

Independent component analysis was applied to human breast cancer proteogenomic data, and pathway-level signatures were further integrated with clinical information. Our results demonstrated that ICA can be used to extract biological relevant signals from multi-omics data in an unsupervised manner.
Keywords: Proteogenomics, Cancer biology, Computational biology, Breast cancer, Mass spectrometry
Graphical Abstract

Highlights
Unsupervised feature extraction from proteogenomics data.
Pathway level integration of multi-omics data based on clinical features.
Abstract
Recent advances in the multi-omics characterization necessitate knowledge integration across different data types that go beyond individual biomarker discovery. In this study, we apply independent component analysis (ICA) to human breast cancer proteogenomics data to retrieve mechanistic information. We show that as an unsupervised feature extraction method, ICA was able to construct signatures with known biological relevance on both transcriptome and proteome levels. Moreover, proteome and transcriptome signatures can be associated by their respective correlation with patient clinical features, providing an integrated description of phenotype-related biological processes. Our results demonstrate that the application of ICA to proteogenomics data could lead to pathway-level knowledge discovery. Potential extension of this approach to other data and cancer types may contribute to pan-cancer integration of multi-omics information.
Breast cancer is the most common cancer among women, and although targeted therapies have helped to significantly reduced breast cancer mortality rate in the past decade, further improvement will require a comprehensive understanding of the molecular mechanisms of the disease (1, 2). Recently, deep mass spectrometry based proteomic characterization of genomically annotated breast cancer samples by the Clinical Proteomic Tumor Analysis Consortium (CPTAC)1 has marked the initial step of a proteogenomic integrative approach, in which recurrent mutations and copy number variations on the genomic level, expression profiles on the transcriptomic level and protein abundance and functional manifestations on proteomic level were measured for the same group of patient samples and examined in the same framework (3–5). The collection of high-quality multi-omics data immediately led to the discovery of concordant gene amplification and protein phosphorylation in key pathways (3). At the same time, there is increasing demand for analytical approaches that could incorporate all data types and extract pathway level signatures. Because in all human patients “-omics” data sets the number of features far exceeds the number of samples, analysis of any single data type is already susceptible to “the curse of dimensionality,” and integration by simple concatenation of multi-omics data would be an even less desirable option. Our previous work has benchmarked the predictive power of multi-omics data sets for classifying breast cancer patients into different survival groups and showed that combined multi-omics data sets produced with data-driven fusion techniques were not able to outperform proteomic data alone (6). This result highlighted the possible redundancy among information contained in different biological levels and motivates us to explore other data fusion techniques that extract both concordant and complementary features from high-dimensional multi-omics data.
In the current study, we applied independent component analysis to proteomic and transcriptomic data of 77 breast cancer samples to extract pathway-level molecular signatures. Independent component analysis (ICA) is an unsupervised learning method widely used in signal processing and has been applied to cancer genomics with notable success (7–9). This approach decomposes the molecular profiles into linear combinations of non-Gaussian independent sources or components, each of which is comprised of weighted contributions from individual genes. Therefore, ICA reduces the dimensionality of original data by representing the molecular profile of each sample as weighted sum of several “meta-genes” or “meta-proteins,” and the weight of specific meta-gene/protein (mixing scores) in one sample reflects the “activity” of that component in the sample. Different from the more conventional dimension reduction method of principal component analysis (PCA), which seeks to find uncorrelated factors that explain the variance among the data, and works the best when the underlying components are normally distributed, ICA are able to discover more informative representations of high-dimensional biological signals, which are usually super-Gaussian and contain more close-to-zero values than a normally-distributed sample (10). As clinical features are also available for the CPTAC samples, molecular signatures can be constructed from clusters of meta-genes/proteins that show activity patterns correlated with these clinical features. Further, taking advantage of a specific clinical feature as an “anchor,” this method may help extract patterns at different biological levels and across different cohorts, which may originate from the same cellular functionality (Fig. 1). The signatures extracted from different data sets were filtered based on their intrinsic stability and association with known clinical features (see Experimental Procedures below) and grouped into modules that showed similar correlation patterns to clinical features. Subsequent gene set enrichment analysis revealed the biological relevance of these modules to pathways such as HER2 signaling, mitosis, and histone modification. Our analysis has demonstrated that ICA was able to blindly extract information in the form of weighted gene combinations, which may be biologically meaningful at pathway level. With input from clinical features or other sample subgrouping indices, these signatures may be further integrated into multilevel models that provide insights into the molecular mechanisms of breast cancer.
Fig. 1.
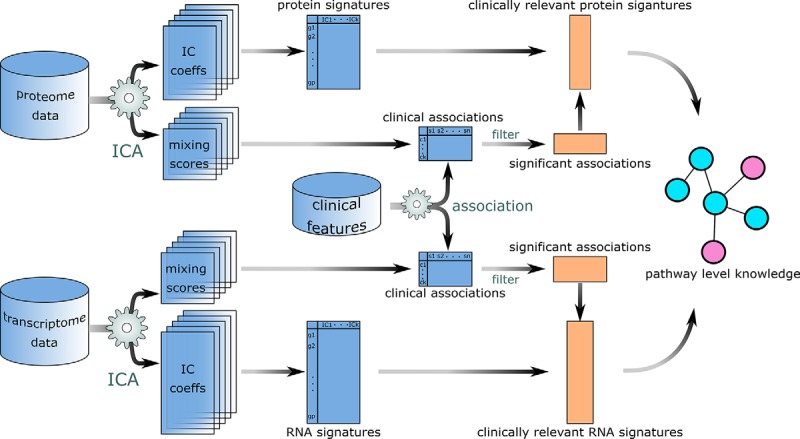
Data analysis workflow. Coefficients of independent components and their corresponding mixing scores are extracted from both proteome and transcriptome data sets for multiple randomly initiated runs. Each independent component is a pathway-level representation. Molecular signatures were identified as cluster centroids of these components, and clinical features were used to select biologically relevant signatures, which were further annotated with pathway analysis.
EXPERIMENTAL PROCEDURES
Data Sets
Transcriptome and proteome data of 77 breast invasive carcinoma (BRCA) patients and 84 ovarian serous cystadenocarcinoma (OV) patients were acquired by the TCGA and CPTAC projects: the transcriptome was characterized using the Agilent mRNA expression array platform (Santa Clara, CA) MS and global protein abundance data were obtained using iTRAQ 4-plex LC-MS/MS (3, 11–13). For OV data, Preprocessing procedure collapsed peptides mapped to the same gene by taking the mean value. Expression levels of genes and abundance of proteins were included in the transcriptome and proteome matrices. Clinical and demographic features were associated with each tumor sample.
Independent Component Analysis
Data were presented in a p × n matrix X with genes in rows and samples in columns. The goal of ICA is to decompose the p × n data matrix into the product of a source matrix S (p × k) and a mixing matrix A (k × n). The ith column of the source matrix represents coefficients of each of the p genes for the ith independent component. The coefficient vector of each component could be considered as p random samples that revealed probability distribution of a specific random variable. The mutual information between any pair of such variables is minimized, and the components are therefore statistically independent. Genes with coefficients of positive or negative values in the same component indicated that their levels may be enhanced or suppressed in the same biological process. The ith row of the mixing matrix represents contributions of the ith component in the source matrix to profiles of each of the n samples. Rows of the mixing matrix therefore record the activity of each components (or meta-genes/meta-proteins) across n samples.
The stochastic nature of most ICA algorithms entails that different randomly initiated runs would give rise to different results, and there is no guarantee that the true structure of the data could be correctly estimated from any single run (10, 14). To assess the statistical reliability of ICA results, stable components were filtered based on an approach adapted from Engreitz et al., which cluster components obtained from different runs with K-medoids method (8). In brief, ICA was run with k = n for 50 times to extract as much information as possible. As the sign of the same component may be flipped during different randomly-initiated runs, we followed the algorithm of Engreitz et al. and took advantage of the fact that the extracted components usually had skewed distributions (different sizes of tails on positive and negative ends) to align flipped components (8). For each individual component, if the larger tail was on the negative side, the component would be flipped to ensure that all larger tails were positive. All 50×n components were then considered as data points in p dimensional space and subjected to K-medoids clustering with Spearman correlation as the dissimilarity measure. For each cluster, the number of different runs that its members were extracted from was documented as a measure for cluster consistency, alongside with the average silhouette width. In general, clusters that with members appeared in more than 50% of all runs (25 out of 50) were considered as likely to contain true biological signals (see Signature Annotation section below).
All computations were carried out on the R platform. Package “fastICA” which implements the iterative FastICA algorithm (15) was used to extract non-Gaussian independent components with logcosh contrast function. Components were subsequently assigned to clusters using the “cluster” package. Clusters were visualized with 2d t-SNE using the R package “tsne.” The number of clusters was determined as equal to number of components extracted at each run of ICA. When the number of samples is small comparing to the number of features, which is usually the case for biological data, it is convenient to retrieve as many as independent signal sources as possible, and the number of components extracted is equal to sample size.
The package “pcaMethods” was used to calculate principal components for comparison with independent components. In permutation tests, for each gene, protein abundance measurements of all samples were randomly shuffled without replacement. One hundred permuted data sets were created to assess the specificity of the ICA method.
Signature Annotation
Component clusters were annotated with GO terms by running Gene Set Enrichment Analysis against centroid coefficients as the pre-ranked gene lists (16, 17). Enrichment map of components were visualized with Cytoscape 3 (18). Each cluster was also associated with clinical features as following: First, 22 clinical features were recoded into ordinal variables (supplemental Table S1). Second, ordinary linear regression models were built with corresponding mixing scores for members in a component cluster to predict each of the ordinal responses. Counts of significant associations between components and clinical features (p value for the slope coefficient < 5.9 × 10−7, Bonferroni correction at α = 0.01 level) were tabulated. Hierarchical clustering with complete linkage was applied to the clinical associations of independent components clusters extracted from both transcriptome and proteome data.
RESULTS
Stable Molecular Signatures Extracted from Proteome and Transcriptome Data
For both the proteome and transcriptome data sets we identified 77 clusters of meta-proteins or meta-genes from independent components obtained from multiple randomly initiated runs. In this study, we chose to run the procedure for 50 times with considerations of both computation time and numeric stability, as further increase the number of runs give rise to similar results (supplemental Fig. S3). Centers of the stable meta-gene and meta-protein clusters, which could be found by averaging gene coefficients within each cluster, could represent pathway-level signatures. The stability of these signatures could be inspected by visualizing all meta-genes and meta-proteins with t-distributed stochastic neighbor embedding (t-SNE), a widely used dimensionality reduction technique (19) (Fig. 2A, 2C, supplemental Fig. S1). A large portion of the meta-gene and meta-protein clusters are compact, whereas the rest formed a visually indistinguishable mixture, consistent with the observation that the average silhouette widths of all clusters followed a bimodal distribution (Fig. 2B, 2D). Another metric that could help evaluate the stability of extracted signatures is the number of different runs that the members of each cluster originated from. It has been proposed that reliable independent components should be highly recurrent, therefore, a cluster the members of which were extracted from different runs would be considered as more stable. Indeed, this metric largely agrees with silhouette width, meaning that compact clusters were most often comprised of meta-proteins or meta-genes extracted from all 50 runs.
Fig. 2.

Evaluation of component consistency. A, C, t-SNE representation of components obtained from 50 randomly initiated runs of ICA workflow on the CPTAC BRCA retrospective data. Colors represent cluster assignments using the partition around medoids algorithm. B, D, average silhouette widths for the 77 clusters were plotted against the number of different individual runs from which the members of each cluster were extracted. Marginal histograms revealed the distribution of the metrics. Large values of individual runs indicate that the cluster is recurrent and larger values of silhouette width indicate that the cluster is more consistent. Both metric could be used to evaluate whether the cluster of components as a signature and more likely to capture true structures in the data.
Mixing Scores Reveal Clinical Relevance of Identified Components
In addition to their numerical stability, the clusters of components representing groups of meta-proteins or meta-genes identified from different ICA runs can also be evaluated with their relevance to known characteristics of breast cancer. For each ICA decomposition, the rows of the mixing score matrices represented the “activity level” of the corresponding independent components in all of the samples. Therefore, it is possible to establish associations between the activity patterns of meta-genes and meta-proteins and clinical features available for TCGA samples, which would in turn reveal the functional relevance of the signatures. We recoded 22 clinical features into ordinal factors and used linear regression to assess their correlation with activity scores of meta-genes or meta-proteins in each signature cluster. To select the most significant associations, Bonferroni correction was applied to control for multiple comparisons at α = 0.01 level, such that a nominal p value of 5.9 × 10−7 was set as the significance threshold (corrected α = 0.01/(77 × 22)). Within each cluster, the percentage of meta-proteins or meta-genes with activity levels that showed significant linear relation with the 22 clinical features was documented. Large portion of significant association between a signature cluster and a clinical feature indicates that the signature may contain pathway-level information about molecular mechanisms underlying the clinical feature. Thirty-three out of the 77 proteome IC clusters showed significant association with 10 clinical features, whereas 60 transcriptome clusters were correlated with 12 clinical features (Fig. 3). Six proteomic signatures and 7 transcriptomic signatures showed strong clinical associations, such that the corresponding mixing scores were significantly correlated with a clinical variable in more than 50% of all runs (counts >25). In contrast, when the same procedure applied to 100 sets of randomly permuted proteome data, no component clusters showed strong clinical associations, as the count of significant linear correlations between mixing scores of a given signature cluster and any of the tested clinical variables was no larger than 2 in all 7700 clusters of the 100 permutation settings.
Fig. 3.

Clinical associations of IC clusters for BRCA proteome and transcriptome data. Percentage of significant associations found between corresponding mixing scores and clinical features for ICs in each cluster extracted from BRCA proteomic and transcriptomic data sets. Only nonzero columns and rows were shown.
To further assess the adequacy and efficacy of the ICA method on the CPTAC breast cancer proteomics data, we compared the results obtained with ICA to that extracted by principal component analysis (PCA), a “standard” and more widely used method of dimension reduction (Fig. 4). With PCA, the BRCA data could be represented by 77 principal components, each of which is a 77-element vector representing scores for the 77 samples. In the context of ICA, because of the stochastic nature of the method, a similar representation was constructed by averaging the activity scores corresponding to the independent component in each of the 77 clusters obtained from the 50 repetitive runs. As shown in Fig. 4, PCA gave rise to signatures that contained mixed information, as the significant associations with clinical features concentrated on Principal Component 2. A total of 6 PCA-extracted signatures showed significant correlation (p < 0.001, linear regression) with one or more clinical variables, whereas 17 ICA-extracted signatures were significant associated with at least one clinical variables. This comparison, consistent with the fact that ICA was designed for “de-mixing” signals of different sources, further demonstrated that ICA could extract more specific information about biological processes underlying high dimensional data.
Fig. 4.

Comparison between PCA and ICA applications on the BRCA proteome data. Significance levels of association between clinical features and dimension-reduced representation of data obtained by PCA and ICA revealed the difference between the two methods. Association between each score and the clinical features are characterized by −log10 p value of linear regression slope coefficient.
The biological relevance of the clusters identified with ICA could be further validated by inspecting the average gene coefficients (the centroids of the clusters). For example, the BRCA proteome signature cluster that contains 29 members that were significantly correlated with the HER2 receptor immuno status and 22 members associated with the Her2 subtype index (pr_02), was heavily weighted on ERBB4 and ERBB2, two tyrosine receptor kinases that mediate the Her2 signaling, and the two proteins were assigned the two largest coefficients (11.13 and 10.68, Table I). Another signature (pr_29) correlated with estrogen receptor (ER) and progesterone receptor (PR) status also exhibited high average scores for PGR (7.73) and ESR1 (4.42) proteins. Interestingly, the signature pr_04, which displayed strong correlation with the Luminal A subtype, is enriched with a group of mitosis-related proteins, which showed low levels of abundance in the Luminal A subtype (Fig. 5). This finding is consistent with the low proliferation level of Luminal A subtype (20). Protein abundance of genes with large weights in the same signatures also showed strong intra-module correlation (supplemental Fig. S2). The aforementioned associations between proteome signatures and known biological processes demonstrated that ICA was able to rediscover clinically relevant information from proteome data in an unsupervised manner.
Table I. Average coefficients of selected BRCA proteome signatures.
| pr_02 |
pr_04 |
pr_29 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive coefficients | Negative coefficients | Positive coefficients | Negative coefficients | Positive coefficients | Negative coefficients | ||||||
| ERBB4 | 11.13 | HBZ | −4.59 | CKS1B | 7.53 | FUCA1 | −4.49 | GFRA1 | 9.42 | TNRC18 | −3.23 |
| ERBB2 | 10.68 | CKB | −4.06 | CDK1 | 7.21 | LTF | −4.25 | PDZK1 | 8.46 | ||
| GRB7 | 10.10 | PIP | −3.96 | PBK | 7.06 | S100A1 | −4.25 | PGR | 7.73 | ||
| MIEN1 | 8.87 | GP2 | −3.91 | UBE2C | 6.75 | FHIT | −3.93 | SCUBE2 | 6.97 | ||
| CASP14 | 8.46 | CKM | −3.56 | ANLN | 6.29 | PPDPF | −3.92 | STC2 | 6.48 | ||
| FABP7 | 8.11 | TNFSF12 | −3.31 | TRIP13 | 6.25 | EFCAB4B | −3.89 | CLEC3A | 5.78 | ||
| PNMT | 6.55 | MAPT | −3.25 | TPX2 | 6.08 | COMP | −3.67 | IGFBP5 | 5.00 | ||
| CLGN | 6.39 | CALB1 | −3.03 | NCAPH | 6.07 | MUC2 | −3.64 | MGP | 4.95 | ||
| AKR1B10 | 6.02 | ECT2 | 5.78 | FABP7 | −3.61 | PKIB | 4.89 | ||||
| PPP1R1B | 5.93 | KIF4B | 5.74 | SUCLA2 | −3.59 | MAPT | 4.84 | ||||
| AKR1B15 | 4.91 | MCM7 | 5.66 | ALDH6A1 | −3.53 | AGR2 | 4.74 | ||||
| ASRGL1 | 4.88 | PRC1 | 5.66 | TNN | −3.38 | AKR7A3 | 4.66 | ||||
| PRODH | 4.71 | MCM6 | 5.51 | C1QTNF3 | −3.37 | AGR3 | 4.51 | ||||
| HMGCS2 | 4.64 | KIF4A | 5.42 | DPP7 | −3.35 | CLIC6 | 4.46 | ||||
| MUCL1 | 4.64 | MCM4 | 5.42 | AZGP1 | −3.34 | IGFBP2 | 4.46 | ||||
| SDR16C5 | 3.96 | NCAPG | 5.34 | ADIRF | −3.24 | ESR1 | 4.42 | ||||
| S100P | 3.91 | ZWILCH | 5.34 | ANO6 | −3.09 | HGD | 4.34 | ||||
| CRISP3 | 3.85 | RACGAP1 | 5.28 | MAMDC2 | −3.02 | CPB1 | 4.34 | ||||
| CRABP1 | 3.72 | TK1 | 5.27 | NAT1 | 4.19 | ||||||
| NCCRP1 | 3.70 | MCM5 | 5.25 | PREX1 | 3.98 | ||||||
| AGR2 | 3.65 | KIFC1 | 5.22 | PEG10 | 3.92 | ||||||
| KRT72 | 3.55 | MCM3 | 5.21 | SYTL4 | 3.82 | ||||||
| STEAP4 | 3.53 | KIF2C | 5.19 | GDAP1 | 3.51 | ||||||
| CDK12 | 3.44 | UHRF1 | 5.18 | SGK3 | 3.31 | ||||||
| ZNF573 | 3.26 | SMC2 | 5.18 | GOLM1 | 3.28 | ||||||
| C19orf33 | 3.25 | UBE2S | 5.10 | NOS1AP | 3.24 | ||||||
| GREM1 | 3.25 | UBE2T | 5.09 | IL6ST | 3.23 | ||||||
| GALNT6 | 3.14 | KIF11 | 5.08 | SEC14L2 | 3.21 | ||||||
| A1CF | 3.10 | SMC4 | 5.05 | BST2 | 3.11 | ||||||
| DHFR | 5.03 | CAP2 | 3.10 | ||||||||
| MCM2 | 5.01 | SEC14L3 | 3.09 | ||||||||
| SPC24 | 4.99 | PLAT | 3.00 | ||||||||
| FANCI | 4.90 | ||||||||||
| TOP2A | 4.89 | ||||||||||
| NCAPD2 | 4.80 | ||||||||||
| CDK3 | 4.79 | ||||||||||
| RRM2 | 4.77 | ||||||||||
| ATAD2 | 4.77 | ||||||||||
| MKI67 | 4.77 | ||||||||||
| HMGCS1 | 4.74 | ||||||||||
| IGFBP5 | 4.71 | ||||||||||
| KIF23 | 4.71 | ||||||||||
| CENPF | 4.59 | ||||||||||
| S100P | 4.54 | ||||||||||
| PLEKHS1 | 4.31 | ||||||||||
| FEN1 | 4.24 | ||||||||||
| MT1M | 4.18 | ||||||||||
| KPNA2 | 4.17 | ||||||||||
| STMN4 | 4.13 | ||||||||||
| CHAF1A | 4.12 | ||||||||||
| TG | 4.03 | ||||||||||
| PHGDH | 4.03 | ||||||||||
| STMN2 | 3.95 | ||||||||||
| RRM1 | 3.93 | ||||||||||
| LIG1 | 3.92 | ||||||||||
| CLEC3A | 3.91 | ||||||||||
| STMN1 | 3.88 | ||||||||||
| DSCC1 | 3.82 | ||||||||||
| NME4 | 3.81 | ||||||||||
| PRIM2 | 3.80 | ||||||||||
| HAT1 | 3.71 | ||||||||||
| NASP | 3.63 | ||||||||||
| GINS2 | 3.61 | ||||||||||
| POLD3 | 3.61 | ||||||||||
| NCAPG2 | 3.60 | ||||||||||
| CHTF18 | 3.58 | ||||||||||
| KNTC1 | 3.46 | ||||||||||
| KRT27 | 3.43 | ||||||||||
| ASNS | 3.42 | ||||||||||
| CHEK2 | 3.42 | ||||||||||
| KIAA1524 | 3.41 | ||||||||||
| WDHD1 | 3.36 | ||||||||||
| PCNA | 3.35 | ||||||||||
| SIGLEC1 | 3.31 | ||||||||||
| MT1X | 3.28 | ||||||||||
| MT2A | 3.26 | ||||||||||
| FAM111B | 3.24 | ||||||||||
| TACC3 | 3.22 | ||||||||||
| MRFAP1 | 3.21 | ||||||||||
| NCAPD3 | 3.20 | ||||||||||
| DHTKD1 | 3.17 | ||||||||||
| EPCAM | 3.15 | ||||||||||
| DUT | 3.14 | ||||||||||
| CETN3 | 3.10 | ||||||||||
| AIM1 | 3.02 | ||||||||||
| TONSL | 3.01 | ||||||||||
Fig. 5.

A BRCA proteome signature enriched with cell cycle related genes is associated with Luminal A subtype. For each signature, genes with coefficient absolute values larger than 3 were identified as important contributors. A, A proteome signature was enriched with genes that play regulatory roles in multiple stages during the cell cycle. B, the abundance of important contributors in the signature in a across 77 samples. Genes with positive (>3) and negative (<−3) coefficients were marked with orange and blue row labels, respectively. The positive contributors of the signature showed consistent low levels of abundance in Luminal A subtype.
Large value coefficients of known marker genes revealed the biological relevance of a subset of the molecular signatures, but more biological information could be exploited on the pathway level. The coefficient vector for each signature is a preranked gene list that can be subjected to gene set enrichment analysis (GSEA) on curated gene sets and GO terms (16), and the collective effect of genes with coefficients of smaller absolute values could contribute to the enrichment metric. GSEA results showed that the clinically related proteome IC clusters retrieved a lot of the breast cancer related gene sets determined by experimental manipulations, as well as gene sets that characterized basic biological processes (Table II). For example, the proteome signatures pr_02 and pr_29, which were strongly associated with Her2 and ER status, also exhibited enrichment of the corresponding gene sets (Table II). The transcriptome signature tx_07, which were correlated with Basal subtype, ER and PR status, showed negative enrichment of targets of LSD1, a histone demethylase of H3K4 sites, which had known links to be poor breast cancer prognostics and ER negative status (21, 22). Interestingly, the same signature cluster also exhibited positive enrichment of genes with H3K27Me3 sites, indicating that epigenetic regulation is highly orchestrated in breast cancer.
Table II. GO terms and curated gene sets enrichment of selected BRCA signatures.
| Top enriched GO terms |
Top enriched curated sets |
|||
|---|---|---|---|---|
| Negative | Positive | Negative | Positive | |
| Clinically relevant signatures (significant total count >25) | ||||
| pr_02 | GO_MYOSIN_II_COMPLEX | GO_ERBB2_SIGNALING_PATHWAY | REACTOME_RESPIRATORY_ELECTRON_TRANSPORT | SMID_BREAST_CANCER_ERBB2_UP |
| pr_04 | GO_BRANCHED_CHAIN_AMINO_ACID_METABOLIC_PROCESS | GO_DNA_REPLICATION | WOO_LIVER_CANCER_RECURRENCE_DN | DUTERTRE_ESTRADIOL_RESPONSE_24HR_UP |
| pr_18 | GO_CILIARY_TIP | GO_OXIDOREDUCTASE_ACTIVITY_ACTING_ON_THE_CH_OH_GROUP_OF_DONORS_NAD_OR_NADP_AS_ACCEPTOR | SMID_BREAST_CANCER_LUMINAL_B_UP | REACTOME_GLUCOSE_METABOLISM |
| pr_29 | GO_MITOCHONDRIAL_TRANSLATION | GO_ANCHORED_COMPONENT_OF_MEMBRANE | REACTOME_LIPOPROTEIN_METABOLISM | DOANE_BREAST_CANCER_ESR1_UP |
| pr_48 | GO_ACTIN_MYOSIN_FILAMENT_SLIDING | GO_CELLULAR_MONOVALENT_INORGANIC_CATION_HOMEOSTASIS | REACTOME_RESPIRATORY_ELECTRON_TRANSPORT_ATP_SYNTHESIS_BY_CHEMIOSMOTIC_COUPLING_AND_HEAT_PRODUCTION_BY_UNCOUPLING_PROTEINS_ | SMID_BREAST_CANCER_BASAL_UP |
| pr_49 | GO_MICROBODY_LUMEN | GO_RIBOSOME_BIOGENESIS | SMID_BREAST_CANCER_BASAL_DN | CAIRO_HEPATOBLASTOMA_CLASSES_UP |
| tx_07 | GO_CYTOSOLIC_LARGE_RIBOSOMAL_SUBUNIT | GO_EMBRYONIC_HINDLIMB_MORPHOGENESIS | WANG_LSD1_TARGETS_DN | MIKKELSEN_NPC_HCP_WITH_H3K27ME3 |
| tx_19 | GO_NECROPTOTIC_PROCESS | GO_OLIGODENDROCYTE_DIFFERENTIATION | KOHOUTEK_CCNT1_TARGETS | NIKOLSKY_BREAST_CANCER_17Q11_Q21_AMPLICON |
| tx_21 | GO_RESPONSE_TO_PH | GO_STEROID_BINDING | SMID_BREAST_CANCER_BASAL_UP | DOANE_BREAST_CANCER_ESR1_UP |
| tx_31 | GO_VASODILATION | GO_MHC_PROTEIN_COMPLEX | BIOCARTA_NO1_PATHWAY | ICHIBA_GRAFT_VERSUS_HOST_DISEASE_D7_DN |
| tx_34 | GO_VOLTAGE_GATED_CALCIUM_CHANNEL_COMPLEX | GO_MUSCLE_CELL_MIGRATION | VANTVEER_BREAST_CANCER_METASTASIS_UP | LIN_SILENCED_BY_TUMOR_MICROENVIRONMENT |
| tx_40 | GO_REGULATION_OF_ENDOCRINE_PROCESS | GO_EPIDERMIS_DEVELOPMENT | NIKOLSKY_BREAST_CANCER_12Q13_Q21_AMPLICON | TURASHVILI_BREAST_DUCTAL_CARCINOMA_VS_DUCTAL_NORMAL_DN |
| tx_46 | GO_NEGATIVE_REGULATION_OF_CALCIUM_ION_TRANSPORT | GO_CHROMOSOME_SEGREGATION | SOTIRIOU_BREAST_CANCER_GRADE_1_VS_3_DN | DUTERTRE_ESTRADIOL_RESPONSE_24HR_UP |
| tx_63 | GO_CALCIUM_DEPENDENT_CELL_CELL_ADHESION_VIA_PLASMA_MEMBRANE_CELL_ADHESION_MOLECULES | GO_CILIUM_ORGANIZATION | NIKOLSKY_BREAST_CANCER_16Q24_AMPLICON | CREIGHTON_ENDOCRINE_THERAPY_RESISTANCE_2 |
| tx_64 | GO_INNER_EAR_RECEPTOR_CELL_DEVELOPMENT | GO_POSITIVE_REGULATION_OF_INTERLEUKIN_2_PRODUCTION | PID_WNT_SIGNALING_PATHWAY | TSUNODA_CISPLATIN_RESISTANCE_DN |
| tx_72 | GO_PROTEIN_LIPID_COMPLEX | GO_NEUROEPITHELIAL_CELL_DIFFERENTIATION | FARMER_BREAST_CANCER_CLUSTER_3 | MASSARWEH_TAMOXIFEN_RESISTANCE_DN |
| Consistent signatures (cluster silhouette >0.9) | ||||
| pr_33 | GO_NUCLEAR_TRANSCRIBED_MRNA_CATABOLIC_PROCESS_NONSENSE_MEDIATED_DECAY | GO_IRON_ION_BINDING | REACTOME_SRP_DEPENDENT_COTRANSLATIONAL_PROTEIN_TARGETING_TO_MEMBRANE | JAATINEN_HEMATOPOIETIC_STEM_CELL_DN |
| pr_45 | GO_TRANSLATIONAL_TERMINATION | GO_LYMPHOCYTE_ACTIVATION | CHARAFE_BREAST_CANCER_LUMINAL_VS_MESENCHYMAL_UP | WALLACE_PROSTATE_CANCER_RACE_UP |
| pr_15 | GO_VOLTAGE_GATED_ION_CHANNEL_ACTIVITY | GO_BLOOD_MICROPARTICLE | REACTOME_PERK_REGULATED_GENE_EXPRESSION | HSIAO_LIVER_SPECIFIC_GENES |
| pr_25 | GO_DNA_PACKAGING_COMPLEX | GO_CONTRACTILE_FIBER | ABRAMSON_INTERACT_WITH_AIRE | REACTOME_MUSCLE_CONTRACTION |
| pr_61 | GO_TUBE_FORMATION | GO_LIPID_PARTICLE | NIKOLSKY_BREAST_CANCER_17Q11_Q21_AMPLICON | NAKAYAMA_SOFT_TISSUE_TUMORS_PCA2_DN |
| pr_08 | GO_SNRNA_METABOLIC_PROCESS | GO_NUCLEAR_OUTER_MEMBRANE_ENDOPLASMIC_RETICULUM_MEMBRANE_NETWORK | KEGG_RNA_DEGRADATION | MILI_PSEUDOPODIA_HAPTOTAXIS_DN |
| pr_54 | GO_FATTY_ACID_BINDING | GO_DEFENSE_RESPONSE_TO_VIRUS | REACTOME_PROCESSING_OF_CAPPED_INTRON_CONTAINING_PRE_MRNA | TAKEDA_TARGETS_OF_NUP98_HOXA9_FUSION_3D_UP |
| pr_63 | GO_GOLGI_VESICLE_TRANSPORT | GO_TRANSFERASE_ACTIVITY_TRANSFERRING_ALKYL_OR_ARYL_OTHER_THAN_METHYL_GROUPS | LEE_LIVER_CANCER_CIPROFIBRATE_UP | KEGG_DRUG_METABOLISM_CYTOCHROME_P450 |
| pr_04 | GO_BRANCHED_CHAIN_AMINO_ACID_METABOLIC_PROCESS | GO_DNA_REPLICATION | WOO_LIVER_CANCER_RECURRENCE_DN | DUTERTRE_ESTRADIOL_RESPONSE_24HR_UP |
| pr_31 | GO_PHOSPHOLIPID_BIOSYNTHETIC_PROCESS | GO_DNA_PACKAGING | KEGG_DRUG_METABOLISM_OTHER_ENZYMES | WAMUNYOKOLI_OVARIAN_CANCER_GRADES_1_2_DN |
| pr_39 | GO_ORGANELLAR_LARGE_RIBOSOMAL_SUBUNIT | GO_NUCLEAR_TRANSCRIBED_MRNA_CATABOLIC_PROCESS_NONSENSE_MEDIATED_DECAY | ZHANG_RESPONSE_TO_IKK_INHIBITOR_AND_TNF_UP | REACTOME_PEPTIDE_CHAIN_ELONGATION |
| tx_46 | GO_NEGATIVE_REGULATION_OF_CALCIUM_ION_TRANSPORT | GO_CHROMOSOME_SEGREGATION | SOTIRIOU_BREAST_CANCER_GRADE_1_VS_3_DN | DUTERTRE_ESTRADIOL_RESPONSE_24HR_UP |
| tx_48 | GO_BICARBONATE_TRANSPORT | GO_PROTEINACEOUS_EXTRACELLULAR_MATRIX | MOOTHA_HUMAN_MITODB_6_2002 | ANASTASSIOU_CANCER_MESENCHYMAL_TRANSITION_SIGNATURE |
| tx_43 | GO_NUCLEOTIDE_PHOSPHORYLATION | GO_ADAPTIVE_IMMUNE_RESPONSE | SMID_BREAST_CANCER_LUMINAL_B_UP | SMID_BREAST_CANCER_NORMAL_LIKE_UP |
| tx_28 | GO_TRANSLATIONAL_INITIATION | GO_SODIUM_CHANNEL_ACTIVITY | HSIAO_HOUSEKEEPING_GENES | GINESTIER_BREAST_CANCER_20Q13_AMPLIFICATION_DN |
| tx_52 | GO_CYTOSOLIC_RIBOSOME | GO_DEFENSE_RESPONSE_TO_VIRUS | REACTOME_PEPTIDE_CHAIN_ELONGATION | HECKER_IFNB1_TARGETS |
| tx_49 | GO_RIBOSOMAL_SUBUNIT | GO_HELICASE_ACTIVITY | REACTOME_PEPTIDE_CHAIN_ELONGATION | GABRIELY_MIR21_TARGETS |
| tx_30 | GO_REGULATION_OF_MESENCHYMAL_CELL_PROLIFERATION | GO_INFLAMMATORY_RESPONSE | SENGUPTA_NASOPHARYNGEAL_CARCINOMA_WITH_LMP1_DN | MCLACHLAN_DENTAL_CARIES_UP |
| tx_63 | GO_CALCIUM_DEPENDENT_CELL_CELL_ADHESION_VIA_PLASMA_MEMBRANE_CELL_ADHESION_MOLECULES | GO_CILIUM_ORGANIZATION | NIKOLSKY_BREAST_CANCER_16Q24_AMPLICON | CREIGHTON_ENDOCRINE_THERAPY_RESISTANCE_2 |
| tx_23 | GO_POSITIVE_REGULATION_OF_MESENCHYMAL_CELL_PROLIFERATION | GO_LIPID_PARTICLE | MUELLER_METHYLATED_IN_GLIOBLASTOMA | NAKAYAMA_SOFT_TISSUE_TUMORS_PCA2_DN |
Although the most clinically relevant clusters (total count of significant association >25) contained proteome and transcriptome signatures that are highly recurrent, these clusters were not very homogeneous (supplemental Table S2). On the other hand, out of the 20 most consistent clusters with average silhouette widths larger than 0.9 and are recurrent in every run for both proteome and transcriptome, only 3 signatures (pr_04, tx_46, tx_63) showed direct clinical relevance. The meta-proteins and meta-genes in the clusters of pr_04 and tx_46 were both significantly associated with Luminal A subtype index and showed enrichment of genes involved in estrogen response (Table II). The signature tx_63 was associated with the Basal subtype and ER status, and its coefficients exhibited significant negative enrichment of genes in 16q24 loci (p < 0.001, FDR < 0.001), a breast cancer risk factor, as well as positive enrichment of genes involved in endocrine therapy response (p < 0.001, FDR < 0.001). The large extent of disagreement between clinical relevance and cluster consistency suggest that ICA may serve as a promising approach to knowledge discovery, as stable clusters that did not show clinical relevance may provide new insights into the molecular mechanisms of breast cancer.
Integrative Pathway Analysis of Proteomic and Transcriptomic Components Guided by Clinical Features
The correlation between mixing scores and clinical features provided a valuable opportunity to find the link between independently extracted components from different data types. For example, in both BRCA proteome and transcriptome analyses, there was a highly consistent signature (pr_04 and tx_46), such that meta-proteins and meta-genes in the two clusters were recurrent in all 50 randomly initiated runs and the corresponding mixing scores all showed significant correlation with index of Luminal A subtype (Figs. 2 and 3). To integrate the proteomic and transcriptomic information with guidance from clinical relevance, we applied the hierarchical clustering algorithm to the vectors of clinical association counts for the most clinically relevant meta-protein and meta-gene clusters. Proteome and transcriptome signatures could therefore be grouped based on their similarity in functional indications, as the metric of direct correlation between gene coefficients of different signatures was limited by noise introduced by high dimensionality (supplemental Fig. S4). Combined network visualization of GO terms enriched in proteome and transcriptome signatures allowed us to examine complementary functional modules on different biological levels. For example, the pr_04 and tx_46 clusters may characterize Luminal A subtype specific processes from protein and RNA perspectives (Fig. 6B). Although majority of the gene set networks consisted of nodes and edges common between proteome and transcriptome, specific networks could be identified for both data modes, with tx_46 showed specific enrichment in a DNA replication-related network, and pr_04 showed specific enrichment in polysaccharide metabolism (Fig. 6C).
Fig. 6.

Integrative analysis of BRCA proteome and transcriptome signatures. Only clinically relevant IC clusters are shown (total count of significant associations >0). A, hierarchical clustering of the clinical association of all signatures. B, GSEA enrichment map of the combined network of cluster centers of pr_04 and tx_46 indicated in A. C, zoomed-in view of a transcriptome-specific (top) and a proteome specific subnetwork highlighted in B. Nodes represent the curated gene sets are colored according to the significance level (1-p value) and the direction of enrichment (positive as red and negative as blue). Displayed gene sets are filtered by a p value cutoff of 0.005 and a FDR cutoff of 0.1. The fill and outline of nodes exhibit enrichment of the proteomic signature and the transcriptomic signature, respectively, and the size of the nodes represents number of genes in the gene set. Thickness of the purple and cyan edges represents the number of shared genes between transcriptomic and proteomic nodes with significant enrichment.
To further validate the ICA workflow, we apply the method to proteomic data of the ovarian cancer CPTAC cohort (4). Signatures found in the data showed association with previously reported transcriptome-based subtypes (supplemental Fig. S5C) (23). Moreover, there were strong correlations between ICA-extracted proteome signatures and protein modules derived with weighted correlation network analysis (supplemental Fig. S5D) (4, 24). To further highlight the potential of ICA workflow as an integrative method, we explored pan-cancer proteomic features by examining the correlation between ICA-extracted signatures obtained from breast cancer and ovarian cancer data sets. Between the 77 breast cancer proteome signatures (Fig. 2–3) and the 84 ovarian cancer proteome signatures (supplemental Fig. S5), four pairs of highly correlated signatures were identified (Spearman correlation larger than 0.2) (supplemental Fig. S6). Notably, the shared signature pairs include BRCA_IC_004/OV_IC_051, which was previously annotated to correlate with Luminal A subtype and show enrichments of mitosis-related genes (Fig. 5). The share signature pair BRCA_IC_014/OV_IC_004, which was associated with the Mesenchymal subtype of ovarian cancer (supplemental Fig. S6), showed consistent enrichments in extracellular-matrix related genes. Taken together, our results demonstrated the ICA workflow could consistently extract biological relevant information from multi-omics data sets.
DISCUSSION
We have used independent component analysis to gain insights into the mechanisms of breast cancer and develop protein/gene modules as clinical signatures. Meta-proteins and meta-genes are extracted blindly from the data, and signatures are further selected based on consistency of meta-gene/protein clusters or the association between their activity scores and known clinical features. Gene set annotation revealed that several selected signatures contained biologically relevant information. A proteome signature (pr_02) characterizing strong activation of the Her2 pathway was recovered as a Her2-related meta-protein cluster. On the transcriptome level, a meta-gene enriched for genes in the 16q24 risk loci (tx_63) formed a stable signature that showed correlation with both ER status and Basal subtype. Stable signatures on both proteome and transcriptome levels (pr_04 and tx_46) were found to be heavily associated with the Luminal A index, suggesting that cell division and growth is specifically regulated in this subtype.
As an unsupervised blind source separation method, independent component analysis has been applied to multiple biological data types. Consistent with previous reports, our results demonstrated that as an unsupervised learning method, ICA can extract biological meaningful information solely based on the intrinsic structures of the transcriptome and proteome data. Because of the underlying assumptions, this method would only yield satisfactory results when the “true” signals in the data do not follow Gaussian distribution, and it would fail to separate any mixture of normally distributed sources. Successful application in the current use case suggested that ICA have captured some aspects of the mechanistic processes underlying proteome and transcriptome profiles. It is reasonable to assume that the observed RNA and protein levels are the sum of several up-regulation and down-regulation modules, in which the distribution of individual gene levels deviate radically from the normal distribution that characterizes a noisy background.
In the current use case, we have designated the number of components to be equal to the number of samples, as is usually assumed in blind source separation applications. It remains an open question whether all extracted signatures are reflective of some “true” biological processes. Moreover, the stochastic nature of the algorithm entails that, although all components were found simultaneously in each run without any component “privileged” over others (25), there is no guarantee that all solutions found at local optima are practically useful features. Indeed, the fact that a lot of the extracted components were neither consistent nor recurrent in multiple runs suggested that they may arise from noise instead of stable signals. However, previous applications of ICA on biological data have showed that overestimating the number of independent sources gave rise to signatures driven by smaller gene groups without affecting the rediscovery of the most stable components, while underestimating the number of sources compromised signal detection (26). It is possible that assuming the number of independent sources to be equal to the number of samples may result in noisy signals that are “unnecessary.” However, in an exploratory analysis setting where the number of samples is relatively small, this problem could be remedied by inspecting the stability and biological relevance of extracted signatures post hoc and extracting more components may lead to better separation of signals (supplemental Fig. S3). In the case of large sample size, the number of components will have to be determined by heuristics, and different choices may be compared in downstream analyses.
Using clinical association (total significant associations >25 across all tested clinical variables) or signature stability (cluster silhouette >0.9) as selection criterion gave rise to two lists of potential signatures (Table II) with only a few overlapping members (pr_04, tx_46, tx_63), suggesting that the most clinically relevant signatures are not very stable under the current method. Other clustering method such as density-based clustering may be used to improve the estimation of stable signatures (14, 27). On the other hand, it is possible that the most stable signatures described the housekeeping processes common in all samples, but they may also help reveal novel molecular mechanisms of breast cancer that are not previously linked to any phenotype. As a feature construction procedure, ICA could also facilitate knowledge integration from multiple data types. In addition to the integration of proteome and transcriptome signatures as demonstrated in this work, future studies could further apply ICA on omics data sets of multiple cancer types. Extracted molecular signatures may be grouped in another round of clustering procedure to reveal pan-cancer and cancer type specific mechanisms.
Data Availability
Proteome data of BRCA and OV are available at the CPTAC data portal (https://cptac-data-portal.georgetown.edu/cptac/s/S015; https://cptac-data-portal.georgetown.edu/cptac/s/S020). Transcriptome data are available at the NCI Genomics Data Commons (https://portal.gdc.cancer.gov) under project name TCGA-BRCA and TCGA-OV. Data tables are also available at the publication page (https://www.nature.com/articles/nature18003; https://www.nature.com/articles/nature10166).
Supplementary Material
Footnotes
* We would like to acknowledge funding by the National Cancer Institute (NCI) through CPTAC award U24 CA210972 and a contract 13XS068 from Leidos Biomedical Research, Inc., and by a grant from the Shifrin-Myers Breast Cancer Discovery Fund.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- CPTAC
- Clinical Proteomic Tumor Analysis Consortium
- ICA
- independent component analysis
- PCA
- principal component analysis
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- FDR
- false discovery rate
- TCGA
- The Cancer Genome Atlas
- BRCA
- breast invasive carcinoma
- OV
- ovarian serous cystadenocarcinoma.
REFERENCES
- 1. Narod S. A., Iqbal J., and Miller A. B. (2015) Why have breast cancer mortality rates declined? J. Cancer Policy 5, 8–17 [Google Scholar]
- 2. Mendes D., Alves C., Afonso N., Cardoso F., Passos-Coelho J. L., Costa L., Andrade S., and Batel-Marques F. (2015) The benefit of HER2-targeted therapies on overall survival of patients with metastatic HER2-positive breast cancer – a systematic review. Breast Cancer Res. 17, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mertins P., Mani D. R., Ruggles K. V., Gillette M. A., Clauser K. R., Wang P., Wang X., Qiao J. W., Cao S., Petralia F., Kawaler E., Mundt F., Krug K., Tu Z., Lei J. T., Gatza M. L., Wilkerson M., Perou C. M., Yellapantula V., Huang K., Lin C., McLellan M. D., Yan P., Davies S. R., Townsend R. R., Skates S. J., Wang J., Zhang B., Kinsinger C. R., Mesri M., Rodriguez H., Ding L., Paulovich A. G., Fenyö D., Ellis M. J., Carr S. A., and NCI CPTAC. (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang H., Liu T., Zhang Z., Payne S. H., Zhang B., McDermott J. E., Zhou J.-Y., Petyuk V. A., Chen L., Ray D., Sun S., Yang F., Chen L., Wang J., Shah P., Cha S. W., Aiyetan P., Woo S., Tian Y., Gritsenko M. A., Clauss T. R., Choi C., Monroe M. E., Thomas S., Nie S., Wu C., Moore R. J., Yu K.-H., Tabb D. L., Fenyö D., Bafna V., Wang Y., Rodriguez H., Boja E. S., Hiltke T., Rivers R. C., Sokoll L., Zhu H., Shih I.-M., Cope L., Pandey A., Zhang B., Snyder M. P., Levine D. A., Smith R. D., Chan D. W., Rodland K. D., CPTAC Investigators SA, Gillette M. A., Klauser K. R., Kuhn E., Mani D. R., Mertins P., Ketchum K. A., Thangudu R., Cai S., Oberti M., Paulovich A. G., Whiteaker J. R., Edwards N. J., McGarvey P. B., Madhavan S., Wang P., Chan D. W., Pandey A., Shih I.-M., Zhang H., Zhang Z., Zhu H., Cope L., Whiteley G. A., Skates S. J., White F. M., Levine D. A., Boja E. S., Kinsinger C. R., Hiltke T., Mesri M., Rivers R. C., Rodriguez H., Shaw K. M., Stein S. E., Fenyo D., Liu T., McDermott J. E., Payne S. H., Rodland K. D., Smith R. D., Rudnick P., Snyder M., Zhao Y., Chen X., Ransohoff D. F., Hoofnagle A. N., Liebler D. C., Sanders M. E., Shi Z., Slebos R. J. C., Tabb D. L., Zhang B., Zimmerman L. J., Wang Y., Davies S. R., Ding L., Ellis M. J. C., and Townsend R. R. (2016) Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M. C., Zimmerman L. J., Shaddox K. F., Kim S., Davies S. R., Wang S., Wang P., Kinsinger C. R., Rivers R. C., Rodriguez H., Townsend R. R., Ellis M. J. C., Carr S. A., Tabb D. L., Coffey R. J., Slebos R. J. C., Liebler D. C., NCI CPTAC S. A., Gillette MA, Klauser K. R., Kuhn E., Mani D. R., Mertins P., Ketchum K. A., Paulovich A. G., Whiteaker J. R., Edwards N. J., McGarvey P. B., Madhavan S., Wang P., Chan D., Pandey A., Shih I.-M., Zhang H., Zhang Z., Zhu H., Whiteley G. A., Skates S. J., White F. M., Levine D. A., Boja E. S., Kinsinger C. R., Hiltke T., Mesri M., Rivers R. C., Rodriguez H., Shaw K. M., Stein S. E., Fenyo D., Liu T., McDermott J. E., Payne S. H., Rodland K. D., Smith R. D., Rudnick P., Snyder M., Zhao Y., Chen X., Ransohoff D. F., Hoofnagle A. N., Liebler D. C., Sanders M. E., Shi Z., Slebos R. J. C., Tabb D. L., Zhang B., Zimmerman L. J., Wang Y., Davies S. R., Ding L., Ellis M. J. C., and Reid Townsend R. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma S., Ren J., and Fenyö D. (2016) Breast cancer prognostics using multi-omics data. AMIA Jt. Summits Transl. Sci. Proc. 2016, 52–59 [PMC free article] [PubMed] [Google Scholar]
- 7. Frigyesi A., Veerla S., Lindgren D., and Höglund M. (2006) Independent component analysis reveals new and biologically significant structures in micro array data. BMC Bioinformatics 7, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engreitz J. M., Daigle B. J., Marshall J. J., and Altman R. B. (2010) Independent component analysis: mining microarray data for fundamental human gene expression modules. J. Biomed. Inform. 43, 932–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biton A., Bernard-Pierrot I., Lou Y., Krucker C., Chapeaublanc E., Rubio-Pérez C., López-Bigas N., Kamoun A., Neuzillet Y., Gestraud P., Grieco L., Rebouissou S., de Reyniès A., Benhamou S., Lebret T., Southgate J., Barillot E., Allory Y., Zinovyev A., and Radvanyi F. (2014) Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 9, 1235–1245 [DOI] [PubMed] [Google Scholar]
- 10. Hyvärinen A. (2012) Independent component analysis: recent advances. Philos. Trans. R. Soc. London A Math. Phys. Eng. Sci. 371, 20110534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koboldt D. C., Fulton R. S., McLellan M. D., Schmidt H., Kalicki-Veizer J., McMichael J. F., Fulton L. L., Dooling D. J., Ding L., Mardis E. R., Wilson R. K., Ally A., Balasundaram M., Butterfield Y. S. N., Carlsen R., Carter C., Chu A., Chuah E., Chun H.-J. E., Coope R. J. N., Dhalla N., Guin R., Hirst C., Hirst M., Holt R. A., Lee D., Li H. I., Mayo M., Moore R. A., Mungall A. J., Pleasance E., Gordon Robertson A., Schein J. E., Shafiei A., Sipahimalani P., Slobodan J. R., Stoll D., Tam A., Thiessen N., Varhol R. J., Wye N., Zeng T., Zhao Y., Birol I., Jones S. J. M., Marra M. A., Cherniack A. D., Saksena G., Onofrio R. C., Pho N. H., Carter S. L., Schumacher S. E., Tabak B., Hernandez B., Gentry J., Nguyen H., Crenshaw A., Ardlie K., Beroukhim R., Winckler W., Getz G., Gabriel S. B., Meyerson M., Chin L., Park P. J., Kucherlapati R., Hoadley K. A., Todd Auman J., Fan C., Turman Y. J., Shi Y., Li L., Topal M. D., He X., Chao H.-H., Prat A., Silva G. O., Iglesia M. D., Zhao W., Usary J., Berg J. S., Adams M., Booker J., Wu J., Gulabani A., Bodenheimer T., Hoyle A. P., Simons J. V., Soloway M. G., Mose L. E., Jefferys S. R., Balu S., Parker J. S., Neil Hayes D., Perou C. M., Malik S., Mahurkar S., Shen H., Weisenberger D. J., Triche Jr, Lai T. P. H., Bootwalla M. S., Maglinte D. T., Berman B. P., Van Den Berg D. J., Baylin S. B., Laird P. W., Creighton C. J., Donehower L. A., Getz G., Noble M., Voet D., Saksena G., Gehlenborg N., DiCara D., Zhang J., Zhang H., Wu C.-J., Yingchun Liu S., Lawrence M. S., Zou L., Sivachenko A., Lin P., Stojanov P., Jing R., Cho J., Sinha R., Park R. W., Nazaire M.-D., Robinson J., Thorvaldsdottir H., Mesirov J., Park P. J., Chin L., Reynolds S., Kreisberg R. B., Bernard B., Bressler R., Erkkila T., Lin J., Thorsson V., Zhang W., Shmulevich I., Ciriello G., Weinhold N., Schultz N., Gao J., Cerami E., Gross B., Jacobsen A., Sinha R., Arman Aksoy B., Antipin Y., Reva B., Shen R., Taylor B. S., Ladanyi M., Sander C., Anur P., Spellman P. T., Lu Y., Liu W., Verhaak R. R. G., Mills G. B., Akbani R., Zhang N., Broom B. M., Casasent T. D., Wakefield C., Unruh A. K., Baggerly K., Coombes K., Weinstein J. N., Haussler D., Benz C. C., Stuart J. M., Benz S. C., Zhu J., Szeto C. C., Scott G. K., Yau C., Paull E. O., Carlin D., Wong C., Sokolov A., Thusberg J., Mooney S., Ng S., Goldstein T. C., Ellrott K., Grifford M., Wilks C., Ma S., Craft B., Yan C., Hu Y., Meerzaman D., Gastier-Foster J. M., Bowen J., Ramirez N. C., Black A. D., Pyatt RE., White P, Zmuda E. J., Frick J., Lichtenberg T. M., Brookens R., George M. M., Gerken M. A., Harper H. A., Leraas K. M., Wise L. J., Tabler T. R., McAllister C., Barr T., Hart-Kothari M., Tarvin K., Saller C., Sandusky G., Mitchell C., Iacocca M. V., Brown J., Rabeno B., Czerwinski C., Petrelli N., Dolzhansky O., Abramov M., Voronina O., Potapova O., Marks J. R., Suchorska W. M., Murawa D., Kycler W., Ibbs M., Korski K., Spychała A., Murawa P., Brzeziński J. J., Perz H., Łaźniak R., Teresiak M., Tatka H., Leporowska E., Bogusz-Czerniewicz M., Malicki J., Mackiewicz A., Wiznerowicz M., Van Le X., Kohl B., Viet Tien N., Thorp R., Van Bang N., Sussman H., Duc Phu B., Hajek R., Phi Hung N., Viet The Phuong T., Quyet Thang H., Zaki Khan K., Penny R., Mallery D., Curley E., Shelton C., Yena P., Ingle J. N., Couch F. J., Lingle W. L., King T. A., Maria Gonzalez-Angulo A., Mills G. B., Dyer M. D., Liu S., Meng X., Patangan M., Waldman F., Stöppler H., Kimryn Rathmell W., Thorne L., Huang M., Boice L., Hill A., Morrison C., Gaudioso C., Bshara W., Daily K., Egea S. C., Pegram M. D., Gomez-Fernandez C., Dhir R., Bhargava R., Brufsky A., Shriver C. D., Hooke J. A., Leigh Campbell J., Mural R. J., Hu H., Somiari S., Larson C., Deyarmin B., Kvecher L., Kovatich A. J., Ellis M. J., King T. A., Hu H., Couch F. J., Mural R. J., Stricker T., White K., Olopade O., Ingle J. N., Luo C., Chen Y., Marks J. R., Waldman F., Wiznerowicz M., Bose R., Chang L.-W., Beck A. H., Maria Gonzalez-Angulo A., Pihl T., Jensen M., Sfeir R., Kahn A., Chu A., Kothiyal P., Wang Z., Snyder E., Pontius J., Ayala B., Backus M., Walton J., Baboud J., Berton D., Nicholls M., Srinivasan D., Raman R., Girshik S., Kigonya P., Alonso S., Sanbhadti R., Barletta S., Pot D., Sheth M., Demchok J. A., Mills Shaw K. R., Yang L., Eley G., Ferguson M. L., Tarnuzzer R. W., Zhang J., Dillon L. A. L., Buetow K., Fielding P., Ozenberger B. A., Guyer M. S., Sofia H. J., and Palchik J. D. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang H., Liu T., Zhang Z., Payne S. H., Zhang B., McDermott J. E., Zhou J.-Y., Petyuk V. A., Chen L., Ray D., Sun S., Yang F., Chen L., Wang J., Shah P., Cha S. W., Aiyetan P., Woo S., Tian Y., Gritsenko M. A., Clauss T. R., Choi C., Monroe M. E., Thomas S., Nie S., Wu C., Moore R. J., Yu K.-H., Tabb D. L., Fenyö D., Bafna V., Wang Y., Rodriguez H., Boja E. S., Hiltke T., Rivers R. C., Sokoll L., Zhu H., Shih I.-M., Cope L., Pandey A., Zhang B., Snyder M. P., Levine D. A., Smith R. D., Chan D. W., Rodland K. D., Carr S. A., Gillette M. A., Klauser K. R., Kuhn E., Mani D. R., Mertins P., Ketchum K. A., Thangudu R., Cai S., Oberti M., Paulovich A. G., Whiteaker J. R., Edwards N. J., McGarvey P. B., Madhavan S., Wang P., Chan D. W., Pandey A., Shih I.-M., Zhang H., Zhang Z., Zhu H., Cope L., Whiteley G. A., Skates S. J., White F. M., Levine D. A., Boja E. S., Kinsinger C. R., Hiltke T., Mesri M., Rivers R. C., Rodriguez H., Shaw K. M., Stein S. E., Fenyo D., Liu T., McDermott J. E., Payne S. H., Rodland K. D., Smith R. D., Rudnick P., Snyder M., Zhao Y., Chen X., Ransohoff D. F., Hoofnagle A. N., Liebler D. C., Sanders M. E., Shi Z., Slebos R. J. C., Tabb D. L., Zhang B., Zimmerman L. J., Wang Y., Davies S. R., Ding L., Ellis M. J. C., and Townsend R. R. (2016) Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bell D., Berchuck A., Birrer M., Chien J., Cramer D. W., Dao F., Dhir R., DiSaia P., Gabra H., Glenn P., Godwin A. K., Gross J., Hartmann L., Huang M., Huntsman D. G., Iacocca M., Imielinski M., Kalloger S., Karlan B. Y., Levine D. A., Mills G. B., Morrison C., Mutch D., Olvera N., Orsulic S., Park K., Petrelli N., Rabeno B., Rader J. S., Sikic B. I., Smith-McCune K., Sood A. K., Bowtell D., Penny R., Testa J. R., Chang K., Dinh H. H., Drummond J. A., Fowler G., Gunaratne P., Hawes A. C., Kovar C. L., Lewis L. R., Morgan M. B., Newsham I. F., Santibanez J., Reid J. G., Trevino L. R., Wu Y.-Q., Wang M., Muzny D. M., Wheeler D. A., Gibbs R. A., Getz G., Lawrence M. S., Cibulskis K., Sivachenko A. Y., Sougnez C., Voet D., Wilkinson J., Bloom T., Ardlie K., Fennell T., Baldwin J., Gabriel S., Lander E. S., Ding L., Fulton R. S., Koboldt D. C., McLellan M. D., Wylie T., Walker J., O'Laughlin M., Dooling D. J., Fulton L., Abbott R., Dees N. D., Zhang Q., Kandoth C., Wendl M., Schierding W., Shen D., Harris C. C., Schmidt H., Kalicki J., Delehaunty K. D., Fronick C. C., Demeter R., Cook L., Wallis J. W., Lin L., Magrini V. J., Hodges J. S., Eldred J. M., Smith S. M., Pohl C. S., Vandin F., Raphael B. J., Weinstock G. M., Mardis E. R., Wilson R. K., Meyerson M., Winckler W., Getz G., Verhaak R. G. W., Carter S. L., Mermel C. H., Saksena G., Nguyen H., Onofrio R. C., Lawrence M. S., Hubbard D., Gupta S., Crenshaw A., Ramos A. H., Ardlie K., Chin L., Protopopov A., Zhang J., Kim T. M., Perna I., Xiao Y., Zhang H., Ren G., Sathiamoorthy N., Park R. W., Lee E., Park P. J., Kucherlapati R., Absher D. M., Waite L., Sherlock G., Brooks J. D., Li J. Z., Xu J., Myers R. M., Laird P. W., Cope L., Herman J. G., Shen H., Weisenberger D. J., Noushmehr H., Pan F., Triche T. Jr., Berman B. P., Van Den Berg D. J., Buckley J., Baylin S. B., Spellman P. T., Purdom E., Neuvial P., Bengtsson H., Jakkula L. R., Durinck S., Han J., Dorton S., Marr H., Choi Y. G., Wang V., Wang N. J., Ngai J., Conboy J. G., Parvin B., Feiler H. S., Speed T. P., Gray J. W., Levine D. A., Socci N. D., Liang Y., Taylor B. S., Schultz N., Borsu L., Lash A. E., Brennan C., Viale A., Sander C., Ladanyi M., Hoadley K. A., Meng S., Du Y., Shi Y., Li L., Turman Y. J., Zang D., Helms E. B., Balu S., Zhou X., Wu J., Topal M. D., Hayes D. N., Perou C. M., Getz G., Voet D., Saksena G., Zhang J., Zhang H., Wu C. J., Shukla S., Cibulskis K., Lawrence M. S., Sivachenko A., Jing R., Park R. W., Liu Y., Park P. J., Noble M., Chin L., Carter H., Kim D., Karchin R., Spellman P. T., Purdom E., Neuvial P., Bengtsson H., Durinck S., Han J., Korkola J. E., Heiser L. M., Cho R. J., Hu Z., Parvin B., Speed T. P., Gray J. W., Schultz N., Cerami E., Taylor B. S., Olshen A., Reva B., Antipin Y., Shen R., Mankoo P., Sheridan R., Ciriello G., Chang W. K., Bernanke J. A., Borsu L., Levine D. A., Ladanyi M., Sander C., Haussler D., Benz C. C., Stuart J. M., Benz S. C., Sanborn J. Z., Vaske C. J., Zhu J., Szeto C., Scott G. K., Yau C., Hoadley K. A., Du Y., Balu S., Hayes D. N., Perou C. M., Wilkerson M. D., Zhang N., Akbani R., Baggerly K. A., Yung W. K., Mills G. B., Weinstein J. N., Penny R., Shelton T., Grimm D., Hatfield M., Morris S., Yena P., Rhodes P., Sherman M., Paulauskis J., Millis S., Kahn A., Greene J. M., Sfeir R., Jensen M. A., Chen J., Whitmore J., Alonso S., Jordan J., Chu A., Zhang J., Barker A., Compton C., Eley G., Ferguson M., Fielding P., Gerhard D. S., Myles R., Schaefer C., Mills Shaw K. R., Vaught J., Vockley J. B., Good P. J., Guyer M. S., Ozenberger B., Peterson J., and Thomson E. (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Himberg J., Hyvärinen A., and Esposito F. (2004) Validating the independent components of neuroimaging time series via clustering and visualization. Neuroimage 22, 1214–1222 [DOI] [PubMed] [Google Scholar]
- 15. Hyvärinen A., and Oja E. (2000) Independent component analysis: algorithms and applications. Neural Networks 13, 411–430 [DOI] [PubMed] [Google Scholar]
- 16. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mootha V. K., Lindgren C. M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., Tamayo P., Spiegelman B., Lander E. S., Hirschhorn J. N., Altshuler D., and Groop L. C. (2003) PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 [DOI] [PubMed] [Google Scholar]
- 18. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Der Maaten L., and Hinton G. (2008) Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 [Google Scholar]
- 20. Coates A. S., Winer E. P., Goldhirsch A., Gelber R. D., Gnant M., Piccart-Gebhart M., Thürlimann B., Senn H.-J., and Panel Members. (2015) Tailoring therapies–improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 26, 1533–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim S., Janzer A., Becker A., Zimmer A., Schule R., Buettner R., and Kirfel J. (2010) Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31, 512–520 [DOI] [PubMed] [Google Scholar]
- 22. Nagasawa S., Sedukhina A. S., Nakagawa Y., Maeda I., Kubota M., Ohnuma S., Tsugawa K., Ohta T., Roche-Molina M., Bernal J. A., Narváez A. J., Jeyasekharan A. D., and Sato K. (2015) LSD1 Overexpression Is Associated with Poor Prognosis in Basal-Like Breast Cancer, and Sensitivity to PARP Inhibition. PLoS ONE 10, e0118002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Verhaak R. G. W., Tamayo P., Yang J.-Y., Hubbard D., Zhang H., Creighton C. J., Fereday S., Lawrence M., Carter S. L., Mermel C. H., Kostic A. D., Etemadmoghadam D., Saksena G., Cibulskis K., Duraisamy S., Levanon K., Sougnez C., Tsherniak A., Gomez S., Onofrio R., Gabriel S., Chin L., Zhang N., Spellman P. T., Zhang Y., Akbani R., Hoadley K. A., Kahn A., Köbel M., Huntsman D., Soslow R. A., Defazio A., Birrer M. J., Gray J. W., Weinstein J. N., Bowtell D. D., Drapkin R., Mesirov J. P., Getz G., Levine D. A., Meyerson M., and Cancer Genome Atlas Research Network. (2013) Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest. 123, 517–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langfelder P., and Horvath S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hyvarinen A. (1999) Fast and robust fixed-point algorithms for independent component analysis. IEEE Trans. Neural Networks 10, 626–634 [DOI] [PubMed] [Google Scholar]
- 26. Kairov U., Cantini L., Greco A., Molkenov A., Czerwinska U., Barillot E., and Zinovyev A. (2017) Determining the optimal number of independent components for reproducible transcriptomic data analysis. BMC Genomics 18, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jahirabadkar S., and Kulkarni P. (2013) Clustering for high dimensional data: density based subspace clustering algorithms. Int. J. Comput. Appl. 63, 29–35 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteome data of BRCA and OV are available at the CPTAC data portal (https://cptac-data-portal.georgetown.edu/cptac/s/S015; https://cptac-data-portal.georgetown.edu/cptac/s/S020). Transcriptome data are available at the NCI Genomics Data Commons (https://portal.gdc.cancer.gov) under project name TCGA-BRCA and TCGA-OV. Data tables are also available at the publication page (https://www.nature.com/articles/nature18003; https://www.nature.com/articles/nature10166).