

Histopathology images are important for cancer diagnosis and prognosis. We extracted quantitative morphological features from breast cancers images and systematically analyzed their relationships with proteins and mRNAs. We observed concordant correlation patterns between image-protein and image-RNA and identified four cancer-related biological processes associated with morphological features related to different tumor components. Further, we observed that proteomic data reveal unique protein-related biological processes associated with morphology. Finally, prognostic morphological features were identified, and their roles are consistent with the underlying biological processes.
Keywords: Proteogenomics, Breast cancer, Tumor microenvironment, Cell cycle, Systems biology, Omics, Computational pathology, Imaging genomics, Immune response, Morphology
Graphical Abstract

Highlights
Consistent correlation patterns between image-protein and image-mRNA at genome level.
Four major biological processes associated with cellular and tissue morphology.
Proteomic data reveal protein-specific biology processes associated with morphology.
Morphological features can predict survival with relevant molecular events.
Abstract
Tumors are heterogeneous tissues with different types of cells such as cancer cells, fibroblasts, and lymphocytes. Although the morphological features of tumors are critical for cancer diagnosis and prognosis, the underlying molecular events and genes for tumor morphology are far from being clear. With the advancement in computational pathology and accumulation of large amount of cancer samples with matched molecular and histopathology data, researchers can carry out integrative analysis to investigate this issue. In this study, we systematically examine the relationships between morphological features and various molecular data in breast cancers. Specifically, we identified 73 breast cancer patients from the TCGA and CPTAC projects matched whole slide images, RNA-seq, and proteomic data. By calculating 100 different morphological features and correlating them with the transcriptomic and proteomic data, we inferred four major biological processes associated with various interpretable morphological features. These processes include metabolism, cell cycle, immune response, and extracellular matrix development, which are all hallmarks of cancers and the associated morphological features are related to area, density, and shapes of epithelial cells, fibroblasts, and lymphocytes. In addition, protein specific biological processes were inferred solely from proteomic data, suggesting the importance of proteomic data in obtaining a holistic understanding of the molecular basis for tumor tissue morphology. Furthermore, survival analysis yielded specific morphological features related to patient prognosis, which have a strong association with important molecular events based on our analysis. Overall, our study demonstrated the power for integrating multiple types of biological data for cancer samples in generating new hypothesis as well as identifying potential biomarkers predicting patient outcome. Future work includes causal analysis to identify key regulators for cancer tissue development and validating the findings using more independent data sets.
The aggregation of large amount of trans-omics data including high-throughput genetic, transcriptomic, proteomic and clinical information has revolutionized disease research in the past decade but also led to a series of new analytical challenges, calling for new approaches and solutions that aim at improving diagnosis, prognosis, and treatment of complex diseases (1–5). Cancer is a disease with complex underlying molecular mechanisms and factors, and researchers have contributed an overwhelmingly large body of data to characterize, diagnose, and ultimately treat patients with greater precision (6–8). This data revolution enabled researchers to identify genetic mutations and gene expression signatures associated with the development of cancers (6, 9, 10). However, despite these considerable progresses in understanding cancer at multiple levels of biological events, a substantial challenge is to link different types of data with cancer cell and tissue morphology, with the latter being essential for diagnosis and prognosis in clinical practice.
Solid tumors are heterogeneous tissues that contain a mixture of malignant and non-malignant cells, such as stromal cells and lymphocytes (11, 12). Distinct molecular differences exist for cells derived from different tissues, reflected in different patterns in multi-omics data including gene and protein expression profiles (9, 13). These molecular differences, in turn, induce changes in biological functions and morphology of tumor tissue and cells, which are often associated with different prognosis of cancers (14). To date, many studies have addressed the close relationship between molecular events and morphological features of tumor tissues. For instance, Baba et al. (15) systematically discussed the association between mitoses and metabolism with nuclear changes and Wang et al. (16) identified genes whose expression levels are associated with multiple morphological features of tumor cells in triple negative breast cancer. Although remarkable achievements have been made, there are still many important questions to be answered. For example, what is the underlying molecular basis for the cellular and tissue heterogeneity in the tumor (17)? How are the transcriptional and proteomic aberrations reflected on cellular morphology? Therefore, studying the correlations between nuclear morphology and molecular data, especially functional data including both transcriptomic and proteomic data will shed light on the molecular basis of various morphological features of cells and tissues, addressing important questions in cancer development.
Pathological diagnosis is critical for clinical oncology where morphological features are extensively used for diagnostics and prognosis (18). Histopathology images derived from hematoxylin and eosin (H&E)-stained cancer tissue slide contain information regarding morphology (e.g. nuclear area, nuclear shape) and spatial context (e.g. cell density) of diverse types of cells coexisting in the tumor microenvironment (12, 14, 19). Although our previous work (16, 20, 21) along with many work by others (17, 19, 22, 23) have successfully demonstrated a connection between cellular and tissue morphology and clinical outcome for cancers, the underlying molecular basis especially key biological processes associated with these morphological features have not been well understood. Therefore, investigating the biological processes underlying the prognostic morphological features is an important issue in cancer biology and outcome prediction.
To address these issues, matched histopathology images and multi-omics datasets for cancers are required. Fortunately, large consortium endeavors, such as The Cancer Genome Atlas (TCGA)1 have accumulated many large datasets to enable such analyses. TCGA aggregates an extensive collection of omics and clinical datasets from large cohorts of patients for more than 30 types of cancers (24). It also archives histopathology images for solid tumor samples from which omics data were sampled. Currently, more than 24,000 histopathology images are available and can be visualized at the Cancer Digital Slide Archive (CDSA, http://cancer.digitalslidearchive.net/). In addition, The NCI Clinical Proteomic Tumor Analysis Consortium (CPTAC) (https://proteomics.cancer.gov/programs/cptac) program also provides high-throughput proteomic data for some of the TCGA tumor specimens such as breast cancer, ovarian cancer, and colorectal cancer based on mass-spectrometry technology. These large-scale experimental datasets make comprehensive integrative and correlative analyses feasible.
In this study we aim to systematic explore the relationship among molecular, morphological, and clinical data for differential cell types in breast cancer. Previously, we developed a quantitative image analysis pipeline that automatically extracts quantitative cellular morphological features such as nuclear size, nuclear shape, and cell density from H&E-stained whole-slide images (25). Based on this pipeline we performed a series of analysis correlating and integrating molecular data, morphological features, and clinical outcome using data from TCGA and CPTAC Breast invasive carcinoma (BRCA) project. Breast cancer is one of the most common malignant cancers (25) and matched histopathology images and omics data including the genome-wide proteomic, transcriptomic, and Reverse Phase Protein Array (RPPA) proteomic data were acquired from CPTAC and TCGA for a subset of 73 patients in BRCA (25, 26). First, we performed a correlative analysis between multi-omics data (including proteomic, transcriptomics data) and morphological features extracted from histopathology images. We observed that proteomic and transcriptomic data shared consistent correlation pattern with various morphological features at genome scale. However, comparing to transcriptomics data, proteomic data can identify specific protein-related biological processes associated with morphological features that otherwise cannot be inferred from transcriptomic data. More comprehensive analysis revealed that four major categories of biology processes related to the hallmarks of cancer (6) are associated with different morphological feature based on the correlated proteomic data. Furthermore, we examined the relationship between nuclear morphology and patient outcome (i.e. survival time). Both prognostically favorable and unfavorable morphological features have been identified. The biological processes associated with these prognostic morphological features were also identified based on proteomic data. The biological processes such as immune responses, cell cycle, and extracellular matrix development have been previously associated with cancer patient outcome. In summary, our work linked molecular data, morphology, and clinical outcome, which led to new insights and hypotheses into the relationships between cancer tissue development and molecular events, thus contributing to a more comprehensive understanding of breast cancer. The entire process and workflow can be applied to other cancers.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
The main objective of this study is to explore the relationship between molecular data and tumor morphology. Firstly, we performed correlation analysis between molecular data (i.e. mRNA-seq data from TCGA and proteomic data from CPTAC respectively) and quantitative morphological features extracted from histopathology images of breast cancers. We examined the distribution patterns of correlation coefficients between image-mRNA and image-protein pairs at genome scale. Secondly, we validated the above distribution patterns of correlation coefficients using proteomic data generated from the RPPA technology and morphological features. Thirdly, we compared the biological processes associated with morphological and spatial features based on the strongly correlated mRNAs and proteins. Finally, we summarized the major biological implications underlying the various morphological features. In addition to the correlation analysis, we explored the relationships among morphology, patient outcome, and the underlying biological processes derived from protein data.
The overall workflow for the analysis is summarized in Fig. 1 with the correlation and other bioinformatics analyses outlined in Fig. 1B. For nuclear morphology-proteomic-transcriptomic correlation analysis, we identified a set of 73 patients whose tumor samples are shared by the TCGA BRAC project and CPTAC breast cancer project with matched H&E-stained whole-slide images, MS-derived proteomic profiles, and RNA-seq transcriptomic data. Specifically, the CPTAC consortium contained 105 breast cancer proteome profiles, of which 77 samples contained non-degraded data (26) that include the 73 selected samples. Proteomic data were accessed and downloaded using the R package “TCGA-Assembler 2” (27, 28) from the CPTAC. Histopathology images were downloaded directly through the GDC TCGA Data Portal, whereas transcriptomic data were downloaded from the UCSC Xena data portal (https://xena.ucsc.edu/public-hubs/) (29). To validate correlation analysis between proteomic and transcriptomic profiles for genes, matched RPPA proteomic data were obtained for each of the 73 samples described above from the Broad GDAC Firehose (https://gdac.broadinstitute.org). The RPPA dataset contains protein expression profiles for 183 genes instead of the entire genome.
Fig. 1.

Study workflow: A, Tissue morphological feature extraction pipeline. B, Schematic diagram for correlation analysis among mRNA, protein abundance, morphology, and clinical prognosis. (Abbreviations: Area: nuclear area; Major_Axis: length major axis of nuclear; Minor_Axis: length of minor axis of a nuclear; Ratio: the ratio between lengths of major and minor axes; Mean_Distance: mean distance to neighboring cells; Max_Distance: maximum distance to neighboring cells; Min_Distance: minimum distance to neighboring cells.)
To understand the relationship between image analysis-derived cell morphological features and patient survival outcomes, 1,057 BRCA-type breast patients with matched 1057 H&E-stained tissue images and corresponding clinical survival information were used. The patient clinical data were obtained from UCSC Xena. Demographic and clinical characteristics of the patients were described in Table I.
Table I. Demographic and clinical characteristics.
| Characteristics | Multi-omics-morphology | Morphology-Clinical analysis |
|---|---|---|
| Patient No. | 73 | 1,057 |
| PAM50 Subtype Patient No.a | ||
| basal-like | 18 | 135 |
| luminal A | 22 | 403 |
| luminal B | 23 | 186 |
| HER2-enriched | 10 | 62 |
| Biomarker Status Patient No.a (Positive/Negative) | ||
| ER Status | 49/24 | 778/232 |
| PR_Status | 41/32 | 674/333 |
| HER2 Status | 16/56 | 158/546 |
| Age (years) | ||
| Range | 30∼88 | 26∼90 |
| Median | 58 | 58 |
| AJCC Stagea | ||
| Stage I | 8 | 182 |
| Stage II | 48 | 610 |
| Stage III | 16 | 245 |
| Stage IV | 1 | 20 |
| Follow-up (days) | ||
| Range | 5∼3316 | 1∼8605 |
| Median | 1148 | 865 |
| Vital_Status | ||
| Living | 66 | 917 |
| Deceased | 7 | 140 |
aSome information is missing for certain patients.
Analysis of Nuclear Morphology from Archived Histopathology Images
As outlined in Fig. 1A, using our previously developed image analysis algorithms and pipeline (30, 31), automated image analysis was carried out and ten types of cell-level features from tissue images were extracted following the three main steps: 1) nuclei segmentation, 2) cell-level feature measurement, and 3) aggregation of cell-level measurements into patient-level statistics. In Step 1, the nuclei of all cells in the image are automatically segmented based on our previous workflow (31). In Step 2, ten types of cell-level features were extracted, including seven types of morphological and spatial traits and three types of pixel traits in the RGB color space. The seven types of morphological and spatial features of cell nuclei were: major axis length (Major_Axis), minor axis length (Minor_Axis), the ratio of major to minor axis length (Ratio), nuclear area (Area), mean distance to neighboring cells (Mean_Distance), maximum distance to neighboring cells (Max_Distance), and minimum distance to neighboring cells (Min_Distance). The seven types of morphological and spatial features of cell nuclei can be summarized as nucleic area (Area), nucleic shape (Major_Axis, Minor_Axis, and Ratio), and cell density (Mean_Distance, Max_Distance and Min_Distance). In Step 3, 5-bin histogram and five distribution statistics (i.e. mean, standard deviation or S.D., skewness, kurtosis, and entropy) were calculated for each of the ten types of morphological features to aggregate the measurements over the whole slide image. Thus for each type of feature, ten measurements (i.e. five histogram bins and five distribution statistics) were generated and 100 image features were generated in total for the ten types of morphological features. The centers of the five bins were determined by clustering each type of cell-level features from all patients instead of a single patient, which ensured that the histogram features are comparable and consistent across the entire patient cohort. The value of each feature based on the five bins of the histogram represented the relative percentage of corresponding image feature over the entire slide for a patient.
To identify distinctive features among the morphological and spatial features, we focused our analysis on the smallest and largest ends of the morphological features for seven types cell-type image features. We designated these features with intuitive names such as Small_Nucleus_Area and Large_Nucleus_Area. The Small_Nucleus_Area is the first bin of the five-bin histogram, thus representing the proportion of very small nuclei. Other feature names are similarly defined. The visual explanation of these morphological features and putative biological interpretations can be found in Table II.
Table II. Examples and interpretation of selected morphological features.

Analysis of MS-based Proteomic Data
We obtained log2-transformed iTRAQ values of protein abundance from CPTAC. The iTRAQ values were calculated as the log2-transformed ratio of spectral counts of target proteins versus a reference. For this analysis, we used iTRAQ data that included peptides that were mapped to multiple proteins. First, to obtain high quality proteomic data, proteins with missing values in more than 20% of the samples were excluded from analyses. Second, expression levels of proteins with missing values in less than 20% of samples were imputed using the Multivariate Imputation tool “mice” package in R (32).
Analysis of RNA-seq Data
Log2-transformed and normalized RSEM gene transcript values of RNA-seq data were obtained and genes with a value of zero in more than 20% of the samples were excluded from analyses.
Image-mRNA and Image-Protein Correlation Analysis
8125 common genes that had both the mRNA and protein expression values were identified. For this subset of genes, the correlations between image features and expression values (protein and mRNA) of a gene were calculated using Spearman's rank correlation (ρ).
Here we chose a relatively loose cutoff of Spearman's rank correlation coefficient ρ, which corresponds to the p value <0.01. Specifically, we designated ρ > 0.3 as correlated whereas ρ ≤ 0.3 was considered uncorrelated.
Validation of the Image-protein Correlation
Besides the CPTAC data, we used the matched proteome data based on RPPA technology of the selected 73 samples for validation. The RPPA proteomic data after normalization were downloaded and the similar processing method with MS-based proteomic data was applied. The Spearman correlation coefficients between image features and protein levels measured using RPPA technology was calculated. The correlations for image-RPPA and image-CPTAC data were then compared.
Biological Process Enrichment Analysis
In order to identify enriched biological processes associated with images features, Gene ontology (GO) analyses were performed using ToppGene (https//topgene.cchmc.org) (33) based on genes whose mRNA or protein product was correlated with morphological features. Here only genes correlated with selected morphological features in Table II were used to perform enrichment analysis. The Fisher's exact test was used to calculate p values for gene set enrichment and a false discovery rate (BH FDR) q value was calculated for multiple test compensation. Only GO terms with q-values less than 0.05 were considered significantly enriched.
Survival Analysis
To assess the association between image features and patient survival information, for each selected image feature, the patient cohort was divided into two groups (high image feature value and low value groups) by applying a cutoff on the image feature values. Then Kaplan-Meier estimator was used for patient stratification and log-rank test was adopted to compare the survival difference between two groups. To choose the best image feature value cut-off with most significant survival difference, we adopted the same procedure as the Human Pathology Atlas, which is part of the Human Proteome Atlas (34). Specifically, all values of the selected feature were ranked and values from the 20th to 80th percentiles were used to identify the cutoff for grouping patients, significant differences in the survival outcomes of the groups were examined and the value yielding the lowest log-rank p value was selected as the cutoff. Features were designated as prognostic image features for those with log-rank p value less than 0.001 for the selected cutoff.
Further, we determined if a prognostic image feature is a “favorable” or “unfavorable” feature by applying the univariate Cox proportional hazards regression analysis with the hazard ratio (HR) larger than 1.2. An unfavorable prognostic image feature was defined as whose higher value is associated with the poor survival whereas favorable prognostic image feature has lower value associated with poor survival.
All analysis above were performed using the “survival” R package (35).
Morphological Features Associated with Clinical Subtype Classification
The associations between each morphological features and clinical subtype classification (i.e. Basal, Luminal A, Luminal B, Her2) were examined using Kruskal-Wallis Test.
Statistical Analysis Software
Except where noted above, all statistical analyses were performed in R version 3.5.1. The analysis scripts that we used for this manuscript are available at GitHub: https://github.com/xiaohuizhan/cor_image_omics_BRCA.
RESULTS
Correlations Analysis Between Multi-omics Data and Morphology
To investigate the relationships between molecular data and histopathology features, we performed correlation analysis between imaging features and mRNA or protein (MS-based global proteomic data) profiles by calculating Spearman's rank correlation coefficients (ρ). A total of 8,125 genes with both mRNA and protein expression and 100 image features for 10 types of cell-level image features extracted from histopathology images were analyzed. As the processes from transcriptome to proteome to morphology were quite complex, in order to be comprehensive in identifying potential molecular basis for different morphological features, correlated image-mRNA or image-protein pairs were designated using a cutoff of ρ > 0.3 to avoid excessive stringency.
The results are summarized in Table III. Among the 100 × 8,125 = 812,500 mage-gene pairs, 5.82% of all image-mRNA pairs and 3.95% of all image-protein pairs were observed to be correlated. In addition, 92.96% showed consistent correlative relationships (either correlated or uncorrelated) in both image-mRNA and image-protein relationships including 0.79% positive correlations, 0.57% negative correlations, and 91.60% no correlation. In contrast, 4.45% image-gene pairs showed correlations only in the image-mRNA relationship, whereas 2.58% were only correlated in image-protein pairs. Opposite image-mRNA and image-protein correlative relationships (i.e. positive correlation in one pair but negative in the other) were observed for only 0.003% of all image-gene pairs. Such globally consistent patterns can also be observed between every image feature and mRNAs and proteins, regardless of positive or negative correlations as demonstrated in supplemental Fig. S1. These results suggested that, at the genome scale, image-mRNA and image-protein shared consistent correlation patterns.
Table III. The statistics of correlation relationships between morphological features and molecular data at the genome scale.
| Image-mRNA | Image-Protein |
||
|---|---|---|---|
| Positive correlation | Noncorrelation | Negative correlation | |
| Positive correlation | 6438 | 17463 | 7 |
| Noncorrelation | 11273 | 744223 | 9707 |
| Negative correlation | 20 | 18702 | 4667 |
Abbreviations: Positive correlation, ρ > 0.3; Negative correlation: ρ < −0.3. Non-correlation: ρ ≤ 0.3.
Next, we compared the distribution of correlation coefficients for image-mRNA and image-protein pairs for individual morphological features (i.e. Area, Major_Axis, Minor_Axis, Ratio, Mean_Distance, Max_Distance and Min_Distance). Specifically, we focused on the selected morphological features in Table II. The correlation for image feature with most of the proteins were consistent with matched mRNA. Supplemental Fig. S2 showed the distribution of correlations for image-mRNA and image-protein for these features. At the individual feature level, the distribution of correlations for individual image features also revealed consistent correlation patterns between image-mRNA and image-protein pairs.
Comparison of Image-protein Correlation Between CPTAC and TCGA Data
In order to test whether the correlation pattern between image-CPTAC measurement overfits the data, we used the matched proteome data based on RPPA technology of these 73 samples for validation. Here we compared the correlation patterns between image features and protein measurements from MS-based technology and RPPA technology. As shown in Fig. 2 for an example for the Large_Nucleus_Area feature. We observed overall consistent results supporting the robustness of correlation between protein profiles and morphological features (correlation coefficients range from 0.472 to 0.597). Results for the rest selected morphological features were displayed in supplemental Fig. S3.
Fig. 2.
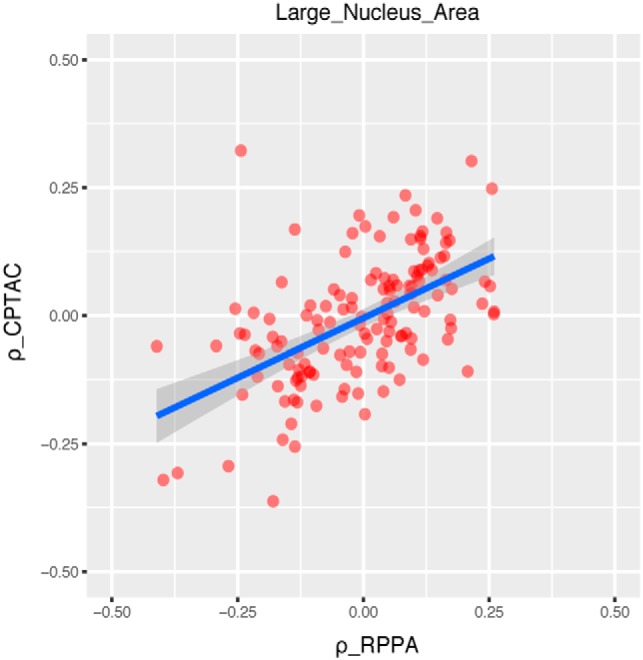
Validating relationships between morphological feature and proteomic data using RPPA data with an example (Large_Nucleus_Area). The correlation coefficient values show consistent distributions for image-RPPA proteomic data and image-CPTAC proteomic data. Each dot represents an image-gene pair with the x axis being the correlation coefficient between image-RPPA measurement (ρ_RPPA) and y axis being the correlation coefficient between image-CPTAC measurement (ρ_CPTAC).
Image-protein Correlation Analysis Reveals Specific Biological Processes Associated with Morphological Features
To test whether proteomics data can reveal biological processes associated with morphological features that cannot be inferred from transcriptomic (mRNA) data alone, we compared enriched gene ontology (GO) terms obtained from mRNAs and proteins that showed strong positive correlation with individual morphological features. After examining these significantly enriched GO Biological Process categories associated with morphological features, we found most of these Biological Process categories identified based on mRNA data can be confirmed based on proteomic data except for Large_Nucleus_Area (implying large nuclei) related biological process. However, some unique Biological Process categories associated with morphological features were found solely based on proteomics data. For instance, for Small_Nucleus_Area (implying small nuclei), protein related Biological processes such as posttranscriptional regulation of gene expression and translation were identified from only proteomics data (supplemental Table S1). Table IV showed the most significantly enriched biological processes terms for individual morphological features based on the positive correlated proteins and mRNAs. In addition, we noticed that for the feature Large_Nucleus_Area (implying large nuclei), mitochondria protein synthesis process involving largely the mitochondrial Ribosomal proteins (MRPs) proteins was significantly enriched based on proteomics data, whereas in contrast it was RNA synthesis process inferred from mRNA data (Fig. 3). Based on these observations, proteomic data can reveal biological processes associated with certain morphological features, which cannot be otherwise identified from transcriptional data alone.
Table IV. The significantly enriched biological process associated with morphological features based on the positive correlated proteins and mRNAs.
| Morphology feature | Protein |
mRNA |
||||
|---|---|---|---|---|---|---|
| GO ID | Name | Adjusted p value | GO_ID | Name | Adjusted p value | |
| Small_Nucleus_Area | GO:0000070 | Mitotic sister chromatid segregation | 3.70E-06 | |||
| GO:0010608 | Posttranscriptional regulation of gene expression | 3.87E-10 | ||||
| Large_Nucleus_Area | GO:0032543 | Mitochondrial translation | 6.12E-33 | GO:0019752 | Carboxylic acid metabolic process | 1.98E-5 |
| Small_Aspect_Ratio | GO:0044419 | Interspecies interaction between organisms | 1.78E-04 | GO:0045087 | Innate immune response | 2.01E-9 |
| GO:0006397 | mRNA processing | 1.78E-04 | ||||
| Large_Aspect_Ratio | GO:0030198 | Extracellular matrix organization | 1.11E-46 | GO:0030198 | Extracellular matrix organization | 2.55E-57 |
| Small_Major_Axis | GO:0006396 | RNA processing | 3.30E-20 | |||
| GO:0000070 | Mitotic sister chromatid segregation | 7.71E-16 | GO:0000819 | Sister chromatid segregation | 1.52E-15 | |
| Large_Major_Axis | GO:0030198 | Extracellular matrix organization | 4.59E-41 | GO:0030198 | Extracellular matrix organization | 2.63E-62 |
| Small_Minor_Axis | GO:0030198 | Extracellular matrix organization | 4.37E-26 | GO:0030198 | Extracellular matrix organization | 1.58E-39 |
| Large_Minor_Axis | GO:0070125 | Mitochondrial translational elongation | 4.47E-83 | GO:0007005 | Mitochondrion organization | 2.48E-7 |
| Small_Max_Distance | GO:0006955 | Immune response | 7.11E-07 | GO:0006955 | Immune response | 2.71E-49 |
| GO:0006397 | mRNA processing | 1.20E-12 | ||||
| Large_Max_Distance | GO:0030198 | Extracellular matrix organization | 2.25E-34 | GO:0030198 | Extracellular matrix organization | 2.891E-18 |
| Small_Min_Distance | GO:0006955 | Immune response | 2.24E-06 | GO:0006955 | Immune response | 2.63E-68 |
| GO:0006397 | mRNA processing | 5.01E-15 | ||||
| Large_Min_Distance | GO:0030198 | Extracellular matrix organization | 3.60E-09 | GO:0030036 | Actin cytoskeleton organization | 1.07E-2 |
| Small_Mean_Distance | GO:0006955 | Immune response | 5.77E-04 | GO:0006955 | Immune response | 3.66E-48 |
| GO:0008380 | RNA splicing | 5.29E-09 | ||||
| Large_Mean_Distance | GO:0030198 | Extracellular matrix organization | 3.49E-35 | GO:0030198 | Extracellular matrix organization | 3.89E-19 |
Fig. 3.

Significantly enriched GO biological processes for large Nuclei Area based on proteomic and Transcriptomic data. x axis lists significant enriched biological processes associated with Large_Nucleus_Area. y axis is -log10(FDR). Orange stands for the mitochondria protein synthesis related biological processes that were identified based on proteomic data. Blue stands for RNA synthesis related biological processes that were identified based on transcriptomic data. Here only top10 enriched biological processes were listed for each category.
Specific Biological Processes Associated with Image Features
As the genes and proteins correlated with the morphological features may shed light on the molecular basis for the cellular and tissue morphology in cancer, gene ontology (GO) enrichment analysis was performed for proteins correlated with each individual morphological feature (i.e. Nuclear Area, Major_Axis, Minor_Axis, Ratio, Mean_Distance, Max_Distance, and Min_Distance) based on if the proteins were positively or negatively correlated. In order to identify biological processes associated with specific types of morphological features, we focused on the selected features listed in Table II.
Overall, the analysis revealed GO terms related to four major categories of biology processes including metabolism, immune process, cell cycle, and extracellular matrix (ECM) were significantly enriched (FDR < 0.05) for morphological features (as shown Fig. 4, supplemental Fig. S4 and Table V). Mitochondrial Ribosomal proteins (MRPs) and mRNA processing related biology processes stood out among metabolic related GO processes. Although for ECM, cell adhesion, cell migration, and vascular system development related GO terms were shared biology processes (Fig. 4 and supplemental Fig. S4). For positive correlations, both Small_Nucleus_Area (implying small nuclei) and Small_Major_Axis (implying small nuclei) were significantly correlated with cell cycle related processes and well-known cell cycle related proteins such as CDCA8, CDC20, NDC80, and BUB1B. In addition, Small_Nucleus_Area (implying small nuclei), Large_Nucleus_Area (implying large nuclei), Small_Aspect_Ratio (implying round nuclei), Small_Major_Axis (implying small nuclei), Large_Minor_Axis (implying large nuclei), Small_Mean_Distance (implying high cell density), Small_Max_Distance (implying high cell density), and Small_Min_Distance (implying high cell density) were all significantly correlated with metabolic processes. Among the proteins associated with these processes, mammalian mitochondrial ribosomal proteins (i.e. MRPL9, MRPL21, MRPL39) showed high correlation, which function in RNA synthesis and processing as well as protein synthesis and translation in cytosol and/or mitochondria and are necessary for the fast growth of tumor cells (36). Although the detailed relationships between cancer cell nuclear size and protein expression as well as metabolism have not been fully investigated, studies based on cancer cell lines suggested protein synthesis rates are positively correlated with cell size, which may be related to nuclear size as well (37).
Fig. 4.

Significantly enriched GO biological processes for morphological features based on proteomic data. Only positive correlations between proteomic data and morphological features were included. Dots represent significantly enriched biological processes based on Benjamini-Hochberg corrected false discovery rate ('F.D.R.B.H') with color coding: purple indicates high enrichment, red indicates low enrichment.
Table V. The summary of significantly enriched biology processes for proteins correlated with morphological features based on proteomic data.
| Morphology feature type | Correlation type | Morphology feature | Significantly enriched biological process |
|---|---|---|---|
| Area | Positive correlation | Small_Nucleus_Area | Cell cycle, Metabolic |
| Large_Nucleus_Area | Metabolic | ||
| Negative correlation | Small_Nucleus_Area | Metabolic | |
| Large_Nucleus_Area | Immune | ||
| Ratio | Positive correlation | Small_Aspect_Ratio | Immune, Metabolic |
| Large_Aspect_Ratio | ECM | ||
| Negative correlation | Small_Aspect_Ratio | ECM | |
| Large_Aspect_Ratio | Metabolic | ||
| Major_axis | Positive correlation | Small_Major_Axis | Cell cycle, Metabolic |
| Large_Major_Axis | ECM | ||
| Negative correlation | Small_Major_Axis | Metabolic | |
| Large_Major_Axis | Metabolic | ||
| Minor_axis | Positive correlation | Small_Minor_Axis | ECM |
| Large_Minor_Axis | Metabolic | ||
| Negative correlation | Small_Minor_Axis | Metabolic | |
| Large_Minor_Axis | TME,Immune | ||
| Max_distance | Positive correlation | Small_Max_Distance | Immune, Metabolic |
| Large_Max_Distance | ECM | ||
| Negative correlation | Small_Max_Distance | ECM | |
| Large_Max_Distance | Immune, Metabolic | ||
| Min_distance | Positive correlation | Small_Min_Distance | Immune, Metabolic |
| Large_Min_Distance | ECM | ||
| Negative correlation | Small_Min_Distance | TME, Metabolic | |
| Large_Min_Distance | Immune, Metabolic | ||
| Mean_distance | Positive correlation | Small_Mean_Distance | Immune, Metabolic |
| Large_Mean_Distance | ECM | ||
| Negative correlation | Small_Mean_Distance | ECM | |
| Large_Mean_Distance | Immune, Metabolic |
Note: ECM related biology processes includes: extracellular matrix (ECM), cell adhesion, cell migration, and vascular system GO functions; Metabolic related biology process mostly include: Mitochondrial Ribosomal proteins (MRPs) and mRNA processing related GO functions.
Moreover, morphological features like Small_Aspect_Ratio (implying round nuclei) and Small_Mean_Distance (implying high cell density), were significantly correlated with immune processes. This is consistent with our knowledge in pathology that many lymphocytes can be identified based on their typical small and round shape (12, 14) as well as densely aggregated patterns. Immune response related proteins such as FCN1, LY75, and major histocompatibility complex (MHC) related proteins such as TAP1, TAP2, B2M were among the ones that are highly correlated with these morphological features.
Further, features such as Large_Aspect_Ratio (implying elongated shape), Large_Major_Axis (implying large or elongated nuclei), Small_Minor_Axis (implying small or elongated nuclei), and Large_Mean_Distance (implying low cell density) correlated with proteins that are significantly enriched with ECM development, which is consistent with the development of tumor stroma in the microenvironment. As stromal cells such as fibroblasts are typically spindle-shaped with elongated nuclei (12) and sparsely scattered in the stroma, they are characterized by long major axes and/or large ratio between major and minor axes and low density compared with epithelial cells. Collagen related proteins such as COL5A1, COL5A2 and COL5A3, which are structural constituent of EMC were identified. From the breast cancer biopsy samples with immunohistochemical staining for COL5A1 in the Human Protein Atlas (HPA) database, we indeed observed a high percentage of stromal cell existing in breast cancer for the ones with high COL5A1 staining (supplemental Fig. S5). Similar results were also observed for MRC2 and COL3A1, which were also highly correlated with morphological features linked to stroma cells (supplemental Fig. S5). Together, the associated biological processes and well-known protein markers support our understanding of the biological basis of different cell type morphological features.
Like the positive correlations between proteomic data and morphological features, such patterns of shared high-level biological processes were also observed in proteins that are negatively correlated with morphological features. Because the values of the image features are relative (i.e. percentages) based on distribution of the values, most of the enriched biology processes associated with the selected extremal features showed inverse enrichment (i.e. the proteins show positive correlations with the large feature values often show negative correlations with the corresponding small aspect). For negative correlations, metabolic process was shown to be significantly associated with features including Small_Nucleus_Area (implying small nuclei), Large_Aspect_Ratio (implying elongated shape), Small_Major_Axis (implying small nuclei), Large_Major_Axis (implying large or elongated nuclei), Small_Minor_Axis (implying small or elongated nuclei), Large_Mean_Distance (implying low cell density), Large_Max_Distance (implying low cell density), Small_Min_Distance (implying high cell density), and Large_Min_Distance (implying low cell density). Immune processes were significantly enriched in proteins negatively correlated with features such as Large_Nucleus_Area (implying large nuclei), Large_Minor_Axis (implying large nuclei), Large_Mean_Distance (implying low cell density), Large_Max_Distance (implying low cell density), and Large_Min_Distance (implying low cell density). The ECM related features were significantly enriched in Small_Aspect_Ratio (implying round nuclei), Large_Minor_Axis (implying large nuclei), Small_Mean_Distance (implying low cell density), Small_Max_Distance (implying low cell density), and Small_Min_Distance (implying low cell density) (Table V and supplemental Fig. S4).
In summary, four major types of biology process including metabolism, immune, cell cycle and ECM development were identified based on proteomic data because of strong associations with morphological features.
Survival Analysis Based on the Morphological Features
Because morphological parameters extracted from histopathology images are essential for breast cancer diagnosis and prognosis by pathologists, we also investigated how well these morphological features are associated with clinical outcome of the patients as described in the Methods section to assess the association between morphological feature and patient overall survival information for all the 1,057 patients in the TCGA BRCA project. Morphological features (p value < 0.001, HR > 1.2) associated with both favorable and unfavorable prognosis have been identified using the workflow described above. In Fig. 5, examples of favorable and unfavorable prognostic morphological features were shown, based on the optimal stratification p value calculated using a similar approach as in (34) (detailed information for other morphological features were provided in supplemental Fig. S6). Five prognostic morphological features that were strongly correlated with patients' overall survival were selected (Fig. 6). After examining these survival-associated variables, we found unfavorable prognostic morphological features including Large_Nucleus_Area (implying large nuclei), Large_Minor_Axis (implying large or elongated nuclei), and Large_Max_Distance (implying low cell density). These morphological features were linked to large cell nuclei or large distances to neighboring cells, which were highly associated with metabolic or ECM related biology processes (Table VI). As for favorable prognostic morphological feature, they tended to be small distances to neighboring cells (Small_Mean_Distance and Small_Min_Distance), which were highly correlated with metabolic or immune related processes (Table VI).
Fig. 5.

Identification of prognostic morphological features based on morphological feature values coupled with survival information for breast cancer. Left: Distribution of p values of log-rank tests against the image feature values with different cutoffs. Right: Kaplan-Meier curves for morphological feature based on the best cutoff. A, Example of an unfavorable prognostic morphological feature; B, example of a favorable prognostic morphological feature.
Fig. 6.
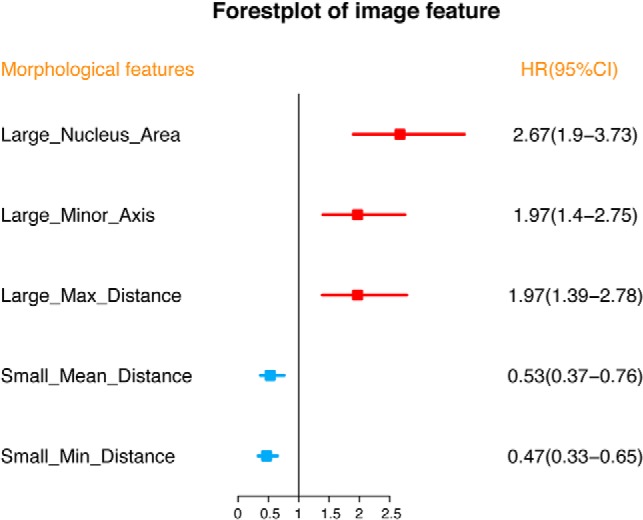
Forest plot for morphological features to predict the overall survival (OS) of breast cancer. Abbreviations: HR = hazard ratio; CI = confidence interval.
Table VI. Summary of survival-associated morphological features.
| Morphology feature | p value | Prognostic type | Associated biology process |
|---|---|---|---|
| Large_Nucleus_Area | 2.96E-09 | unfavorable prognostic | Metabolic |
| Large_Minor_Axis | 6.11E-05 | unfavorable prognostic | Metabolic |
| Large_Max_Distance | 9.71E-05 | unfavorable prognostic | ECM |
| Small_Mean_Distance | 0.51E-03 | favorable prognostic | Immune, Metabolic |
| Small_Min_Distance | 6.55E-06 | favorable prognostic | Immune, Metabolic |
Note: p value formatting as the following example: 2.96E-09 is 0.00000000296.
Morphological Features Significantly Associated with Clinical Subtype Classification
As breast patients can be categorized into subtypes including Basal, Luminal A, Luminal B, Her2 type based on histology and molecular signatures, we performed Kruskal-Wallis Test for each selected image feature to test if they have significant associations with these clinical subtypes. All the image features exhibit significant differences between breast cancer subtypes except for Small_Minor_Axis (supplemental Table S2). These results are consistent with the fact that morphological features are critical to guide diagnosis and treatment.
DISCUSSION
Solid tumors such as breast cancer are highly heterogeneous, with multiple types of cells such as epithelial cells, immune cells and other stromal cells. Given the importance of tumor morphological features in diagnosis and prognosis, investigating the relationship between the molecular data and morphology can lead to potential new insight on the molecular basis underlying cancer development and prognosis. Taking advantage of the computational pathology workflow we established for processing whole slide images, we were able to extract quantitative morphological features from histopathology slides of breast cancer tissues, thus enabling investigating relationships between tumor tissue morphology and omics data. In addition, because mRNA and protein data contain related but different levels of molecular information, integrating both data with morphological features can lead to discovery of different biological events associated with cancer tissue morphology.
Based on the correlation analysis between morphological features derived from whole slide images of tissue samples and molecular data (mRNA or proteomic data), four major types of biology processes, namely metabolic, cell cycle, immune, and ECM development processes have been identified. These processes have all been strongly associated with cancer hallmarks (6). Morphological features enriched with metabolic and cell cycle processes were associated with cancer (epithelial) cells. Among these metabolic processes, we observed strong signals for mitochondria related biology processes, the protein translational process related to mitochondrial Ribosomal proteins (MRPs). Kim et al. (36) previously demonstrated the important function of MRPs in regulating apoptosis, cell cycle, and cell proliferation. As for cell cycle processes, Yuan et al. have highlighted its close relationship with cancer morphologic features (12). However, although it is often anticipated that active cell cycle progress may be associated with large nuclei because of chromosome duplication and mitosis, our results suggest that they may also lead to more smaller cells in breast cancer possibly because of active division even though the detailed mechanism calls for future in-depth investigation. In addition, ECM development and immune response processes for tumor microenvironment were associated with stromal cells and tumor infiltrating lymphocytes respectively (38–41). We found that these stromal cells related features are most strongly associated with tumor microenvironment (TME) development (e.g. ECM, cell adhesion, cell migration). Previous studies have demonstrated that the interaction between stromal cells (such as cancer-associated fibroblasts, a typical stroma cell) and ECM has a crucial role in tumor initiation, progression, and metastasis (42–44), which is an important hallmark of cancers. Beck et al. previously demonstrated the importance of TME related morphological features in breast cancer prognosis (22) and our results linked related features to the potential underlying genes. In addition, cancer-associated fibroblast (CAF) is a typical stromal cell and can recruit and bind collagen fibers (key components of ECM) thus convert a loose stroma into a dense stromal network (43, 44), this network acts as a barrier to drug flow, thereby increasing chemoresistance. Lastly, Yuan et al. also identified that immune related pathways were correlated with the lymphocyte morphologic features (12), which is consistent with our observation. Taken together, our approach can identify the specific biological process associated with individual morphological features. These results not only confirm our understanding of the molecular basis of morphology, but also offer new insights and hypotheses regarding the development of cancer tissues for future investigation.
When comparing the significantly enriched biological processes associated with morphological features based on mRNA and protein, we found that although most of the significantly enriched biological process categories were similar, some unique biological processes associated with morphological features were identified only based on proteomics data (e.g. posttranscriptional related biological processes). In addition, the mitochondria-related metabolism processes also stood out based on proteomic data. Latonen et al. recently showed that post-transcriptional events take important roles in the mitochondria during cancer progression (45). These results strongly suggest that proteomic data are important in fully characterizing the molecular events associated with morphological changes at cellular and tissue levels and are important for understand the development of cancers.
Because histopathology images are essential for cancer diagnosis and prognosis, we also identified favorable and unfavorable prognostic morphological features and the corresponding biological process associated with them. Among these unfavorable predictors, large values of long distance to adjacent nuclei imply a high percentage of stromal components in the in whole-slide images. Yuan et al. and Beck et al. both demonstrated that stromal morphologic structure is an important prognostic factor in breast cancer, patients with higher stromal proportions had worse prognosis than other patients (12, 22). In addition, we also observed that large nuclear area is associated with poor survival. Previous studies have highlighted that cancer cells with enlarged nuclei almost always indicate more aggressive outcomes (46). Currently, anti-estrogen therapy to decreased nuclear size in tumors are used for preoperative treatment of breast cancer patients (46). As for favorable predictors, most of them were related to immune responses, suggesting that activation of immune system plays critical roles in fighting cancer, which are consistent with many recent studies on cancer immunology and immunotherapy (12, 47, 48).
Despite the extensive observations and results generated from our analysis, some limitations of this study should be noticed as well. First, the key molecular regulators for the cell type morphology features were still unknown, even though the associated biological processes were inferred because our current study focuses on correlation analysis instead of causal analysis. Deeper analysis for the regulatory and driver genes and proteins using more sophisticated statistical methods combined with experimental validation will be carried out soon. Second, we only included 73 breast cancer patients for the correlation analysis between molecular data and morphology phenotypes in this study because of the limitation of available data. The image-protein and image-mRNA relationships identified here may not represent all breast cancer subtypes. Despite that the correlative relationships between proteomic data and morphology were validated using matched RPPA data, further confirmation using independent datasets is still needed despite the lack of such data at the meantime. Last but not the least, even though we showed that the cell nucleic features suggested stromal or tumor cells, it is difficult to distinguish different cell types accurately just based on the nucleic morphology alone.
In summary, we carried out a unique systematic study on the relationship between tumor tissue morphology and transcriptomic as well as proteomic data in breast cancer. We observed concordant distribution patterns of correlation coefficients between image-mRNA and image-protein at the genome scale. Four major types of important biological processes related to cancers have been associated with various morphological features. Importantly, proteomic data are critical in identifying protein related biological processes associated with morphological features, which cannot be captured by transcriptomic data. In addition, morphological features associated with patient survival have been identified and their underlying molecular processes based on the associated proteins can link these morphological features to different hallmarks of cancers.
In conclusion, our analysis demonstrated the potential for integrating morphological information and molecular data for generating new biological hypothesis for cancer research. The algorithmic development for computational pathology unleashes the potential for similar large-scale studies for different cancers. More sophisticated modeling and integration methods will lead to deeper understanding of the regulation of the tissue morphology and importance of protein in this process, contributing to the generation of new insights for cancer biology and outcome prediction.
Data Availability
The data used for this study are downloaded from various public sources. Proteomic data were accessed from the NCI CPTAC Data Portal. Histopathology images were downloaded directly through the NCI GDC TCGA Data Portal, whereas transcriptomic data were downloaded from the UCSC Xena data portal (https://xena.ucsc.edu/public-hubs/). Matched RPPA proteomic data were obtained from the Broad GDAC Firehose (https://gdac.broadinstitute.org). cell morphological features and patient survival outcomes, 1,057 BRCA-type breast patients with matched 1057 H&E-stained tissue images and corresponding clinical survival information were used. The patient clinical data were obtained from UCSC Xena. The analysis scripts that we used for this manuscript are available at GitHub: https://github.com/xiaohuizhan/cor_image_omics_BRCA.
Supplementary Material
Acknowledgments
We thank Mrs. Megan Metzger for editing the manuscript.
Footnotes
* This work was partially supported by the Shenzhen Peacock Plan (KQTD2016053112051497) to X.Z., J.C., T.-F.W., and D.N., NCI ITCR U01CA188547 to J.Z. and K.H., and Indiana University Precision Health Initiative to K.H., J.Z., Z. Han, B.H., and Z. Huang.
 This article contains supplemental Figures and Tables. No potential conflicts of interest were disclosed.
This article contains supplemental Figures and Tables. No potential conflicts of interest were disclosed.
1 The abbreviations used are:
- TCGA
- The Cancer Genome Atlas
- CPTAC
- Clinical Proteomic Tumor Analysis Consortium
- RPPA
- reverse phase protein array
- TME
- tumor microenvironment
- GO
- Gene ontology
- BP
- biological process
- ECM
- extracellular matrix
- MRPs
- mitochondrial ribosomal proteins.
REFERENCES
- 1. Stratton M. R., Campbell P. J., and Futreal P. A. (2009) The cancer genome. Nature 458, 719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lynch C. (2008) How do your data grow? Nature 455, 28–29 [DOI] [PubMed] [Google Scholar]
- 3. Tatonetti N. P. (2019) Translational medicine in the Age of Big Data. Briefings in Bioinformatics 20, 457–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murdoch T. B., and Detsky A. S. (2013) The inevitable application of Big Data to Health Care. JAMA 309, 1351–1352 [DOI] [PubMed] [Google Scholar]
- 5. Gomez-Cabrero D., Abugessaisa I., Maier D., Teschendorff A., Merkenschlager M., Gisel A., Ballestar E., Bongcam-Rudloff E., Conesa A., and Tegnér J. (2014) Data integration in the era of omics: current and future challenges. BMC Systems Biol. 8, I1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 7. Bertram J. S. (2000) The molecular biology of cancer. Mol. Aspects Med. 21, 167–223 [DOI] [PubMed] [Google Scholar]
- 8. Hassanpour S. H., and Dehghani M. (2017) Review of cancer from perspective of molecular. J. Cancer Res. and Practice 4, 127–129 [Google Scholar]
- 9. Hoadley K. A., Yau C., Hinoue T., Wolf D. M., Lazar A. J., Drill E., Shen R., Taylor A. M., Cherniack A. D., Thorsson V., Akbani R., Bowlby R., Wong C. K., Wiznerowicz M., Sanchez-Vega F., Robertson A. G., Schneider B. G., Lawrence M. S., Noushmehr H., Malta T. M., Cancer Genome Atlas, N., Stuart J. M., Benz C. C., and Laird P. W. (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Michor F., Iwasa Y., and Nowak M. A. (2004) Dynamics of cancer progression. Nat. Rev. Cancer 4, 197–205 [DOI] [PubMed] [Google Scholar]
- 11. Balkwill F. R., Capasso M., and Hagemann T. (2012) The tumor microenvironment at a glance. J. Cell Sci. 125, 5591–5596 [DOI] [PubMed] [Google Scholar]
- 12. Yuan Y., Failmezger H., Rueda O. M., Ali H. R., Gräf S., Chin S. F., Schwarz R. F., Curtis C., Dunning M. J., Bardwell H., Johnson N., Doyle S., Turashvili G., Provenzano E., Aparicio S., Caldas C., Markowetz F. (2012) Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci. Transl. Med. 4, 157ra143. [DOI] [PubMed] [Google Scholar]
- 13. Wang M. N., Zhao J. Z., Zhang L. S., Wei F., Lian Y., Wu Y. F., Gong Z. J., Zhang S. S., Zhou J. D., Cao K., Li X. Y., Xiong W., Li G. Y., Zeng Z. Y., and Guo C. (2017) Role of tumor microenvironment in tumorigenesis. J/ Cancer 8, 761–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heindl A., Nawaz S., and Yuan Y. Y. (2015) Mapping spatial heterogeneity in the tumor microenvironment: a new era for digital pathology. Lab. Invest. 95, 377–384 [DOI] [PubMed] [Google Scholar]
- 15. Baba A. I., and Câtoi C. (2007) Tumor cell morphology. Comparative Oncology, The Publishing House of the Romanian Academy; [PubMed] [Google Scholar]
- 16. Wang C., Pécot T., Zynger D. L., Machiraju R., Shapiro C. L., and Huang K. (2013) Identifying survival associated morphological features of triple negative breast cancer using multiple datasets. J. Am. Med. Inform. Assoc. 20, 680–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cooper L. A., Kong J., Gutman D. A., Wang F., Gao J., Appin C., Cholleti S., Pan T., Sharma A., Scarpace L., Mikkelsen T., Kurc T., Moreno C. S., Brat D. J., and Saltz J. H. (2012) Integrated morphologic analysis for the identification and characterization of disease subtypes. J. Am. Med. Inform. Assoc. 19, 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patey D. H., and Scarff R. W. (1928) The Position of Histology in the Prognosis of Carcinoma of the Breast. Lancet 211, 801–804 [Google Scholar]
- 19. Yuan Y. Y. (2016) Spatial heterogeneity in the tumor microenvironment. Cold Spring Harbor Perspect. Med. 6, pii: a026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng J., Mo X., Wang X., Parwani A., Feng Q., and Huang K. (2018) Identification of topological features in renal tumor microenvironment associated with patient survival. Bioinformatics 34, 1024–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang C., Machiraju R., and Huang K. (2014) Breast cancer patient stratification using a molecular regularized consensus clustering method. Methods 67, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beck A. H., Sangoi A. R., Leung S., Marinelli R. J., Nielsen T. O., van de Vijver M. J., West R. B., van de Rijn M., and Koller D. (2011) Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci. Transl. Med. 3, 108–113 [DOI] [PubMed] [Google Scholar]
- 23. Yuan Y. Y., Failmezger H., Rueda O. M., Ali H. R., Graf S., Chin S. F., Schwarz R. F., Curtis C., Dunning M. J., Bardwell H., Johnson N., Doyle S., Turashvili G., Provenzano E., Aparicio S., Caldas C., and Markowetz F. (2012) Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci. Trans. Med. 4, 157ra143. [DOI] [PubMed] [Google Scholar]
- 24. Grossman R. L., Heath A. P., Ferretti V., Varmus H. E., Lowy D. R., Kibbe W. A., and Staudt L. M. (2016) Toward a shared vision for cancer genomic data. New Engl. J. Med. 375, 1109–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. The Cancer Genome Atlas, N., Koboldt D. C., Fulton R. S., McLellan M. D., Schmidt H., Kalicki-Veizer J., McMichael J. F., Fulton L. L., Dooling D. J., Ding L., Mardis E. R., Wilson R. K., Ally A., Balasundaram M., Butterfield Y. S. N., Carlsen R., Carter C., Chu A., Chuah E., Chun H.-J. E., Coope R. J. N., Dhalla N., Guin R., Hirst C., Hirst M., Holt R. A., Lee D., Li H. I., Mayo M., Moore R. A., Mungall A. J., Pleasance E., Gordon Robertson A., Schein J. E., Shafiei A., Sipahimalani P., Slobodan J. R., Stoll D., Tam A., Thiessen N., Varhol R. J., Wye N., Zeng T., Zhao Y., Birol I., Jones S. J. M., Marra M. A., Cherniack A. D., Saksena G., Onofrio R. C., Pho N. H., Carter S. L., Schumacher S. E., Tabak B., Hernandez B., Gentry J., Nguyen H., Crenshaw A., Ardlie K., Beroukhim R., Winckler W., Getz G., Gabriel S. B., Meyerson M., Chin L., Park P. J., Kucherlapati R., Hoadley K. A., Todd Auman J., Fan C., Turman Y. J., Shi Y., Li L., Topal M. D., He X., Chao H.-H., Prat A., Silva G. O., Iglesia M. D., Zhao W., Usary J., Berg J. S., Adams M., Booker J., Wu J., Gulabani A., Bodenheimer T., Hoyle A. P., Simons J. V., Soloway M. G., Mose L. E., Jefferys S. R., Balu S., Parker J. S., Neil Hayes D., Perou C. M., Malik S., Mahurkar S., Shen H., Weisenberger D. J., Triche Jr, Lai T. P. H., Bootwalla M. S., Maglinte D. T., Berman B. P., Van Den Berg D. J., Baylin S. B., Laird P. W., Creighton C. J., Donehower L. A., Getz G., Noble M., Voet D., Saksena G., Gehlenborg N., DiCara D., Zhang J., Zhang H., Wu C.-J., Yingchun Liu S., Lawrence M. S., Zou L., Sivachenko A., Lin P., Stojanov P., Jing R., Cho J., Sinha R., Park R. W., Nazaire M.-D., Robinson J., Thorvaldsdottir H., Mesirov J., Park P. J., Chin L., Reynolds S., Kreisberg R. B., Bernard B., Bressler R., Erkkila T., Lin J., Thorsson V., Zhang W., Shmulevich I., Ciriello G., Weinhold N., Schultz N., Gao J., Cerami E., Gross B., Jacobsen A., Sinha R., Arman Aksoy B., Antipin Y., Reva B., Shen R., Taylor B. S., Ladanyi M., Sander C., Anur P., Spellman P. T., Lu Y., Liu W., Verhaak R. R. G., Mills G. B., Akbani R., Zhang N., Broom B. M., Casasent T. D., Wakefield C., Unruh A. K., Baggerly K., Coombes K., Weinstein J. N., Haussler D., Benz C. C., Stuart J. M., Benz S. C., Zhu J., Szeto C. C., Scott G. K., Yau C., Paull E. O., Carlin D., Wong C., Sokolov A., Thusberg J., Mooney S., Ng S., Goldstein T. C., Ellrott K., Grifford M., Wilks C., Ma S., Craft B., Yan C., Hu Y., Meerzaman D., Gastier-Foster J. M., Bowen J., Ramirez N. C., Black A. D., Pyatt R. E., White P., Zmuda E. J., Frick J., Lichtenberg T. M., Brookens R., George M. M., Gerken M. A., Harper H. A., Leraas K. M., Wise L. J., Tabler T. R., McAllister C., Barr T., Hart-Kothari M., Tarvin K., Saller C., Sandusky G., Mitchell C., Iacocca M. V., Brown J., Rabeno B., Czerwinski C., Petrelli N., Dolzhansky O., Abramov M., Voronina O., Potapova O., Marks J. R., Suchorska W. M., Murawa D., Kycler W., Ibbs M., Korski K., Spychała A., Murawa P., Brzeziński J. J., Perz H., Łaźniak, Teresiak R. M., Tatka H., Leporowska E., Bogusz-Czerniewicz M., Malicki J., Mackiewicz A., Wiznerowicz M., Van Le X., Kohl B., Viet Tien N., Thorp R., Van Bang N., Sussman H., Duc Phu B., Hajek R., Phi Hung N., Viet The Phuong T., Quyet Thang H., Zaki Khan K., Penny R., Mallery D., Curley E., Shelton C., Yena P., Ingle J. N., Couch F. J., Lingle W. L., King T. A., Maria Gonzalez-Angulo A., Mills G. B., Dyer M. D., Liu S., Meng X., Patangan M., Waldman F., Stöppler H., Kimryn Rathmell W., Thorne L., Huang M., Boice L., Hill A., Morrison C., Gaudioso C., Bshara W., Daily K., Egea S. C., Pegram M. D., Gomez-Fernandez C., Dhir R., Bhargava R., Brufsky A., Shriver C. D., Hooke J. A., Leigh Campbell J., Mural R. J., Hu H., Somiari S., Larson C., Deyarmin B., Kvecher L., Kovatich A. J., Ellis M. J., King T. A., Hu H., Couch F. J., Mural R. J., Stricker T., White K., Olopade O., Ingle J. N., Luo C., Chen Y., Marks J. R., Waldman F., Wiznerowicz M., Bose R., Chang L.-W., Beck A. H., Maria Gonzalez-Angulo A., Pihl T., Jensen M., Sfeir R., Kahn A., Chu A., Kothiyal P., Wang Z., Snyder E., Pontius J., Ayala B., Backus M., Walton J., Baboud J., Berton D., Nicholls M., Srinivasan D., Raman R., Girshik S., Kigonya P., Alonso S., Sanbhadti R., Barletta S., Pot D., Sheth M., Demchok J. A., Mills Shaw K. R., Yang L., Eley G., Ferguson M. L., Tarnuzzer R. W., Zhang J., Dillon L. A. L., Buetow K., Fielding P., Ozenberger B. A., Guyer M. S., Sofia H. J., and Palchik J. D. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mertins P., Mani D. R., Ruggles K. V., Gillette M. A., Clauser K. R., Wang P., Wang X., Qiao J. W., Cao S., Petralia F., Kawaler E., Mundt F., Krug K., Tu Z., Lei J. T., Gatza M. L., Wilkerson M., Perou C. M., Yellapantula V., Huang K. L., Lin C., McLellan M. D., Yan P., Davies S. R., Townsend R. R., Skates S. J., Wang J., Zhang B., Kinsinger C. R., Mesri M., Rodriguez H., Ding L., Paulovich A. G., Fenyo D., Ellis M. J., and Carr S. A. (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wei L., Jin Z. L., Yang S. J., Xu Y. X., Zhu Y. T., and Ji Y. (2018) TCGA-assembler 2: software pipeline for retrieval and processing of TCGA/CPTAC data. Bioinformatics 34, 1615–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu Y. T., Qiu P., and Ji Y. (2014) TCGA-Assembler: open-source software for retrieving and processing TCGA data. Nat Methods 11, 599–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goldman M., Craft B., Brooks A. N., Zhu J., and Haussler D. (2018) The UCSC Xena Platform for cancer genomics data visualization and interpretation. bioRxiv 326470 [Google Scholar]
- 30. Shao W., Cheng J., Sun L., Han Z., Feng Q., Zhang D., and Huang K. (2018) Ordinal multi-modal feature selection for survival analysis of early-stage renal cancer. Int. Conference Med. Image Computing Computer-Assisted Intervention, pp. 648–656, Springer, New York City [Google Scholar]
- 31. Cheng J., Zhang J., Han Y., Wang X., Ye X., Meng Y., Parwani A., Han Z., Feng Q., and Huang K. (2017) Integrative analysis of histopathological images and genomic data predicts clear cell renal cell carcinoma prognosis. Cancer Res. 77, e91–e100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Buuren S., and Groothuis-oudshoorn K. (2011) mice: Multivariate Imputation by Chained Equations in R. J. Stat. Softw. 45, 1–67 [Google Scholar]
- 33. Chen J., Bardes E. E., Aronow B. J., and Jegga A. G. (2009) ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uhlen M., Zhang C., Lee S., Sjostedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z. T., Edfors F., Sanli K., von Feilitzen K., Oksvold P., Lundberg E., Hober S., Nilsson P., Mattsson J., Schwenk J. M., Brunnstrom H., Glimelius B., Sjoblom T., Edqvist P. H., Djureinovic D., Micke P., Lindskog C., Mardinoglu A., and Ponten F. (2017) A pathology atlas of the human cancer transcriptome. Science 357, pii: eaan2507. [DOI] [PubMed] [Google Scholar]
- 35. Borgan Ø. (2001) Modeling survival data: extending the Cox Model. Statistics Med. 20, 2053–2054 [Google Scholar]
- 36. Kim H. J., Maiti P., and Barrientos A. (2017) Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 47, 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dolfi S. C., Chan L. L.-Y., Qiu J., Tedeschi P. M., Bertino J. R., Hirshfield K. M., Oltvai Z. N., and Vazquez A. (2013) The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metabolism 1, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bhowmick N. A., and Moses H. L. (2005) Tumor-stroma interactions. Curr. Opin. Genet. Dev. 15, 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Savas P., Salgado R., Denkert C., Sotiriou C., Darcy P. K., Smyth M. J., and Loi S. (2016) Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat. Rev. Clin. Oncol. 13, 228–241 [DOI] [PubMed] [Google Scholar]
- 40. Binnewies M., Roberts E. W., Kersten K., Chan V. A.-O., Fearon D. F., Merad M., Coussens L. M., Gabrilovich D. I., Ostrand-Rosenberg S. A.-O., Hedrick C. C., Vonderheide R. H., Pittet M. J., Jain R. K., Zou W., Howcroft T. K., Woodhouse E. C., Weinberg R. A., and Krummel M. A.-O. (2018) Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bonnans C., Chou J., and Werb Z. (2014) Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 5, 786–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Selam B., Kayisli U. A., Garcia-Velasco J. A., and Arici A. (2002) Extracellular matrix-dependent regulation of Fas ligand expression in human endometrial stromal cells. Biol. Reprod. 66, 1–5 [DOI] [PubMed] [Google Scholar]
- 43. Valkenburg K. C., de Groot A. E., and Pienta K. J. (2018) Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 15, 366–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Desmouliere A., Guyot C., and Gabbiani C. (2004) The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int. J. Dev. Biol. 48, 509–517 [DOI] [PubMed] [Google Scholar]
- 45. Latonen L., Afyounian E., Jylha A., Nattinen J., Aapola U., Annala M., Kivinummi K. K., Tammela T. T. L., Beuerman R. W., Uusitalo H., Nykter M., and Visakorpi T. (2018) Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 9, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Edens L. J., White K. H., Jevtic P., Li X. Y., and Levy D. L. (2013) Nuclear size regulation: from single cells to development and disease. Trends Cell Biol. 23, 151–159 [DOI] [PubMed] [Google Scholar]
- 47. Emens L. A. (2018) Breast cancer immunotherapy: facts and hopes. Clin. Cancer Res. 24, 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmid P., Adams S., Rugo H. S., Schneeweiss A., Barrios C. H., Iwata H., Dieras V., Hegg R., Im S. A., Shaw Wright G., Henschel V., Molinero L., Chui S. Y., Funke R., Husain A., Winer E. P., Loi S., and Emens L. A. (2018) Atezolizumab and Nab-Paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used for this study are downloaded from various public sources. Proteomic data were accessed from the NCI CPTAC Data Portal. Histopathology images were downloaded directly through the NCI GDC TCGA Data Portal, whereas transcriptomic data were downloaded from the UCSC Xena data portal (https://xena.ucsc.edu/public-hubs/). Matched RPPA proteomic data were obtained from the Broad GDAC Firehose (https://gdac.broadinstitute.org). cell morphological features and patient survival outcomes, 1,057 BRCA-type breast patients with matched 1057 H&E-stained tissue images and corresponding clinical survival information were used. The patient clinical data were obtained from UCSC Xena. The analysis scripts that we used for this manuscript are available at GitHub: https://github.com/xiaohuizhan/cor_image_omics_BRCA.