

Reverse-phase protein arrays represent a powerful functional proteomics approach. Using this platform, we have characterized ∼8,000 patient samples of 32 cancer types through The Cancer Genome Atlas and built a widely used, open-access bioinformatic resource, The Cancer Proteome Atlas (TCPA). Here we have developed a new module called “TCGA Pan-Cancer Analysis,” which provides comprehensive protein-centric, pan-cancer analyses in rich context of TCGA data. This upgraded TCPA (v3.0) provides a more valuable tool for studying functional proteomics and making translational impacts.
Keywords: Bioinformatics Software, Proteogenomics, Cancer Biology, Biomarker: Prognostic, Pathway Analysis, Gene Expression, Functional Proteomics, Pan-Cancer Analysis, Protein Array, TCGA
Graphical Abstract

Highlights
A new protein-centric pan-cancer analytic module for multi-omic TCGA data.
Systematic assessment of correlations of molecular features with protein expression.
Perform a co-expression network analysis of proteins and protein-based pathways.
Identify clinically relevant patterns of protein markers across cancer types.
Abstract
Reverse-phase protein arrays represent a powerful functional proteomics approach to characterizing cell signaling pathways and understanding their effects on cancer development. Using this platform, we have characterized ∼8,000 patient samples of 32 cancer types through The Cancer Genome Atlas and built a widely used, open-access bioinformatic resource, The Cancer Proteome Atlas (TCPA). To maximize the utility of TCPA, we have developed a new module called “TCGA Pan-Cancer Analysis,” which provides comprehensive protein-centric analyses that integrate protein expression data and other TCGA data across cancer types. We further demonstrate the value of this module by examining the correlations of RPPA proteins with significantly mutated genes, assessing the predictive power of somatic copy-number alterations, DNA methylation, and mRNA on protein expression, inferring the regulatory effects of miRNAs on protein expression, constructing a co-expression network of proteins and pathways, and identifying clinically relevant protein markers. This upgraded TCPA (v3.0) will provide the cancer research community with a more powerful tool for studying functional proteomics and making translational impacts.
Functional proteomics is a powerful approach to characterizing cell signaling pathways and understanding their phenotypic effects on cancer development. Reverse-phase protein arrays (RPPAs)1 represent a cutting-edge proteomics technology that can quantitatively assess a large number of protein markers in thousands of samples in a cost-effective, sensitive, and high-throughput manner (1–3). Using the RPPA platform, we have characterized ∼8,000 patient samples across 32 cancer types through The Cancer Genome Atlas (TCGA) project and >650 independent cancer cell lines of 19 cell lineages (4–6). To better utilize the RPPA data and serve a broad biomedical research community, we developed an open-access bioinformatics resource, The Cancer Proteome Atlas (TCPA). This web-based platform not only releases the most updated data that are generated by our RPPA platform but also provides a user-friendly interface allowing users to analyze and visualize RPPA data in a rich context (7), which has substantially reduced the computational barriers to analyzing complex RPPA data in large-scale sample sets.
However, analytic modules in the previous TCPA versions have focused on the protein-based analyses in individual cancer types only and do not provide integrative analyses of RPPA data with other types of molecular data. Using those modules, it is difficult to explore the similarities and differences among cancer types, which limits the full potential of TCPA. To maximize its utility, we have updated TCPA by adding a newly developed module called “TCGA Pan-Cancer Analysis.” This module provides comprehensive protein-centric analyses that integrate association studies between protein data and other types of molecular and clinical data from TCGA. These data include somatic mutations, somatic copy-number alterations (SCNAs), DNA methylation, mRNA expression and miRNA expression, as well as patient survival, tumor subtype, and disease stage. With the new module, users can easily identify protein markers that show interesting patterns across cancer types. Overall, the current TCPA (v3.0) represents a comprehensive, cutting-edge, protein-centric pan-cancer analytic platform that is freely available at http://tcpaportal.org.
MATERIALS AND METHODS
RPPA Data
The RPPA data were first quantified in the samples collected from TCGA project, which included 7,694 patient samples across 32 cancer types (6, 8–10). In total, 258 protein markers (including total and phosphorylated proteins) were assayed. The raw RPPA data were further standardized and normalized by SuperCurve, median polish, and replicate-based normalization (10, 11) for downstream analyses.
TCGA Pan-Cancer Atlas Data
All the clinical and molecular data were obtained from the TCGA Pan-Cancer Atlas project consisting of (i) DNA data: somatic mutations, DNA methylation, and SCNAs; (ii) RNA data: RNA-seq and miRNA-seq-based gene expression data; and (iii) clinical data: stage, tumor grade, overall survival, and progression-free survival data. Specifically, SCNA data were downloaded from Synapse (https://www.synapse.org/) with accession number syn5049520.1. All the other data were retrieved from the TCGA Pan-Cancer Atlas website (https://gdc.cancer.gov/about-data/publications/pancanatlas). In addition, all the tumor subtype information were collected from the TCGA marker publications (12).
Statistical Analysis
Multiple statistical methods were used to examine pan-cancer associations between RPPA data and other types of molecular and clinical data: (i) for continuous data (including DNA methylation, SCNA, mRNA expression, miRNA expression, RPPA, and pathway scores), Spearman's rank correlation analyses were performed; (ii) for dichotomous variables (e.g. wild type versus mutated), t-tests were performed; and (iii) ANOVA tests were applied to those with more than two levels (e.g. tumor subtype, stage, and grade). For survival analysis, both Cox proportional-hazards model and log-rank test were used to calculate the correlations between the levels of protein markers and patient survival times. All the p values derived from multiple comparisons were adjusted by false discovery rate (FDR). We reported significant results at FDR < 0.1.
Pathway Score Calculation
Eleven core cancer pathway scores were calculated based on (signed) average of the RPPA levels for each cancer type before being applied in the network analysis (13). The protein members and their functional directions (i.e. up- or down-regulation) within each pathway were defined in Akbani et al. (10). The 11 pathways included were apoptosis, core reactive, cell cycle, DNA damage response, epithelial-mesenchymal transition, hormone a, hormone b, PI3K/Akt, Ras/MAPK, RTK, and TSC/mTOR.
Bipartite Network Analysis
To show the correlations between protein levels and mutated genes, a bipartite network was generated based on the mutational status of significantly mutated genes (SMGs) and the 11 pathway scores. In total, 299 SMGs were obtained from Bailey et al. (14), which were used to define the mutated and wild-type sample groups in each cancer type. The correlations between pathways and SMGs were assessed by t-tests with FDR < 0.1. In the bipartite network, the thickness of each edge represents the number of cancer types with a significant correlation (FDR < 0.1), and the color of each edge represents the direction of correlations.
Analysis of Protein Expression with SCNA, DNA Methylation, and mRNA Expression
The total proteins were first extracted from the RPPA data for the correlation analysis. Among 191 total proteins, 187 had matched genes in SCNA, DNA methylation, and mRNA data. When multiple methylation probes were mapped to the same gene, the one showing the most negative correlation with the mRNA expression was selected. The matched protein-gene pairs were named “cis-pairs” and others were “trans-pairs.” For each cancer type, Spearman's rank correlation analysis was performed to assess both the cis- and trans-pairs. All the p values were then adjusted by FDR. The correlation coefficients of cis- and trans-pairs were plotted in histograms with corresponding density curves. The difference between cis- and trans- distributions were tested by Student's t test.
Identification of Common miRNA Regulators Across Cancer Types
We first obtained predicted miRNA targets from TargetScanHuman 7.2 (http://www.targetscan.org/vert_72/) (15). Only 191 total proteins were used in this analysis. Next, the RPPA-associated genes were matched to the miRNA-target list, which generated 1,345 miRNA-gene pairs including 223 mature miRNAs and 179 coding genes. The miRNAs with at least five target genes were included in the further analysis (119 miRNAs retained). For each miRNA-gene pair, Spearman's rank correlations were calculated. To evaluate the miRNA effects in different cancer types, the difference between the distributions of miRNA—target-protein and miRNA—nontarget-protein correlations were examined by the Kolmogorov-Smirnov test. The difference of the means was calculated to determine the regulation direction (repression or activation) in a cancer type. The miRNAs were then ranked by the number of cancer types showing negative direction, and only those with negative directions in at least two cancer types were displayed in the heatmap.
Network Analysis of RPPA Pathways
A nested network visualization was generated to show the co-expression relationships among protein markers and RPPA pathways. In the networks, each node represents a pathway/protein, and each edge represents the number of cancer types in which the Spearman's rank correlations between two nodes were significant (FDR < 0.1). For simplicity, the edges with significance in <10 cancer types were filtered out in the network. The complete graphs were further identified by the “igraph” package in R.
Implementation of the TCGA Pan-cancer Analytic Module
All the analytic results were first precomputed by univariate statistical analyses. The result data, along with both molecular and clinical data, were further converted to JSON format and curated into CouchDB. The pan-cancer analyses were conducted in R. The web interface of the pan-cancer analytic module was implemented in JavaScript. All the table results were displayed by DataTables, and all the nested plots were generated by HighCharts.
RESULTS
A Protein-centric Pan-cancer Analytic Module
Here we present TCPA v3.0, a user-friendly, interactive platform with a newly integrated protein-centric analytic module for researchers to visualize and analyze RPPA data from a pan-cancer multi-omic perspective (Fig. 1, i). This new release includes RPPA pan-cancer data with 258 protein markers quantified from 7,694 TCGA patient samples across 32 cancer types (7,436 primary tumors and 258 metastatic samples) (Fig. 1, ii). The protein markers (include both total and phosphorylated proteins) cover all major signaling pathways relevant to human cancer, such as PI3K/Akt, Ras/MAPK, and Hippo signaling pathways. To maximize the utility of the RPPA data, we collected other types of molecular and clinical data from TCGA Pan-Cancer Atlas (Fig. 1, iii) to allow multidimensional analyses.
Fig. 1.

Overview of TCPA v3.0. KIRC, kidney renal clear cell carcinoma; LGG, low-grade glioma; MESO, mesothelioma; PFI, progression-free survival interval; BRCA, breast invasive carcinoma; UCEC, uterine corpus endometrial carcinoma; Meth., DNA methylation.
The pan-cancer analytic module (Fig. 1, iv) is protein-centric, which enables users to examine the associations between protein expression and all other types of clinical and molecular features (i.e. clinical, DNA, RNA, and protein data) (Fig. 1, v-viii). The pan-cancer analytic module consists of four submodules: (i) the “Clinical relevance” submodule shows how protein expression correlates with patient clinical data, including patient survival, tumor subtype, stage, and grade. (ii) “Protein-DNA correlation” evaluates the associations between protein expression and gene mutational status, SCNAs, and DNA methylation. (iii) In the “Protein-RNA correlation” submodule, the correlations between protein expression and mRNA/miRNA expression are examined; and (iv) the “Protein-protein correlation” submodule assesses protein correlations. All the analytic results can be visualized by interactive tables under separated tabs. Users can easily sort and search any column or search any keyword globally in a search box. Because the results of all the cancer types are integrated into a single table, it is very simple for users to test their pan-cancer hypotheses directly. In addition, each row in the result table has a nested plot showing the original data points, which can be easily exported in either PNG or PDF format. For example, Kaplan-Meier plots, box plots, and scatter plots are generated to help visualize survival analysis, protein-mutation, and protein-mRNA correlations, respectively.
Specifically, with the “Clinical relevance” submodule, users can examine whether a protein marker correlates with patient overall or progression-free survival time (as tested by univariate Cox proportional-hazards model and log-rank test; visualized through a Kaplan-Meier plot), or whether a protein shows differential expression among tumor stages, grades, or subtypes (as tested by ANOVA; visualized through box plots). This submodule can help discover potential prognostic markers across different cancer types. For example, patients with high PTEN protein expression show better overall survival than those with low expression in kidney renal clear cell carcinoma, low-grade glioma, and mesothelioma, while it shows better progression-free survival in kidney renal clear cell carcinoma alone (Fig. 1, v). The “Protein-DNA correlation” submodule enables users to examine whether the expression of a protein marker is affected by somatic mutations in a specific gene (as tested by Student's t test among the mutated and wild-type sample groups; visualized through box plots) or whether its expression correlates with DNA methylation and SCNAs (as tested by Spearman's rank correlation; visualized through scatter plots). For example, mutated PTEN leads to significantly lower protein expression in breast invasive carcinoma, low-grade glioma, and uterine corpus endometrial carcinoma (Fig. 1, vi). The “Protein-RNA correlation” submodule allows users to examine whether a protein marker shows correlations with RNA abundance of its own or other genes, including coding or miRNA genes (both as tested by Spearman's rank correlation; visualized through scatter plots). For example, CYCLINB1 (protein marker) and KIF2C have a positive correlation in breast invasive carcinoma and other cancer types, except for cholangiocarcinoma, thyroid carcinoma, and uveal melanoma (Fig. 1, vii). Finally, the “Protein correlation” submodule (Fig. 1, viii) provides an effective way for users to examine whether the expression levels of any two protein markers correlate with each other across cancer types (as tested by Spearman's rank correlation; visualized through scatter plots). For each submodule, these pairwise associations are presented in a table view. The first column is the cancer types observed for the associations, followed by the protein markers and their relevant clinical or molecular features, and then the corresponding statistic and p value. All these precomputed significant results can be directly downloaded through the portal. To demonstrate the utility of this newly developed pan-cancer analytic module, we further examined the significant findings of the pan-cancer results in TCPA v3.0.
Correlations of Protein Expression with Significantly Mutated Genes
To assess the associations between protein expression and somatic mutations, we focused on 299 SMGs identified in a recent study (14) because SMGs are usually cancer driver genes and the sample sizes of their mutated groups are sufficiently large for statistical tests. First, we examined whether SMGs were associated with the levels of their corresponding proteins. As shown in Fig. 2A, p53 was the most differentially expressed protein between TP53 wild-type and mutated patient samples across cancer types, showing a significant difference in 11 cancer types. Intriguingly, in all the 11 cancer types, p53 protein expression was up-regulated in the mutated group, suggesting a potential feedback-loop regulation involved in p53-related pathways. ARID1A, PTEN, and ATM showed decreased expression in the mutated group, which might be partly due to nonsense-mediated RNA degradation (16). Next, we investigated how cancer-related pathways were associated with the mutational status of SMGs. As in Fig. 2B, TP53 again showed the strongest correlations among all the SMGs. Most of the pathways surveyed were highly correlated with TP53 mutation status in many cancer types, suggesting that TP53 serves as a master regulator in signaling cascades of cancer cells. Specifically, DNA damage response, TSC/mTOR, apoptosis, and cell cycle were highly up-regulated in multiple cancer types in TP53 mutated samples. Among all the pathways, RTK was regulated by the largest number of SMGs (in total seven SMGs, such as BRAF, EPAS1, and KIT) across many cancer types. Interestingly, all seven SMGs were associated with down-regulation of RTK pathway. These results provide an overall view of potential effects of somatic mutations on their protein and pathway activities.
Fig. 2.

Protein expression affected by significantly mutated genes. (A) Protein expression change of the 299 SMGs across cancer types. Student's t test followed by multiple testing correction (FDR) was used to identify differentially expressed proteins among the mutated and wild-type groups defined by the mutational status of a gene. Only the differentially expressed proteins with FDR < 0.1 are shown. The circle size indicates the level of differential expression based on the t statistic values. The red/blue color indicates high/low protein expression in the mutated group. (B) Pathway activity impacted by the SMGs. Nodes are shaped and colored according to the data types. The yellow circles indicate the SMGs, and the green squares indicate the cancer pathways. The links between nodes are colored in red/blue to represent the up-/down-regulation of the pathway in the mutated group of an SMG. The line thickness represents how many cancer types show a significant correlation, and only those relationships observed in at least two cancer types are shown.
Predictive Power of mRNA, SCNA, and DNA Methylation on Protein Expression
Protein expression is affected by a variety of genomic and transcriptomic features, where SCNAs, DNA methylation, and mRNA expression are expected to associate with protein abundance change directly. Thus, we conducted a comparative analysis to see which feature contributes most to protein expression. As shown in Fig. 3A, all the three features showed correlations in cis pairs significantly different from trans pairs (all three t test p values < 2.2e-16). Specifically, mRNA expression showed much stronger positive correlations with protein abundance (the averaged ρ = 0.30; same to the average ρ computed from our previous TCGA pan-cancer by Akbani et al. (10)); SCNAs also showed positive effects but to a lesser extent than mRNA (the averaged ρ = 0.11); and DNA methylation showed more negative correlations (the averaged ρ = −0.09), consistent with their gene-silencing role. We observed similar patterns within particular cancer types, although the correlation strength varied from protein to protein (Fig. 3B). We next wanted to identify the best predictor for the protein abundance given a specific protein. Thus, for the samples with all the four data types available, we collected all the significant correlations (FDR < 0.1 and ρ ≥ 0.3) and selected a feature from SCNA, methylation, or mRNA as the best predictor if its correlation dominated the other two. As shown in Fig. 3C, the expression of most proteins can be best predicted by mRNA in almost all cancer types, except for cholangiocarcinoma and uveal melanoma. Only for a small number of proteins, the protein abundance was better inferred by DNA methylation (such as testicular germ cell tumors or SCNAs (such as sarcoma), which may be due to potential noise of gene expression quantification, especially for those genes with close paralogs or complicated splicing isoforms. These results help assess the relative importance of different cis-elements on protein expression.
Fig. 3.

Correlations of protein expression with mRNA, SCNA, and DNA methylation. (A) Histogram of Spearman's rank correlation (ρ values) between protein abundance and the three features (mRNA, SCNA, and DNA methylation) across cancer types. The red bars and curve represent the cis-pairs of protein–feature that are associated with the same gene. The green bars and curve represent the background of ρ values constructed from the trans-pairs in the same dataset. (B) Box plots of the ρ values for the three features across cancer types. The dotted lines indicate a correlation magnitude of 0.3 (sign independent). (C) Bar plots for the percentages of proteins with predicted abundances that can be explained by at least one of the three features (FDR < 0.1 and ρ ≥ 0.3). Different colors represent the percentages of proteins that can be best predicted by SCNA (orange), methylation (red), or mRNA (blue). Gray represents the percentage of proteins that cannot be predicted by any of the three features.
Regulatory Effects of miRNAs on Protein Expression
To better understand the miRNA regulation on protein expression, we performed an integrative analysis to investigate their correlations in different cancer contexts. As shown in Fig. 4A, by comparing the correlations of predicted target genes versus nontarget genes, we identified 19 miRNAs as potential key regulators across cancer types by using the KS-test-based approach (see Methods). The top two frequently observed miRNAs is hsa-miR-18a-5p (in seven cancer types), which has been reported to function as an oncogene in renal cell carcinoma (17) and lung cancer (18) and hsa-miR-532-5p (in five cancer types), a tumor suppressor in liver cancer (19). These results supported their regulatory roles in kidney renal clear cell carcinoma, lung squamous cell carcinoma, and hepatocellular carcinoma (Fig. 4A). To pinpoint the high-confidence miRNA target proteins, we inferred the regulatory networks across cancer types (Figs. 4B and 4C) using those pairs with a significantly negative Spearman's rank correlation (FDR < 0.1). The miRNA, hsa-miR-18a-5p, appeared to have a strong effect on ATM, c-KIT, HER2, ERα, and Gab2, while hsa-miR-532-5p showed significant effects on protein targets, ETS-1, ACRL1, and N-Cadherin. Because miRNAs can regulate gene expression either through mRNA decay or translational repression (20), we next examined which regulatory mechanisms these miRNAs may involve by performing parallel miRNA-mRNA correlation analysis. Interestingly, in some cases, only proteins but not mRNAs showed significantly negative correlations, suggesting a dominating effect of translational inhibition. One such example is the case of hsa-miR-532–5p (Fig. 4D). The results suggest that the RPPA data provide additional information gain to help identify miRNA direct targets and elucidate the corresponding regulatory mechanisms.
Fig. 4.
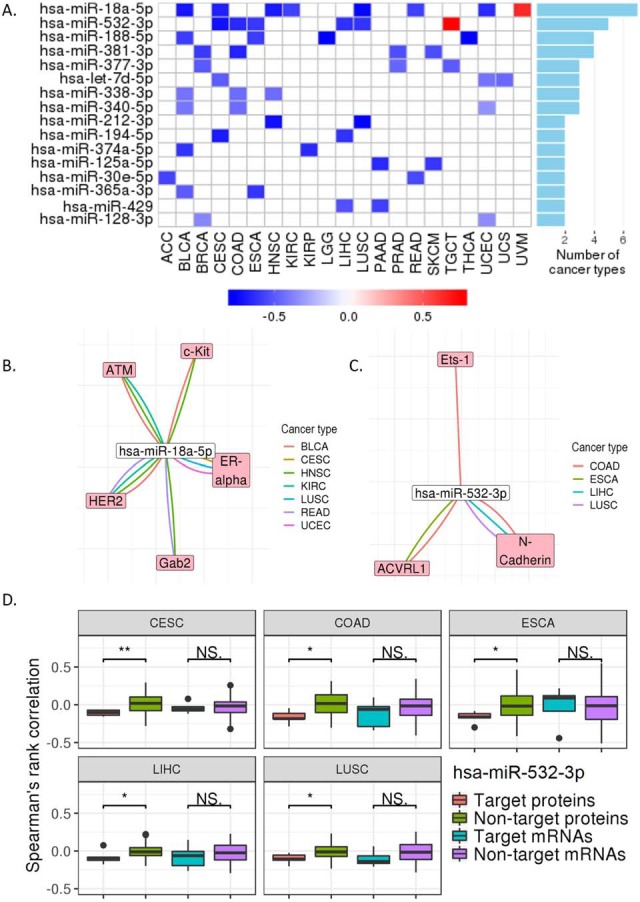
Protein expression regulated by miRNAs. (A) The miRNAs associated with protein expression. The regulatory signals from repression to activation are indicated on a low-to-high scale (blue-white-red) based on the KS test (p < 0.05). The bar plot on the right panel shows the numbers of cancer types that have repression observed. (B) and (C) Regulatory networks of the top-1 and top-2 miRNAs from (A). The negative correlations with significance are shown (Spearman's rank correlation, FDR < 0.1). (D) Box plots of Spearman's rank correlation coefficients of target proteins versus nontarget proteins at the mRNA and protein levels for has-miR-532–3p. KS test was used to assess the differences. *, p < 0.05; **, p < 0.01.
Co-expression Network Analysis of Proteins and Pathways
To better characterize the functional associations among proteins, we first calculated pathway scores and built a nested co-expression network based on the correlations between pathways and proteins (Fig. 5). The network consists of two parts: (i) the inner network shows how different pathways are correlated among cancer types, in which each node represents a pathway and each edge represents the recurrence of significant correlations in different cancer types; (ii) the outer network shows the zoom-in relationships between protein members in each pathway, in which each node is a protein marker with the linked pathway and each edge represents the recurrence of significant correlations. Among the pathways, we found two complete graphs (cliques): one consisted of PI3K/Akt, TSC/mTOR, RTK, and Ras/MAPK pathways, showing all positive correlations with each other in at least 20 cancer types, and the other consisted of apoptosis, epithelial-mesenchymal transition, and core reactive with all positive correlations in at least 15 cancer types. The clique patterns highlight the coordination of these pathways. Interestingly, these two cliques were linked by another clique consisting of apoptosis, TSC/mTOR, and RTK where apoptosis was negatively correlated with the other two pathways. These results present a summary of interactions among pathway members and potential crosstalk between pathways in cancer.
Fig. 5.

A nested co-expression network for proteins and cancer pathways. This nested co-expression network consists of an inner and an outer network, which, respectively, represents the connections between and within the 11 cancer pathways. The links between nodes are colored in red/green to indicate positive/negative correlations. The thickness of the links represents how many cancer types showing statistical significance (Spearman's rank correlation, FDR < 0.1). For simplicity, only the links supported by at least 10 cancer types are shown.
Clinically Relevant Patterns of RPPA Protein Markers
Finally, we examined the clinical relevance of RPPA protein markers by assessing their correlations with patient survival time, tumor subtype, and disease stage across different cancer types. As shown in Fig. 6A, we found that 21 protein markers correlated with overall patient survival times in at least five cancer types (FDR < 0.1, log-rank test or Cox proportional-hazards model). Among the 21 protein markers, the best prognostic marker fibronectin (FN1) showed a consistent and significant prognostic pattern in nine cancer types (Fig. 6B): high fibronectin expression correlated with worse patient survival (Fig. 6C). Consistent with the previously reported patterns in breast, colorectal, and head and neck cancer types (21–23), these results suggest that fibronectin could serve as a robust prognostic marker across cancer types. As for correlations with established tumor subtypes, we found that many of the 258 protein markers were differentially expressed among the established tumor subtypes (e.g. PAM50 for breast invasive carcinoma), suggesting the power of RPPA protein markers in defining tumor heterogeneity within a particular cancer type (Fig. 6D). Also, we observed some proteins showing the expression patterns correlated with tumor stages in a monotonic manner (continuously increased/decreased expression along with tumor stage), suggesting their involvement in tumor progression (Fig. 6E). Together, these results demonstrate the translational potential of protein markers in improving prognosis and defining tumor heterogeneity.
Fig. 6.

Clinical relevance of protein markers. (A) Bar plot for the number of proteins that are associated with overall patient survival times. (B) The protein associated with overall patient survival times observed in at least five cancer types. The circles represent an association showing significance in log-rank test or Cox proportional-hazards model (FDR < 0.1). The circle colors indicate that high protein expression is associated with better (red) or worse (blue) overall survival times than that with low expression based on the hazard ratios of Cox proportional-hazards model. (C) Kaplan Meier curves of fibronectin (FN1) in the nine cancer types. (D) Bar plot for the numbers of differentially expressed proteins among known tumor subtypes. (E) Bar plot for the numbers of differentially expressed proteins along with tumor stage and the red bars show the numbers of proteins with a monotonic change.
DISCUSSION
Pan-cancer analyses using multi-omic TCGA data have demonstrated tremendous potentials to identify biologically and clinically meaningful patterns. Here we present TCPA v3.0 as a user-friendly, interactive platform to explore and analyze TCGA pan-cancer RPPA-based protein expression data. The new pan-cancer analytic module provides a unique opportunity for biomedical researchers to test their protein-driven multi-omic hypotheses across a broad range of cancer types. Based on the analytic results obtained from this new module, we have identified many molecular and clinical features that show significant associations with protein markers in diverse cancer contexts. These findings further demonstrate the utility of the pan-cancer analytic module by helping users confirm known mechanisms, reveal novel biological insights, and test/refine specific hypotheses. We recognize one major limitation that the current pan-cancer RPPA data only cover ∼260 protein markers, which limits the scope of using functional proteomics to elucidate cancer-related mechanism. For example, when searching for cis-regulators, only a small number of known miRNA targets were identified. We are now in the process of expanding the list to 500 proteins covering all major cancer pathways. We expect this new version of TCPA to be a valuable bioinformatic resource for the cancer research community.
Data Availability
All the raw data are available at TCGA Pan-Cancer Atlas website (https://gdc.cancer.gov/about-data/publications/pancanatlas). The data and results are also available at TCPA website (http://tcpaportal.org).
Acknowledgments
We gratefully acknowledge contributions from the TCGA Research Network.
Footnotes
* This study was supported by the National Institutes of Health (CA098258, CA217842, and CA210950 to G.B.M.; CA175486 to H.L.; CA209851 to H.L. and G.B.M.; and CCSG grant CA016672), the Lorraine Dell Program in Bioinformatics for Personalization of Cancer Medicine, and the Adelson Medical Research Foundation (to G.B.M.). G.B.M. has sponsored research support from AstraZeneca, Critical Outcomes Technology, Karus, Illumina, Immunomet, Nanostring, Tarveda, and Immunomet and is on the Scientific Advisory Board for AstraZeneca, Critical Outcomes Technology, ImmunoMet, Ionis, Nuevolution, Symphogen, and Tarveda. H.L. is a shareholder and scientific advisor of Precision Scientific Ltd., (Beijing, China) and Eagle Nebula Inc.
1 The abbreviations used are:
- RPPA
- reverse-phase protein arrays
- TCGA
- The Cancer Genome Atlas
- TCPA
- The Cancer Proteome Atlas
- FDR
- false discovery rate
- SCNAs
- somatic copy-number alterations
- SMGs
- significantly mutated genes.
REFERENCES
- 1. Sheehan K. M., Calvert V. S., Kay E. W., Lu Y., Fishman D., Espina V., Aquino J., Speer R., Araujo R., Mills G. B., Liotta L. A., Petricoin E. F. 3rd, Wulfkuhle J. D. (2005) Use of reverse phase protein microarrays and reference standard development for molecular network analysis of metastatic ovarian carcinoma. Mol. Cell. Proteomics 4, 346–355 [DOI] [PubMed] [Google Scholar]
- 2. Spurrier B., Ramalingam S., and Nishizuka S. (2008) Reverse-phase protein lysate microarrays for cell signaling analysis. Nat. Protoc. 3, 1796–1808 [DOI] [PubMed] [Google Scholar]
- 3. Lu Y., Ling S., Hegde A. M., Byers L. A., Coombes K., Mills G. B., and Akbani R. (2016) Using reverse-phase protein arrays as pharmacodynamic assays for functional proteomics, biomarker discovery, and drug development in cancer. Semin. Oncol. 43, 476–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li J., Lu Y., Akbani R., Ju Z., Roebuck P. L., Liu W., Yang J. Y., Broom B. M., Verhaak R. G., Kane D. W., Wakefield C., Weinstein J. N., Mills G. B., and Liang H. (2013) TCPA: A resource for cancer functional proteomics data. Nat. Methods 10, 1046–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J., Zhao W., Akbani R., Liu W., Ju Z., Ling S., Vellano C. P., Roebuck P., Yu Q., Eterovic A. K., Byers L. A., Davies M. A., Deng W., Gopal Y. N., Chen G., von Euw E. M., Slamon D., Conklin D., Heymach J. V., Gazdar A. F., Minna J. D., Myers J. N., Lu Y., Mills G. B., and Liang H. (2017) Characterization of human cancer cell lines by reverse-phase protein arrays. Cancer Cell 31, 225–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoadley K. A., Yau C., Hinoue T., Wolf D. M., Lazar A. J., Drill E., Shen R., Taylor A. M., Cherniack A. D., Thorsson V., Akbani R., Bowlby R., Wong C. K., Wiznerowicz M., Sanchez-Vega F., Robertson A. G., Schneider B. G., Lawrence M. S., Noushmehr H., Malta T. M., Cancer Genome Atlas, N., Stuart J. M., Benz C. C., and Laird P. W. (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J., Akbani R., Zhao W., Lu Y., Weinstein J. N., Mills G. B., and Liang H. (2017) Explore, visualize, and analyze functional cancer proteomic data using the Cancer Proteome Atlas. Cancer Res. 77, e51–e54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tibes R., Qiu Y., Lu Y., Hennessy B., Andreeff M., Mills G. B., and Kornblau S. M. (2006) Reverse phase protein array: Validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 5, 2512–2521 [DOI] [PubMed] [Google Scholar]
- 9. Hennessy B. T., Lu Y., Gonzalez-Angulo A. M., Carey M. S., Myhre S., Ju Z., Davies M. A., Liu W., Coombes K., Meric-Bernstam F., Bedrosian I., McGahren M., Agarwal R., Zhang F., Overgaard J., Alsner J., Neve R. M., Kuo W. L., Gray J. W., Borresen-Dale A. L., and Mills G. B. (2010) A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin. Proteomics 6, 129–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akbani R., Ng P. K., Werner H. M., Shahmoradgoli M., Zhang F., Ju Z., Liu W., Yang J. Y., Yoshihara K., Li J., Ling S., Seviour E. G., Ram P. T., Minna J. D., Diao L., Tong P., Heymach J. V., Hill S. M., Dondelinger F., Städler N., Byers L. A., Meric-Bernstam F., Weinstein J. N., Broom B. M., Verhaak R. G., Liang H., Mukherjee S., Lu Y., and Mills G. B. (2014) A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 5, 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ju Z., Liu W., Roebuck P. L., Siwak D. R., Zhang N., Lu Y., Davies M. A., Akbani R., Weinstein J. N., Mills G. B., and Coombes K. R. (2015) Development of a robust classifier for quality control of reverse-phase protein arrays. Bioinformatics 31, 912–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J., Han L., Roebuck P., Diao L., Liu L., Yuan Y., Weinstein J. N., and Liang H. (2015) TANRIC: An interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 75, 3728–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y., Xu X., Maglic D., Dill M. T., Mojumdar K., Ng P. K., Jeong K. J., Tsang Y. H., Moreno D., Bhavana V. H., Peng X., Ge Z., Chen H., Li J., Chen Z., Zhang H., Han L., Du D., Creighton C. J., Mills G. B., Cancer Genome Atlas Research Network, Camargo F., and Liang H. (2018) Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep. 25, 1304–1317.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bailey M. H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M. C., Kim J., Reardon B., Kwok-Shing Ng P., Jeong K. J., Cao S., Wang Z., Gao J., Gao Q., Wang F., Liu E. M., Mularoni L., Rubio-Perez C., Nagarajan N., Cortés-Ciriano I., Zhou D. C., Liang W. W., Hess J. M., Yellapantula V. D., Tamborero D., Gonzalez-Perez A., Suphavilai C., Ko J. Y., Khurana E., Park P. J., Van Allen E. M., Liang H., MC3 Working Group, Cancer Genome Atlas Research Network, Lawrence M. S., Godzik A., Lopez-Bigas N., Stuart J., Wheeler D., Getz G., Chen K., Lazar A. J., Mills G. B., Karchin R., and Ding L. (2018) Comprehensive characterization of cancer driver genes and mutations. Cell 174, 1034–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agarwal V., Bell G. W., Nam J. W., and Bartel D. P. (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang C., Asthagiri A. R., Iyer R. R., Lu J., Xu D. S., Ksendzovsky A., Brady R. O., Zhuang Z., and Lonser R. R. (2011) Missense mutations in the NF2 gene result in the quantitative loss of merlin protein and minimally affect protein intrinsic function. Proc. Natl. Acad. Sci. U.S.A. 108, 4980–4985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou L., Li Z. W., Pan X., Lai Y., Quan J., Zhao L., Xu J., Xu W., Guan X., Li H., Yang S., Gui Y., and Lai Y. (2018) Identification of miR-18a-5p as an oncogene and prognostic biomarker in RCC. Am. J. Transl. Res. 10, 1874–1886 [PMC free article] [PubMed] [Google Scholar]
- 18. Liang C., Zhang X., Wang H. M., Liu X. M., Zhang X. J., Zheng B., Qian G. R., and Ma Z. L. (2017) MicroRNA-18a-5p functions as an oncogene by directly targeting IRF2 in lung cancer. Cell Death Dis. 8, e2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han J., Wang F., Lan Y., Wang J., Nie C., Liang Y., Song R., Zheng T., Pan S., Pei T., Xie C., Yang G., Liu X., Zhu M., Wang Y., Liu Y., Meng F., Cui Y., Zhang B., Liu Y., Meng X., Zhang J., and Liu L. (2019) KIFC1 regulated by miR-532–3p promotes epithelial-to-mesenchymal transition and metastasis of hepatocellular carcinoma via gankyrin/AKT signaling. Oncogene 38, 406–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bartel D. P. (2009) MicroRNAs: Target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernandez-Garcia B., Eiró N., Marín L., Gonzalez-Reyes S., González L. O., Lamelas M. L., and Vizoso F. J. (2014) Expression and prognostic significance of fibronectin and matrix metalloproteases in breast cancer metastasis. Histopathology 64, 512–522 [DOI] [PubMed] [Google Scholar]
- 22. Gopal S., Veracini L., Grall D., Butori C., Schaub S., Audebert S., Camoin L., Baudelet E., Radwanska A., Beghelli-de la Forest Divonne S., Violette S. M., Weinreb P. H., Rekima S., Ilie M., Sudaka A., Hofman P., and Van Obberghen-Schilling E. (2017) Fibronectin-guided migration of carcinoma collectives. Nat. Commun. 8, 14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yi W., Xiao E., Ding R., Luo P., and Yang Y. (2016) High expression of fibronectin is associated with poor prognosis, cell proliferation and malignancy via the NF-kappaB/p53-apoptosis signaling pathway in colorectal cancer. Oncol. Rep. 36, 3145–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the raw data are available at TCGA Pan-Cancer Atlas website (https://gdc.cancer.gov/about-data/publications/pancanatlas). The data and results are also available at TCPA website (http://tcpaportal.org).