

Abstract
Attempts to create novel ligand-binding proteins often focus on formation of a binding pocket with shape complementarity against the desired ligand (particularly for compounds that lack distinct polar moieties). Although designed proteins often exhibit binding of the desired ligand, in some cases they display unintended recognition behavior. One such designed protein, that was originally intended to bind tetrahydrocannabinol (THC), was found instead to display binding of 25-hydroxy-cholecalciferol (25-D3) and was subjected to biochemical characterization, further selections for enhanced 25-D3 binding affinity and crystallographic analyses. The deviation in specificity is due in part to unexpected altertion of its conformation, corresponding to a significant change of the orientation of an α-helix and an equally large movement of a loop, both of which flank the designed ligand-binding pocket. Those changes led to engineered protein constructs that exhibit significantly more contacts and complementarity towards the 25-D3 ligand than the initial designed protein had been predicted to form towards its intended THC ligand. Molecular dynamics simulations imply that the initial computationally designed mutations may contribute to the movement of the helix. These analyses collectively indicate that accurate prediction and control of backbone dynamics conformation, through a combination of improved conformational sampling and/or de novo structure design, represents a key area of further development for the design and optimization of engineered ligand-binding proteins.
Keywords: affinity versus specificity, crystal structure, ligand binding, protein engineering
Introduction
The appropriate balance of ligand-binding affinity and specificity is a fundamental feature of most biological processes, including immune recognition, cellular metabolism, gene expression and cell signaling. The ability to accurately predict and recapitulate the basis for ligand affinity and specificity is a crucial part of understanding and manipulating such biological phenomena. It also represents a critical technical requirement in the reciprocal fields of drug design and protein engineering.
The creation of novel ligand-binding proteins that display tight-binding affinity to their desired target and that can also discriminate between closely related targets is an important goal of protein engineering. Purely computational approaches for such tasks are hindered by a somewhat poor ability to accurately calculate binding affinities (even when armed with high resolution structures of the relevant protein–ligand complexes) (Ashtawy and Mahapatra, 2012; Ross et al., 2013; Ballester et al., 2014) and by the challenge of adequately sampling variation of both the protein sequence and the protein conformation during the design process (MacDonald and Freemont, 2016). As a result, the creation of tight-binding, highly specific ligand-binding proteins usually requires screening a large number of computationally designed proteins to identify a construct that displays measurable binding activity, which is then further optimized in the laboratory (Stoddard, 2016). Nevertheless, a variety of studies have demonstrated that engineered ligand-binding proteins can in fact be created that perform as desired, even for highly demanding in vivo applications (recently reviewed in Yang and Lai, 2017).
We have recently reported a series of protein engineering studies in which computational approaches were employed for structure-based design of novel ligand-binding proteins. These projects included the creation of (i) a highly specific binding protein that binds the cardiac drug digoxigenin (Tinberg et al., 2013); (ii) a protein that binds 17-α-hydroxyprogesterone (17-OHP) (Dou et al., 2017); (iii) a protein that binds fentanyl (Bick et al., 2017) and (iv) a protein that binds the small molecular fluorescent ligand (Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one (DFHBI) (Dou et al., 2018). That final engineered protein was created from a de novo designed protein scaffold (instead of a pre-existing, naturally evolved protein) and enforced a defined binding mode that constrains the bound ligand to a unique conformation required for fluorescence.
In those studies, a variety of strategies were employed to balance requirements of (i) enough flexibility to facilitate binding function, (ii) sufficient structural pre-organization in the unbound state to reduce unfavorable entropic penalties upon ligand binding and (iii) enough shape complementarity in the bound complex between the designed binding pocket and the ligand to enforce specificity. Each study included the determination of ligand-bound crystal structures of the final ‘optimized’ constructs, as well as ‘intermediate’ constructs that were generated along the pathway of the overall design and engineering process. These analyses enabled several observations important for the improvement of computational algorithms for the design of ligand-binding proteins:
Initial computationally designed constructs often display mediocre affinity corresponding to micromolar dissociation constants (KD). Subsequent rounds of mutagenesis and selection for enhanced affinity can generate changes to the protein sequence and structure that appear to influence the structure and function of the binding pocket, often from a distance, that are difficult to predict computationally (Tinberg et al., 2013; Dou et al., 2017, 2018).
Engineered proteins intended to bind 17-OHP were found to capture that ligand as desired, but in a binding mode rotated 180° around a pseudo-two-fold axis in the compound, while still interacting with all the designed residues in the engineered site (Dou et al., 2017). Subsequent analyses suggested that the difference between the designed protein–ligand complex versus that which was observed in the crystal structure results from insufficient conformational sampling and energy function inaccuracies (in particular, because of under-estimation of the cost to desolvate the ligand’s hydrophilic groups). These issues were exacerbated by the near twofold symmetry of the molecule and a lack of polar groups that might distinguish between competing binding orientations.
The practice of iterating between computational steps of ligand docking and accompanying protein backbone movements, followed by alterations in protein sequence, can lead to inadequate sampling of each, that may produce molecular models and interactions trapped in local energetic minima. Such issues might be addressed by more sophisticated search algorithms that utilize ensemble docking and search approaches. This concept was exploited with a new computational search strategy to create fluorescence-activating binding proteins that bind the chromophore DHFBI and constrain it in a well-defined photoactive conformation (Dou et al., 2018).
Design of proteins with novel functions and properties is not limited to the geometries and properties of naturally occurring proteins and structures, but can also be accomplished, perhaps more efficiently, as part of a de novo protein fold design effort (Dou et al., 2018).
In each of the examples cited above, the initial engineered protein constructs were observed to bind the intended molecular ligand preferentially, over and above binding of alternative molecules that were also being employed as ligand targets for other purposes. However, in some cases (particularly when the target ligand lacks defining polar groups and potential hydrogen-bond partners) computational design efforts have been found to generate protein constructs that display preference for binding of a ligand other than the intended target. One such example arose during the course of designing two separate groups of ligand-binding proteins, intended to recognize either tetrahydrocannabinol (THC; the primary bioactive ingredient in the cannabis plant) or 25-hydroxy-cholecalciferol (25-D3; the hormonally active form of vitamin D3) (Fig. 1).
Fig. 1.

ChemDraw and space fill representations of (a) 25-hydroxy-cholecalciferol (25-D3) and (b) tetrahydrocannabinol (THC).
Here we report the initial design, subsequent characterization, lab-based maturation and X-ray crystallographic analyses of constructs from that project. These studies suggest that in this particular case, ligand mistargeting is due to a significant and unanticipated alteration of backbone structure.
Methods and materials
Computational protein engineering
The computational protocol used in this study to generate protein binders for hydrophobic small molecules focused primarily on generation of high surface complementarity (SC) between the small molecule and the protein scaffold, as well as the relative interface energy (IFE) for each docked model (Supplementary Fig. 1).
A PatchDock (Schneidman-Duhovny et al., 2005) constraints file that defines the receptor binding pocket was generated for each scaffold. Various conformers were docked into the defined pocket in parallel for all the scaffolds. PatchDock scores were used to rank the docking solutions and top 100 docked configuration from each scaffold were selected and filtered for highest SC scores. After this initial round of docking, the scaffold set was then expanded to allow docking of the same ligand into additional crystal structures that exhibit similar folded topologies to the top scoring designs. Because initial designed protein constructs were intended to subsequently be tested for binding via protein surface display on yeast (Gai and Wittrup, 2007) coupled with flow cytometric staining (using a probe corresponding to the intended ligand covalently linked to a fluorescent moiety) the predicted orientation of the small molecule in the binding pocket was also filtered to remove any designs where the linker would be unable to escape the pocket.
The ligand position was then systematically sampled by spatial perturbations of its initial placement within a cartesian grid in the binding pocket, by generating translations and rotations, filtering for shape complementarity and packing interactions in an iterative fashion and applying a Monte Carlo search algorithm to changes in binding pocket amino acid identity in an iterative manner with movements of each modeled ligand conformer. This resulted in the optimization of physicochemical interactions and led to discrete amino acid identity changes during exploration of the complex protein energy landscape.
The interactions between the ligand and protein were then optimized using the Rosetta energy function (Leaver-Fay et al., 2011). The potential designs were filtered primarily on ‘SC’, calculated ‘IFE’ and solvent accessible surface area. Lastly, the computational designs were manually inspected and rational substitutions were tested using Rosetta. This protocol was implemented for the design of two hydrophobic ligands: 25-D3 and THC.
To evaluate the method’s ability to generate designs with predicted high affinity ligand binding, we used RosettaDock (Lyskov and Gray, 2008) to compare calculated binding energies of each ligand engaged within the final designed protein scaffold, versus corresponding energies calculated after docking the same compound to the original wild-type protein crystal structures that gave rise to each designed construct. Fig. 2 shows IFE vs SC of the 20 lowest docked poses for each design and native structure. Designed proteins have on average a more favorable IFE compared with the random set of protein folds, especially for 25-D3. The variance in SC scores was higher for the native set compared to 25-D3, and there was a tendency for the designed proteins to cluster in a narrower area and to have an overall higher SC. The non-designed set of wild-type protein structures included a native vitamin D binder (PDB ID code 1DB1) (Rochel et al., 2000), which was the only native protein to score among the top IFE predictions. For THC, there was less separation between the design and native populations, although designs tended to have higher IFE scores on average. This may be due to the different sizes and chemical groups of the ligands: THC is smaller and therefore has fewer potential interactions available for creating a favorable energy.
Fig. 2.
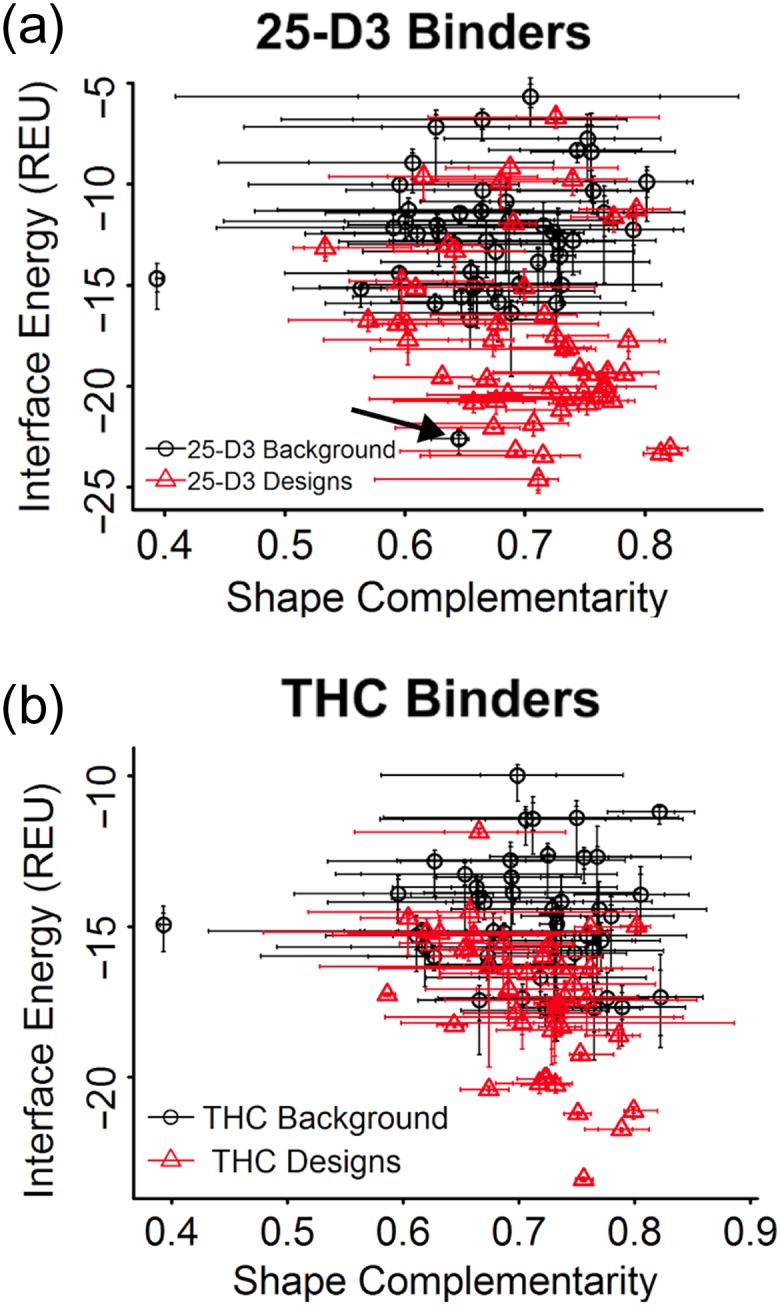
Score comparison of designs vs a representative set of randomized scaffolds. Shape complementarity (x-axis) and Rosetta interface energy (y-axis) for all ordered designs targeting the ligands (a) 25-hydroxy-cholecalciferol (25-D3) and (b) tetrahydrocannabinol (THC). Each plot compares the top twenty dock energies for the generated designs with each ligand (red) vs a random set of native protein structures (black). A naturally occuring 25-D3 binder (PDB ID: 1DB1) is also included in the random set of wild-type protein scaffolds (indicated by arrow). Its predicted energy places it among the best 25-D3 designs and helps validate the design metrics.
Yeast surface display and mutagenesis
Synthetic genes with 5′ and 3′ vector-overlapping sequences were synthesized (Gen9 Inc.) with codon usage optimized for Escherichia coli expression. They were first cloned between the NdeI and XhoI sites of pETCON for yeast surface display as an Aga2p-fusion protein (Boder and Wittrup, 1997). EBY100 yeast cells were treated and induced for protein display according to the published protocol (Boder and Wittrup, 2000). Mutagenesis (SSM) libraries were generated using an overlapping PCR method (Procko et al., 2013), with synthetic DNA oligos containing degenerate codons (Integrated DNA Technologies). Libraries were transformed as linear PCR product together with linear cut pETCON (digested with NdeI and XhoI) into EBY100 yeast cells by electroporation (Benatuil et al., 2010).
Flow cytometric analyses of ligand binding
Yeast cells displaying the designed protein were pre-coated by 0.01% BSA in 100uL phosphate buffered saline with 0.1% bovine serum albumin (PBSF) buffer (Chao et al., 2006). In a 1.5-ml Eppendorf tube, pre-coated cells were incubated with 10 μg/ml (0.33 μM) streptavidin–phycoerythrin (PE) (Invitrogen), 1.65 μM biotinylated probes and 5 μg/ml fluorescein isothiocyanate (FITC)-conjugated chicken anti-c-Myc (Immunology Consultants Laboratory) in 50 μl PBSF for 1 hr on a benchtop rotator at room temperature. Cells were spun down and washed twice by 50 μl cold PBSF before running through a Accuri C6 flow cytometer (BD Biosciences) or sorted with a BD Influx cell sorter(BD Biosciences).
Expression and purification of individual protein constructs
Functional designs were cloned between the NdeI and XhoI sites of pET29b (Novagen), placing a 6His-tag on the protein’s C-terminus. Plasmids were transformed into E. coli BL21(DE3) cells for protein expression. Cells were grown in LB at 37°C to OD600 ~0.6–0.9 and induced with 0.5 mM IPTG overnight at 18°C. Cells were lysed in phosphate-buffered saline (PBS) (140 mM NaCl, 1 mM KCl, 12 mM Na2HPO4 and 1.2 mM KH2PO4, pH 7.4) containing 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and 0.05 mg/ml DNase by sonication. Cleared lysate was loaded on NiNTA resin (Qiagen) and washed with 30 column volumes of wash buffer (PBS, 20 mM imidazole). Protein were eluted with elution buffer (PBS, 200 mM imidazole) and concentrated by centrifugal ultrafiltration before dialyzing overnight at 4°C against PBS. Protein concentration was determined by absorbance at 280 nm using calculated extinction coefficients.
X-ray crystallographic structure determinations
Purified proteins were complexed to 25-D3 by diluting protein to 22 μM, adding stock 25-D3 dissolved in 100% DMSO to 25 μM (keeping DMSO at <1%) and concentrating the complex. Complexes were initially tested for crystallization via sparse matrix screens in 96-well sitting drops using a mosquito (TTP LabTech). Crystallization conditions were then optimized in larger 24-well hanging drops.
CDL2.2 + 25-D3 crystallized in 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) sodium pH 7.5 and 1.4 M Sodium citrate at a concentration of 12.2 mg/ml. The crystal was transferred to a solution containing 75% mother liquor plus 25% glycerol and flash frozen in liquid nitrogen.
CDL2.3b + 25-D3 crystallized in 100 mM Sodium acetate, 100 mM sodium cacodylate pH 6.5 and 32% (w/v) polyethylene glycol 8000 at a concentration of 7.5 mg/ml. The crystal was transferred to a solution containing 75% mother liquor plus 25% ethylene glycol and flash frozen in liquid nitrogen.
CDL2.3a + 25-D3 crystallized in 100 mM HEPES sodium pH 7.5 and 1.25 M sodium citrate at a concentration of 4.8 mg/ml. The crystal was transferred to a solution containing 75% mother liquor plus 25% ethylene glycol and flash frozen in liquid nitrogen.
Data was collected on an in-house rotating anode generator and Four++ Imaging Plate Area Detector (Rigaku USA Inc.) and processed using the HKL2000 crystallographic software suite (Otwinowski and Minor, 1997). The structures were solved by Molecular Replacement using program Phaser in the PHENIX program suite (McCoy et al., 2007) using the original scaffold 3HX8 coordinates as a structural query. The structures were then rebuilt and refined using Coot (Emsley et al., 2007).
Molecular dynamics simulations
MD simulations were run for the original protein scaffold (PDB 3HX8) and for the ensuing initial Rosetta design (CDL2, which was produced via mutation of 3HX8 while restraining the protein backbone during the design process). We used the online server MDWEB (http:// http://mmb.irbbarcelona.org/MDWeb/index.php) (Hospital et al., 2012) to conduct the MD setup and computations using GROMACS (Pronk et al., 2013) with desolvated protein models and the AMBER-99SB force field (Salomon-Ferrer et al., 2013). An isothermal-isobaric (NPT) ensemble preparation (Uline and Corti, 2013) was chosen as the design constraint, producing a 2.5 picosecond simulation with 2.5 fs steps at 300 K. PDB snapshots and conformational parameters were produced every 50 steps.
Results
The amino acid sequences of all constructs described below are provided in Supplemental data.
Creation of 25-D3 binding proteins
Using suggestions from the computational protocol, genes encoding 28 separate designed proteins were ordered that target the ligand 25-D3. The designed constructs were generated from 17 different PDB scaffolds that span six different protein folds (Table 1). Seven of the 28 designs displayed a measurable binding signal in assays that combine yeast surface display of the designed protein with flow cytometric staining using a fluorescently labeled version of the intended ligand. Of these designs, the tightest binder (named VD1) was generated from hypothetical bacterial protein with a nuclear transport factor-2 (NTF2) folded topology (PDB ID: 1Z1S). This protein family is known to bind a variety of small molecules (Eberhardt et al., 2013). VD1 harbored nine mutations relative to the wild-type protein (which itself did not display any binding for 25-D3 under the highly avid initial screening concentrations). The designed amino acid substitutions were all located in the binding pocket and primarily served to increase the volume of the pocket, improve SC and make the pocket more hydrophobic.
Table 1.
Designed protein constructs intended to bind Vitamin D3 (“VD#”) or THC (“THC#”)
| Name | Native scaffold | Binds? | Mutations encorporated | Native scaffold | Number of designs | Number of binders |
|---|---|---|---|---|---|---|
| VD1 | 1Z1S | Yes | S21A, L25V, W33F, L43M, W52Y, V75I, Y112F, Y126S, D128I | |||
| VD2 | 1DMM | 3D9R | 2 | 0 | ||
| VD3 | 1DMM | Yes | M12A, Y15M, V19L, V37L, D39A, M83A, M89T, D102S, M104S, M115T, W119Y | 1DMM | 2 | 1 |
| VD4 | 1E3R | Yes | Y15L, Y31F, N39A, Y56F, V87I, D102S, M104I, I112V, W119F, L124M | 1E3R | 1 | 1 |
| VD5 | 1IWM | 1IWM | 1 | 0 | ||
| VD6 | 1WUB | 1WUB | 3 | 0 | ||
| VD7 | 1WUB | 1Z1S | 2 | 1 | ||
| VD8 | 1WUB | 2BNG | 2 | 0 | ||
| VD9 | 2BNG | 2F98 | 1 | 0 | ||
| VD10 | 2BNG | 3EN8 | 1 | 0 | ||
| VD11 | 2F98 | 3FKA | 1 | 0 | ||
| VD12 | 3D9R | 3HX8 | 6 | 2 | ||
| VD13 | 3D9R | 3LYG | 1 | 0 | ||
| VD14 | 3EN8 | 3ROB | 1 | 1 | ||
| VD15 | 3FKA | 1OHO | 1 | 1 | ||
| VD16 | 3HX8 | 2GEY | 1 | 0 | ||
| VD17 | 3HX8 | 3GZR | 1 | 0 | ||
| VD18 | 3HX8 | 3NHX | 1 | 0 | ||
| VD19 | 3HX8 | |||||
| VD20 | 3HX8 | Yes | A37M, I63A, L66V, L68I, L88A, A90V, A100I | |||
| VD21 | 3LYG | |||||
| VD22 | 3ROB | Yes | W26F, L27V, L49Y, C65A, A76M, A78V, L96V, A115I, D132A, A133Y, N134A | |||
| VD23 | 1OHO | Yes | V19M, Y31F, N39L, Y56F, G59M, A67M, V87A, M89Y, D102A, M115S, W119F | |||
| VD24 | 1Z1S | |||||
| VD25 | 2GEY | |||||
| VD26 | 3GZR | |||||
| VD27 | 3HX8 | Yes | N12T, I63A, L66A, L68A, V106A, D121L | |||
| VD28 | 3NHX | |||||
| THC1 | 3AKR | Yes | S16L, Y17F, N44L, V46I, E86A, Y88F, R122A, I128M, Q136M, Y171F, E177G | |||
| THC2 | 1JYH | |||||
| THC3 | 1N9L | |||||
| THC4 | 1QV1 | |||||
| THC5 | 1UYG | 3F0L | 1 | 0 | ||
| THC6 | 1YWC | 1JYH | 1 | 0 | ||
| THC7 | 2BVV | Yes | 5YF, Q7L, N35A, V37A, F69Y, Y80F, R112M, S117A, I118A, D119N, D120S, A165S, R172I | 1N9L | 1 | 0 |
| THC8 | 2GKP | 1QV1 | 1 | 0 | ||
| THC9 | 2OVD | Yes | L33I, V36I, T53F, V66A, T68F, R70V, Y83F, L94N, R100Q, H104I, V105A, L118V, L120T, L129Q, Y131F | 1UYG | 1 | 0 |
| THC10 | 2V1B | 1YWC | 1 | 0 | ||
| THC11 | 2WC5 | 2BVV | 1 | 1 | ||
| THC12 | 2WC5 | 2GKP | 1 | 0 | ||
| THC13 | 2WEX | 2OVD | 1 | 1 | ||
| THC14 | 3F0L | 2V1B | 1 | 0 | ||
| THC15 | 3F44 | 2WC5 | 2 | 0 | ||
| THC16 | 3I94 | 2WEX | 1 | 0 | ||
| THC17 | 3TGC | 3AKR | 1 | 1 | ||
| THC18 | 4F6B | Yes | Q44V, K47Q, T48Q, T49F, D51A, Y52F, A62F, L65A, R68A, H69S, R72L, V74I, I102V, I105V | 3F44 | 1 | 0 |
| 3I94 | 1 | 0 | ||||
| 3TGC | 1 | 0 |
To increase the binding affinity of the initial computational design, VD1 was then artificially evolved via error prone polymerase chain reaction (epPCR) to create a new variant named VD1.1 containing four additional mutations (P46S, R55A, H68P and G136V). The P46S and H68P mutations are located near the entrance of the binding site, while the two other mutations were distal to the binding pocket. These substitutions were not sampled in the initial computational design runs due to their positions being rather distant from the binding pocket.
Using the same yeast surface display and flow cytometric binding assay mentioned above, the initial design VD1 displayed an apparent KD of ~2 μM; in contrast the evolved variant VD1.1 had an apparent KD of 230 nanomolar (a ninefold enhancement). The initial design VD1 did not show a preference for 25-D3 over D3, while the evolved variant VD1.1 displayed improved (approximately twofold) specificity for 25-D3 (Fig. 3a).
Fig. 3.
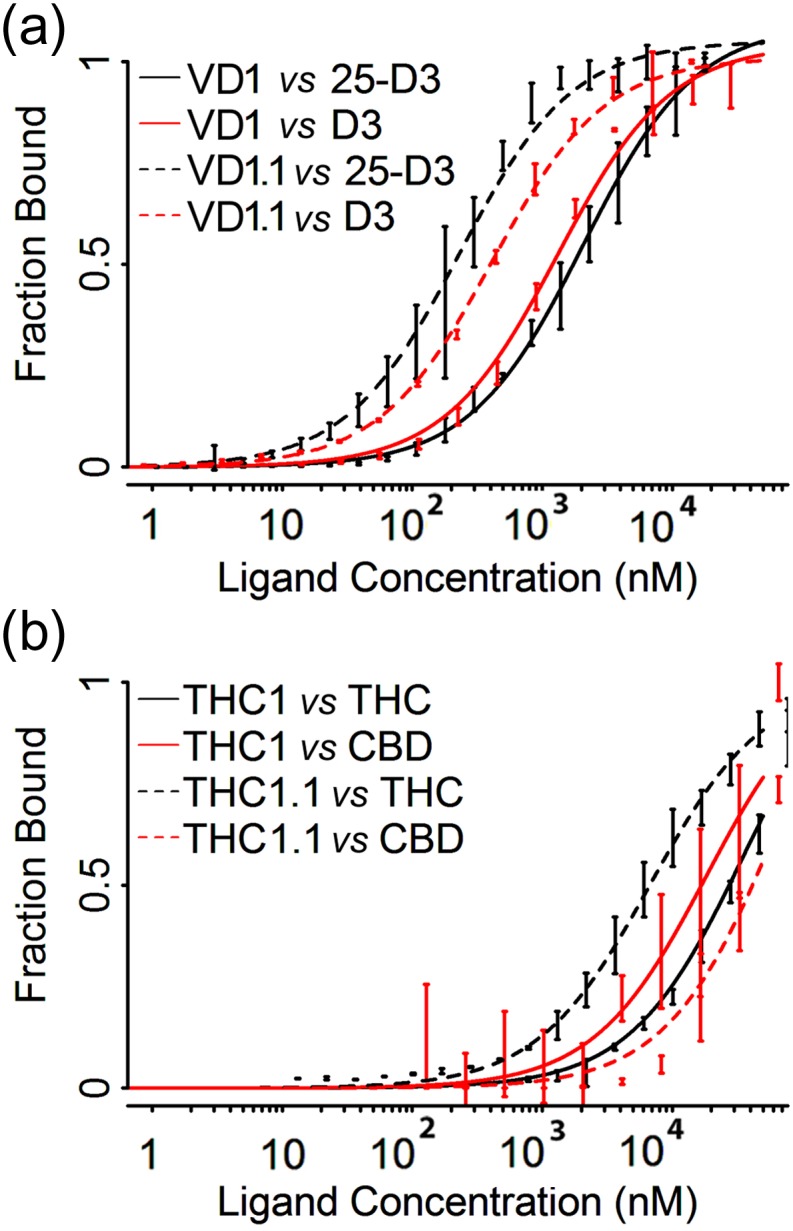
Yeast surface display titrations for affinity and specificity estimation of designed binders CDL1 and THC1. Yeast surface display titrations for both initial designs (solid lines) and evolved variants (dashed lines). (a) Designs CDL1 and CDL1.1 targeting 25-D3 (black lines) were tested for specificity against similar ligand D3 (red lines). (b) Designs THC1 and THC1.1 targeting THC (black lines) were tested for specificity against similar ligand CBD (red lines). Approximate KD values are 2 μM for CDL1 versus 25-D3, 1 μM for CDL1 versus D3, 200 nM for CDL1.1 versus 25-D3, 400 nM for CDL1.1 versus D3, ~30 μM for THC1 versus THC, >10 μM for THC1 versus CBD, ~5 μM for THC1.1 versus THC and >10 μM for THC1.1 versus CBD.
Design of THC binding proteins
Genes encoding 18 designed proteins were obtained that target the ligand THC (generated from originating from 17 different PDB scaffolds that span 16 different protein folds) (Table 1). Three of the 18 designs displayed a binding signal in assays that again combined yeast surface display of the engineered constructs and flow cytometric measurements of binding. Of these designs, the tightest (‘THC1’; KD ~ 35 micromolar) was derived from a xylanase enzyme (PDB ID: 3AKR) (Sugahara et al., 2011) containing a ‘Jelly Roll’ folded topology; it contained 11 mutations relative to the wild-type sequence. The designed mutations altered the native binding pocket into a more hydrophobic environment by replacing charged/polar residues with apolar residues. To optimize the initial design’s binding affinity, we again used epPCR and flow cytometric selections for enhanced affinity, thereby reducing the KD to ~7 micromolar. The evolved variant, THC1.1, contained six mutations beyond the original design: D20H, N69Y, I85F, C108V, M128I and M136I. Three of the seven mutations, I85F, M128I and M136I, were located in the binding pocket of the protein. The rest were distributed at several more distal positions on the protein scaffold. The initial ‘THC1’ design did not display specificity towards the intended ligand (THC) over its close analog cannabidiol (‘CBD’). In contrast, the selected ‘THC1.1’ construct displayed binding discrimination towards THC over CBD of approximately one order of magnitude (Fig. 3b).
Identification, analysis and laboratory optimization of an unintended, off-target 25-D3 binder
During additional studies of binding specificity (in which we examined the relative ability of many designed proteins to bind each member of an extended panel of ligands that were being employed as targets in the lab) we discovered an unintended binder of 25-D3 that was initially designed to bind THC (which it did, with an estimated high micromolar affinity) but displayed a stronger binding signal in our assays against 25-D3 (with an affinity later determined to correspond to a KD value of ~2 μM). This construct, named ‘CDL2’, corresponds to a putative ketosteroid isomerase and displays an ‘NTF2’ fold and topology (PDB ID: 3HX8) which. The designed construct contained 10 mutations compared with the wild-type protein, to which additional mutations were added during subsequent rounds of design and selection for enhanced binding (see Fig. 4 for an alignment of all constructs and corresponding structural illustrations of their positions within the protein fold).
Fig. 4.

(a) Sequence alignment of the wild-type protein scaffold used for engineering (PDB ID: 3HX8), the original computationally redesigned variant of that protein scaffold intended to bind THC, but instead displayed binding signal against 25-D3 (‘CDL2’), and a series of subsequent variants produced through a combination of epPCR and redesign steps that iteratively display enhanced binding of 25-D3 (‘CDL2.1’, ‘CDL2.2’, ‘CDL2.3a’ and ‘CDL2.3b’). The individual mutations relative to the starting protein scaffold that found in each step of design and selection are listed below the alignment. (b) Cartoon representation of the wild-type protein scaffold with the residues subjected to mutagenesis indicated by side chain sticks (colored corresponding to the highlighted residue positions in the sequence alignment, indicating their first appearance during the engineering process). The position of bound 25-D3, extracted from the crystal structure of the engineered CDL2.3a construct, is shown in spheres to illustrate the location and size of the designed ligand-binding pocket.
Binding of 25-D3 by the designed protein was then further optimized by (i) an initial round of epPCR using the designed scaffold as a starting protein sequence and then (ii) generation and screening of a computationally guided library that was used to further evolve improved affinity. The initial epPCR step identified a single point mutation located in the binding pocket (V106E) that improved affinity and increased protein expression (construct ‘CDL2.1’). The subsequent computationally guided protein library was then designed after docking 25-D3 (rather than THC) into a model of the CDL2.1 binding site and computationally optimizing the interactions between 25-D3 and the protein. To increase the sampling of motions across the protein fold and of potential protein–ligand contacts, short molecular dynamics simulations were performed to make small perturbations of the surrounding backbone. Several independent runs of this type generated a small list of suggested mutations that might further improve 25-D3 binding. We produced a protein library harboring these mutations in various random combinations, passed the library through epPCR and screened for improved binding. The tightest variant from these efforts, ‘CDL2.2’, incorporated an additional four mutations distributed throughout the protein scaffold. Two of the mutations in that construct (V100I and S123A) were introduced within the computational library, while another three (V106E, L66P and F86I) were introduced via epPCR. After purification of the original designed construct (CDL2) and the designed and evolved CDL2.2 construct, in vitro analyses of 25-D3 binding produced values for the ligand dissociation constant (KD) of ~2 μM and 300 nM, respectively (Fig. 5). Final rounds of mutagenesis resulted in an additional pair of constructs (named ‘CDL2.3a’ and ‘CDL2.3b’) that displayed slightly tighter binding, with KD values for each of ~100 nM.
Fig. 5.
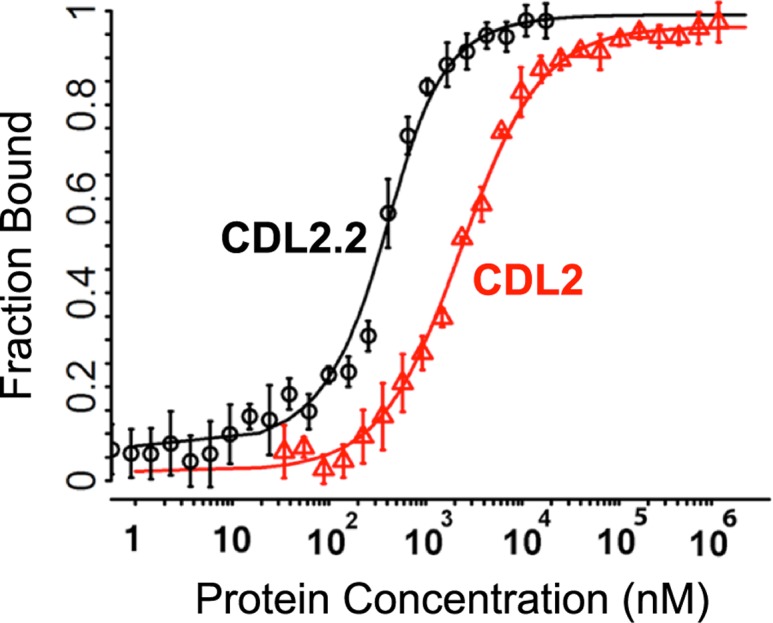
Comparison between design model CDL2 and its evolved variant CDL2.1. Fluorescence polarization binding data for 25-D3 binder CDL2 (red) versus its evolved variant CDL2.1 (black) binding a fluorescently labeled 25-D3 molecule. CDL2 has an approximate KD = 2 μM. CDL2.1 has an approximate KD = 200 nM
The crystal structures of the final three constructs from the experimental campaign (CDL2.2, CDL2.3a and CDL2.3b) were each determined in complex with 25-D3, at resolutions ranging from 2.09 to 1.85 Å (Table 2 and Fig. 6). The final structures were deposited at the protein structure database with PDB ID codes 5IEN (CDL2.2), 5IEO (CDL2.3a) and 5IEP (CDL2.3b). Despite considerable effort, crystals could not be grown of these constructs in the absence of bound ligand; neither could crystals be grown of the original computational design (CDL2) or the first variant of that design (CDL2.1, containing a single additional V106E mutation) in the presence or absence of ligand.
Table 2.
Crystallographic Data and Refinement Statistics
| CDL2.2 | CDL2.3a | CDL2.3b | |
|---|---|---|---|
| PDB ID | 5IEN | 5IEO | 5IEP |
| Data collection | |||
| Space group | P 21 21 21 | P 41 21 2 | P 41 21 2 |
| Unit cell | |||
| a, b, c | 48.8 60.4 93.9 | 71.1 71.1 62.1 | 71.8 71.8 60.8 |
| alpha, beta, gamma | 90.0 90.0 90.0 | 90.0 90.0 90.0 | 90.0 90.0 90.0 |
| Wavelength (Å) | 1.54 | 1.54 | 1.54 |
| Resolution range (Å) | 37.1–2.09 (2.16–2.09) | 31.8–1.85 (1.92–1.85) | 32.1–1.9 (2.0–1.9) |
| R-merge | 0.078 (0.532) | 0.059 (0.236) | 0.053 (0.242) |
| R-meas | 0.082 (0.566) | 0.063 (0.249) | 0.056 (0.261) |
| CC1/2 | (0.941) | (0.984) | (0.979) |
| I/sigma(I) | 32.0 (3.78) | 37.9 (12.0) | 49.2 (7.1) |
| Chi square | 1.216 | 1.211 | 1.14 |
| Multiplicity | 10.8 (7.8) | 10.3 (10.4) | 12.9 (6.9) |
| Completeness (%) | 99.7 (97.5) | 99.9 (100.0) | 99.6 (97.2) |
| Refinement | |||
| R-work | 0.2058 | 0.1606 | 0.2277 |
| R-free | 0.2408 | 0.1913 | 0.2445 |
| Number of non-hydrogen atoms | 1991 | 1019 | 978 |
| Macromolecules | 1844 | 898 | 889 |
| Ligands | 64 | 45 | 29 |
| Water | 83 | 76 | 60 |
| Protein residues | 253 | 118 | 121 |
| RMS (bonds) | 0.005 | 0.012 | 0.009 |
| RMS (angles) | 0.81 | 1.26 | 0.88 |
| Ramachandran favored (%) | 97 | 99 | 99 |
| Ramachandran allowed (%) | 2.59 | 1 | 1 |
| Ramachandran outliers (%) | 0.41 | 0 | 0 |
| Clashscore | 1.61 | 1.63 | 2.25 |
| Average B-factor | 42.9 | 23.5 | 40.5 |
| Macromolecules | 42.8 | 22 | 40.2 |
| Ligands | 42.9 | 36.9 | 41.1 |
| Solvent | 44.4 | 33.7 | 44.7 |
Fig. 6.

Crystallographic structures of engineered constructs. Electron density are unbiased omit maps. Left: Fo–Fc difference maps calculated in the absence of modeled ligand. Right: 2Fo–Fc difference maps contoured across the bound ligand and nearest contacting side chains. (a) CDL2.2, (b) CDL2.3a and (c) CDL2.3b.
The density for the bound ligand and surrounding side chains was well resolved in all three structures and consistently indicated a single bound conformation (Fig. 6). Comparison of the original designed model of the intended THC binding protein (CDL2) against these crystal structures indicate that the protein backbone has undergone significant movement that was not sampled or predicted in the original computations (Fig. 7). Whereas the modeled conformation of the computationally designed CDL2 construct is closely related to the starting wild-type protein scaffold (backbone RMSD ~0.1 Å), the crystal structures of CDL2.2, CDL2.3a and CDL2.3b display significantly larger RMSD values against the same wild-type protein (0.7, 1.1 and 1.1 Å, respectively).
Fig. 7.

Superposition of starting wild-type protein scaffold against the original computationally designed model (CDL2) and against the crystallographic structure of CDL2.2. The wild-type (non-engineered) starting protein is blue in all superpositions. The original computationally designed model (CDL2) and the crystal structure of the first laboratory-evolved variant of that design model (CDL2.2) are light green and dark green, respectively. (a) Superposition of the starting protein and CDL2 design model and corresponding fit of the intended THC ligand into the designed binding pocket in that computational model. (b) Superposition of the same starting protein and the CDL2.2 crystal structure and corresponding fit of the observed 25-D3 ligand into the binding pocket in that structure. The largest backbone differences between the original protein scaffold and the engineered and laboratory-evolved construct are indicated with highlighted arrows in the upper (helix motion) and lower (loop motion) panels.
These structural differences are the product of two large-scale backbone rearrangements (indicated with arrows in Fig. 7b): a rigid body rotation of ~15° exhibited by the protein’s N-terminal helix (spanning the first 21 residues of the protein chain) and a large motion of a six-residue loop (residues 39–44 in Fig. 4). These two structural elements each contribute contacts to the bound 25-D3 ligand (L12 and F15 from the helix; M42 from the loop) and multiple positions that were altered during the engineering process (N12L from the helix; P40L and R44P from the loop). In combination with smaller structural differences between the original design model and the crystal structures, the net effect is a significant remodeling of the ligand-binding pocket surface and shape, ultimately accommodating the bound 25-D3 ligand (Fig. 7, bottom).
The difference between the starting protein structure and the structures of the three engineered constructs are not attributable to crystallographic packing artifacts: the CDL2.3a and CDL2.3b constructs both display precisely the same backbone structural changes and conformation as CDL2.2; however, those latter constructs were crystallized in a completely different crystal form than the former construct, with unique unit cell dimensions and lattice contacts (Table 2).
Molecular dynamics simulations of the starting scaffold (PD 3HX8) and the initial Rosetta designed construct (CDL2) indicated a movement of the N-terminal helix, of greater magnitude in the CDL2 construct in the same direction as that observed in our crystal structures (Fig. 8). That analysis implies that motion may be induced (at least in part) by the incorporation of mutations in that first designed construct. In particular, the introduction of L12 in the helix is quite close in space to residue V106 (eventually mutated to E in the first selected CDL2.1 variant) and overpacking between those positions might have led to a slight shift in backbone conformation.
Fig. 8.

Molecular dynamics simulations of PDB 3HX8 and design CDL2. A small shearing movement of the N-terminal helix, in the same direction as that observed in our crystal structures (Fig. 8) is observed for the CDL2 starting model, with peaks in RMSD backbone shifts corresponding to residues near the two ends of the helix.
To examine whether the computational algorithms used in this study (if provided the experimentally determined structures) could produce binding energy predictions that reflect the results of these structural studies, we next examined whether RosettaDock would retrospectively recognize that the sequence changes and structural differences described above are both a necessary component of high affinity 25-D3 binding (Fig. 9). Docking of the 25-D3 ligand into the original designed model of CDL2 does not produce a favorable energy funnel (Fig. 9a) (i.e. the calculated interface energy, or ‘IFE’, does not become more favorable as the root mean square deviation (RMSD) of the docked ligand approaches the observed position of the same ligand in the crystal structure). When the five amino acid substitutions corresponding to the CLD2.2 construct are introduced into that design model (while maintaining the original backbone conformation, but allowing the ligand and surrounding side chains to move) the energy funnel visibly improves in shape but still does not produce a unambiguous energy well corresponding to the observed position of the ligand (Fig. 9b). In contrast, when 25-D3 is docked into the protein backbone conformation corresponding to the CDL2.2 crystal structure (and again allowed to move, along with motions of the surrounding side chains) the analysis displays a clearly favorable and predictive docking funnel corresponding to the observed ligand position (Fig. 9c). Thus, the identity of the newly introduced side chains in the ligand-binding pocket appears to partially contribute to binding affinity and their full contribution to that affinity is realized via the conformational rearrangement of the surrounding protein backbone.
Fig. 9.

Calculated energetic docking funnels. For all docking plots, the y-axis represents the calculated Rosetta interface energy and the x-axis represents the root mean squared deviation (RMSD) of various docked ligand positions within the protein model binding site. (a) 25-D3 docked in the original CDL2 design model. (b) 25-D3 docked in the original CDL2 design model, after the addition of five amino acid substitutions found in the CDL2.2 construct. The surrounding backbone conformation is unchanged from the original design. (c) 25-D3 docked in the actual CDL2.2 crystal structure.
A similar analysis, docking THC into each model, produced the inverse result: the calculated IFE for THC when docked into CDL2.2 is less favorable by ~3 Rosetta energy units than when docked into the original CDL2 design. The reason for this difference appears to be a small degree of clash between that ligand and various points throughout the binding pocket, which has been considerably enhanced for 25-D3 binding relative to the starting computational design.
The model of the original designed CDL2 complex (bound to THC) and the structure of the CDL2.2 construct (bound to 25-D3) were further examined, using the PLIP protein–ligand structure analysis webserver (https://projects.biotec.tu-dresden.de/plip-web/plip/) (Salentin et al., 2015) to compare the number of contacts and overall complementarity in each. The difference (Fig. 10) was striking. The original design of the CDL2 construct incorporated a total of eight hydrophobic van der Waals interactions with distances of 4 Å or less between THC and surrounding side chains (plus one additional π-stacking interaction to Phe 86 and a single H-bond by Ser 123). In contrast, the crystal structure of CDL2.2 bound to 25-D3 displayed a total of 15 contacts (all hydrophobic van der Waals interactions) of 4 Å or less between the ligand and surrounding side chains, plus one water-mediated H-bond between Thr 121 and the tertiary hydroxyl oxygen on 25-D3. An additional analysis of the intermediate computationally docked models of CDL2.1 bound to 25-D3 also indicated an improved set of contacts and complementarity relative to the original design model versus THC.
Fig. 10.

Computed versus observed ligand-binding side chain contacts. (a) Computationally designed CDL2/THC complex. (b) Crystallographic structure of CDL2.2/25-D3 complex.
Discussion
This study demonstrates that a computationally engineered protein construct established an effective binding site for an unintended ligand (hence its identification as an unexpected 25-D3 binder in our original analyses) and that the affinity for that ligand could be further improved via additional rounds of computational modeling and selections. The structural basis for this observation appears to be that the protein scaffold was capable (or rapidly became capable, during the earliest steps of protein engineering) of adopting a conformation that differs substantially from its previously observed crystallographic structure.
There are several possible causes, that are not exclusive of one another, for these results:
The starting protein scaffold itself may possess an inherent flexibility that was not evident or predicted from its crystallographic structure, that facilitated an unanticipated alteration of binding specificity at the earliest stages of engineering. Given that the initial scaffold is hypothesized to correspond to an enzyme (an isomerase), but the structure does not contain a bound substrate or substrate analog, it seems possible that those coordinates (which were constrained during computational design to maintain the crystallographically observed backbone conformation) are capable of motions related to its natural function, that may be partially recapitulated in the binding of 25-D3.
The incorporation of the first computationally designed mutations may have altered the protein conformation in an unexpected manner. Molecular dynamics simulations conducted on the crystal structure of the 3HX8 protein scaffold, alongside similar simulations of the initial designed CDL2 variant of that scaffold, imply that one or more of the computationally suggested mutations (possibly L13 on the N-terminal helix) may have contributed to motion of that secondary structure element. However, such simulations are always merely suggestive and the magnitude of the helix motion in those computations is smaller than that observed in the crystal structures.
The subsequent addition of additional mutations via additional laboratory-based evolution of the designed protein may have caused (or further contributed) to altered protein conformation. We note that the E106 residue that was then incorporated into the CDL2.1 constructs during the first round of epPCR and selections is close in space to L13; the two residues may have conspired to further push the protein conformation towards that which we observe in the crystallographic structures.
The binding of the ligand itself induced the conformational changes via an induced fit mechanism or conformational selection in a manner unique to the shape and chemical properties of that ligand.
Because we were unable to crystallize either the original CDL2 designed construct (in the presence or absence of either ligand), or any of the constructs in the absence of bound ligand, we cannot easily discriminate between the possible causes of the observed protein remodeling listed above. However, regardless of the relative contributions of each of these factors, the conformation of the protein backbone was obviously too tightly constrained to maintain its original starting conformation during the original computational design process.
It is possible that the unexpected conformational motions and changes observed in this study were encouraged by the inherent structural and dynamic properties of the initial protein scaffold (corresponding to a predicted ketosteroid isomerase enzyme from Mesorhizodium loti; PDB 3HX8) related to its putative catalytic behavior. It is certainly well-established that the evolutionary optimization of enzymatic catalysts involves not only recognition of substrate, but also the corresponding selection and fine-tuning of dynamic behaviors that facilitate transition-state stabilization and minimal energy barriers along the reaction trajectory, many of which are difficult to visualize using conventional structural analyses (Klinman and Kohen, 2014; Campbell et al., 2016; Gonzalez et al., 2016). In addition, many evolved proteins (both enzymes and non-enzymes) display complex relationships between sequence and structure, due to both their tendency to (i) exist in a folded state that is no more stable than necessary (thereby ensuring a balance of function versus stability and turnover) and (ii) their ability to form folded states that can exchange (reversibly or irreversibly) with an alternative conformation via small alterations in sequence or environment. As a result, it can be quite challenging to predict what changes in backbone conformation a given set of mutations will give rise to in an evolved, wild-type protein scaffold. These factors may also contribute to the consistent observation in this and related studies that a large fraction of initial computational designs, all derived from wild-type evolved proteins, fail to exhibit desired binding activity, despite computational metrics that appear relatively favorable (recall that in this study, only 10 of the 46 (22%) of the initial designed constructs were found to display measurable binding affinity towards their intended ligands).
If it remains true that the conformational behaviors of naturally occurring protein scaffolds (particularly large-scale backbone rearrangements) are often too unpredictable to be easily modeled or rationally altered during computational protein engineering, then an alternative strategy might be to pursue de novo protein fold design, coupled from an early stage of engineering to a desired biochemical function. There are two advantages of de novo scaffolds: (i) the sequence–structure relationship is user-defined; therefore investigators may have a better understanding of how sequence changes will produce structural changes as compared to native proteins and (ii) investigators can (at least in principle) generate very large numbers of scaffolds in silico and choose those with the best shape for binding the desired ligand.
We have recently described such an effort, in which the de novo design of an novel beta-barrel protein fold was accomplished, followed by the use of a ‘Rotamer Interaction Field’ docking method to generate a highly specific ligand-binding site and function (Dou et al., 2018). We believe that the type of computational approach in that study (design of a protein fold that facilitates a binding site geometry more closely matched to the ligand of interest, and optimization of complementarity to the desired ligand by concurrent sampling of protein sequence and the binding mode of the ligand) might allow investigators to more effectively predict and control potential backbone rearrangements during the design process. When evaluated in combination, the outcome of attempts to redesign naturally existing protein scaffolds for new functions (such as in this study) versus attempts to design entirely new protein folds and function in a more unified manner, may provide considerable insight to further improve choices of approach and algorithms for future protein engineering efforts.
Supplementary Material
Acknowledgments
We thank the members of the Baker and Stoddard Laboratories for advice and assistance with this project and Betty Shen for advice and assistance with the crystallographic analyses. B.L.S. and L.D. were supported by the NIH (R01 GM115545).
Author contributions
A.D. contributed to the development of the computational design methods, engineering of protein designs, yeast experiments involving surface display and FACS, library design, design optimization and characterization assays. P.G. developed computational methods for designing small molecule binders using shape complementarity for hydrophobic molecule (vitamin D, THC and cannabidiol) and tested design protocols. A.S. synthesized the biotinylated and the fluorescent probes used for initial activity determinations. N.S. provided cannabinoid compounds and guidance in protocol design. L.D. performed all crystallographic analyses. B.L.S. and D.B. conceived and directed the research project and critically assessed all data generated during its execution. All authors contributed to the preparation of the manuscript, approved its final form and content and took responsibility for the individual results reported as listed above.
References
- Ashtawy H. and Mahapatra N. (2012) IEEE/ACM Trans. Comput. Biol. Bioinformatics, 9, 1301–1312. [DOI] [PubMed] [Google Scholar]
- Ballester P., Schreyer A. and Blundell T. (2014) J. Chem. Inf. Model., 54, 944–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatuil L., Perez J.M., Belk J. and Hsieh C.-M. (2010) Protein Eng. Des. Sel., 23, 155–159. [DOI] [PubMed] [Google Scholar]
- Bick M.J., Greisen P.J., Morey K.J., Antunes M.S., La D., Sankaran B., Reymond L., Johnsson K., Medford J.I. and Baker D. (2017) Elife, 6, e28909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boder E.T. and Wittrup K.D. (1997) Nat. Biotechnol., 15, 553–557. [DOI] [PubMed] [Google Scholar]
- Boder E.T. and Wittrup K.D. (2000) Methods Enzymol., 328, 430–444. [DOI] [PubMed] [Google Scholar]
- Campbell E., Kaltenbach M., Correy G.J., Carr P.D., Porebski B.T., Livingstone E.K., Afriat-Jurnou L., Buckle A.M., Weik M., Hollfelder F. et al. (2016) Nat. Chem. Biol., 12, 944–950. [DOI] [PubMed] [Google Scholar]
- Chao G., Lau W.L., Hackel B.J., Sazinsky S.L., Lippow S.M. and Wittrup K.D. (2006) Nat. Protoc., 1, 755–768. [DOI] [PubMed] [Google Scholar]
- Dou J., Doyle L. Jr, Greisen P., Schena A., Park H., Johnsson K., Stoddard B.L. and Baker D. (2017) Protein Sci., 26, 2426–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou J., Vorobieva A.A., Sheffler W., Doyle L.A., Park H., Bick M.J., Mao B., Foight G.W., Lee M.Y., Gagnon L.A. et al. (2018) Nature, 561, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt R.Y., Chang Y., Bateman A., Murzin A.G., Axelrod H.L., Hwang W.C. and Aravind L. (2013) BMC Bioinformatics, 14, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G. and Cowtan K. (2007) Acta Crystallogr., D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai S.A. and Wittrup K.D. (2007) Curr. Opin. Struct. Biol., 17, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M.M., Abriata L.A., Tomatis P.E. and Vila A.J. (2016) Mol. Biol. Evol., 33, 1768–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital A., Andrio P., Fenollosa C., Cicin-Sain D., Orozco M. and Gelpi J.L. (2012) Bioinformatics, 28, 1278–1279. [DOI] [PubMed] [Google Scholar]
- Klinman J.P. and Kohen A. (2014) J. Biol. Chem., 289, 30205–30212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A., Tyka M., Lewis S.M., Lange O.F., Thompson J., Jacak R., Kaufman K., Renfrew P.D., Smith C.A., Sheffler W. et al. (2011) Methods Enzymol., 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S. and Gray J.J. (2008) Nucleic Acids Res., 36, W233–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald J.T. and Freemont P.S. (2016) Biochem. Soc. Trans., 44, 1523–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C. and Read R.J. (2007) J. Appl. Cryst., 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. (1997) Carter C.W. and Sweet R.M. (eds),. Methods in Enzymology. Academic Press, New York, pp. 307–326. [Google Scholar]
- Procko E., Hedman R., Hamilton K., Seetharaman J., Fleishman S.J., Su M., Aramini J., Kornhaber G., Hunt J.F., Tong L. et al. (2013) J. Mol. Biol., 425, 3563–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D. et al. (2013) Bioinformatics, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochel N., Wurtz J.M., Mitschler A., Klaholz B. and Moras D. (2000) Mol. Cell, 5, 173–179. [DOI] [PubMed] [Google Scholar]
- Ross G., Morris G. and Biggin P. (2013) J. Chem. Theory Comput., 9, 4266–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin S., Schreiber S., Haupt V.J., Adasme M.F. and Schroeder M. (2015) Nucleic Acids Res., 43, W443–W447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R., Case D.A. and Walker R.C. (2013) WIREs Comput. Mol. Sci., 3, 198–210. [Google Scholar]
- Schneidman-Duhovny D., Inbar Y., Nussinov R. and Wolfson H.J. (2005) Nucleic Acids Res., 33, W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard B.L. (2016) Stoddard B.L. (ed),. Computational Design of Ligand Binding Proteins. Humana Press, New York, pp. v–ix. [Google Scholar]
- Sugahara M., Kageyama-Morikawa Y. and Kunishima N. (2011) Cryst. Growth Des., 11, 110–120. [Google Scholar]
- Tinberg C.E., Khare S.D., Dou J., Doyle L., Nelson J.W., Schena A., Jankowski W., Kalodimos C.G., Johnsson K., Stoddard B.L. et al. (2013) Nature, 501, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uline M.J. and Corti D.S. (2013) Entropy, 15, 3941–3969. [Google Scholar]
- Yang W. and Lai L. (2017) Curr. Opin. Struct. Biol., 45, 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.