

metaQuantome enables quantitative analysis of the taxonomic and functional state of a microbiome. Leveraging quantitative mass spectrometry data generated from metaproteomic samples along with taxonomic and functional annotations, metaQuantome unravels the complex and hierarchical data structure of taxonomic and functional ontologies. As a result, metaQuantome enables data exploration, tests hypotheses, and generates high-quality visualizations. metaQuantome deciphers the contribution of taxa to a functional process and vice versa. Its accessibility will pave the way for advanced multi-omic analysis of diverse microbiomes.
Keywords: Microbiome, Functional Inference, Quantification, Bioinformatics Software, Computational Biology, Mass Spectrometry, Microbiology, Multiomics, Taxonomy
Graphical Abstract

Highlights
The new tool metaQuantome analyzes the taxonomic and functional state of microbiomes.
Benchmarking against ground-truth datasets demonstrates metaQuantome's accuracy.
Using a case study, we showcase metaQuantome's publication-ready visualizations.
metaQuantome will enable advanced multi-omic microbiome analysis.
Abstract
Microbiome research offers promising insights into the impact of microorganisms on biological systems. Metaproteomics, the study of microbial proteins at the community level, integrates genomic, transcriptomic, and proteomic data to determine the taxonomic and functional state of a microbiome. However, standard metaproteomics software is subject to several limitations, commonly supporting only spectral counts, emphasizing exploratory analysis rather than hypothesis testing and rarely offering the ability to analyze the interaction of function and taxonomy - that is, which taxa are responsible for different processes.
Here we present metaQuantome, a novel, multifaceted software suite that analyzes the state of a microbiome by leveraging complex taxonomic and functional hierarchies to summarize peptide-level quantitative information, emphasizing label-free intensity-based methods. For experiments with multiple experimental conditions, metaQuantome offers differential abundance analysis, principal components analysis, and clustered heat map visualizations, as well as exploratory analysis for a single sample or experimental condition. We benchmark metaQuantome analysis against standard methods, using two previously published datasets: (1) an artificially assembled microbial community dataset (taxonomy benchmarking) and (2) a dataset with a range of recombinant human proteins spiked into an Escherichia coli background (functional benchmarking). Furthermore, we demonstrate the use of metaQuantome on a previously published human oral microbiome dataset.
In both the taxonomic and functional benchmarking analyses, metaQuantome quantified taxonomic and functional terms more accurately than standard summarization-based methods. We use the oral microbiome dataset to demonstrate metaQuantome's ability to produce publication-quality figures and elucidate biological processes of the oral microbiome. metaQuantome enables advanced investigation of metaproteomic datasets, which should be broadly applicable to microbiome-related research. In the interest of accessible, flexible, and reproducible analysis, metaQuantome is open source and available on the command line and in Galaxy.
Microbiome analysis has enabled the understanding of the effect of microorganisms on diverse biological systems (1–4). The microbiome can be studied using a variety of methods, including metagenomics (5–7), metatranscriptomics (8), and metaproteomics (9). Metaproteomics studies detect the presence and abundance of microbial peptides and proteins, offering a more direct understanding of the processes being catalyzed by the microbiome than metatranscriptomics and metagenomics (9–16). Furthermore, metaproteomics allows the analysis of both taxonomic abundance and functional state from the same mass spectrometry data.
Although metaproteomics is an important component of microbiome research and a complement to other 'omics analyses, limitations in current software restrict the range of methods and accuracy of analyses that can be carried out. First, metaproteomics studies have traditionally quantified peptides with spectral counts, based on counting the number of tandem mass (MS/MS)1 spectra assigned to peptides or proteins (17). Accordingly, many available metaproteomics tools only offer amenability to spectral counting-based quantification, including MEGAN (18), metaGOmics (19), and Unipept (20). However, research has shown that spectral counts offer a less accurate estimate of peptide abundance than the spectral intensity of the precursor peptide (which is typically done by either integrating the MS1 peak or by recording the apex intensity) (21).
Second, some available bioinformatics tools that intend to support microbiome analysis follow a “gene list” approach and require explicit protein or gene inference, such as DAVID (22). In metaproteomics, however, it is sometimes difficult to unambiguously assign a parent protein to a detected peptide because proteins between and within species can be highly homologous (23). Other tools only support certain types of microbiota in a small number of organisms, such as iMetaLab (24), which only supports mouse and human gut microbiome analysis.
Furthermore, metaproteomics tools rarely offer the ability to directly compare many samples or multiple experimental conditions. Some, such as Unipept, focus on detailed exploratory analysis of a single sample. Others, such as metaGOmics, allow comparison between only two samples. However, as metaproteomics is marked by large datasets and many thousands of functional terms and dozens of taxa, it is essential to compare larger numbers of samples to distinguish true effects from random variation. In addition, available metaproteomics tools rarely offer methods to filter out redundant annotations, leading to less informative conclusions from the data.
Finally, while both the taxonomic origin and functional role of peptides (more specifically, of their parent protein) can be determined, few metaproteomics software tools are able to explore the function-taxonomy interaction, that is, the contribution of different taxa to a given functional process and vice versa.
In this manuscript, we present a new software suite called metaQuantome, which is composed of several complementary functionalities developed with the intent to fill some of the aforementioned gaps in metaproteomic bioinformatics tools. metaQuantome is free and open source and is available via GitHub, Bioconda (25), and Galaxy (26). To our knowledge, metaQuantome is the only software to enable fully quantitative differential abundance analysis of the functional and taxonomic profile of a metaproteome and one of only a few software tools to enable function-taxonomy interaction analysis. metaQuantome is amenable to data quantified using peptide-level MS1 intensity values, as well as data quantified by more traditional spectral counting methods. It also utilizes functional annotation and taxonomic annotation—generated from any software—to carry out a multifaceted analysis of a metaproteomics dataset, without requiring the use of a specific database or explicit protein inference. Importantly, it provides novel and powerful functionality for analyzing function-taxonomy interactions, enabling users to determine microbe-specific contributions to the functional profile or the profile of microbes contributing to a specific functional protein class—and visualize the results from these investigations.
We evaluate the accuracy of metaQuantome in quantifying abundance measures of taxa and biochemical functions indicated from peptide abundance data, compared with standard summarization-based methods. First, we benchmark taxonomic abundance estimation using a mock microbial community dataset (27). We also benchmark functional abundance estimation with a dataset consisting of the Universal Proteomics Standards 1 and 2 (UPS1 and UPS2, Sigma-Aldrich) spiked into an E. coli background (21). Finally, we demonstrate the analysis and visualization capabilities of the software on a previously published oral microbiome dataset (28). Our results demonstrate the value of metaQuantome for quantitative analysis of metaproteomics data and advanced exploration of these datasets for microbiome characterization.
EXPERIMENTAL PROCEDURES
Software Structure
metaQuantome is a software suite developed in Python using an object-oriented framework and has a command-line interface divided into several modules (Fig. 1A). The modular structure allows for efficient workflows and examination of the data files at each stage of analysis. In the design of the software, we have leveraged the similarities between different functional and taxonomic annotation types to reduce code duplication. metaQuantome is open source under the Apache 2.0 license, and the source code is available for examination at https://github.com/galaxyproteomics/metaquantome. A detailed description of each module follows. Throughout the text, we use “intensity” to refer to the measured spectral intensity from the mass spectrometer and “abundance” to refer to the relative presence of a peptide, taxon, or functional term in the sample.
Fig. 1.

(A) Outline of metaQuantome program structure. Note that the viz module can be used on results from either filter or stat. (B) The first step in the expand module. The set of all “direct” annotations (those provided by the annotation tool) is expanded to include all of the ancestors of the direct annotations. (C) The second step in the expand module. Abundances are calculated for each term in the expanded hierarchy.
Database Module
The database (db) module downloads the reference databases: Gene Ontology (GO) terms (29), Enzyme Commission (EC) numbers (30), and the the National Center for Biotechnology Information (NCBI) taxonomy database (31). We have leveraged existing Python libraries to facilitate the use of these databases: ete3 (32) (for taxonomy), GOATOOLS (33) (for GO terms), and Biopython (34) (for the ENZYME database).
Expand Module
After downloading the databases, the next module in the metaQuantome analysis is expand, in which we expand the set of all directly annotated functional or taxonomy terms to include all terms implied by the original annotations (Fig. 1B). We use the term “implied” because many domains of biological knowledge are organized hierarchically, where more specific annotations imply more general annotations above them in the hierarchy, also known as “parents” (one level above in the hierarchy) or “ancestors” (any number of levels above in the hierarchy). For example, the taxonomic annotation “Streptococcus genus” is a parent term to “Streptococcus mutans species.” Similarly, hierarchical functional ontologies include GO terms and EC numbers, both of which are supported in metaQuantome. Often, taxonomic and functional annotation tools only provide the most specific term or terms associated with a peptide, for example, Unipept annotates peptides with their lowest common ancestor (LCA), the most specific taxon that is consistent with all potential parent proteins for that peptide (35). Therefore, the information returned by annotation tools such as Unipept is often not the full set of information associated with that annotation.
In metaQuantome, we expand the set of original annotations to include all the ancestors of the direct annotations. To do this, we have defined several custom Python classes that mirror the structure of the annotation hierarchies. Specifically, each term is defined as an instance of the class AnnotationNode, which contains variables specifying the precursor intensity, the number of unique peptides annotated with that term, and other data (for each experimental sample). The AnnotationNodes are collected into an AnnotationHierarchy, which propagates observed intensities for a term up to each of the term's ancestors. That is, the total abundance of a taxon or functional term is calculated as the sum of the abundances of all peptides annotated with the term and/or any of its descendants (see Fig. 1C), an approach that was also used with spectral counts in metaGOmics (19). This allows the user to examine his or her data at different levels of generality—for example, while many peptides may not be specific to a species, examining a taxonomic family allows for estimating the abundance of all species-specific peptides and those specific to the relevant genus and family.
The expand process for function-taxonomy interaction analysis is slightly different (Fig. 2). First, taxonomic annotations are “mapped” to the desired rank—that is, a genus is mapped to the associated family. The annotations that have a lower rank than the desired rank are removed. The directly annotated GO terms are used without modification unless the user selects the “map to slim” option. In that case, each GO term is mapped to its closest relative in the GO slim, which is a smaller set of more general GO terms. Finally, the total abundance for a taxon/GO term combination is calculated as the sum of peptide abundances annotated with the taxon/GO term pair.
Fig. 2.
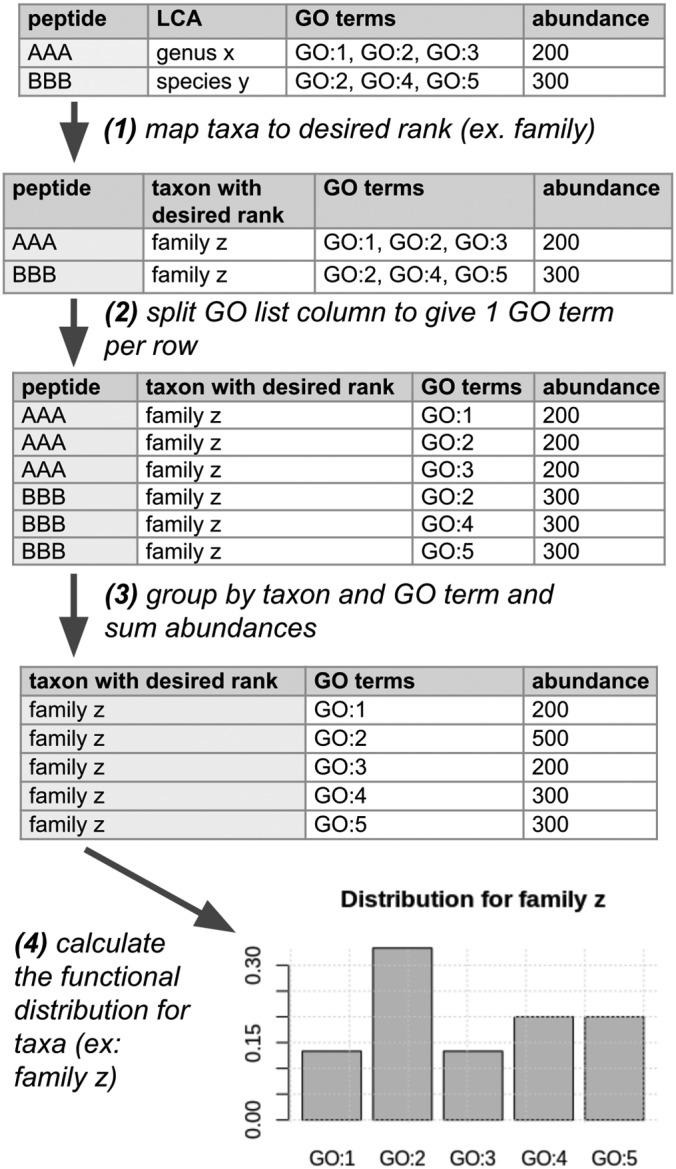
An illustration of the function-taxonomy analysis process. The user must provide a taxonomic rank at which they wish to analyze the dataset, and currently only GO terms are supported. In addition, before the process shown in the figure, metaQuantome ensures that the GO term annotations for each peptide are nonredundant—i.e. that no term in the list is an ancestor of another term in the list. Then, metaQuantome performs the following four actions: (1) The LCA for each peptide is “mapped” to the taxon at the desired rank. In this example, species y is a member of genus x, and genus x is a member of family z. (2) The list of GO terms is split so that there is a single GO term per row. This assumes that each GO term gets the full peptide intensity. (3) Sum to get the total peptide intensity for each combination of taxa and GO terms. This intensity is an estimate of the abundance for each taxon-GO term pair. (4) The viz module calculates either the distribution of taxonomic abundance for a selected GO term or the distribution of GO term abundance for a selected taxon. In this example, we see the function distribution for family z.
The required input for the expand module is:
Quantitative information: a tabular file with peptide sequences and the associated intensities. The values can be calculated using any accepted label-free methods, such as MS1 intensity measurements or spectral counting. Prior to use in metaQuantome, the values should be normalized (36).
Functional and/or taxonomic information: tabular files with peptide sequences and associated functional terms (either GO terms, EC numbers, or COG categories (37), for functional analysis) and/or taxonomic LCA assignments (for taxonomic analysis).
The databases downloaded by metaQuantome db module (described earlier)
Aside from the databases, the quantitative information and the functional and/or taxonomic annotations utilized by this module may be derived from any software. Therefore, metaQuantome can always be used with the most up-to-date quantification and annotation tools. The output of the expand module is a tabular file with columns for the term identifiers (IDs), associated descriptive information, aggregated precursor intensities, number of unique peptides annotated, and number of sample children (described below). The filter module should be used before carrying out any visualization or statistics on the output file.
Filter Module
Because the analysis of many datasets results in many thousands of functional and taxonomic terms, quality control is essential to ensure that spurious term assignments do not mask true term detections. We employ three strategies to ensure that detected terms are well-supported by the data and are nonredundant (see Fig. 3).
Fig. 3.

Filtering methods. The circles indicate terms, the gray arrows indicate “is a” relationships, and the blue arrows indicate metaQuantome filtering procedures. (A) Filtering results by number of unique peptides. The numbers inside each term indicate the hypothetical number of peptides giving evidence to each term. (B) Filtering by the number of sample children. The number inside each term indicates the number of children (direct descendants) that term has within the sample. metaQuantome filters out terms that are neither leaves nor meet the user-specified criterion for minimum sample children (here, two, which is the default). (C) Filtering by the number of samples in which the term was quantified.
First, the user may specify that a term must be supported by a minimum number of distinct peptide sequences (different peptide sequences annotated with the term in question) (Fig. 3A). This allows for filtering out spurious taxonomic or functional terms in which we have lower confidence due to relatively low amounts of supporting data. To enable this filtering, metaQuantome calculates the number of peptides giving evidence to the presence of this term, which is the number of unique peptides directly annotated with this term and/or any of its descendants. Note the difference in the term “children” and “descendants” that has been used here. Descendants for a term A are those terms that are any number of levels below A in the hierarchy and are instances of A, while children of A are descendants that are exactly one level below A.
Next, metaQuantome allows for filtering out redundant terms, which we define in this case as terms that carry the exact same quantitative information as a child—that is, if it has exactly one child term in the data. To filter out these redundant terms, metaQuantome calculates the “sample children” (children in the dataset) of each term in the expanded hierarchy then keeps only those with no sample children or at least the number of sample children set by the user (Fig. 3B). The term “sample children” is used to distinguish between a term's children in the database and the term's children in the sample. For example, the GO term “biological adhesion” (GO:0022610) has four children in the GO database as of February 25, 2019 (multicellular organism adhesion, adhesion of symbiont to host, cell adhesion, intermicrovillar adhesion). However, for a given sample, the term “biological adhesion” may only have two children observed in the sample (i.e. detected peptides might be annotated with “multicellular organism adhesion” and “cell adhesion” and not the others). In this case, biological adhesion would have two sample children. When multiple samples are being analyzed, the user is able to select the minimum number of samples per experimental condition for which the criteria must be met for both number of peptides and number of sample children.
Finally, metaQuantome can filter terms down to those that are quantified in a minimum number of samples per experimental condition (Fig. 3C). This is especially useful in processing multireplicate datasets for statistical analysis, where, for a given term, a minimum of three replicates per experimental condition is necessary.
The output of the filter module is a tabular file with the same columns as that from the expand module, with rows (annotations) that do not fit the specified criteria removed. This file may be used in the stat or viz modules, depending on the researcher's question.
Stat Module
The stat module offers methods for the analysis of differential functional abundance and differential taxonomic abundance between two experimental conditions, using validated statistical analysis functions from the statsmodels Python package (38). The user may choose a standard parametric t test or a nonparametric rank sum test for unpaired samples and may also choose a parametric paired t test or a nonparametric Wilcoxon signed-rank test for paired samples (39). The resulting p values are corrected for multiple tests using the false discovery rate procedure (40). The results from the stat module may be displayed in a volcano plot, available within the viz module.
Viz Module
The viz module of metaQuantome produces a variety of high-quality, publication-ready visualizations: barplots for the analysis of a single sample or experimental condition and differential abundance analysis, volcano plots, heatmaps, and principal components analysis for comparisons between two or more experimental conditions. The visualizations and some of the statistical operations are carried out by linking to R (41) code, due to R's unparalleled visualization capabilities. The visualizations are demonstrated in the Case Study subsection of the Results section. Beyond the built-in visualizations, the filter and stat modules generate a standard tabular file, which permits the user to utilize any preferred statistical or visualization software to analyze the metaQuantome results. Generally, viz should be used after quality control filtering (see Fig. 1A).
Barplot
The viz module offers barplots for descriptive visualization of taxonomic analysis, functional analysis, and function-taxonomy analysis. For taxonomic or functional analysis barplots, the N (default = 5) highest-abundance terms are plotted ranked by abundance. In the function-taxonomy interaction analysis, the user has two options: they can specify a NCBI taxonomy ID (taxID) and obtain the functional distribution of peptide abundances assigned to that taxID, or they can specify a functional term and obtain the taxonomic distribution of peptide abundances annotated with that function. In both cases, the abundances are normalized to one so that the proportion of peptide abundance is obtained.
Principal Components Analysis
metaQuantome carries out a standard principal components analysis, using the prcomp function available within the R stats package. First, any missing values are imputed with 1/1000 times the minimum value in the data. Then, metaQuantome uses prcomp to project the samples onto the principal components and plot the first two principal components with their associated proportion of variance explained. In addition, to obtain a quantitative measure of how well the points are separated in principal component space, we take the ratio of the between-cluster variance to the sum of within-cluster variance, where larger values indicate a better separation, and return this value in the title of the principal component analysis plot. In the case of more than two experimental conditions, the “between cluster” variance is the average of distances between all combinations of cluster centers. In mathematical notation, let pcj be the jth point of the cth cluster, tc be the center of the cth cluster, n being the number of clusters (i.e. the number of experimental conditions), and lc be the number of points within the cth cluster. Then, we define the separation, sep, as
| (1) |
Clustered Heatmap
Like the principal component analysis plot, the hierarchically clustered heatmap analysis may be used for two or more samples. We impute missing values with 1/1000 times the minimum value in the data, use one minus the correlation as our distance measure, and the Ward method of hierarchical clustering (hclust(x, method = “ward.D”) in version 3.4.4 of the stats package in R), all choices suggested by Key (42). If differential abundance analysis has been done, the user may choose to filter the rows to only those terms significantly differentially abundance at a prespecified significance threshold—otherwise, every term present after filtering is included in the heatmap.
Benchmarking
In order to benchmark our methods, we used datasets of known taxonomic and functional composition to evaluate the accuracy of metaQuantome compared with a standard “summarization” method. The summarization method amounts to summing up the abundance of all peptides directly annotated with each taxon or function. In contrast, metaQuantome uses the hierarchical structure of the annotation ontologies to assign abundance to taxonomy or functional categories, including those not present in the set of original annotations. We performed two separate benchmarking analyses. First, we used a dataset of known taxonomic composition (“mock microbial community”) to evaluate metaQuantome's accuracy in estimating taxonomic composition (27). Second, we used a dataset of known functional composition (“spiked-in universal protein standard”) to evaluate metaQuantome's accuracy in estimating functional abundance (21). All metaQuantome analyses were run on a Lenovo ThinkPad T460 with a two-core, four-thread Intel Core i7-6600U 2.6 GHz processor and 32 GB of RAM. metaQuantome is software with relatively low computational demand and can be run on modern laptop computers.
Mock Microbial Community
The objective of using the mock microbial community was to evaluate the accuracy of taxonomic quantitation with metaQuantome versus a standard summarization-based method. We used publicly available proteomic data acquired from an artificial microbial community composed of 32 species and strains (ProteomeXchange accession: PXD006118). The data that were specifically used for our benchmarking were the “equal protein amount” mock community, which was composed of a mixture of equal protein amounts of each of the 32 species and strains, except for bacteriophage proteomes, which were included at 10x lower concentrations than the other proteomes. The dataset consisted of four biological replicates and two technical replicates of each biological replicate. We identified peptides by searching against the protein sequence database provided by Kleiner et al. (27) with SearchGUI (version 3.2.13) (43) and PeptideShaker (1.16.9) (44). To generate quantitative input for metaQuantome, identified peptides were quantified with FlashLFQ (Version 0.1.108) to generate MS1-level precursor intensity values (45). The peptide intensity values were normalized using the “quantile” method within the R package limma (46). The LCA of each identified peptide was obtained by using Unipept 4.0 (20). In Supplementary Document 1, we have included full details on software parameters, the Peptide Report from PeptideShaker, the quantitative information from FlashLFQ, and the Unipept taxonomic annotations.
Next, the true abundance of each taxon was obtained by using the nopep mode of metaQuantome, which calculates the abundance of each taxon in the full taxonomic tree by summing up the protein amounts in the input sample (in micrograms) for each taxon and all of their descendants observed in the sample. In the summarization method, the total abundance of each taxon was obtained by summing up the MS1 intensities of all peptides with that taxon as their LCA. In the metaQuantome method, we estimated the total abundance of each taxon by summing up the MS1 intensities of all peptides with that taxon or a lower taxon as their LCA. In both cases, we averaged the eight replicates and calculated the base-2 logarithm of the resulting average.
As the true abundances and estimated abundances were on different scales (micrograms of protein concentrations versus log2 abundance), we scaled the vector of abundances for each method to have a mean of zero and a standard deviation of one. This allowed us to directly compare true abundance to estimated abundance. Finally, we calculated the mean squared error (MSE) for each estimation method, using all N observed taxa for that method:
| (2) |
That is, MSE is the average squared difference between the estimated abundance and true abundance. It is a measure of the quality of an estimation method, and values closer to zero are better. In our study, the outcomes of interest included the number of taxa quantified and the MSE obtained via the metaQuantome method and the summarization method.
Spiked-in Universal Proteomic Standard
The objective of using the spiked-in Universal Proteomic Standard in this analysis was to compare the accuracy of metaQuantome functional quantitation to that of a summarization approach. To do so, we used a publicly available dataset consisting of the Sigma-Aldrich Universal Proteomic Standard (UPS1 and UPS2) spiked into an E. coli background (21) (ProteomeXchange accession: PXD000279). There were four biological replicates of each of the two conditions. UPS1 consists of an equimolar (5000 fmol) mixture of 48 human proteins, while UPS2 consists of the same 48 proteins mixed at concentrations ranging from 50,000 fmol to 0.5 fmol. The measure of interest for our study was the log2 fold change (L2FC) in functional abundance between UPS2 and UPS1 for the GO term annotations of the 48 spiked-in human proteins.
The UniProt GO annotations for each of the UPS proteins were obtained by querying the UniProt “Retrieve/ID Mapping” tool available on the UniProt web site (accessed November 1, 2018). Then, the metaQuantome nopep mode within the expand module was used to obtain the true L2FC for each direct GO annotation and all of their ancestors.
In order to generate peptide inputs for metaQuantome, we used SearchGUI (version 3.2.13) and PeptideShaker (version 1.16.9) to search the spectrum files against the FASTA database provided by Cox. et al. (21). Then, we used FlashLFQ (version 0.1.108) to obtain the precursor MS1 intensity to estimate abundance for the identified peptides and Unipept 4.0 to obtain GO term annotations for identified peptides. The peptide intensity values were normalized using the “quantile” method within the R package limma (46). In Supplementary Document 2, we have included an Excel sheet with software parameters, the Peptide Report from PeptideShaker, the quantitative information from FlashLFQ, and the Unipept taxonomic annotations.
To estimate the L2FC in the summarization analysis, we simply summed the total abundance of all peptides annotated directly with each GO term, took the average across replicates, calculated the log of the average, and then, for each term, subtracted the average UPS1 log2 abundance from the average UPS1 log2 abundance. To estimate the L2FC in the metaQuantome analysis, we followed a similar method but instead summed the total abundance of all peptides annotated with each GO term and any of their descendants. The outcomes of interest were the total number of GO terms identified and the MSE of the estimate of L2FC over all N GO terms:
| (3) |
Case Study: Bioreactor Model of Oral Dysbiosis
The objective for the case study was to demonstrate the analysis and visualization capabilities of metaQuantome in the context of a full experiment, representative of large-scale metaproteomic studies carried out by microbiome researchers. Full details of data collection are available in the original article (28) (ProteomeXchange accession: PXD003151). Briefly, plaque samples were collected from 12 children with high risk of dental caries. The samples were grown in pairs of biofilm reactors containing hog gastric mucin as the primary carbohydrate source. One of the reactors was pulsed with sucrose five times daily (with sucrose, or WS) and the other was used as a control containing only the mucin-rich medium (no sucrose, or NS). Proteins were extracted from the samples and digested peptides were subjected to LC fractionation and MS/MS analysis on a Velos Orbitrap system (Thermo Fisher Scientific; Waltham, MA). We used SearchGUI (version 3.2.13) and PeptideShaker (version 1.16.9) to search the spectrum files against the Human Oral Microbiome Database (47). Peptide intensity values were obtained with FlashLFQ (version 0.1.108), and the values were normalized using the “quantile” method within the R package limma (46). Further parameter details are available at http://doi.org/10.5281/zenodo.2652530, along with the PeptideShaker Peptide Report, the MS1 intensities determined by FlashLFQ, and the Unipept taxonomic and functional annotations.
RESULTS
Benchmarking
The three benchmarking analyses below took ∼8 min, 2.5 min, and 30 min to run to completion, respectively, while requiring no more than 1–2% of memory. The required databases occupy ∼500 MB of disk space.
Mock Microbial Community
The results from the mock microbial benchmarking analysis are shown in Table I. The ability of metaQuantome to expand the set of direct annotations resulted in an increase in the number of taxa quantified: 36 taxa with metaQuantome versus 33 taxa with the summarization method. In addition, the metaQuantome analysis resulted in a 33% lower MSE than the summarization method, which indicates that using metaQuantome provides a more accurate overall estimate of taxonomic composition than the summarization method.
Table I. Mock microbial community benchmarking results. The “ground truth” indicates the true number of taxa present in the mock microbial community. The MSE reflects the error in the estimate provided by each method (lower is better) and is defined in Equation 2.
| Method | Number of unique taxa quantified | MSE |
|---|---|---|
| Ground truth | 47 | — |
| metaQuantome | 36 | 0.64 |
| Summarization | 33 | 0.95 |
Spiked-In Universal Proteomic Standard
In the functional analysis benchmarking, the capability of metaQuantome to expand the set of direct annotations once again led to a higher number of quantified GO terms (Table II). In this case, metaQuantome quantified more than twice as many terms as the summarization method. In addition, metaQuantome provided a lower MSE, which indicates that it is a better estimator of the overall functional term abundance than the summarization method.
Table II. Spiked-in Universal Protein Standard benchmarking results. The “ground truth” indicates the total number of unique GO terms with which the UPS proteins are annotated. The MSE reflects the error in the estimate provided by each method (lower is better) and is defined in Equation 3.
| Method | Number of unique go terms quantified | MSE |
|---|---|---|
| Ground truth | 3,130 | — |
| metaQuantome | 1,716 | 25.2 |
| Summarization | 712 | 26.8 |
Case Study
The objective of the case study was to demonstrate the visualization capabilities of metaQuantome using a full-fledged metaproteomic experimental dataset, representative of those that would benefit from our software's capabilities. Hence, we show a selection of visualizations for the functional, taxonomic, and function-taxonomy interaction analysis (Fig. 4). We emphasize that this is a demonstration of the use of metaQuantome on an earlier published dataset (28) and do not stress the biological implications of the results.
Fig. 4.

A sampling of metaQuantome visualizations for the oral microbiome dataset. (A) The five most abundant genera in the WS (sugar-pulsed) condition. (B) A principal component analysis on functional abundance separates NS (blue) and WS samples (orange), with some outliers. The separation between the clusters can be seen in the title and is defined in Equation 1. (C) Proportion of total peptide abundance in WS attributed to genera contributing to carbohydrate metabolism (GO:0005975). (D) A volcano plot representing the results of the taxonomic differential abundance analysis, with the fold change reported as abundance in WS over abundance in NS. Taxa with a statistically significant fold change at a user-defined alpha (here, 0.05) are shown with green dots and labeled (some labels removed to reduce overplotting). (E) A hierarchically clustered heatmap of functional annotations separates NS (blue) and WS (orange) samples.
We demonstrate the barplot visualization in Fig. 4A, which shows the five most abundant genera in the WS (sugar-pulsed) experimental condition. The total peptide abundance is on the y axis, and genera are on the x axis. In the barplot visualization, the user can select the number of terms to display, and the terms are automatically sorted in order of decreasing abundance from left to right. For reference, the total abundance assigned to each genus in WS is provided in Supplementary Document 3.
In Fig. 4B, we show the functional principal components analysis visualization. In this example, the separation between the NS and WS clusters is included in the title (see Equation 2 for how this is calculated), but the user has the option to omit it.
We demonstrate a function-taxonomy interaction analysis visualization in Fig. 4C, which is a plot of the taxonomic distribution at the genus level of peptide intensities annotated with the carbohydrate metabolic process (GO term GO:0005975) in WS. As a further demonstration, we provide the full results for taxonomic distribution of carbohydrate metabolism in Supplementary Document 3. After a function-taxonomy analysis is performed, the user may plot the functional distribution of any taxon included in the dataset, as well as the taxonomic distribution of any functional term in the dataset. We anticipate that this will enable in-depth and illuminating exploration of a metaproteomics dataset.
In Fig. 4D, we show metaQuantome's taxonomic differential abundance volcano plot. The user may select the significance threshold (0.05 by default), and terms with statistically significant fold changes are colored green and labeled. For reference, we have also included the output of the stat module that was used to create this plot in Supplementary Document 3.
Finally, we demonstrate a hierarchically clustered heatmap of the functional analysis results in Fig. 4E. The samples are indicated by text labels below each column, and the experimental condition to which each sample belongs is indicated by the color at the top of each column. If stat was previously run, the user also has the option to restrict the heatmap plot to the statistically significant terms (not shown).
DISCUSSION
metaQuantome is a novel and multifunctional bioinformatics software suite that leverages quantitative information and functional and taxonomic annotations to describe the multidimensional state of a microbiome. Among the novel features of metaQuantome are: the multifaceted quality control filtering process, which reduces redundancy and spurious annotations; amenability to either label-free MS1-based intensity or spectral counting quantification methods; the support for differential abundance and clustering analysis across multiple experimental conditions; the use of a peptide-centric approach to mitigate the protein inference problem; and the combination of functional and taxonomic information to elucidate their interaction in a microbiome. As we demonstrate, metaQuantome leads to more complete and accurate estimates of functional and taxonomic abundance than more basic summarization methods. It also provides a variety of visualizations of results that should prove valuable to users for biological interpretation and publication. Collectively, these attributes distinguish metaQuantome from other available software for advanced analysis of metaproteomic data.
An important and unique capability of metaQuantome is its support of function-taxonomy interaction analysis, which allows investigation of how taxa contribute to metabolic pathways, and how the “roles” of the members of a microbial community change due to perturbations of the system. metaQuantome allows investigation of this phenomenon from two directions: the distribution of functional processes for a given taxon and the taxonomic distribution of a certain functional process. As an illustrative example, in the case study, metaQuantome identified a dramatic change in the taxonomic contribution to carbohydrate metabolism: in WS, the Streptococcus genus accounts for a disproportionately higher share of carbohydrate metabolism (82.6% in WS versus 19.7% in NS), while Fusobacteria are responsible for the greatest share of carbohydrate metabolism in NS (66.1%) and hardly any carbohydrate metabolism in WS (1.2%). The identification of such important effects is uniquely facilitated by metaQuantome, through its ability to analyze function and taxonomy at once.
There are some limitations and challenges that should be noted, which we look forward to addressing in the future. First, in its current version, metaQuantome is only able to work with peptides that can be annotated with functional and taxonomic information and automatically discards peptides of unknown function or organismal source. Peptides and proteins of unknown function and taxonomy are often identified in metaproteomics studies (14). Because the interrogation of peptides and proteins of unknown function and/or taxonomy will be an important part of future metaproteomics studies, we look forward to incorporating the ability to analyze these peptides and proteins via metaQuantome. Second, metaQuantome currently provides static visualizations, which are ideal for publication but less ideal for data exploration. In the future, we anticipate developing an interactive visualization application to allow for easier data exploration, as was recently done for another Galaxy-based tool for proteogenomic data analysis (48). Thirdly, we also realize that the outputs generated from metaQuantome are largely dependent on the quality of input datasets. However, as a flexible component of a modular workflow, metaQuantome can always be used with the most cutting-edge quantitation, normalization, functional, and taxonomic assignment tools.
We also see an opportunity to integrate metaQuantome into existing metaproteomics workflows, including those that have been developed within the Galaxy platform (49). Implementation in Galaxy also provides a user interface for the software, in addition to potential for integration with other Galaxy-based tools and workflows. We have designed metaQuantome to take inputs in a standard tabular format, such that it is agnostic to the upstream software used for generating peptide sequence matches from MS/MS data, assigning taxa/function, and quantifying peptides based on label-free methods (MS1-based intensity or spectral counting methods). As such, we envision metaQuantome to fit into a variety of metaproteomic workflows, Galaxy-based or otherwise. It also offers a chance for comparison to, or potentially integration with, other multi-omic workflows for microbiome characterization, such as existing quantitative metatranscriptomics workflows (50). metaQuantome should offer new possibilities and empower users to perform much deeper and advanced multi-omic studies.
In the interest of accessibility, we have made metaQuantome available on GitHub (https://github.com/galaxyproteomics/metaquantome), Bioconda, and on Galaxy, and metaQuantome is supported on macOS and Linux environments. All software is freely available and published following the Apache license. An introduction to using metaQuantome on Galaxy, and details on how to install and analyze data via metaQuantome on the command line is provided at https://galaxyproteomics.github.io/metaquantome_mcp_analysis/, as is the full set of analysis scripts for all three datasets discussed here.
In conclusion, we look forward to the use of metaQuantome in a variety of metaproteomics studies. We have developed the software with an eye toward flexibility and integration with other software tools, and we anticipate further collaborations with others to advance the cause of metaproteomic software development aimed at enabling robust, reproducible, and transparent science. The novel features offered by metaQuantome, combined with usability by bench scientists, should provide a powerful tool to advance our understanding of the role of microbiomes in diverse contexts, from studies related to human health, including clinical applications, to those of environmental and industrial importance.
Data Availability
In Supplementary Documents 1 and 2, and a Zenodo repository at http://doi.org/10.5281/zenodo.2652530, we have provided an Excel document containing the peptide reports with accession numbers, the FlashLFQ reports (with MS1 intensity values) and the Unipept outputs (taxonomy and function) for each of the datasets. In Supplementary Document 3, we have included some of the metaQuantome outputs from the oral microbiome case study. The original datasets are available via ProteomeXchange identifiers PXD006118 (mock microbial community), PXD000279 (spiked-in Universal Proteomic Standard), and PXD003151 (oral microbiome case study). The full set of metaQuantome commands for each of the three analyses is available in the GitHub repository associated with this manuscript (https://github.com/galaxyproteomics/metaquantome_mcp_analysis).
Supplementary Material
Acknowledgments
We would like to thank Bjoern Gruening and the Galaxy community for the help in the support during Galaxy implementation. We would also like to thank Brook Nunn (University of Washington, Seattle, Washington), Alessandro Tanca (Porto Conte Ricerche, Italy), Carolin Kolmeder (University of Helsinki, Finland), and Nadia Szeinbaum (Georgia Tech, Atlanta, Georgia) for discussion during the development of metaQuantome. We thank Emma Leith for proofreading the manuscript.
Footnotes
* We acknowledge funding for this work from the grant National Cancer Institute - Informatics Technology for Cancer Research (NCI-ITCR) grant 1U24CA199347 and National Science Foundation (U.S.) grant 1458524 to T.G. We would also like to acknowledge the Extreme Science and Engineering Discovery Environment (XSEDE) research allocation BIO170096 to P.D.J. and use of the Jetstream cloud-based computing resource for scientific computing (https://jetstream-cloud.org/) maintained at Indiana University. We also acknowledge the support from the Minnesota Supercomputing Institute for maintenance and update of the Galaxy instances.
1 The abbreviations used are:
- MS/MS
- tandem mass spectrometry
- MS1
- precursor mass spectrum
- UPS1
- UPS2, Universal Proteomics Standards 1 and 2
- GO
- Gene Ontology
- EC
- enzyme commission
- NCBI
- National Center for Biotechnology Information
- LCA
- lowest common ancestor
- MSE
- mean squared error
- L2FC
- logarithm base 2 of the fold change
- WS
- with sucrose
- NS
- no sucrose.
REFERENCES
- 1. Gilbert J. A., Blaser M. J., Caporaso J. G., Jansson J. K., Lynch S. V., and Knight R. (2018) Current understanding of the human microbiome. Nat. Med. 24, 392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moran M. A. (2015) The global ocean microbiome. Science 350, aac8455. [DOI] [PubMed] [Google Scholar]
- 3. Fierer N. (2017) Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590 [DOI] [PubMed] [Google Scholar]
- 4. Hörmannsperger G., Schaubeck M., and Haller D. (2015) Intestinal microbiota in animal models of inflammatory diseases. ILAR J. 56, 179–191 [DOI] [PubMed] [Google Scholar]
- 5. Kuczynski J., Costello E. K., Nemergut D. R., Zaneveld J., Lauber C. L., Knights D., Koren O., Fierer N., Kelley S. T., Ley R. E., Gordon J. I., and Knight R. (2010) Direct sequencing of the human microbiome readily reveals community differences. Genome Biol. 11, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quince C., Walker A. W., Simpson J. T., Loman N. J., and Segata N. (2017) Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844 [DOI] [PubMed] [Google Scholar]
- 7. Human Microbiome Project Consortium. (2012) Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bashiardes S., Zilberman-Schapira G., and Elinav E. (2016) Use of Metatranscriptomics in microbiome research. Bioinform. Biol. Insights 10, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilmes P., Heintz-Buschart A., and Bond P. L. (2015) A decade of metaproteomics: Where we stand and what the future holds. Proteomics 15, 3409–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verberkmoes N. C., Russell A. L., Shah M., Godzik A., Rosenquist M., Halfvarson J., Lefsrud M. G., Apajalahti J., Tysk C., Hettich R. L., and Jansson J. K. (2009) Shotgun metaproteomics of the human distal gut microbiota. ISME J. 3, 179–189 [DOI] [PubMed] [Google Scholar]
- 11. Xiong W., Giannone R. J., Morowitz M. J., Banfield J. F., and Hettich R. L. (2015) Development of an enhanced metaproteomic approach for deepening the microbiome characterization of the human infant gut. J. Proteome Res. 14, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heyer R., Schallert K., Zoun R., Becher B., Saake G., and Benndorf D. (2017) Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 261, 24–36 [DOI] [PubMed] [Google Scholar]
- 13. Kolmeder C. A., and de Vos W. M. (2014) Metaproteomics of our microbiome—Developing insight in function and activity in man and model systems. J. Proteomics 97, 3–16 [DOI] [PubMed] [Google Scholar]
- 14. Heintz-Buschart A., and Wilmes P. (2018) Human gut microbiome: Function matters. Trends Microbiol. 26, 563–574 [DOI] [PubMed] [Google Scholar]
- 15. Wilmes P., and Bond P. L. (2004) The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 6, 911–920 [DOI] [PubMed] [Google Scholar]
- 16. Zhang X., and Figeys D. (2019) Perspective and guidelines for metaproteomics in microbiome studies. J. Proteome Res. 18, 2370–2380 [DOI] [PubMed] [Google Scholar]
- 17. Lundgren D. H., Hwang S.-I., Wu L., and Han D. K. (2010) Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics 7, 39–53 [DOI] [PubMed] [Google Scholar]
- 18. Huson D. H., Beier S., Flade I., Górska A., El-Hadidi M., Mitra S., Ruscheweyh H.-J., and Tappu R. (2016) MEGAN community edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLOS Comput. Biol. 12, e1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Riffle M., May D. H., Timmins-Schiffman E., Mikan M. P., Jaschob D., Noble W. S., and Nunn B. L. (2017) MetaGOmics: A web-based tool for peptide-centric functional and taxonomic analysis of metaproteomics data. Proteomes 6, E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gurdeep Singh R., Tanca A., Palomba A., Van der Jeugt F., Verschaffelt P., Uzzau S., Martens L., Dawyndt P., and Mesuere B. (2019) Unipept 4.0: Functional analysis of metaproteome data. J. Proteome Res. 18, 606–615 [DOI] [PubMed] [Google Scholar]
- 21. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang D. W., Sherman B. T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M. W., Lane H. C., and Lempicki R. A. (2007) DAVID bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35, W169–W175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muth T., Behne A., Heyer R., Kohrs F., Benndorf D., Hoffmann M., Lehtevä M., Reichl U., Martens L., and Rapp E. (2015) The MetaProteomeAnalyzer: A powerful open-source software suite for metaproteomics data analysis and interpretation. J. Proteome Res. 14, 1557–1565 [DOI] [PubMed] [Google Scholar]
- 24. Liao B., Ning Z., Cheng K., Zhang X., Li L., Mayne J., and Figeys D. (2018) iMetaLab 1.0: A web platform for metaproteomics data analysis. Bioinformatics 34, 3954–3956 [DOI] [PubMed] [Google Scholar]
- 25. Grüning B., Dale R., Sjödin A., Chapman B. A., Rowe J., Tomkins-Tinch C. H., Valieris R., Köster J., and Bioconda Team. (2018) Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B. A., Guerler A., Hillman-Jackson J., Hiltemann S., Jalili V., Rasche H., Soranzo N., Goecks J., Taylor J., Nekrutenko A., and Blankenberg D. (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kleiner M., Thorson E., Sharp C. E., Dong X., Liu D., Li C., and Strous M. (2017) Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 8, 1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rudney J. D., Jagtap P. D., Reilly C. S., Chen R., Markowski T. W., Higgins L., Johnson J. E., and Griffin T. J. (2015) Protein relative abundance patterns associated with sucrose-induced dysbiosis are conserved across taxonomically diverse oral microcosm biofilm models of dental caries. Microbiome 3, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gene Ontology Consortium. (2015) Gene Ontology Consortium: Going forward. Nucleic Acids Res. 43, D1049–D1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bairoch A. (2000) The ENZYME database in 2000. Nucleic Acids Res. 28, 304–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Federhen S. (2012) The NCBI taxonomy database. Nucleic Acids Res. 40, D136–D143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huerta-Cepas J., Serra F., and Bork P. (2016) ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klopfenstein D. V., Zhang L., Pedersen B. S., Ramírez F., Vesztrocy A. W., Naldi A., Mungall C. J., Yunes J. M., Botvinnik O., Weigel M., Dampier W., Dessimoz C., Flick P., and Tang H. (2018) GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 8, 10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cock P. J., Antao T., Chang J. T., Chapman B. A., Cox C. J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., and de Hoon M. J. (2009) Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mesuere B., Devreese B., Debyser G., Aerts M., Vandamme P., and Dawyndt P. (2012) Unipept: Tryptic peptide-based biodiversity analysis of metaproteome samples. J. Proteome Res. 11, 5773–5780 [DOI] [PubMed] [Google Scholar]
- 36. Välikangas T., Suomi T., and Elo L. L. (2018) A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 19, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Galperin M. Y., Kristensen D. M., Makarova K. S., Wolf Y. I., and Koonin E. V. (2017) Microbial genome analysis: The COG approach. Brief. Bioinform. 10.1093/bib/bbx117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seabold S., and Perktold J. (2010) Statsmodels: Econometric and Statistical Modeling with Python, in Proceedings of the 9th Python in Science Conference (Scipy), p. 61 [Google Scholar]
- 39. Ewens W. J., and Grant G. R. (2005) Statistical methods in bioinformatics: An introduction, Springer, New York, 2nd ed. [Google Scholar]
- 40. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 [Google Scholar]
- 41. RCore Team. (2018) R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 42. Key M. (2012) A tutorial in displaying mass spectrometry-based proteomic data using heat maps. BMC Bioinformatics 13, S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barsnes H., and Vaudel M. (2018) SearchGUI: A highly adaptable common interface for proteomics search and de novo engines. J. Proteome Res. 17, 2552–2555 [DOI] [PubMed] [Google Scholar]
- 44. Vaudel M., Burkhart J. M., Zahedi R. P., Oveland E., Berven F. S., Sickmann A., Martens L., and Barsnes H. (2015) PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 33, 22–24 [DOI] [PubMed] [Google Scholar]
- 45. Millikin R. J., Solntsev S. K., Shortreed M. R., and Smith L. M. (2018) Ultrafast peptide label-free quantification with FlashLFQ. J. Proteome Res. 17, 386–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen T., Yu W.-H., Izard J., Baranova O. V., Lakshmanan A., and Dewhirst F. E. (2010) The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database J. Biol. Databases Curation 2010, baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sajulga R., Mehta S., Kumar P., Johnson J. E., Guerrero C. R., Ryan M. C., Karchin R., Jagtap P. D., and Griffin T. J. (2018) Bridging the chromosome-centric and biology/disease-driven human proteome projects: Accessible and automated tools for interpreting the biological and pathological impact of protein sequence variants detected via proteogenomics. J. Proteome Res. 17, 4329–4336 [DOI] [PubMed] [Google Scholar]
- 49. Blank C., Easterly C., Gruening B., Johnson J., Kolmeder C. A., Kumar P., May D., Mehta S., Mesuere B., Brown Z., Elias J. E., Hervey W. J., McGowan T., Muth T., Nunn B., Rudney J., Tanca A., Griffin T. J., and Jagtap P. D. (2018) Disseminating metaproteomic informatics capabilities and knowledge using the Galaxy-P framework. Proteomes 6, E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Batut B., Gravouil K., Defois C., Hiltemann S., Brugère J.-F., Peyretaillade E., and Peyret P. (2018) ASaiM: A Galaxy-based framework to analyze microbiota data. GigaScience 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In Supplementary Documents 1 and 2, and a Zenodo repository at http://doi.org/10.5281/zenodo.2652530, we have provided an Excel document containing the peptide reports with accession numbers, the FlashLFQ reports (with MS1 intensity values) and the Unipept outputs (taxonomy and function) for each of the datasets. In Supplementary Document 3, we have included some of the metaQuantome outputs from the oral microbiome case study. The original datasets are available via ProteomeXchange identifiers PXD006118 (mock microbial community), PXD000279 (spiked-in Universal Proteomic Standard), and PXD003151 (oral microbiome case study). The full set of metaQuantome commands for each of the three analyses is available in the GitHub repository associated with this manuscript (https://github.com/galaxyproteomics/metaquantome_mcp_analysis).