B: representative immunostainings for BKα (green; left), desmin (red; middle), and merged image (right) with DAPI (blue) for nuclear staining (N = 4 per group). Original scale bars = 10 µm. DOX, doxorubicin.
DISCUSSION
Various malignancies and their requisite treatments are known to induce LUTD, which compromises the long-term quality of life of survivors of cancer (14, 19, 22, 31, 41). DOX has been regarded as one of the most effective chemotherapy drugs and is used to treat a wide variety of cancers, not only in adults but also in children. As such, this agent has greatly contributed to the substantial improvement in survival rates, particularly for many childhood cancers. The improvement in survival from pediatric cancer has shifted some focus toward the recognition of the cancer treatment’s late effects (41). The present study provides new and important information regarding the effects of systemic DOX administration on urinary bladder function. First, our data from VSA demonstrated a progressive increase in frequency of small voids, suggesting a decline in the bladder function along the course of DOX exposure (29, 41). The moderate voiding dysfunction observed in this study would explain the fact that the effects of DOX on bladder function have been underrecognized, partly due to the difficulty in clinical assessment of bladder function, especially as the dysfunction can be occult in the absence of urinary retention. Second, our findings support the hypothesis that in vivo DOX exposure induces an impairment of DSM function in the urinary bladder. Our data provide strong evidence for an increasing recognition of DOX-induced myotoxicity on skeletal and smooth muscle tissues besides its well-known cardiotoxicity (20, 25, 35, 47).
It has been established that oxidative stress is the predominant cause of DOX-induced cardiotoxicity (15, 43, 45). Oxidative stress occurs when the generation of free radicals (RONS) overwhelms the capacity of endogenous antioxidant defense systems in the tissues and organs. The accumulated free radicals readily react with DNA, lipids, and proteins, which cause a range of cellular damages (e.g., DNA breaks, lipid peroxidation, and loss of enzyme activity), ultimately leading to cell death. The most important endogenous sources of RONS are enzymes of the mitochondrial respiratory chain, NOX and NOS. Previous studies have demonstrated the expression of at least four isoforms of NOX enzymes (Nox1, Nox2, Nox4, and Duox1) among seven isoforms (NOX1−NOX5, DUOX1, and DUOX-2) and two types of NOS (endothelial and neuronal) in the urinary bladder, mainly in the urothelium (4, 12, 16, 42). On the other side, SOD comprises the major antioxidant defense systems, which consists of three isoforms (SOD1−SOD3) (40). Our data revealed no changes in the expression level of any of those three types of enzymes in the bladders between the groups, implying that systemic DOX administration promoted neither RONS production nor downregulation of antioxidant systems in the bladder. Furthermore, we demonstrated that DOX did not induced proinflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α in the bladder. Studies have shown that oxidative stress induces those proinflammatory cytokines and vice versa (15, 18, 46). Collectively, the present data indicated that in vivo, DOX exposure did not induce oxidative stress in the bladder. As such, unlike in cardiomyocytes, a minimal, if any, contribution of oxidative stress to DSM dysfunction was caused by DOX exposure. This aligns with the bladder histology data from the DOX-treated group that showed no signs of muscle atrophy, fibrosis, or inflammation, which are reliably induced by oxidative stress in DOX-induced cardiotoxicity (15). Several lines of evidence demonstrated that DOX-induced skeletal muscle dysfunction is also mediated by oxidative stress-independent mechanisms, e.g., a dysregulation of Ca2+ homeostasis in the sarcoplasmic reticulum and myotubes, a suppression of protein synthesis, an interference of actin-myosin interactions, and a decrease in Ca2+ sensitivity (1, 36, 47). It has been previously shown that mitochondria typically comprise ~35% and 3–5% of the volume of cardiac and smooth muscle tissues, respectively. Furthermore, the intrinsic mitochondrial respiratory function in smooth muscle has been shown to be lowest among all three types of muscle (37). Accordingly, we consider that the distinct contribution of oxidative stress to the muscular dysfunction reflects the relatively small number of mitochondria and lower intrinsic mitochondrial respiratory functions in DSM cells compared with those in cardiac or skeletal muscle.
In the effort to identify the oxidative stress-independent molecular mechanism underlying DOX-induced DSM dysfunction, we identified no changes in the expression level of genes involved in DSM contractile function except for a downregulation of BKβ1 in the bladder after in vivo DOX exposure. BK channels are composed of a pore-forming α-subunit and tissue-specific regulatory β-subunits (β1−β4). Bladders express two β isoforms (β1- and β4-subunits) that are known to be predominantly expressed in smooth muscle and in neurons, respectively (38, 48). The finding that DOX did not change the expression levels of neuronal markers, as well as the BK β4-subunit in the bladder, is in good agreement with our data of DSM responses to different types of stimuli, implying that DOX-induced DSM dysfunction is primarily myogenic with a minimal impact on the neuronally mediated mechanism. BK channels have been proposed to play a role in the control of DSM excitability and contractility by maintaining the resting membrane potential (38). The present data that the DSM cell-specific decrease in cell surface-localized BKα protein in the DOX-treated group indicates that BKβ1 plays a role in the cell surface trafficking of BKα, therefore facilitating channel activity in DSM cells. Furthermore, studies have demonstrated that BKβ1 alters the gating kinetics to favor a more open state and increases Ca2+ sensitivity of the channel (2). Therefore, a decrease of BKβ1 would cause depolarization and hyperexcitability in DSM cells and, consequently, lead to detrusor overactivity and a related, overactive bladder and/or constrictive, low-compliant bladder through an increased muscle tone. This notion appears to, at least partly, explain the increased frequency of small voids observed in the DOX-treated group similar to that previously observed in BKα/Slo knockout mice (33). It has been shown that the BK channel extrudes K+ from cells, leading to the membrane potential restoration upon activation by both a local (known as Ca2+ sparks/puffs) and global increase in intracellular Ca2+ concentration, which facilitates DSM relaxation (38). As such, the attenuation of relaxation velocity of DSM, presented in the DOX-treated group, also implies the downregulation of BK channel activity in the DSM after in vivo DOX exposure. The reduction of relaxation velocity has also been previously reported in skeletal muscle after DOX exposure, suggesting a possible involvement of the BK channel in DOX-induced skeletal muscle impairment (47). Increasing evidence suggests that the BK channel also plays a major role in β-adrenoceptor-mediated bladder relaxation (13, 38). Our data showed that in vivo DOX exposure induced statistically insignificant but relatively large decreases in the expression level of β2- and β3-adrenoceptors (Adrb2 and Adrb3) in the bladders (Fig. 5B). Collectively, we consider the attenuation of BK channel activity as a primary cause of DOX-induced impairment of DSM relaxation responses, which resulted from at least two mechanisms: a reduced activation probability with a declined stimulation by β-adrenoceptors and reduced cell surface expression.
A primary mechanism for the activation of DSM contraction involves an increase in intracellular Ca2+ concentration, leading to myosin regulatory light-chain phosphorylation, cross-bridge cycling, and force development. Phosphorylation of Ser19 on myosin light chain 20 (MLC20) via MLCK allows subsequent interaction with actin filaments, leading to initiation of contraction (24). Therefore, our data that DOX induced altered smMlck activity and consequent decrease in the phosphorylation of MLC20, Myl9, in the bladder offers a compelling explanation for the attenuation of DSM contractility after in vivo DOX exposure as well as additional evidence that DOX-induced DSM dysfunction is mainly myogenic. A schematic presentation of our proposed mechanism underlying DOX-induced DSM dysfunction is shown in Fig. 7.
Fig. 7.

Schematic diagram of the proposed mechanisms underlying doxorubicin (DOX)-induced detrusor smooth muscle (DSM) dysfunction. An activation of the muscarinic acetylcholine receptor (mAChR) triggers an increase in intracellular Ca2+ concentration ([Ca2+]i), leading to Ca2+/calmodulin (CaM)-dependent activation of myosin light chain kinase (MLCK*). MLCK* phosphorylates myosin light chain 20 (MLC20), which enables the forming of actin-myosin cross-bridges, leading to muscle contraction. DOX causes a decrease in MLC20 phosphorylation, resulting in a reduction of muscle contractility (left). Increased Ca2+, cell depolarization, and β-adrenoceptor (β-AR) pathway activate the large-conductance Ca2+-activated K+ (BK) channel, which causes K+ efflux and hyperpolarizes membrane potential, blocking voltage-gated Ca2+ channels, leading to a decrease in [Ca2+]i. A decreased level of phosphorylated MLC20 by myosin light-chain phosphatase (MLCP) and deactivation of myosin light-chain kinase (MLCK) via the β-AR/PKA pathway and a low level of intracellular Ca2+ lead to muscle relaxation (right). DOX causes a downregulation of BK channel activity, leading to an impairment of muscle relaxation responses. DSMC, DSM cell; encircled P, phosphorylation of the proteins MLC20 and MLCK.
The present study sheds light on the previously underappreciated aspect of DOX toxicity on visceral smooth muscle, specifically, DSM in the urinary bladder, and provided evidence that DOX treatment causes bladder dysfunction. However, the DOX-induced bladder dysfunction may be manifested by only mild symptoms, at least in the short term, as the DOX-induced DSM hypertrophy and slower DSM relaxation after contractions could compensate for insufficient DSM contractile force to some extent and, therefore, can be overlooked in a clinical setting. Since many of the cancer late effects occur years after therapy, it is important for physicians to be aware that survivors of childhood cancer who were treated with DOX are at a potentially increased risk of bladder dysfunction, and, accordingly, those patients must be monitored closely. Future studies, both in the clinic and in the laboratory, concerning longer-term effects of DOX on the bladder function, as well as a possible prevention and reversibility/progression, will allow for deeper understanding of these understudied side effects.
GRANTS
This work was supported by a Colorado Clinical and Translational Sciences Institute Research Grant (National Center for Advancing Translational Sciences Colorado Clinical and Translational Science Awards Grant KL2-TR-001080, to N. G. Cost) and the Ponzio Family Endowment Fund (to D. T. Wilcox). Support for the histological study was provided by the University of Colorado Denver Research Histology Shared Resource, funded by a Cancer Center Support Grant (P30-CA-046934).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.I. and N.G.C. conceived and designed research; N.I. and A.C. performed experiments; N.I. and R.H.P. analyzed data; N.I., D.T.W., and N.G.C. interpreted results of experiments; N.I. prepared figures; N.I. and M.İ.D. drafted manuscript; N.I., D.T.W., A.P.M., and N.G.C. edited and revised manuscript; N.I., M.İ.D., A.C., D.T.W., R.H.P., A.P.M., and N.G.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Leah Crawford (University of Colorado Denver) for assistance with this study.
REFERENCES
- 1.Abramson JJ, Buck E, Salama G, Casida JE, Pessah IN. Mechanism of anthraquinone-induced calcium release from skeletal muscle sarcoplasmic reticulum. J Biol Chem 263: 18750–18758, 1988. [PubMed] [Google Scholar]
- 2.Bao L, Cox DH. Gating and ionic currents reveal how the BKCa channel’s Ca2+ sensitivity is enhanced by its beta1 subunit. J Gen Physiol 126: 393–412, 2005. doi: 10.1085/jgp.200509346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beamish JA, He P, Kottke-Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev 16: 467–491, 2010. doi: 10.1089/ten.teb.2009.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birder LA, Nealen ML, Kiss S, de Groat WC, Caterina MJ, Wang E, Apodaca G, Kanai AJ. Beta-adrenoceptor agonists stimulate endothelial nitric oxide synthase in rat urinary bladder urothelial cells. J Neurosci 22: 8063–8070, 2002. doi: 10.1523/JNEUROSCI.22-18-08063.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisazza A. Social organization and territorial behaviour in three strains of mice. Ital J Zool (Modena) 48: 157–167, 1981. [Google Scholar]
- 6.Brodkin ES. BALB/c mice: low sociability and other phenotypes that may be relevant to autism. Behav Brain Res 176: 53–65, 2007. doi: 10.1016/j.bbr.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Bi D, Lu ZH, McClafferty H, Shipston MJ. Distinct domains of the β1-subunit cytosolic N terminus control surface expression and functional properties of large-conductance calcium-activated potassium (BK) channels. J Biol Chem 292: 8694–8704, 2017. doi: 10.1074/jbc.M116.769505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Citak EC, Oguz A, Karadeniz C, Karaoglan B, Tan MO, Okur V, Cansu A. Vincristine-induced peripheral neuropathy and urinary bladder paralysis in a child with rhabdomyosarcoma. J Pediatr Hematol Oncol 30: 61–62, 2008. doi: 10.1097/MPH.0b013e318158343b. [DOI] [PubMed] [Google Scholar]
- 9.Clayburgh DR, Rosen S, Witkowski ED, Wang F, Blair S, Dudek S, Garcia JG, Alverdy JC, Turner JR. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J Biol Chem 279: 55506–55513, 2004. doi: 10.1074/jbc.M408822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox G, Kable E, Jones A, Fraser I, Manconi F, Gorrell MD. 3-Dimensional imaging of collagen using second harmonic generation. J Struct Biol 141: 53–62, 2003. doi: 10.1016/S1047-8477(02)00576-2. [DOI] [PubMed] [Google Scholar]
- 11.Desai VG, Herman EH, Moland CL, Branham WS, Lewis SM, Davis KJ, George NI, Lee T, Kerr S, Fuscoe JC. Development of doxorubicin-induced chronic cardiotoxicity in the B6C3F1 mouse model. Toxicol Appl Pharmacol 266: 109–121, 2013. doi: 10.1016/j.taap.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 12.Donkó A, Ruisanchez E, Orient A, Enyedi B, Kapui R, Péterfi Z, de Deken X, Benyó Z, Geiszt M. Urothelial cells produce hydrogen peroxide through the activation of Duox1. Free Radic Biol Med 49: 2040–2048, 2010. doi: 10.1016/j.freeradbiomed.2010.09.027. [DOI] [PubMed] [Google Scholar]
- 13.Frazier EP, Peters SL, Braverman AS, Ruggieri MR Sr, Michel MC. Signal transduction underlying the control of urinary bladder smooth muscle tone by muscarinic receptors and beta-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol 377: 449–462, 2008. doi: 10.1007/s00210-007-0208-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gargiulo P, Arenare L, Pisano C, Cecere SC, Falivene S, Greggi S, Tambaro R, Facchini G, De Palma G, Scaffa C, Della Pepa C, Pignata S, Di Napoli M. Long-term toxicity and quality of life in patients treated for locally advanced cervical cancer. Oncology 90: 29–35, 2016. doi: 10.1159/000441226. [DOI] [PubMed] [Google Scholar]
- 15.Ghigo A, Li M, Hirsch E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim Biophys Acta 1863: 1916–1925, 2016. doi: 10.1016/j.bbamcr.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 16.Gillespie JI, Markerink-van Ittersum M, de Vente J. Expression of neuronal nitric oxide synthase (nNOS) and nitric-oxide-induced changes in cGMP in the urothelial layer of the guinea pig bladder. Cell Tissue Res 321: 341–351, 2005. doi: 10.1007/s00441-005-1151-3. [DOI] [PubMed] [Google Scholar]
- 17.Godoy LY, Fukushige J, Igarashi H, Matsuzaki A, Ueda K. Anthracycline-induced cardiotoxicity in children with malignancies. Acta Paediatr Jpn 39: 188–193, 1997. doi: 10.1111/j.1442-200X.1997.tb03579.x. [DOI] [PubMed] [Google Scholar]
- 18.Guichard C, Pedruzzi E, Dewas C, Fay M, Pouzet C, Bens M, Vandewalle A, Ogier-Denis E, Gougerot-Pocidalo MA, Elbim C. Interleukin-8-induced priming of neutrophil oxidative burst requires sequential recruitment of NADPH oxidase components into lipid rafts. J Biol Chem 280: 37021–37032, 2005. doi: 10.1074/jbc.M506594200. [DOI] [PubMed] [Google Scholar]
- 19.Hasanov E, Hasanov M, Kuria IM, Hasanov R, Rzazade R, Jonasch E, Altundag K. Effects of tamoxifen on urinary incontinence: Case report and review of literature. Medicine (Baltimore) 96: e6785, 2017. doi: 10.1097/MD.0000000000006785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayward R, Hydock D, Gibson N, Greufe S, Bredahl E, Parry T. Tissue retention of doxorubicin and its effects on cardiac, smooth, and skeletal muscle function. J Physiol Biochem 69: 177–187, 2013. doi: 10.1007/s13105-012-0200-0. [DOI] [PubMed] [Google Scholar]
- 21.Hecht S, Brazell A, Holbrook S, Wilcox DT, Cost NG. Clinical survey of voiding dysfunction in childhood cancer survivors after vincristine and/or doxorubicin chemotherapy. American Urological Association 2019 Annual Meeting Chicago, IL, May 3–6, 2019. http://www.spuonline.org/abstracts/2019/P23.cgi. [Google Scholar]
- 22.Heesakkers J, Farag F, Bauer RM, Sandhu J, De Ridder D, Stenzl A. Pathophysiology and contributing factors in postprostatectomy incontinence: a review. Eur Urol 71: 936–944, 2017. doi: 10.1016/j.eururo.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 23.Hershman DL, McBride RB, Eisenberger A, Tsai WY, Grann VR, Jacobson JS. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell non-Hodgkin’s lymphoma. J Clin Oncol 26: 3159–3165, 2008. doi: 10.1200/JCO.2007.14.1242. [DOI] [PubMed] [Google Scholar]
- 24.Hill WG. Control of urinary drainage and voiding. Clin J Am Soc Nephrol 10: 480–492, 2015. doi: 10.2215/CJN.04520413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodjat M, Haller H, Dumler I, Kiyan Y. Urokinase receptor mediates doxorubicin-induced vascular smooth muscle cell senescence via proteasomal degradation of TRF2. J Vasc Res 50: 109–123, 2013. doi: 10.1159/000343000. [DOI] [PubMed] [Google Scholar]
- 27.Iguchi N, Dönmez MI, Malykhina AP, Carrasco A Jr, Wilcox DT. Preventative effects of a HIF inhibitor, 17-DMAG, on partial bladder outlet obstruction-induced bladder dysfunction. Am J Physiol Renal Physiol 313: F1149–F1160, 2017. doi: 10.1152/ajprenal.00240.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iguchi N, Hou A, Koul HK, Wilcox DT. Partial bladder outlet obstruction in mice may cause E-cadherin repression through hypoxia induced pathway. J Urol 192: 964–972, 2014. doi: 10.1016/j.juro.2014.03.037. [DOI] [PubMed] [Google Scholar]
- 29.Iguchi N, Malykhina AP, Wilcox DT. Early life voiding dysfunction leads to lower urinary tract dysfunction through alteration of muscarinic and purinergic signaling in the bladder. Am J Physiol Renal Physiol 315: F1320–F1328, 2018. doi: 10.1152/ajprenal.00154.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maruniak JA, Wysocki CJ, Taylor JA. Mediation of male mouse urine marking and aggression by the vomeronasal organ. Physiol Behav 37: 655–657, 1986. doi: 10.1016/0031-9384(86)90300-8. [DOI] [PubMed] [Google Scholar]
- 31.Matz EL, Hsieh MH. Review of advances in uroprotective agents for cyclophosphamide- and ifosfamide-induced hemorrhagic cystitis. Urology 100: 16–19, 2017. doi: 10.1016/j.urology.2016.07.030. [DOI] [PubMed] [Google Scholar]
- 32.McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther 31: 63–75, 2017. doi: 10.1007/s10557-016-6711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem 279: 36746–36752, 2004. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- 34.Merten KE, Jiang Y, Feng W, Kang YJ. Calcineurin activation is not necessary for doxorubicin-induced hypertrophy in H9c2 embryonic rat cardiac cells: involvement of the phosphoinositide 3-kinase-Akt pathway. J Pharmacol Exp Ther 319: 934–940, 2006. doi: 10.1124/jpet.106.108845. [DOI] [PubMed] [Google Scholar]
- 35.Millington KA, Pascasio JM, Halligan GE, de Chadarévian JP. Chemotherapy-related leiomyopathy: a suggested morphological explanation for the intestinal dysmotility affecting patients treated with anthracyclines. Mod Pathol 25: 289–294, 2012. doi: 10.1038/modpathol.2011.115. [DOI] [PubMed] [Google Scholar]
- 35a.National Cancer Institute Late Effects of Treatment for Childhood Cancer (PDQ®)−Health Professional Version (Online). https://www.cancer.gov/types/childhood-cancers/late-effects-hp-pdq [accessed 29 April 2019].
- 36.Nissinen TA, Degerman J, Räsänen M, Poikonen AR, Koskinen S, Mervaala E, Pasternack A, Ritvos O, Kivelä R, Hulmi JJ. Systemic blockade of ACVR2B ligands prevents chemotherapy-induced muscle wasting by restoring muscle protein synthesis without affecting oxidative capacity or atrogenes. Sci Rep 6: 32695, 2016. doi: 10.1038/srep32695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park SY, Gifford JR, Andtbacka RH, Trinity JD, Hyngstrom JR, Garten RS, Diakos NA, Ives SJ, Dela F, Larsen S, Drakos S, Richardson RS. Cardiac, skeletal, and smooth muscle mitochondrial respiration: are all mitochondria created equal? Am J Physiol Heart Circ Physiol 307: H346–H352, 2014. doi: 10.1152/ajpheart.00227.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petkov GV. Central role of the BK channel in urinary bladder smooth muscle physiology and pathophysiology. Am J Physiol Regul Integr Comp Physiol 307: R571–R584, 2014. doi: 10.1152/ajpregu.00142.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poperechnaya A, Varlamova O, Lin PJ, Stull JT, Bresnick AR. Localization and activity of myosin light chain kinase isoforms during the cell cycle. J Cell Biol 151: 697–708, 2000. doi: 10.1083/jcb.151.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci 38: 592–607, 2017. doi: 10.1016/j.tips.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 41.Ritchey M, Ferrer F, Shearer P, Spunt SL. Late effects on the urinary bladder in patients treated for cancer in childhood: a report from the Children’s Oncology Group. Pediatr Blood Cancer 52: 439–446, 2009. doi: 10.1002/pbc.21826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts M, Amosah J, Sui G, Wu R, Archer S, Ruggieri M, Wu C. Identifying the functional subtypes of nadph oxidases (nox) from the urothelium: implication for ros-controlling targets. J Urol 199: e1061, 2018. doi: 10.1016/j.juro.2018.02.2631. [DOI] [Google Scholar]
- 43.Rochette L, Guenancia C, Gudjoncik A, Hachet O, Zeller M, Cottin Y, Vergely C. Anthracyclines/trastuzumab: new aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol Sci 36: 326–348, 2015. doi: 10.1016/j.tips.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 44.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 69: 7–34, 2019. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 45.Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, Jones A. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer 10: 337, 2010. doi: 10.1186/1471-2407-10-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: 44–84, 2007. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 47.van Norren K, van Helvoort A, Argilés JM, van Tuijl S, Arts K, Gorselink M, Laviano A, Kegler D, Haagsman HP, van der Beek EM. Direct effects of doxorubicin on skeletal muscle contribute to fatigue. Br J Cancer 100: 311–314, 2009. doi: 10.1038/sj.bjc.6604858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu RS, Marx SO. The BK potassium channel in the vascular smooth muscle and kidney: α- and β-subunits. Kidney Int 78: 963–974, 2010. doi: 10.1038/ki.2010.325. [DOI] [PubMed] [Google Scholar]