

Abstract
Vascular stiffening and its sequelae are major causes of morbidity and mortality in the elderly. The increasingly accepted concept of “smooth muscle cell (SMC) stiffness syndrome” along with matrix deposition has emerged in vascular biology to account for the mechanical phenotype of arterial aging, but the molecular targets remain elusive. In this study, using an unbiased proteomic analysis, we identified lysyl oxidase-like 2 (LOXL2) as a critical SMC mediator for age-associated vascular stiffening. We tested the hypothesis that loss of LOXL2 function is protective in aging-associated vascular stiffening. We determined that exogenous and endogenous nitric oxide markedly decreased LOXL2 abundance and activity in the extracellular matrix of isolated SMCs and LOXL2 endothelial cells suppress LOXL2 abundance in the aorta. In a longitudinal study, LOXL2+/− mice were protected from age-associated increase in pulse-wave velocity, an index of vascular stiffening, as occurred in littermate wild-type mice. Using isolated aortic segments, we found that LOXL2 mediates vascular stiffening in aging by promoting SMC stiffness, augmented SMC contractility, and vascular matrix deposition. Together, these studies establish LOXL2 as a nodal point for a new therapeutic approach to treat age-associated vascular stiffening.
NEW & NOTEWORTHY Increased central vascular stiffness augments risk of major adverse cardiovascular events. Despite significant advances in understanding the genetic and molecular underpinnings of vascular stiffening, targeted therapy has remained elusive. Here, we show that lysyl oxidase-like 2 (LOXL2) drives vascular stiffening during aging by promoting matrix remodeling and vascular smooth muscle cell stiffening. Reduced LOXL2 expression protects mice from age-associated vascular stiffening and delays the onset of isolated systolic hypertension, a major consequence of stiffening.
Keywords: aging, lysyl oxidase-like 2, pulse-wave velocity, vascular stiffness
INTRODUCTION
Central vascular stiffening occurs during aging and often precedes the onset of hypertension (2). Increased vascular stiffness is a strong and independent predictor for adverse cardiovascular events such as perioperative myocardial infarctions, heart failure, stroke, and death (27). Key components of the multifactorial process that leads to vascular stiffening include inflammation, endothelial dysfunction, remodeling of the composition and architecture of the extracellular matrix (ECM), and changes in vascular smooth muscle cell (VSMC) content and function (7). Although the contributions of endothelial dysfunction and matrix remodeling to increases in vascular stiffness are clearly evident, they fail to fully capture the alterations observed in in vivo models of aging and hypertension.
The knowledge that VSMC stiffening and VSMC interaction with and adhesion to the ECM are critically important has led to the introduction of a concept known as “smooth muscle cell stiffness syndrome,” which has now considerably extended our mechanistic understanding of the vascular stiffness phenotype (20, 21). Moreover, it is increasingly evident that VSMC tone can compensate for compromised matrix mechanics (8, 25). The changes observed with vascular stiffening thereby span from the endothelium to the adventitia, as they encompass modifications to the composition of the vessel and changes in the interactions between the cellular and structural elements of the vessel (14). Discovery and characterization of molecular targets that promote matrix deposition and VSMC stiffness can thus assist in the identification of novel targets that mediate vascular stiffening.
Using an unbiased proteomic analysis, we identified lysyl oxidase-like 2 (LOXL2) as a potential VSMC mediator for age-associated vascular stiffening. Recently, LOXL2 emerged as one of the candidate genes in genome-wide association studies focused on hypertension and arterial stiffness (15, 19). Additional bioinformatics analysis also identified LOXL2 as a putative marker gene in hypertension (10). LOXL2 is a member of the copper-dependent amine oxidase gene family. Because of these genetic associations and our preliminary data, here, we examined the role of LOXL2 in the development of age-associated vascular stiffening, a primary contributor to systolic hypertension and determined the role of LOXL2 in modulating matrix composition and stiffness as well as VSMC stiffness and function.
METHODS
Animal model.
LOXL2+/− mice were generated by using cryopreserved sperm (Taconic), in which the LOXL2 gene was disrupted by inserting a long terminal repeat (LTR)-trapping cassette in exon 1. The mice were bred in-house and genotyped by standard PCR as follows: the LTR-trapping cassette (LOXL2 gene knockout) was identified using LTR-2 forward primer (5′-AAATGGCGTTACTTAAGCTAGCTTGC-3′) and TF1717 lower primer: (5′-GTGATTGTTCACCTTGATCCCAGG-3′), that yielded a 157-bp amplicon. The wild-type (WT) LOXL2 gene was TF1717 upper (5′-CTCCCAAATTGAGTTGAGCT-3′) and TF1717 lower primers, which yielded a 259-bp amplicon. Both PCR reactions were performed for each sample. The presence of both amplicons indicates a heterozygous mouse, and a WT amplicon only indicates a WT mouse. Male and female LOXL2+/− and WT littermate mice were used in this study. Animals were maintained in the Johns Hopkins University School of Medicine animal care facility. Animals were fed and watered ad libitum and maintained on a 12-h:12-h light-dark cycle. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University.
Cell culture.
Human aortic smooth muscle cells (HASMCs; Thermo Fisher) were cultured in SMC media (ScienCell Research Laboratories) containing 2% FBS, antibiotics, and SMC growth supplement. Human aortic endothelial cells (HAECs; Thermo Fisher) were cultured in endothelial cell media (ScienCell Research Laboratories) containing 5% FBS, antibiotics, and endothelial cell growth supplement. Mouse aortic SMCs were isolated as previously described (13, 25) and maintained in DMEM containing 10% FBS, an antibiotic, and an antimycotic.
Smooth muscle cell secretome analysis by iTRAQ.
HASMCs were seeded into six-well plates at 80% confluence, and HAECs were seeded in hanging cell culture inserts (Millipore) in their respective media. After 24 h of culture, both HASMCs and HAECs were serum starved for 18 h. The HAEC inserts were next transferred to the HASMCs to initiate coculture. Control HASMCs received inserts without any HAECs. A total of six HASMC + HAEC and six HASMC-only wells were prepared. After 4 days, the conditioned medium from the HASMC layers was collected. The medium from three replicates was combined to yield four samples (2 with HAECs and 2 without HAECs). The samples were concentrated and examined using 4-Plex iTRAQ at the Johns Hopkins Proteomics Core as follows: proteins were digested with LysC and trypsin and then labeled with isobaric mass tags (114, 115, 116, and 117; Thermo Fisher) followed by HPLC-MS/MS for identification and quantification of proteins.
Peptide sequences were analyzed using Proteome Discoverer v1.4 (Thermo Fisher) software for protein identification. Protein identities and their corresponding primary iTRAQ reporter ion areas were exported from Proteome Discoverer v1.4 (Thermo Fisher) software and imported into Partek Genomics Suite v6.6 (Partek) for further analysis. Mass spectroscopy data underwent normalization to minimize the technical differences between iTRAQ lanes and illuminate relative differences in protein abundance across the four biologic samples. To ensure unambiguous nomenclature and ease of evaluation, we mapped all spectras' NCBI protein gi identifiers to their cognate genes in the NCBI Entrez database.
All peptide spectra first underwent a quality check, wherein only Proteome Discoverer Isolation Interference values <30% were retained for evaluation. For each sample, the median signal values of all spectra were determined for each peptide and were deemed to represent that peptide, yielding 1 value per sample for each of the resulting 1,499 peptides. These median signal values were then quantile normalized to minimize potential differences across the four iTRAQ lanes and transformed to log2 notation using Partek Genomics Suite v6.6. We applied a two-tailed, one-way ANOVA, using a Partek’s t-test, to evaluate each protein’s relative concentration as fold change between the two biological classes; each class is represented by two biologic replicates. These values were then exported for further evaluation and graphical representation to the Spotfire DecisionSite with Functional Genomics v9.1.2 (TIBCO Spotfire).
LOX activity assay.
LOX activity was examined using the Amplite LOX-activity assay kit (AAT Bioquest) in conditioned cell culture media, isolated cell-derived matrix, and tissue homogenates. In this assay, the release of H2O2 by the oxidation of a proprietary substrate by LOX is quantified by horseradish peroxidase-coupled oxidation of Amplex Red to resorufin. The assay detects total LOX activity in cell/tissue homogenates.
Nitric oxide production in the aorta.
Aortic rings from young (3 mo old) and old (18+ mo old) WT mice were weighed, transected, and pinned en face endothelium facing up in a Silastic-coated petri dish. Samples were maintained in Krebs-HEPES buffer containing the nitric oxide (NO)-sensitive fluorescent dye 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA; 5 μM; Molecular Probes, Eugene, OR). The vessel strips were incubated in a cell culture incubator for 20 min in the dark.
Control rings were incubated without DAF-FM DA to subtract autofluorescence in the sample.
Four distinct regions were imaged per aortic specimen using a Nikon 80i Epifluorescence microscope, equipped with a CoolSnapHQ2 camera.
Effect of endothelium on LOXL2 abundance in the vascular matrix.
Aortas from young (3 mo old) and old (18+ mo old) WT mice were isolated and cut into 2-mm rings. The endothelium was removed by mechanical scraping in half of the rings, and the other half was maintained intact. All samples were incubated in a cell culture incubator in ECM media for 24 h. Samples were then decellularized using previously described methods (13, 23, 24). Briefly, aortic rings were incubated in decellularization solution 1 (8 mM CHAPS, 1 M NaCl, and 25 mM EDTA in PBS) for 18 h. Samples were next washed three times in PBS and incubated in decellularization solution 2 (1.8 mM SDS, 1 M NaCl, and 25 mM EDTA in PBS) for 24 h. Samples were then washed three times with PBS to remove the detergents. All these steps were conducted at room temperature, with continuous shaking under sterile conditions. Finally, samples were placed in a cell culture incubator in endothelial cell media (ScienCell Research Laboratories) followed by three 15-min washes with PBS to obtain the decellularized specimen. Removal of cells was confirmed by the absence of DNA assayed using the Pico Green assay kit (Invitrogen), and absence of GAPDH (cytosolic protein) was confirmed by Western blot analysis. Decellularization was also confirmed by confocal microscopy FITC-conjugated phalloidin and DAPI-stained segments. LOXL2 abundance in the decellularized samples was then determined by Western blot analysis.
Aging study.
WT and LOXL2+/− littermate mice were used. The WT cohort comprised 10 mice of which 5 were female and 5 were male. The LOXL2+/− cohort comprised 12 mice of which 7 were female and 5 were male. Blood pressure (BP) of awake animals was measured noninvasively with the tail-cuff method (Kent Scientific), and pulse-wave velocity (PWV) was measured noninvasively by using high-frequency Doppler (Indus Instruments) as previously described (11). Five-week-old LOXL2+/− and WT mice were acclimatized to the BP cone for 1 wk. Starting at 6 wk of age, PWV, BP (systolic, mean, and diastolic), and heart rate were measured every 2–4 wk until 20 mo of age. Measurements were grouped into age groups of 1–3, 3–8, 8–12, 12–15, and 15–20 mo for statistical evaluation. For histochemical, immunofluorescence, and Western blot analyses, aortas from young adult (3–4 mo-old) and old (20–22 mo old) mice were used.
Pressure myography.
The carotid arteries of mice were dissected out and cleaned. The sample was mounted in the chamber of a pressure myograph (DMT) filled with oxygenated calcium-free Krebs buffer at 37°C. After the buffer was briefly passed through the vessel to evacuate blood remaining in the lumen, the vessel was equilibrated at 35 mmHg for 5 min followed by 50 mmHg for 15 min. The pressure was next increased by 10 mmHg every 2 min from 50 to 120 mmHg. Outer and inner diameter and wall thicknesses were recorded for each pressure level in calcium-free Krebs buffer. The lumen diameter was measured at each inflating pressure. Compliance over the physiologic pulse pressure was determined pre- and postremodeling with the following equation:
where V is volume, P is pressure, and Di is lumen diameter.
Wire myography.
Vasoreactivity of isolated aortic rings was studied as previously described (13, 23, 24). Vasocontractile response was determined with a phenylephrine concentration response (10−9 to 10−5 mol/l) and normalized to a potassium chloride response (60 mM). Vessels were next preconstricted with phenylephrine (10−6 mol/l; Sigma-Aldrich) and endothelium-dependent and -independent vasorelaxation responses were determined with acetylcholine (10−9 to 10−5 mol/l) and sodium nitroprusside (10−9 to 10−5 mol/l; Sigma-Aldrich), respectively.
Tensile testing.
The elastic properties of intact and decellularized aortic rings were analyzed by tensile testing as previously described (13, 23, 24). Briefly, the thoracic aortas from 15- to 20-mo-old WT and LOXL2+/− littermates were harvested and cut into 2-mm rings. Two intact and two decellularized rings from each animal were tested. Transverse and longitudinal images of the sample were obtained to calculate vessel dimensions [lumen diameter (Di), wall thickness (t), and length (L)]. Samples were then mounted on an electromechanical puller (DMT). After calibration and alignment, the pins were moved apart using an electromotor and displacement and force were recorded continuously. Engineering stress (S) was calculated by normalizing force (F) to the initial stress-free area of the specimen (S = F/2t × L; where t = thickness and L = length of the sample). Engineering strain (λ) was calculated as the ratio of displacement to the initial stress-free diameter. The stress-strain relationship was represented by the equation S = α exp (βλ), where α and β are constants. α and β were determined by nonlinear regression for each sample and used to generate stress-strain curves by treating the x-axis as a continuous variable.
Live cell micromechanical methods.
Material properties of VSMCs freshly isolated from mouse aortas were assessed by spontaneous and forced bead motions using magnetic twisting cytometry, as described previously (1, 4). For these experiments, we used RGD-coated ferrimagnetic microbeads (~4.5 μm in diameter) anchored to the cytoskeleton through cell surface integrin receptors of the adherent living cell. The beads were subjected to a vertically aligned magnetic field at a frequency of 0.75 Hz. Lateral bead displacements were measured optically, and the ratio of specific torque to bead displacement was computed and expressed as the cell stiffness in units of Pascals per nanometers. For spontaneous bead motions, we computed mean square displacements as an index of cytoskeletal remodeling dynamics.
Cell motility.
Cell motility was examined with a fluorescence-based SMC motility kit (Millipore; Boyden chamber-based assay) in which 10% FBS was used as the chemoattractant. Cell migration to the bottom chamber was quantified by the PicoGreen assay.
Deadhesion.
Cells were grown to 80% confluence and used to monitor deadhesion as previously described (22, 25). After briefly rinsing the cell monolayer with sterile PBS, we severed the cell-matrix adhesive contacts with 0.05% trypsin. The cellular retraction that precedes cell detachment was monitored by imaging the cell monolayer every 10 s for 2 min at ×10 magnification. Total area occupied by cells was then calculated for each image with ImageJ software (NIH). The time-dependent normalized area was quantified as:
where Ainitial is initial spread area, A(t) is spread area at time t, and Afinal is final spread area. The normalized area versus time data were then fit to the following Boltzmann sigmoidal curve:
where τ1 and τ2 are time constants of deadhesion, and lower values represent rapid detachment of cells. τ1 Is the time at which 50% retraction in cell spread area has occurred, and τ2 describes the steepness (slope) of the curve.
Fourier transform traction microscopy.
Cells were seeded on inert elastic gel blocks (Young’s modulus ~8 kPa with Poisson’s ratio 0.48) that were coated with type I collagen with embedded green-fluorescent microbeads. The contractile stress arising at the interface between each adherent cell and its substrate was measured with traction microscopy as previously described (26, 28). Briefly, both fluorescence and phase contrast images of the cells were acquired. Trypsin was next added to remove the cells, and a traction-free image of the gel was obtained, which served as a reference image. The displacement field was determined by an image correlation method between a paired traction-free and cell image.
Western blot analysis.
Proteins from conditioned cell culture media (CCM) were enriched using StrataClean Resin (6, 9). Briefly, 10 μl of StrataClean resin were added to CCM (1.5 ml), and samples were vortexed for 1 min. The resin was recovered by centrifugation at 12,000 rpm for 5 min. Cells were lysed with mammalian protein extraction reagent (M-PER; Thermo Fisher) containing protease inhibitors (Roche) and centrifuged. The concentration of soluble proteins was determined with the Bio-Rad Protein assay reagent (Bio-Rad). The StrataClean resin pellet and M-PER insoluble pellet were resuspended in 35 μl of 2× Laemmli buffer and used for Western blotting. Proteins from the M-PER soluble fraction (25 μg), ~5 μg of CCM proteins, and the entire volume of M-PER insoluble fraction were used for Western blot analysis. For the cell-derived matrix (ECM) and conditioned media blots, total protein loading was examined by Ponceau S staining before exposure to antibodies and used to normalize Western blot analysis densitometry data. Tissue samples were homogenized in SDS lysis buffer (1× RIPA buffer supplemented with 1% wt/vol SDS) to facilitate solubilization of ECM proteins. For cytosolic proteins, GAPDH was used as loading control. For Western blot analysis, the following antibodies were used: LOXL2 (rabbit monoclonal, 1:1,000; Abcam), fibronectin (mouse monoclonal, 1:2,500; Abcam), GAPDH (mouse monoclonal, 1:5,000; Novus Bio), LOX (rabbit polyclonal, 1:1,000; Santa Cruz Biotechnology), and LOXL3 (rabbit polyclonal, 1:1,000; Abcam) by Western blot analysis.
Immunofluorescence assay.
For immunofluorescent imaging, aortas were dissected, cleaned free of connective tissue, and fixed in 4% formaldehyde for 24 h. The samples were then rinsed and permeabilized (4% paraformaldehyde + 0.1% Triton X-100) for 20 min. After rinsing was completed, LOXL2 was labeled by treating samples with rabbit-anti-LOXL2 primary antibody (1:100; 2 h) followed by AlexaFluor 546-conjugated goat anti-rabbit secondary (1:500) antibody. Nuclei were labeled with DAPI, samples were mounted and imaged en face endothelium facing up by confocal microscopy.
Histology and immunohistochemistry.
Mouse thoracic aortic rings were cut at 2-mm length and then formalin fixed and paraffin embedded. These formalin-fixed and paraffin-embedded tissues were sectioned at 6 μm and mounted onto charged (plus) slides. Hematoxylin and eosin, Masson trichrome, and Movat pentachrome stains were performed in the Department of Pathology Reference Histology laboratory using standard methods. Images were digitally captured with a Laxco microscope at ×10. Aortas from different mice were blindly, qualitatively evaluated by a cardiovascular pathologist for histopathologic changes by hematoxylin and eosin, Masson trichrome, or Movat pentachrome stains on a scale of 1 (healthy) to 4 (maximal loss of nuclei, medial fibrosis, elastic fiber fracture).
Statistical analysis.
Cell micromechanical data are presented as geometric means ± SE. To satisfy the assumption of normal distributions associated with ANOVA, we converted micromechanical data to log scale before analysis. Student’s t-test was used to compare two means. All other data are presented as arithmetic means ± SE. Sample size (n) is indicated for each reported value. For statistical evaluation, two means were compared by the nonparametric Wilcoxon rank-sum test and groups were compared with the Kruskal-Wallis nonparametric test with Dunn’s comparison. For multiple comparisons, two-way ANOVA with Bonferroni post hoc analysis was used. Means were considered to be statistically different at P < 0.05.
RESULTS
Identification of LOXL2 as a novel target.
The proteins secreted by HASMCs in the presence or absence of HAECs in coculture inserts was examined (Fig. 1A). A total of 16,243 peptides composing 1,499 proteins were identified and classified based on function in the HASMC secretome (Fig. 1B). Data were normalized to the samples without HAECs. A pseudo P value was calculated to generate a volcano plot (Fig. 1C), and proteins whose secretion was either increased or decreased in the presence of HAECs were identified. Within this subset of proteins, given the central importance of matrix remodeling in vascular stiffness, we focused our attention on enzymes that catalyze matrix deposition. The secretion of LOX family proteins was decreased when HASMCs were cocultured with HAECs [LOX (4.3-fold), LOXL2 (3.6-fold), and LOXL3 (1.2-fold)]. In the literature, genome-wide association studies and bioinformatics analysis linked LOXL2 to vascular remodeling in human aging and hypertension (10, 15, 19). However, the specific role of LOXL2 in central vascular stiffening has not been studied to date. Therefore, we selected LOXL2 as the target in this study.
Fig. 1.

Analysis of the human aortic smooth muscle cell (HASMC) secretome. A: HASMCs were cocultured with or without human aortic endothelial cells (HAECs) in cell-culture inserts. The conditioned cell culture medium from the HASMC layer was collected and used for iTRAQ analysis. B: proteins secreted by HASMCs were classified based on function. C: volcano plot showing proteins with 2-fold or greater increase or decrease in secretion by the HASMC layer with P < 0.05 vs. control.
We first verified that LOXL2 secretion is altered by coculture of HASMCs with HAECs on a candidate basis by repeating the coculture assay and using Western blot analysis to probe for LOXL2 secretion to the HASMC media and cell-derived matrix. Both LOXL2 expression and activity (Fig. 2A) were diminished in the HASMC media and cell-derived matrix from HASMCs when cocultured with HAECs when compared with those from HASMCs cultured alone.
Fig. 2.

Endogenous and exogenous nitric oxide (NO) regulate extracellular lysyl oxidase-like 2 (LOXL2) abundance and activity in vascular smooth muscle cells (VSMCs). Ai–Aiv: representative Western blot (Ai), densitometry analysis of LOXL2 protein in the cell-derived extracellular matrix (ECM; Aii), conditioned cell culture media (CCM; Aiii), and LOXL2 activity in the CCM (Aiv) in the human aortic smooth muscle cells (HASMCs) in the presence or absence of human aortic endothelial cell (HAEC) coculture and the endothelial nitric oxide synthase inhibitor nitro-l-arginine methyl ester (l-NAME; 100 μM; n = 6, *P < 0.05, **P < 0.01 by Kruskal Wallis test with Dunn’s post hoc test). IB, immunoblot. Bi and Bii: representative Western blots of LOXL2 protein in the aortic matrix of intact and endothelium-denuded young (3–4 mo old) and old (18+ mo old) wild-type (WT) mice (Bi; n = 5 in each group; *P < 0.05, **P < 0.01, and ***P < 0.001 by 2-way ANOVA); basal NO production was lower in the aorta of old WT mice when compared with young (Bii; n = 5 in each group, *P < 0.05, **P < 0.01 by Wilcoxon rank sum test). Ci–Civ: representative Western blot (Ci) of LOXL2 protein abundance and densitometry analysis in the ECM (Cii), CCM (Ciii), and LOX activity in the CCM (Civ) of HASMCs with increasing concentration of the nitric oxide donor S-nitrosoglutathione (GSNO; n = 6, *P < 0.05, **P < 0.01 by Kruskal Wallis test with Dunn’s post hoc test). RFU, relative fluorescence units.
Endothelial cells regulate LOXL2 secretion by VSMCs.
HAEC effectively suppressed LOXL2 secretion by HASMCs in our coculture system. We next examined whether endothelial cells exert a similar effect in the aorta using thoracic aortic specimens from young (3–4 mo old) and old (18+ mo old) WT mouse aorta. Specimens from old WT mice had significantly higher LOXL2 abundance than those from young mice (Fig. 2Bi). Mechanical removal of the endothelium in aortic rings from young and old WT mice resulted in a marked increase in LOXL2 abundance. Basal NO production was significantly lower in the aortas of old WT mice when compared with those from young mice (Fig. 2Bii). Because endothelium-derived NO bioavailability is a hallmark of aging, we next determined the role of NO in secretion of LOXL2 by HASMCs. In the coculture experiments, treatment of the HAEC layer with the NO synthase inhibitor nitro-l-arginine methyl ester partially reversed LOXL2 secretion and activity in the HASMC layer toward that of untreated controls (Fig. 2, Ai–Aiii). Moreover, exposure to increasing concentrations of the NO donor S-nitrosoglutathione resulted in declined LOXL2 abundance and activity in the cell-derived ECM (Fig. 2, Ci–Ciii).
LOXL2+/− mouse model.
Germline deletion of LOXL2 results in high perinatal lethality (18). We therefore generated LOXL2+/− mice to study the role of LOXL2 in vivo. LOXL2+/− mice bred normally, and offspring were predominantly LOXL2+/− (~75%) with the remainder being WT (~25%). The expression of the lysyl oxidase family was evaluated in the aorta, heart, lung, liver, kidney, uterus, and endothelial and SMCs isolated from the aorta. Cells and tissue from LOXL2+/− mice showed significant loss of LOXL2 protein abundance. The expression of LOX and LOXL3 was markedly elevated in the uterus of LOXL2+/− mice but not in other organs (Fig. 3). In the aorta, LOXL3 expression was modestly elevated in the LOXL2+/− mice, but LOX expression was similar.
Fig. 3.

Expression of lysyl oxidase (LOX) proteins in LOXL2+/− mice. Representative Western blots comparing the expression of LOX, LOXL2, and LOXL3 in the heart, lung, uterus, aorta, and liver of wild-type (WT) and littermate LOXL2+/− mice with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was as a loading control. (n = 6 mice). ND, not detected.
LOXL2+/− mice are protected from age-associated increases in aortic stiffness.
We examined the role of LOXL2 in aging by measuring PWV, an index of in vivo vascular stiffness, in LOXL2+/− and littermate WT mice every 2–4 wk from 5 wk of age to 20 mo of age. In WT mice, PWV increased rapidly with age, and by 8–12 mo was significantly greater than that of young (1–3 mo old) mice (P < 0.05, Fig. 4A). When compared with young WT adults (4–8 mo old), the older WT mice (12+ mo old) had significantly higher PWV. Increase in PWV preceded the onset of systolic hypertension in WT mice, as age-associated increase in systolic pressure reached statistical significance in the oldest mice (15–20 mo old; Fig. 4B). On the other hand, LOXL2+/− mice were protected from the age-associated increase in PWV that was observed in WT mice (Fig. 4A), with significantly lower PWV when compared with their littermate WT controls in the oldest mice (15–20 mo old). The LOXL2+/− mice were further protected from age-associated increase in systolic pressure that was observed in their WT littermates. LOXL2 protein abundance was examined in the aortas of young (3–4 mo old) and old (20+ months old) aortas of LOXL2+/− mice and littermate WT controls. First, we used Western blot analysis to detect LOXL2, LOX, LOXL3, and fibronectin in aortic homogenates (Fig. 4C). A remarkable increase in LOXL2 protein abundance was observed in the old WT mouse aortas when compared with young WTs. Also, aged WT mice had significantly higher LOXL2 protein abundance than did LOXL2+/− littermate mice. Aging also resulted in a significant increase in fibronectin content in the aorta, with aged WT mice showing a markedly higher fibronectin abundance than LOXL2+/− littermates. LOX expression increased with age in both WT and LOXL2+/− mice. LOXL3 expression increased with age in LOXL2+/− mice. We next evaluated LOXL2 expression and localization in intact aortas from old WT and LOXL2+/− littermates by immunofluorescent staining and confocal microscopy. Again, LOXL2 abundance was markedly higher in WT mice than in LOXL2+/− mice and predominantly present in the extracellular space and elastic lamellae (Fig. 4D). Finally, in the aging study, we used histochemical analysis to compare the aortas of young (3–4 mo old) and old (20+ mo old) WT and LOXL2+/− littermates. Aging resulted in a significant loss of nuclei (Fig. 5, A and D) and a marked increase in wall fibrosis (Fig. 5, B–D) to a similar extent in both the WT and LOXL2+/− mice.
Fig. 4.

Lysyl oxidase-like 2 (LOXL2) knockdown is protective against age-associated vascular stiffening in mice. A: pulse-wave velocity (PWV) with increasing age in the LOXL2+/− mice (n = 12; 7 female, 5 male) and littermate wild-type (WT) mice (n = 10; 5 female, 5 male; n = 20–35 measures per age for each cohort; *P < 0.05, **P < 0.01 vs. WT 1–4 mo old; ‡P < 0.05 vs. WT 4–8 mo old; #P < 0.05, ##P < 0.01 vs. age-matched LOXL2+/− littermates by 2-way ANOVA). B: systolic pressure with increasing age in WT (n = 10; 5 female, 5 male) and LOXL2+/− littermate mice (n = 12; 7 female, 5 male) (n = 25–35 measurements per age group per genotype; **P < 0.01 vs WT 1–4 mo old; ###P < 0.001 vs. age-matched LOXL2 littermates by 2-way ANOVA). C: representative Western blots for LOXL2 protein expression in the aortic matrix of young (Y) and old (O) LOXL2+/− mice and WT littermates (images are representative of n = 5 in each group; old = 20–22 mo old, young = 3–4 mo old; *P < 0.05 vs. old WT by 2-way ANOVA). ECM, extracellular matrix. D: representative confocal microscopy images of intact aortas from 15+-mo-old WT and LOXL2+/− mice (n = 5) stained for LOXL2 (red) and nuclei (blue). Scale bars = 50 μm.
Fig. 5.

Immunohistochemical analysis of young (3–4 mo old) and old (22 mo old) wild-type (WT) and lysyl oxidase-like 2 (LOXL2) littermate mouse aorta. All images shown are representative of images from 6 independent mouse aortas (×10 magnification) as whole rings. Insets: ×40 for each specimen. A–V: hematoxylin and eosin (H&E) stain (A), Masson trichrome stain (B), and Movat pentachrome stain (C). Scale bars = 50 μm. D: aging-related changes in nuclear content and wall thickness of aortas of WT and LOXL2+/− mice. *P < 0.05, **P < 0.01, and ***P < 0.001.
LOXL2 promotes vascular stiffening by promoting matrix stiffness, VSMC stiffness, and VSMC contractility.
Increased stiffness of both the matrix and VSMC contributes to overall aging driven vascular stiffening in vivo. Thus, we next examined if depletion of LOXL2 confers protection in vascular aging by reduced matrix remodeling or by reduced VSMC stiffness and contractility. In these experiments, the old cohort was composed of mice 15 mo or older, where significant vascular stiffening has already occurred, as reflected in the significantly higher PWV (Fig. 4A). We first measured passive stiffness of carotid arteries isolated from young (3–4 mo old) and old (15+ mo old) LOXL2+/− and littermate WT mice by pressure myography. Aging resulted in a large loss of passive compliance in WT mice but not in LOXL2+/− mice (Fig. 6A). We next measured the mechanical properties of intact and decellularized aortic rings isolated from 15-mo-old littermate mice. The intact WT aorta was significantly stiffer than the LOXL2+/− aorta (Fig. 6Bi) over the entire range of strain. The decellularized WT samples were also stiffer than LOXL2+/− mice but to a lesser magnitude than the intact samples (Fig. 6Bii). Moreover, an overall change in the nonlinear viscoelastic properties of the decellularized samples was noted, wherein the WT decellularized samples are stiffer at low strains but less stiff at higher strains when compared with LOXL2+/− decellularized samples. This suggests that the VSMCs, one of the central load-bearing elements in the blood vessel, are themselves altered. Therefore, we next evaluated the stiffness of SMCs isolated from the aortas of old (15 mo old) WT and LOXL2+/− littermates. When compared with WT VSMCs, there was a marked decrease in LOXL2 protein abundance in the extracellular space (conditioned cell culture media and cell-derived ECM) in the LOXL2+/− VSMC (Fig. 6C). WT cells were significantly stiffer than LOXL2+/− cells over five decades of frequency (Fig. 6D) and demonstrated strikingly lower cytoskeletal remodeling dynamics (Fig. 6E). Additionally, depletion of LOXL2 resulted in delayed VSMC deadhesion dynamics (Fig. 6F) and reduced VSMC motility (Fig. 6G) when compared with WT cells.
Fig. 6.

Lysyl oxidase-like 2 (LOXL2) promotes aortic matrix stiffness and vascular smooth muscle cell (VSMC) stiffness in aging. A: passive compliance of the carotid artery, measured by pressure myography, aged [old (O)] wild-type (WT; 15 mo old, n = 7; 3 female, 4 male), young (Y) WT (3–4 mo old, n = 10; 5 female, 5 male), young LOXL2+/− (3-4 mo old, n = 12; 7 female, 5 male) and aged LOXL2+/− (15 mo old, n = 12; 7 female, 5 male) mice (*P < 0.05 by Kruskal-Wallis test with Dunn’s correction). Bi and Bii: tensile testing of intact (Bi) and decellularized (Bii) aortic segments of WT and littermate LOXL2+/− mice. Data are shown as means (solid line) ± SE (broken lines) (n = 12 LOXL2+/− and n= 8 WT mice; ***P < 0.0001 by Wilcoxon rank sum test). C: LOXL2 expression in the conditioned media and cell-derived ECM of endothelial and smooth muscle cells isolated from LOXL2+/− and littermate WT mouse aorta; GAPDH is shown as a reference control. Blot is representative of 5 independent experiments. IB, immunoblot. D: stiffness of mouse aortic smooth muscle cells isolated from old (15 mo old) WT and LOXL2+/− littermate mice measured by magnetic torsion cytometry (n = 543 WT cells; 165 LOXL2+/− cells; *P < 0.05 by Student’s t-test). E: cytoskeletal remodeling dynamics of aortic smooth muscle cells from old (15 mo old) WT and littermate LOXL2+/− mice (n = 599 WT cells, 586 LOXL2+/− cells; ****P < 0.05 by Student’s t-test). MSD, mean square displacement. F: motility of WT- and LOXL2-depleted VSMC examined by transwell migration assay using 10% serum as chemoattractant (n = 6; **P < 0.01 by 2-way ANOVA). RFU, relative fluorescence units. G: deadhesion dynamics of WT- and LOXL2-depleted VSMCs using 0.05% trypsin (n = 5; ****P < 0.0001 by Wilcoxon rank sum test).
We next characterized the role of LOXL2 in VSMC function in both isolated cells and intact tissue. Isolated aortic VSMCs from LOXL2+/− mice exerted significantly lower traction stress than did those from WT mice (Fig. 7, A and Bi–Biv). Consistent with this observation, LOXL2+/− mouse aortic rings exhibited a diminished vasoconstriction response to phenylephrine than those from WT littermates (Fig. 7C). Endothelium-dependent (acetylcholine induced) and endothelium-independent (sodium nitroprusside induced) vasorelaxation were similar in phenylephrine preconstricted rings from WT and LOXL2+/− littermates (Fig. 7, D and E).
Fig. 7.

Lysyl oxidase-like 2 (LOXL2) depletion decreases vascular smooth muscle cell (VSMC) contractility. A: representative phase contrast and traction map images of VSMCs from old (15 mo old) wild-type (WT) and LOXL2+/− cells as measured by Fourier transform traction microscopy. The white lines show the cell boundary, and the colors show the magnitude of the traction force in Pascals indexed to the color bar at the right. Bi–Biv: computed projected area (Bi), traction stress (Bii), and derived strain energy (Biii), and prestress (Biv) of isolated VSMCs. For these studies, cells were plated onto an inert elastic gel (8 kPa) coated with type I collagen (WT, n = 18; LOXL2+/−, n = 19 individual cell measurements). For data that were not normally distributed [root mean squares (RMS) traction, total strain energy, and prestress], statistical analyses were performed on transformed data (*P < 0.05, **P < 0.01 by Student’s t-test). C: constriction response of LOXL2+/− and WT mouse aortic rings to increasing concentrations of phenylephrine (n = 8 per group; *P < 0.05 vs. WT at same phenylephrine concentration by Kruskal-Wallis test with Dunn’s post hoc correction). D and E: endothelial-independent relaxation of vessels pre-constricted with phenylephrine with increasing concentrations of sodium nitroprusside (n = 8; D) and endothelial-dependent relaxation of vessels preconstricted with phenylephrine with increasing concentrations of acetylcholine (n = 8; E) in LOXL2+/− and WT mouse aortic rings.
DISCUSSION
In this study, we examined the role of LOXL2 in the stiffening of the conduit vasculature that occurs with aging. Our in vitro studies showed that LOXL2 itself is regulated by NO bioavailability. Importantly, LOXL2 knockdown prevents age-associated vascular stiffening in mice when compared with littermate WT mice. This protective effect occurs due to lower VSMC stiffness, VSMC contractility, and vascular matrix stiffness in the LOXL2+/− mice when compared with littermate WT mice.
LOX and LOXL2 are of critical importance to organogenesis; indeed, germline deletion of LOX or LOXL2 is lethal in mice (17, 18). The high embryonic and neonatal mortality in these mice stems from severe cardiac and vascular defects. In our study, we noted <1% of the pups from breeding LOXL2+/− mice were homozygous LOXL2−/−, which is in good agreement with the prior studies. Several studies have now shown that the expression and activation of LOX/LOXL2 later in the life span of the animal plays a causal role in fibrosis (5, 12, 16, 29) and promotes tumor angiogenesis (3, 30). These studies underscore the central importance of the LOX enzyme family to vascular health and disease. In this study, we report for the first time, a specific role for LOXL2 in central vascular stiffening during aging.
At the cellular and molecular levels, endothelial dysfunction and loss of NO bioavailability are hallmarks of the aging vasculature and are associated with activation of profibrotic pathways that result in matrix deposition and remodeling. Here we show that augmented NO bioavailability from exogenous NO (chemical NO donor S-nitrosoglutathione) and endogenous NO (from HAECs) reduces LOXL2 abundance in the extracellular space of VSMCs. We note that the low serum conditions used in the coculture studies could, by itself, activate endothelial stress response, and alter LOXL2 synthesis and secretion in vitro. We found a marked increase in extracellular LOXL2 protein abundance in the aging aorta, and denuding the endothelium resulted in a significant increase in LOXL2 abundance in the vascular matrix. These findings together implicate endothelial dysfunction with aging in promoting augmented LOXL2 abundance and activity in the vascular ECM and, consequently, vascular remodeling, VSMC dysfunction, and stiffening.
The central goal of this study was to determine whether targeting LOXL2 ameliorates vascular stiffening observed in aging. In vivo, LOXL2 knockdown prevented age-associated increase in PWV a well-accepted index of aortic stiffness that occurred in WT littermates. Furthermore, in WT mice, the elevation in PWV with age occurred early at 8–12 mo of age and preceded the onset of significant isolated systolic hypertension, which occurred only at 15–20 mo. Because of the pressure dependence of PWV, elevated PWV must be corrected for increases in BP. In our study, the increased PWV in the 8- to 12-mo-old WT cohort represents an authentic increase in vascular stiffness, as BP is not elevated in this age group. In the older cohorts, where BP was also elevated, we employed additional stiffness measures to further elucidate changes in vascular mechanics. Specifically, we examined the mechanical properties of the aorta from young and old mice ex vivo. Our studies showed that passive compliance of conduit vessels was maintained in aged LOXL2+/− mice but was diminished in WT littermates, as evaluated by pressure myography. Taken together with the PWV values and Western blotting data, this study showed that while both LOX and LOXL2 abundance are augmented with age in the vascular matrix, suppression of LOXL2 expression alone is sufficient to provide a significant protection against vascular stiffening in aging. This finding is intriguing, particularly as prior studies focused on germline deletion of LOX have postulated that the prototypical LOX is responsible for ~60–70% of total amine oxidase activity occurring in the vascular matrix and that other members, specifically LOXL2, contribute only 30–40% of total amine oxidase activity. Indeed, after comparison of the decellularized aortic rings from aged WT and LOXL2+/− littermates, it is evident that while LOXL2 depletion imparts only a modest benefit in lowering the elastic modulus of the vascular matrix in aged mice. In fact, while decellularized samples from old WT mice are stiffer than those from LOXL2+/− mice at low magnitudes of strain, at high strain there is no statistically significant difference between the two. This is supported by histological analysis, wherein we noted similar age-related alterations in the WT and LOXL2+/− mice; i.e, LOXL2 depletion did not prevent age-driven loss of VSMCs or medial thickening. On the other hand, two complementary measures, tensile testing and passive pressure-dimension analysis, show that intact vessels from aged LOXL2+/− mice are significantly less stiff (i.e., more compliant) when compared with their WT littermates. Concurrently, we also note that isolated VSMCs from LOXL2+/− mice are significantly less stiff and depletion of LOXL2 protein results in lower VSMC traction forces in vitro in isolated cells and ex vivo in intact tissue. When taken together, these data show that augmented VSMC stiffness and contractility are a significant mechanism by which LOXL2 promotes vascular stiffening in aging.
In conclusion, the protective effects of LOXL2 knockdown in the aging vasculature are due to improved VSMC stiffness and tone in the aged LOXL2+/− mice compared with the WT littermates (Fig. 8), along with attenuated matrix deposition. Furthermore, because of the bidirectional communication between the VSMCs and the vascular matrix, the two effects combined (i.e., lower matrix stiffness and lower VSMC stiffness) likely delay the deterioration of vascular mechanical properties during aging and confer a substantial benefit in overall vascular stiffness and function. Further studies are warranted to assess the specific mechanism by which LOXL2 ablation preserves vascular mechanics during aging.
Fig. 8.
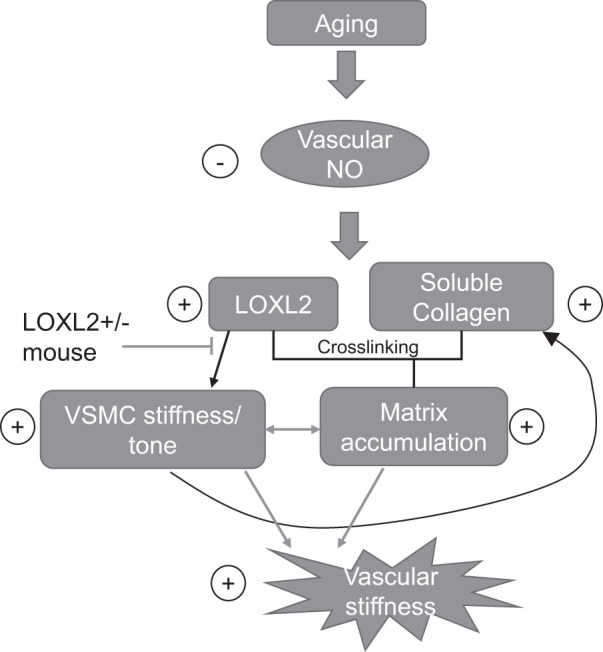
Lysyl oxidase-like 2 (LOXL2) promotes vascular stiffening in aging. Aging results in endothelial dysfunction that in turn results in increased LOXL2 and soluble collagen in the vascular matrix. This leads to both increased matrix cross linking and vascular smooth muscle cell (VSMC) stiffness/tone. Together, these changes result in increased vascular stiffness. LOXL2 knockdown limits this process and prevents vascular stiffening with age, thus interrupting the onset of isolated systolic hypertension. NO, nitric oxide.
GRANTS
This work was supporting by two Stimulating and Advancing ACCM Research (StAAR) grants from the Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University (JHU; to L. Santhanam and J. Steppan), a JHU Discovery Award (to S. S. An and L. Santhanam), National Heart, Lung, and Blood Institute Grant R01-HL-105296 (to D. E. Berkowitz), the Maryland Cigarette Restitution Fund (to S. S. An), a JHU Catalyst Award (to S. S. An), and a Foundation for Anesthesia Education and Research (FAER) Mentored Research Training Grant–Basic Science (to J. Steppan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.S. conceived and designed research; J.S., H.W., Y.B., M.R., S.T., S.J., K.N., S.B.O., R.N.C., T.N.B., W.Z., and L.S. performed experiments; J.S., H.W., M.R., S.T., J.S., M.K.H., S.S.A., and L.S. analyzed data; S.S.A., D.E.B., J.S., and L.S. interpreted results of experiments; J.S., H.W., S.T., S.S.A., and L.S. prepared figures; L.S. drafted manuscript; M.K.H., J.S., and L.S. edited and revised manuscript; L.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Karen Fox-Talbot for technical assistance with histology.
Present address of D. E. Berkowitz: University of Alabama at Birmingham, Birmingham, AL.
REFERENCES
- 1.An SS, Fabry B, Trepat X, Wang N, Fredberg JJ. Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am J Respir Cell Mol Biol 35: 55–64, 2006. doi: 10.1165/rcmb.2005-0453OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bia D, Zócalo Y, Farro I, Torrado J, Farro F, Florio L, Olascoaga A, Brum J, Alallón W, Negreira C, Lluberas R, Armentano RL. Integrated evaluation of age-related changes in structural and functional vascular parameters used to assess arterial aging, subclinical atherosclerosis, and cardiovascular risk in uruguayan adults: CUiiDARTE project. Int J Hypertens 2011: 1–12, 2011. doi: 10.4061/2011/587303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bignon M, Pichol-Thievend C, Hardouin J, Malbouyres M, Bréchot N, Nasciutti L, Barret A, Teillon J, Guillon E, Etienne E, Caron M, Joubert-Caron R, Monnot C, Ruggiero F, Muller L, Germain S. Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood 118: 3979–3989, 2011. doi: 10.1182/blood-2010-10-313296. [DOI] [PubMed] [Google Scholar]
- 4.Bursac P, Fabry B, Trepat X, Lenormand G, Butler JP, Wang N, Fredberg JJ, An SS. Cytoskeleton dynamics: fluctuations within the network. Biochem Biophys Res Commun 355: 324–330, 2007. doi: 10.1016/j.bbrc.2007.01.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng T, Liu Q, Zhang R, Zhang Y, Chen J, Yu R, Ge G. Lysyl oxidase promotes bleomycin-induced lung fibrosis through modulating inflammation. J Mol Cell Biol 6: 506–515, 2014. doi: 10.1093/jmcb/mju039. [DOI] [PubMed] [Google Scholar]
- 6.Debeer P, Schoenmakers EF, Twal WO, Argraves WS, De Smet L, Fryns JP, Van De Ven WJ. The fibulin-1 gene (FBLN1) is disrupted in a t(12;22) associated with a complex type of synpolydactyly. J Med Genet 39: 98–104, 2002. doi: 10.1136/jmg.39.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Lazzaro V, Pilato F, Dileone M, Tonali PA, Ziemann U. Dissociated effects of diazepam and lorazepam on short-latency afferent inhibition. J Physiol 569: 315–323, 2005. doi: 10.1113/jphysiol.2005.092155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferruzzi J, Murtada SI, Li G, Jiao Y, Uman S, Ting MY, Tellides G, Humphrey JD. Pharmacologically improved contractility protects against aortic dissection in mice with disrupted transforming growth factor-β signaling despite compromised extracellular matrix properties. Arterioscler Thromb Vasc Biol 36: 919–927, 2016. doi: 10.1161/ATVBAHA.116.307436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fong SF, Dietzsch E, Fong KS, Hollosi P, Asuncion L, He Q, Parker MI, Csiszar K. Lysyl oxidase-like 2 expression is increased in colon and esophageal tumors and associated with less differentiated colon tumors. Genes Chromosomes Cancer 46: 644–655, 2007. doi: 10.1002/gcc.20444. [DOI] [PubMed] [Google Scholar]
- 10.Gao Y, Qi GX, Jia ZM, Sun YX. Prediction of marker genes associated with hypertension by bioinformatics analyses. Int J Mol Med 40: 137–145, 2017. doi: 10.3892/ijmm.2017.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartley CJ, Taffet GE, Michael LH, Pham TT, Entman ML. Noninvasive determination of pulse-wave velocity in mice. Am J Physiol 273: H494–H500, 1997. doi: 10.1152/ajpheart.1997.273.1.H494. [DOI] [PubMed] [Google Scholar]
- 12.Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, Mikels-Vigdal A, Smith V, Schuppan D, Popov YV. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 66: 1697–1708, 2017. doi: 10.1136/gutjnl-2016-312473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung SM, Jandu S, Steppan J, Belkin A, An SS, Pak A, Choi EY, Nyhan D, Butlin M, Viegas K, Avolio A, Berkowitz DE, Santhanam L. Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice. Am J Physiol Heart Circ Physiol 305: H803–H810, 2013. doi: 10.1152/ajpheart.00103.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: causes and consequences. Front Genet 6: 112, 2015. doi: 10.3389/fgene.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy D, Larson MG, Benjamin EJ, Newton-Cheh C, Wang TJ, Hwang SJ, Vasan RS, Mitchell GF. Framingham Heart Study 100K Project: genome-wide associations for blood pressure and arterial stiffness. BMC Med Genet 8, Suppl 1: S3, 2007. doi: 10.1186/1471-2350-8-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu SB, Ikenaga N, Peng ZW, Sverdlov DY, Greenstein A, Smith V, Schuppan D, Popov Y. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J 30: 1599–1609, 2016. doi: 10.1096/fj.14-268425. [DOI] [PubMed] [Google Scholar]
- 17.Mäki JM, Sormunen R, Lippo S, Kaarteenaho-Wiik R, Soininen R, Myllyharju J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am J Pathol 167: 927–936, 2005. doi: 10.1016/S0002-9440(10)61183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin A, Salvador F, Moreno-Bueno G, Floristán A, Ruiz-Herguido C, Cuevas EP, Morales S, Santos V, Csiszar K, Dubus P, Haigh JJ, Bigas A, Portillo F, Cano A. Lysyl oxidase-like 2 represses Notch1 expression in the skin to promote squamous cell carcinoma progression. EMBO J 34: 1090–1109, 2015. doi: 10.15252/embj.201489975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parmar PG, Taal HR, Timpson NJ, Thiering E, Lehtimäki T, Marinelli M, Lind PA, Howe LD, Verwoert G, Aalto V, Uitterlinden AG, Briollais L, Evans DM, Wright MJ, Newnham JP, Whitfield JB, Lyytikäinen LP, Rivadeneira F, Boomsma DI, Viikari J, Gillman MW, St Pourcain B, Hottenga JJ, Montgomery GW, Hofman A, Kähönen M, Martin NG, Tobin MD, Raitakari O, Vioque J, Jaddoe VW, Jarvelin MR, Beilin LJ, Heinrich J, van Duijn CM, Pennell CE, Lawlor DA, Palmer LJ; Early Genetics and Lifecourse Epidemiology Consortium . International genome-wide association study consortium identifies novel loci associated with blood pressure in children and adolescents. Circ Cardiovasc Genet 9: 266–278, 2016. doi: 10.1161/CIRCGENETICS.115.001190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE, Meininger GA, Vatner SF. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res 107: 615–619, 2010. doi: 10.1161/CIRCRESAHA.110.221846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sehgel NL, Vatner SF, Meininger GA. “Smooth Muscle Cell Stiffness Syndrome”–revisiting the structural basis of arterial stiffness. Front Physiol 6: 335, 2015. doi: 10.3389/fphys.2015.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sen S, Kumar S. Cell-matrix de-adhesion dynamics reflect contractile mechanics. Cell Mol Bioeng 2: 218–230, 2009. doi: 10.1007/s12195-009-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steppan J, Bergman Y, Viegas K, Armstrong D, Tan S, Wang H, Melucci S, Hori D, Park SY, Barreto SF, Isak A, Jandu S, Flavahan N, Butlin M, An SS, Avolio A, Berkowitz DE, Halushka MK, Santhanam L. Tissue transglutaminase modulates vascular stiffness and function through crosslinking-dependent and crosslinking-independent functions. J Am Heart Assoc 6: e004161, 2017. doi: 10.1161/JAHA.116.004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steppan J, Sikka G, Jandu S, Barodka V, Halushka MK, Flavahan NA, Belkin AM, Nyhan D, Butlin M, Avolio A, Berkowitz DE, Santhanam L. Exercise, vascular stiffness, and tissue transglutaminase. J Am Heart Assoc 3: e000599, 2014. doi: 10.1161/JAHA.113.000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steppan JB, Bergman Y, Viegas K, Armstrong D, Tan S, Wang H, Melucci S, Hori D, Park SY, Barreto SF, Isak A, Jandu S, Flavahan NA, Butlin M, An SS, Avolio A, Berkowitz DE, Halushka MK, Santhanam L. Tissue transglutaminase modulates vascular stiffness and function through crosslinking-dependent and crosslinking-independent functions. J Am Heart Assoc 6: e004161, 2017. doi: 10.1161/JAHA.116.004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tolić-Nørrelykke IM, Butler JP, Chen J, Wang N. Spatial and temporal traction response in human airway smooth muscle cells. Am J Physiol Cell Physiol 283: C1254–C1266, 2002. doi: 10.1152/ajpcell.00169.2002. [DOI] [PubMed] [Google Scholar]
- 27.Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol 55: 1318–1327, 2010. doi: 10.1016/j.jacc.2009.10.061. [DOI] [PubMed] [Google Scholar]
- 28.Wang N, Tolić-Nørrelykke IM, Chen J, Mijailovich SM, Butler JP, Fredberg JJ, Stamenović D. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am J Physiol Cell Physiol 282: C606–C616, 2002. doi: 10.1152/ajpcell.00269.2001. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, Barry V, Mikels-Vigdal A, Karpinski S, Kornyeyev D, Adamkewicz J, Feng X, Zhou Q, Shang C, Kumar P, Phan D, Kasner M, López B, Diez J, Wright KC, Kovacs RL, Chen PS, Quertermous T, Smith V, Yao L, Tschöpe C, Chang CP. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun 7: 13710, 2016. doi: 10.1038/ncomms13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaffryar-Eilot S, Marshall D, Voloshin T, Bar-Zion A, Spangler R, Kessler O, Ghermazien H, Brekhman V, Suss-Toby E, Adam D, Shaked Y, Smith V, Neufeld G. Lysyl oxidase-like-2 promotes tumour angiogenesis and is a potential therapeutic target in angiogenic tumours. Carcinogenesis 34: 2370–2379, 2013. doi: 10.1093/carcin/bgt241. [DOI] [PubMed] [Google Scholar]