

Abstract
The metabolic reprogramming associated with malignant transformation has led to growing appreciation of the nutrients required to support anabolic cell growth. Less well studied is how cancer cells satisfy those demands in vivo, where they are dispersed within a complex microenvironment. Tumor-associated stromal components can support tumor growth by providing nutrients that supplement those provided by the local vasculature. These non-malignant stromal cells are phenotypically similar to those that accumulate during wound healing. Owing to their immediate proximity, stromal cells are inevitably affected by the metabolic activity of their cancerous neighbors. Until recently, a role for tumor cell metabolism in influencing the cell fate decisions of neighboring stromal cells has been underappreciated. Here, we propose that metabolites consumed and released by tumor cells act as paracrine factors that regulate the non-malignant cellular composition of a developing tumor by driving stromal cells toward a regenerative response that supports tumor growth.
eTOC blurb
CELL-METABOLISM-D-18-00936R1
The concept of tumor stromal cells supporting tumor growth by providing nutrients has gained growing appreciation in recent years. In this review, Schwörer et al. discuss how the metabolic activity of cancer cells contributes to converting various stromal cell types into promoting a regenerative response that supports tumor growth.
Introduction
Organs contain four major cell types: parenchymal cells performing the primary organ-specific function, mesenchymal cells providing extracellular matrix (ECM) for mechanical support, immune cells surveying organ integrity, and vascular cells controlling oxygen and nutrient supply. Delegating supportive functions to accessory cells enables parenchymal cells to optimize their primary function (Okabe and Medzhitov, 2016). Nevertheless, a proper ratio of parenchymal and supportive cell types is essential to achieve optimal organ performance (Adler et al., 2018; Raff, 1996). Tumors are derived from organ-specific parenchymal cells that acquire oncogenic mutations (Hanahan and Weinberg, 2000). The resulting cellular transformation and cell growth causes disruption of normal tissue architecture which can compromise normal organ function (Egeblad et al., 2010).
While the progressive accumulation of cancer cells disrupts the normal organization of the adjacent stroma, tumor growth is nevertheless dependent on stromal support (Hanahan and Coussens, 2012). This was first appreciated nearly thirty years ago, when several groups demonstrated that co-injection of cancer-associated fibroblasts (CAFs) with cancer cells enhanced the growth of xenograft tumors (Camps et al., 1990; Gleave et al., 1991; Olumi et al., 1999). Strikingly, when immortalized but nonmalignant prostate epithelial cells were grafted with fibroblasts isolated from cancerous tissue, gross tumors were formed, and the epithelial cells were able to form tumors on their own in a second round of grafting (Olumi et al., 1999), suggesting that stromal cells could enhance malignant transformation. Of note, fibroblasts isolated from non-malignant tissues were not able to enhance tumor growth, suggesting that the capacity of stromal cells to facilitate tumorigenesis is acquired during tumor development. In some cancers such as gastric, mammary or pancreatic tumors, stromal cells may account for up to 90% of cells within a tumor mass (Casazza et al., 2014; Dvorak, 1986).
How alterations in stromal composition and function support tumor growth is now the subject of intense study. For example, both mesenchymal cells themselves and the matrix proteins they produce provide an additional nutrient source in poorly vascularized regions where nutrient delivery is compromised (Commisso et al., 2013; Loo et al., 2015; Olivares et al., 2017; Sousa et al., 2016). Similarly, while immune cells are thought normally to recognize and eliminate parenchymal cells that have acquired somatic mutations (Taylor and Odili, 1970), the immune cells found within tumors are primarily subtypes that suppress anti-tumor immunity, and accordingly their presence is associated with poor prognosis in multiple malignancies (Leek et al., 1996; Steidl et al., 2010; Wolf et al., 2005) (Figure 1).
Figure 1. The tumor microenvironment.
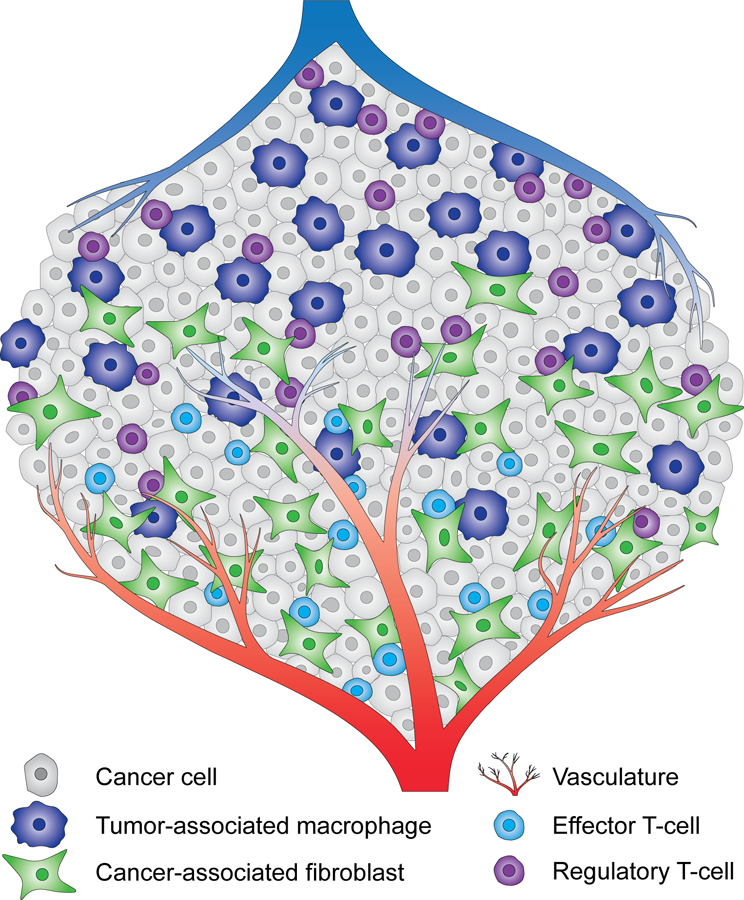
Cancer cells are dispersed within a complex microenvironment. Multiple nonmalignant cell types are found in a bulk tumor, including fibroblasts, T-cells and macrophages. While cancer-associated fibroblasts and effector T-cells are preferentially found in the proximity of the vasculature, tumor-associated macrophages and regulatory T-cells accumulate in hypovascular tumor regions. Other non-malignant cell types are present in tumor stroma but not depicted here to aid visualization.
While the recruitment of stromal cells to tumors, largely by chemotactic cues, has been studied (Balkwill, 2004), how tumor cells directly instruct microenvironmentresident stromal cells to facilitate tumor growth is not fully understood. Of note, the environment of a tumor is reminiscent of a healing wound, where a variety of distinct cell types, including fibroblasts as well as immune cells, play a coordinated role in tissue regeneration (Dvorak, 2015; Hagemann et al., 2008) (Figures 1 and 2). Mimicking a regenerative environment may therefore be a mechanism by which tumor cells can redirect surrounding stromal cells to enhance tumor growth. We have previously proposed the metabolic interaction of cancer cells with the tumor microenvironment as a hallmark of cancer metabolism (Pavlova and Thompson, 2016). Here, we consider whether cancer cell metabolism can be sufficient to induce and maintain the stromal cell fate changes required to establish a continuous regenerative phenotype. While extracellular nutrients provide building blocks for cell growth, the resulting waste products serve as ideal paracrine factors to instruct cell fate decisions in surrounding cells. Accumulation or depletion of metabolites within the extracellular environment can exert direct paracrine effects by activating signal transduction cascades in neighboring stromal cells, as seen in both immune cells (Oh et al., 2010; Suganami et al., 2005) and endothelial cells (Ignarro et al., 1988; Moncada et al., 1976). In addition, tumor cell-driven changes in nutrient availability can activate nutrient-sensitive signaling pathways that alter the phenotypic state of stromal cells, at least in part through influencing chromatin organization and gene expression (Wellen et al., 2009). In this review, we will consider how metabolic alterations in the tumor microenvironment resulting from the metabolic activity of cancer cells can promote stromal cell evolution to support tumor development and progression.
Figure 2. The wound healing response.
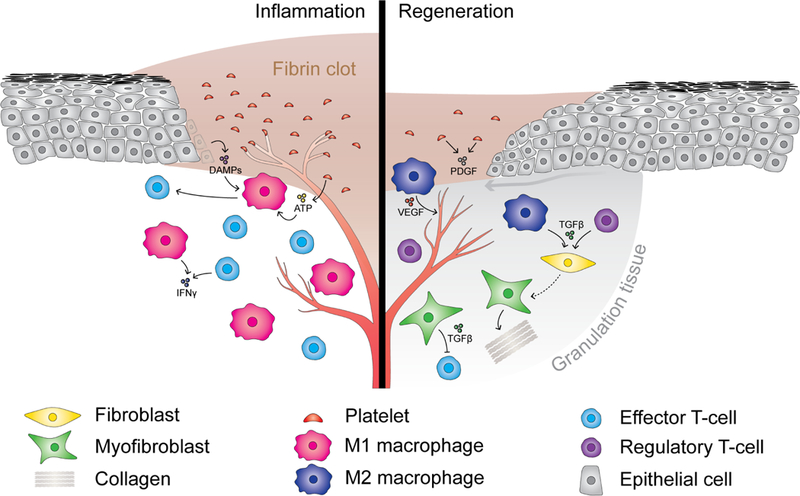
The inflammatory phase of the wound healing response (left) is characterized by accumulation of pro-inflammatory immune cells, including M1 macrophages and effector T-cells. Damage-associated molecular patterns (DAMPs) including ATP released from dying epithelial cells and platelets initially recruit monocytes to the site of tissue damage where they differentiate into pro-inflammatory M1 macrophages. M1 macrophages clear necrotic tissue and secrete cytokines that recruit effector T-cells. Following clearance of cell debris, M2 macrophages and regulatory T-cells accumulate (right), secreting cytokines and growth factors such as TGFβ that induce activation and differentiation of fibroblasts into myofibroblasts. Myofibroblasts contribute to the formation of granulation tissue and wound repair by secretion of ECM proteins such as collagens, and by secretion of immunosuppressive factors including TGFβ. Angiogenesis is stimulated by VEGF secreted by M2 macrophages.
Tumors co-opt the wound healing response
The concept that tumors co-opt the normal wound healing response is intriguing given Virchow’s hypotheses on cancer and irritation more than 150 years ago (reviewed in Balkwill and Mantovani, 2001), which led Dvorak to coin his famous description of tumors as “wounds that do not heal” (Dvorak, 1986). Wound healing has four phases: hemostasis, inflammation, tissue formation, and tissue remodeling (Singer and Clark, 1999). Tumor development shares many mechanistic similarities with this process and, with the exception of platelets, involves the same cell types (Dvorak, 2015). The wound repair process is conserved across tissue types and injuries; thus, co-opting this process allows cancer cells to recruit and alter the stroma by similar means across a wide variety of tissue settings.
The regenerative response to tissue damage is often initiated by cells of the innate immune system (Adler et al., 2018; Coussens and Werb, 2002) (Figure 2). Monocytes are recruited to the site of tissue injury in large part by the release of small molecules that act in a paracrine fashion. Many of these damage-associated molecular patterns (DAMPs) are metabolites, including ATP, uric acid, and nucleotides (Bianchi et al., 2007; Ishii et al., 2001; Matzinger, 2002; Shi et al., 2003). Monocytes arriving at the site of damaged tissue differentiate into activated macrophages, which initially adopt a pro-inflammatory “M1” phenotype in order to facilitate the clearance of necrotic tissue (Arnold et al., 2007; Dai et al., 2002; Nahrendorf et al., 2007; Perdiguero et al., 2011). As the necrotic debris is cleared from the site of damage, macrophages undergo a cell fate transition towards an “M2” phenotype. These cells produce anti-inflammatory cytokines and growth factors such as transforming growth factor beta (TGFβ) that initiate wound resolving and tissue regeneration. The transition of macrophages from a pro-inflammatory M1 to a regenerative M2 phenotype is facilitated by regulatory T-cells, which coordinate both the resolution of inflammation and activation of parenchymal progenitor cells to begin the process of tissue regeneration (Arnold et al., 2007; Burzyn et al., 2013; Kuswanto et al., 2016).
During the regenerative response in a healing wound, fibroblasts are recruited primarily by chemotactic factors released by innate immune cells, and rapidly become activated and undergo differentiation into myofibroblasts displaying enhanced contractile activity (Eming et al., 2014; Gabbiani et al., 1971; Hinz et al., 2001). This cell fate change is marked by increased expression of alpha smooth muscle actin (αSMA) and can be initiated by growth factors such as TGFβ and platelet-derived growth factor (PDGF), by reactive oxygen species (ROS), or by mechanical properties of the ECM (Calvo et al., 2013; Desmoulière et al., 1993; Rønnov-Jessen and Petersen, 1993; Toullec et al., 2010). Myofibroblasts produce and secrete growth factors and ECM proteins that support the proliferation and survival of the remaining parenchymal cells (Desmoulière and Gabbiani, 1988).
The stromal signature of a growing tumor displays many overlapping features of the regenerative phase of the wound healing response. For example, the immune milieu of tumors is characterized not by inflammatory clearance but rather by unopposed immunosuppression featuring a prevalence of M2-like macrophages, regulatory T-cells and myeloid-derived suppressor cells (Corzo et al., 2010; Liyanage et al., 2002; De Palma and Lewis, 2013; Solinas et al., 2009) (Figure 1). Sustained growth factor and cytokine stimulation results in irreversible activation and differentiation of fibroblasts into myofibroblast-like CAFs, which are the most abundant cell type in many tumors and support growth, survival and metastasis of malignant parenchymal cells (Kalluri, 2016) (Figure 1).
This aberrant regenerative response is a widely generalizable feature of human cancers; the serum-induced response of fibroblast cultures reveals a “wound response signature”, and enrichment of this signature in tumors is predictive of poor outcomes in multiple epithelial cancers (Chang et al., 2004, 2005). Furthermore, manipulation of the extracellular environment by cancer cells is sufficient to induce this regenerative response, as culturing fibroblasts in cancer cell-conditioned medium is sufficient to induce their activation (Rønnov-Jessen and Petersen, 1993). This suggests that cancer cells can direct a regenerative response in stromal cells by manipulating the extracellular milieu.
Cancer cell metabolism alters the extracellular environment to activate a wound healing response
How might cancer cells reprogram the activity of their surrounding stroma? One approach which has been widely pursued by the research community is the study of tumor-produced cytokines and growth factors; these studies have been reviewed elsewhere (Coussens and Werb, 2002; Turley et al., 2015). An additional mechanism by which cancer cells might direct the fate of stromal cells within the tumor is by altering the metabolic composition of the extracellular environment through the consumption of available nutrients and secretion of metabolic “waste” products. During tumorigenesis, oncogenic mutations in growth factor-responsive signaling proteins such as PIK3CA, Akt, and KRas induce high levels of glucose uptake, the majority of which is excreted as lactate. This shift drives the accumulation of metabolic intermediates that are required for anabolic cell growth, including ribose sugars, fatty acids, and reducing equivalents (Elstrom et al., 2004; Fan et al., 2014; Vander Heiden et al., 2009; Hosios et al., 2016; Keibler et al., 2016). The increased glucose consumption and lactate excretion within tumors has been well described for almost a century (Warburg et al., 1927). This metabolic behavior also radically alters the composition of the extracellular environment. As a result of constitutive glucose uptake and lactate excretion, glucose is rapidly depleted from the tumor microenvironment (Ho et al., 2015; Kamphorst et al., 2015), while lactate accumulates and is exported via the monocarboxylate transporter system (MCT) in cotransport with H+ ions, which causes acidification of the local environment (Schornack and Gillies, 2003) (Figure 3).
Figure 3. Metabolic gradients in tumors.
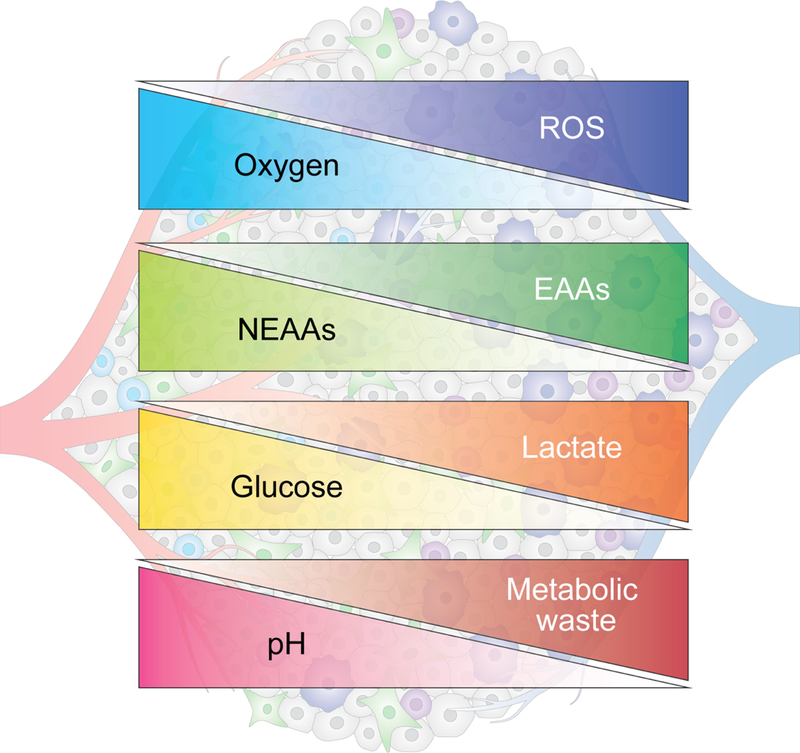
The metabolic activity of cancer cells and stromal cells as well as proximity to the vasculature contribute to the formation of metabolic gradients within tumors. In hypovascularized tumor core regions (right), glucose, glutamine and other nonessential amino acids (NEAAs) are depleted, while certain essential amino acids (EAAs) and metabolic waste products such as lactate accumulate. These areas are also characterized by elevated levels of reactive oxygen species (ROS) and acidic pH.
In addition to glucose, amino acids are the most highly consumed source of carbon for proliferating cells, and indeed comprise the majority of cellular biomass during proliferation (Hosios et al., 2016). As such, cancer cells engage in high levels of amino acid uptake and catabolism, driven in many cases by oncogenes such as c-Myc (Wise et al., 2008). Glutamine is the most heavily consumed amino acid by proliferating cells and serves numerous critical functions that support cellular proliferation. It can be utilized to regenerate TCA cycle intermediates, particularly during aerobic glycolysis (known as anaplerosis) (Deberardinis et al., 2007). It is also a key intermediate in the biosynthesis of nucleotides, non-essential amino acids (NEAA) and glutathione, and acts as exchange substrate for the import of essential amino acids (EAAs) (Conrad and Sato, 2012; Liu et al., 2008; Nicklin et al., 2009). As a result, glutamine and other NEAAs such as cystine and serine are consumed and depleted from the tumor microenvironment, while byproducts of amino acid catabolism, such as ammonia, accumulate (Kamphorst et al., 2015; Spinelli et al., 2017) (Figure 3). This effect is further augmented by a relative depletion of functional vasculature, which reduces both nutrient and oxygen delivery as well as the removal of waste products (Vaupel et al., 1989) (Figure 3).
Cancer cells have evolved multiple mechanisms to survive, and even proliferate, under the harsh conditions present within the tumor microenvironment. They can use a number of alternative substrates to fuel growth, including branched chain amino acids for de novo NEAA and nucleotide biosynthesis (Mayers et al., 2016), acetate for acetyl-CoA and fatty acid synthesis (Schug et al., 2015), scavenging of extracellular lysophospholipids to bypass de novo lipogenesis (Kamphorst et al., 2013), and macropinocytotic uptake and degradation of extracellular protein to maintain amino acid supply and bioenergetics (Commisso et al., 2013; Kamphorst et al., 2015). Thus, tumor cells retain a high level of metabolic plasticity, allowing them to both establish and subsequently adapt to the harsh extracellular environment of a developing tumor. The same cannot be said of the non-malignant components of a tumor. As these cells are not oncogenically transformed, they remain highly sensitive to both growth factors and available nutrients to dictate their growth and cell fate decisions. Interestingly, the inhospitable conditions that stromal cells face within tumors bear striking similarities to that seen during wound repair. Healing wounds have high rates of glucose uptake and lactate excretion (Im and Hoopes, 1970; Trabold et al., 2003). Similarly, extracellular glutamine availability drops profoundly following trauma (Ardawi, 1988; Caldwell, 1989). Finally, healing wounds are generally hypoxic before the onset of angiogenesis (Gurtner et al., 2008).
Thus, the metabolic environment created by tumor cells appears to mimic the microenvironment in which wound repair occurs. In the following sections, we will describe how specific cancer-driven nutrient fluctuations in the tumor microenvironment may help direct the phenotype and function of different stromal cell types.
Glucose
Tumor cells harbor frequent mutations in the PI3K/Akt signaling pathway that drive high rates of glucose uptake and catabolism; this rate is sustained by regenerating NAD+ through LDHA-mediated conversion of pyruvate to lactate (Elstrom et al., 2004). High glycolytic flux, coupled with a decreased vascular supply, results in profound glucose depletion within tumors, with intratumoral glucose concentrations measuring less than one tenth of that seen in interstitial fluid of normal organs (Gullino et al., 1964; Ho et al., 2015).
Glucose depletion has a profound impact on the growth and function of surrounding immune cells. The maintenance of inflammatory effector T-cells depends on glucose availability; high levels of glycolytic flux result in phosphoenolpyruvate-mediated inhibition of SERCA activity, leading to enhanced calcium signaling and nuclear NFAT translocation (Ho et al., 2015). Additionally, glycolytic flux sustains T-cell effector function by sequestering GAPDH, thus preventing it from binding interferon gamma (IFNγ) mRNA and inhibiting its translation (Chang et al., 2013). When glucose availability is compromised, reductions in histone acetylation impair IFNγ expression, preventing CD4 T-cell differentiation towards an effector Th1 subtype (Peng et al., 2016). While T-cell effector function is highly sensitive to decreases in glucose availability (Chang et al., 2015; Ho et al., 2015; Jacobs et al., 2008), depletion of available glucose favors the growth and differentiation of regulatory T-cells, which feature higher AMPK activity, decreased glucose oxidation, and increased fatty acid oxidation to support energy homeostasis (Angelin et al., 2017; Gualdoni et al., 2016; Michalek et al., 2011).
The above findings suggest that glucose depletion in the tumor microenvironment serves as a potential ‘metabolic checkpoint’ in the suppression of anti-tumor T-cell responses. This checkpoint likely synergizes with the inhibitory effects of programmed cell death-1 (PD-1), which profoundly suppresses glucose uptake in activated T-cells (Parry et al., 2005), likely by suppressing CD28 signaling (Hui et al., 2017a; Kamphorst et al., 2017), which is required for Akt activation and aerobic glycolysis (Frauwirth et al., 2002). Indeed, blockade of PD-1/PD-L1 interactions upregulates GLUT1 on tumor-infiltrating T-cells, making them more effective scavengers of the remaining glucose in the tumor microenvironment (Chang et al., 2015). However, this benefit is restricted to conditions in which glucose remains available and may explain the high responsiveness to checkpoint inhibitors in tumors of highly vascularized tissues such as lung, skin, kidney, and lymph nodes (Topalian et al., 2012). It may also explain the recent finding that highly glycolytic tumors are more resistant to adoptive T-cell therapy (Cascone et al., 2018).
The impact of intratumoral glucose depletion on immunity is not restricted to T-cells. Aerobic glycolysis is a hallmark of inflammatory macrophages and is induced by endotoxin stimulation (Fukuzumi et al., 1996; Rodriguez-Prados et al., 2010). Pyruvate dehydrogenase kinase expression, which limits entry of glucose into the TCA cycle, is critical for macrophage polarization into an inflammatory M1 phenotype (Tan et al., 2015), and enhancing glycolytic flux has been shown to promote M1 polarization (Freemerman et al., 2014). Conversely, depletion of available glucose suppresses inflammatory macrophage differentiation (Venter et al., 2014).
The depletion of glucose within the tumor microenvironment may also help explain the regional hypovascularization often observed in tumors. Endothelial cells are largely dependent on glycolysis for bioenergetic homeostasis in a manner that is dependent on 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3)-mediated activation of phosphofructokinase-1 (De Bock et al., 2013). This highly glycolytic phenotype is further augmented in tumor-associated endothelial cells, likely due to high levels of VEGF in tumors, which upregulate PFKFB3 (Cantelmo et al., 2016). While endothelial cells within the mature vasculature have unlimited access to glucose from the blood, sprouting endothelial cells might be susceptible to glucose depletion during neovascularization. Indeed, decreased activity of phosphofructokinase-1 due to inhibition of PFKFB3 resulted in decreased proliferation and migration of sprouting endothelial cells (Cantelmo et al., 2016). Thus, reduced glucose availability might contribute to hypovascularization in certain regions of tumors.
Finally, stromal fibroblasts can also be reprogrammed by glucose depletion to support tumor growth. Glucose depletion and subsequent AMPK activation in fibroblasts might activate autophagy, driving secretion of NEAAs into the tumor microenvironment. As the tumor microenvironment is significantly depleted of NEAAs, stromal autophagy-dependent NEAA secretion can selectively promote tumor cell growth in vivo (Katheder et al., 2017; Sousa et al., 2016).
The effects of glucose depletion on the multiple non-malignant cell types of a tumor may be derived from their physiologic roles in wound healing. Depletion of glucose in healing wounds can appropriately limit macrophage and effector T-cell-driven inflammation and concomitantly initiate M2 macrophage and regulatory T-cell-dependent TGFβ secretion, leading to fibroblast activation. This process may be in part dependent on activation of AMPK signaling in response to glucose deprivation, which drives both regulatory T-cell function and M2 macrophage polarization to support tissue regeneration (Angelin et al., 2017; Mounier et al., 2013), suggesting that during normal wound healing, glucose depletion triggers immune cells to activate a regenerative response. Similarly, activated myofibroblasts depend on high glycolytic rates for proliferation and matrix production (Andrianifahanana et al., 2016; Bernard et al., 2015). Low glucose availability might therefore limit excessive fibrosis and scar formation during wound healing. In fact, the transition from inflammation to regeneration may actually require glucose depletion, as evidenced by the enhanced tissue repair seen in mice upon glycolysis inhibition (Shyh-Chang et al., 2013).
Lactate
The combination of high rates of aerobic glycolysis and diminished waste clearance due to inadequate vascularization leads to an accumulation of lactate in the tumor microenvironment. Up to 40 mM lactate can be found inside tumors, and high intratumoral lactate concentrations are associated with an increased risk of metastasis and death (Walenta et al., 2000). Once regarded solely as a metabolic waste product, lactate is now understood to fuel the bioenergetic needs of tissues under both physiologic and pathologic conditions. Indeed, mitochondrial oxidation of circulating lactate is seen in virtually every tissue in the body (Hui et al., 2017b). The fact that most tissues engage in some level of lactate excretion as well as lactate uptake and oxidation suggests that lactate can be shuttled between different cells. This phenomenon is known to occur in the Cori cycle, in which lactate produced by muscles is converted to glucose via gluconeogenesis in the liver, and then supplied back to muscles (Cori, 1981). In addition, fast-twitch glycolytic fibers and slow-twitch oxidative fibers exchange lactate in working skeletal muscle and heart (Baker et al., 1998; Bergman et al., 1999; Dubouchaud et al., 2000), and astrocytes shuttle lactate to neurons in the brain (Pellerin et al., 1998).
Lactate shuttling is facilitated by MCT proteins which catalyze the proton-coupled transport of lactate and other monocarboxylates along a concentration gradient (Halestrap and Price, 1999). While MCT1 is ubiquitously expressed in most tissues, MCT4 expression is confined to highly glycolytic cell types such as fast-twitch muscle fibers (Dimmer et al., 2000) and is induced under hypoxia in a HIF-1α dependent manner (Ullah et al., 2006), highlighting its key role in promoting lactate efflux under conditions when glycolysis is the major source of ATP. In contrast, MCT1 can promote either lactate uptake or secretion, depending on the metabolic state of a cell and its environment (Halestrap and Wilson, 2012). For example, MCT1 facilitates lactate influx into liver parenchymal cells or slow-twitch muscle fibers under conditions in which lactate accumulates, such as during exercise (Bonen, 2001). Conversely, MCT1 drives lactate export in highly glycolytic cells, such as rapidly proliferating T-cells (Murray et al., 2005).
Increasing evidence supports the existence of a lactate shuttle within tumors (Faubert et al., 2017; Sonveaux et al., 2008). MCT4 is expressed by tumor cells engaging in high rates of aerobic glycolysis, while MCT1 is expressed by tumor cells primarily displaying oxidative metabolism and particularly in cells that take up and oxidize lactate (Baek et al., 2014; Sonveaux et al., 2008). This suggests that tumor cells can variably excrete and utilize lactate in a manner that may depend on the extracellular environment; the importance of this possibility is underscored by metabolic tracing studies suggesting that both glucose and lactate are significant nutrient sources in vivo (Faubert et al., 2017; Hensley et al., 2016). An essential area of future investigation will be in defining whether distinct tumor cell subtypes primarily excrete or utilize lactate within the tumor microenvironment. One intriguing hypothesis is that cancer stem cells may preferentially be able to utilize lactate to maintain bioenergetic homeostasis. Lactate would be a particularly useful bioenergetic substrate for quiescent cancer stem cells, as it can be oxidized for ATP production but cannot independently drive proliferation due to the lack of a glyoxylate cycle in mammalian cells. Consistent with this hypothesis, cancer stem cells rely on mitochondrial metabolism (Janiszewska et al., 2012; Sancho et al., 2015; Viale et al., 2014; Vlashi et al., 2011), and are relatively insensitive to glucose deprivation (Pastò et al., 2014). Furthermore, chemotherapy resistant cancer cells are able to oxidize lactate (Ippolito et al., 2016), and inhibition of lactate uptake by targeting MCT1 reduced mammosphere formation by breast cancer cells (Lamb et al., 2014), suggesting that lactate uptake and oxidation can promote the long-term survival and tumor initiating capacity of cancer stem cells.
In addition to paracrine effects between tumor cell subtypes, lactate accumulation has significant paracrine effects on non-malignant cells in the tumor microenvironment. For example, during co-culture with tumor-cells, fibroblasts have been shown to activate aerobic glycolysis and MCT4-dependent lactate excretion, while tumor cells activate MCT1-mediated lactate uptake and oxidation (Fiaschi et al., 2012; Whitaker-Menezes et al., 2011). Tumor-promoting effects of CAFs are dependent on MCT4-mediated lactate excretion (Knudsen et al., 2016), and stromal MCT4 expression has been correlated with poor prognosis in patients with triple-negative breast cancer (Witkiewicz et al., 2012), suggesting that CAFs promote tumor growth by contributing to tumoral lactate production (Figure 4). CAFs may also provide support of tumor cell homeostasis by serving as a pH buffer; CAFs express the acid transporter SLC4A2, which confers a high capacity to absorb extracellular acid, thereby potentially buffering lactate-dependent acidification in the tumor microenvironment (Hulikova et al., 2016).
Figure 4. Lactate’s effects on tumor and stromal cells.
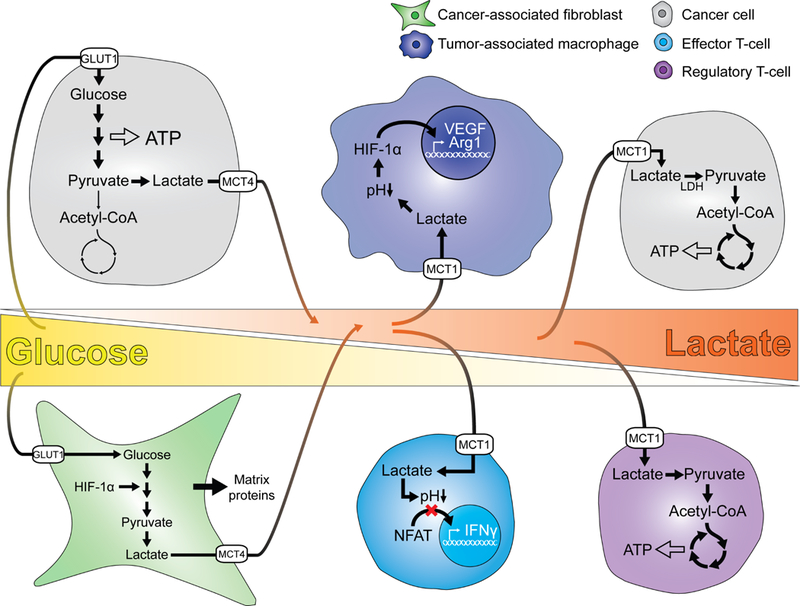
Glucose is present in high levels in proximity to the vasculature (left) and is consumed by tumor cells and cancer-associated fibroblasts. Glucose consumption is coupled to lactate secretion, resulting in lactate accumulation with increasing distance from the efferent vasculature. In poorly vascularized regions (right), cancer cells and regulatory T-cells are able to metabolize lactate to sustain energy homeostasis. In contrast, lactate exerts paracrine effects on tumor-associated macrophages and effector T-cells, stimulating differentiation of macrophages into an immunosuppressive M2-like phenotype and suppressing T-cell effector function.
Lactate also has important effects on the immune cells infiltrating a tumor. Lactate accumulation suppresses T-cell glycolytic flux by opposing lactate export, impairing T-cell effector function (Brand et al., 2016; Fischer et al., 2015; Haas et al., 2015). Conversely, regulatory T-cells, which are potently able to take up and oxidize lactate, are metabolically favored by high lactate concentrations, particularly when glucose is limiting (Angelin et al., 2017) (Figure 4). How lactate exerts its effects on T-cells remains an area of active study. Lactate-dependent intracellular acidification has been shown to prevent nuclear NFAT translocation in activated T-cells, resulting in diminished IFNγ production (Brand et al., 2016) (Figure 4). In addition, intracellular acidification may also affect the substrate specificity of metabolic enzymes. For example, LDHA increases its promiscuous activity against α-ketoglutarate to form the oncometabolite 2-hydroxyglutarate as intracellular pH decreases (Intlekofer et al., 2017), which can suppress T-cell function by perturbing nuclear NFAT translocation and activation of NFAT target genes (Bunse et al., 2018).
The paracrine effects of intratumoral lactate extend beyond T-cells. Lactate inhibits monocyte migration and secretion of pro-inflammatory cytokines, alters antigenpresentation by monocyte-derived dendritic cells, and drives macrophages into an immunosuppressive M2-like state, particularly in the setting of hypoxia (Colegio et al., 2014; Dietl et al., 2010; Goetze et al., 2011) (Figure 4). Furthermore, lactate can trigger tube formation in endothelial cells via HIF-1α-dependent NF-kB activation (Carmona-Fontaine et al., 2017; Végran et al., 2011). Given that sprouting endothelial cells depend on glucose uptake and metabolism (De Bock et al., 2013), it is possible that neovascularization is driven by glucose availability, while local accumulation of lactate drives blood vessel maturation. In support of this hypothesis, extracellular acidification, which is a direct consequence of lactate accumulation in the tumor microenvironment, prevents apoptosis and induces an ER stress response in endothelial cells (D’Arcangelo et al., 2002; Dong et al., 2013), which might contribute to survival of the newly formed vasculature.
The importance of lactate as a paracrine factor is underscored by its emerging role in wound healing. Similar to tumors, the concentration of lactate in healing wounds rises significantly, from 1.8–2 mM to 10–15 mM (Gladden, 2004). Lactate serves to support tissue regeneration, as evidenced by the fact that exogenous lactate supplementation stimulated neovascularization (Hunt et al., 2007) and improved reperfusion and healing of ischemic hindlimb wounds (Porporato et al., 2012). On a molecular level, increasing lactate concentration acts as a chemoattractant for mesenchymal cells and enhances collagen deposition (Rattigan et al., 2012; Savolainen et al., 1984; Trabold et al., 2003). Furthermore, treating wounds with lactate results in increased VEGF secretion by macrophages, inducing angiogenesis (Constant et al., 2000; Trabold et al., 2003). These findings indicate that lactate may serve as a potent paracrine signal and selective energetic substrate, permitting survival of tumor cells while reprogramming the stromal environment to promote a regenerative response, including angiogenesis and immunosuppression.
Amino acids
Oncogene-driven activation of cell growth is associated with an increased rate of amino acid uptake (Eagle, 1955a; Eagle, 1955b; Edinger and Thompson, 2002), resulting in depletion of several critical NEAAs including glutamine, serine and cystine from tumors compared to adjacent benign tissue (Kamphorst et al., 2015) (Figure 3). In addition to glutamine’s critical role for cell growth and proliferation discussed above, serine provides essential one-carbon units for nucleotide synthesis (Maddocks et al., 2016), while cystine is essential for maintaining redox homeostasis by serving as a precursor for glutathione biosynthesis. The uptake of amino acids is critical for cell growth, as amino acids acquired from the extracellular environment comprise the majority of the biomass of proliferating cells (Hosios et al., 2016).
How do cancer cells sustain proliferation as these nutrients become depleted from the extracellular space? First, they express high surface transporter levels to enhance uptake from existing nutrient sources. Second, they utilize alternative methods to acquire amino acids, such as macropinocytosis of extracellular proteins (Commisso et al., 2013; Kamphorst et al., 2015). For example, cancer cells are able to catabolize ECM components to maintain energy homeostasis under conditions where extracellular glucose and amino acids are depleted (Muranen et al., 2017; Olivares et al., 2017).
In contrast to cancer cells, stromal cells cease to proliferate and instead activate autophagy in response to nutrient deprivation. The resultant catabolism of intracellular proteins results in the release of nonessential amino acids (NEAAs) into the extracellular space, which can contribute to tumor growth (Katheder et al., 2017; Sousa et al., 2016). For example, in response to glutamine deficiency, CAFs sustain tumor growth by activating autophagy to supply cancer cells with asparagine and other NEAAs (Linares et al., 2017; Yang et al., 2016). Activation of autophagy within stromal fibroblasts can supply alanine to tumor cells, providing an alternate anaplerotic substrate to maintain TCA cycle homeostasis when glucose and glutamine are depleted (Sousa et al., 2016). These studies suggest that autophagy is a key element driving the metabolic exchange between CAFs and cancer cells.
How are fibroblasts and other stromal cells induced to secrete amino acids that support tumor growth? The secretion of NEAAs by fibroblasts has been shown to depend on stabilization of the stress responsive transcription factor ATF4 in response to downregulation of the negative autophagy regulator p62 (Linares et al., 2017). P62 is degraded by autophagy, and its downregulation in the stromal compartment has been documented in a number of cancers (Valencia et al., 2014). Autophagy might be activated in stromal cells by depletion of available nutrients, including glucose. Another mechanism by which autophagy can be activated in stromal fibroblasts is through oxidative stress, which is detected upon co-culture with cancer cells (Martinez-Outschoorn et al., 2010a). Oxidative stress can be driven by depletion of available glutamine, cystine, and glycine, which are required for glutathione biosynthesis, and can be further enhanced by the relative hypoxia seen in poorly vascularized tumors. Generation of ROS in stromal fibroblasts promotes autophagy and leads to metabolic reprogramming (Valencia et al., 2014). Indeed, ROS accumulation in response to nutrient stress in the tumor stroma promoted tumor growth via autophagy-dependent amino acid transfer (Katheder et al., 2017). The role of oxidative stress in promoting fibroblast activation and metabolic reprogramming both under physiological conditions and in cancer has been highlighted by numerous studies and may involve HIF-1α stabilization (Fu et al., 2008; Jain et al., 2013; Ono et al., 2009; Toullec et al., 2010). Given that ROS also play a critical role in the normal wound healing response (Dunnill et al., 2017), induction of oxidative stress through depletion of extracellular nutrients may serve as an additional mechanism by which cancer cells can orchestrate a stromal regenerative response.
The depletion of critical NEAAs from the extracellular environment by cancer cells also contributes to the suppression of anti-tumor immune responses. Like most rapidly proliferating cells, T-cells engage in high amounts of Myc-dependent glutamine uptake and utilization (Nakaya et al., 2014; Wang et al., 2011). As a result, T-cells are dependent on exogenous glutamine to sustain proliferation (Carr et al., 2010). Decreasing glutamine availability not only suppresses T-cell growth but actually drives differentiation of T-cells into the regulatory T-cell lineage (Klysz et al., 2015), suggesting that depletion of glutamine within the tumor microenvironment may convert anti-tumor effector T-cells into regulatory T-cells.
The amino acid requirements for T-cell proliferation and effector function extend beyond glutamine; arginine, cysteine, serine and tryptophan have all been shown to be critical for effective T-cell responses (Angelini et al., 2002; Fletcher et al., 2015; Geiger et al., 2016; Rodriguez et al., 2007). Serine donates essential one-carbon units for de novo nucleotide biosynthesis, and its depletion within tumors may contribute to impaired intratumoral T-cell proliferation (Ma et al., 2017). Arginine and tryptophan are depleted primarily by the inducible expression of enzymes that facilitate their degradation. Indoleamine-2,3-dioxygenase (IDO1) and tryptophan-2,3dioxygenase (TDO2) are induced primarily in tumor cells in response to IFNγ but are also expressed by CAFs (Hsu et al., 2016). These enzymes suppress T-cell proliferation by catabolizing tryptophan into kynurenine, which can induce regulatory T-cell differentiation through the aryl hydrocarbon receptor (Munn, 2006; Opitz et al., 2011). Hydrolysis of arginine by Arg1, which is produced by M2 macrophages under conditions of hypoxia or high lactate (Colegio et al., 2014), also limits T-cell proliferation (Geiger et al., 2016). The degradation of both tryptophan and arginine are two examples of how the metabolic manipulation of the extracellular environment is an adaptive mechanism by which tumor and stromal cells exert non-cell autonomous effects on other cell types within the tumor microenvironment.
Inflammatory macrophages are also highly sensitive to depletion of key NEAAs. To neutralize ROS generated during respiratory bursts, inflammatory M1 macrophages are highly dependent on both NADPH produced by the pentose phosphate pathway (Freemerman et al., 2014; Haschemi et al., 2012) and maintenance of intracellular reduced glutathione pools. Intratumoral depletion of cystine and serine, whose metabolism is essential for maintaining NADPH and glutathione, promotes accumulation of ROS to toxic levels in inflammatory macrophages during a chronic anti-tumor response (Nabeyama et al., 2010). Intriguingly, healing wounds also utilize amino acid depletion to limit the inflammatory response and initiate tissue regeneration; IFNγ triggers IDO1 expression in macrophages and fibroblasts, depleting regional tryptophan and enhancing local regulatory T-cell differentiation to promote the repair process (Curran et al., 2014; Opitz et al., 2011).
Finally, endothelial cells also depend on NEAAs. Endothelial cells require glutamine for TCA cycle anaplerosis and asparagine biosynthesis, and glutamine deprivation prevents endothelial cell proliferation, resulting in impaired vessel sprouting (Huang et al., 2017; Kim et al., 2017). In addition, serine is critical for mitochondrial function (Vandekeere et al., 2018), and glycine is required for the angiogenic effects of VEGF in endothelial cells (Guo et al., 2017). Thus, depletion of NEAAs might also contribute to vascular defects observed in some tumors.
Hypoxia
The growth of cancer cells frequently outstrips the local blood supply, leading to regional hypoxia (Dewhirst et al., 1994; Graeber et al., 1996; Helmlinger et al., 1997) (Figure 3). In non-transformed cells, growth is suppressed under low oxygen tension (Lum et al., 2007). In contrast, many cancer cells are able to continue to grow and proliferate at oxygen tensions that normally suppress growth. This is in part due to synergy between oncogenic activation of glucose uptake and hypoxic stabilization of HIF-1α. This synergy enables tumor cells to outcompete stromal cells for the minimal available glucose in hypovascularized, hypoxic tumor regions.
Stromal cells within the tumor microenvironment are therefore subject to depletion of both available oxygen and glucose. While the impact of hypoxia alone on immune cells within the tumor environment is controversial, the combination of hypoxia and glucose depletion clearly exerts an immunosuppressive effect. For example, stabilization of HIF-1α alone can enhance cytotoxic T-cell function and tumor control (Doedens et al., 2013), however, T-cell activation is impaired under hypoxic conditions (Lukashev et al., 2006), and this effect is enhanced when glucose availability becomes limiting (Zhang et al., 2017). Similarly, M1 macrophages rely on HIF-1α-dependent glycolytic metabolism for production of pro-inflammatory cytokines (Cramer et al., 2003). However, under the combination of glucose depletion, low oxygen tension, and lactate accumulation, macrophages adopt a tolerogenic M2-like phenotype with marked Arg1 expression (Carmona-Fontaine et al., 2017; Movahedi et al., 2010). This combination of hypoxia and glucose depletion may explain the profound accumulation of immunosuppressive macrophages within hypoxic tumor regions (Murdoch et al., 2004).
The complexity of hypoxia’s effects within the tumor microenvironment extends to fibroblasts. Hypoxia promotes HIF-1α-driven expression of profibrotic genes (Norman et al., 2000; Orphanides et al., 1997), production of TGFβ (Ammirante et al., 2014; Modarressi et al., 2010), and autophagy in fibroblasts (Martinez-Outschoorn et al., 2010b), all of which can promote tumor growth. However, hypoxic tumor regions are notably devoid of αSMA-positive fibroblasts (Madsen et al., 2015). Given the established dependence of activated fibroblasts on aerobic glycolysis (Bernard et al., 2015; Zhang et al., 2015), it is reasonable to propose that hypoxia and glucose deprivation individually drive fibroblast function, while the concurrent presence of hypoxia and glucose deprivation leads to impaired survival of CAFs in the tumor core.
Metabolic changes mediated by secreted factors and receptor-ligand interactions
As mentioned previously, in addition to changing the metabolic microenvironment, tumor cells also influence stromal cells through the secretion of soluble factors such as cytokines, growth factors and exosomes, or through ligand-receptor interactions. While these have been well-reviewed by others, it is worth mentioning that many of these paracrine factors may exert their effects by altering stromal cell metabolism. An example of how cellular metabolism contributes to the effects of tumor-derived soluble factors is TGFβ, which is produced by many cancer subtypes and is associated with an inferior prognosis (Robson et al., 1996; Saito et al., 1999). Moreover, TGFβ is known to have potent effects on multiple non-malignant cell types. TGFβ alone can drive differentiation of normal fibroblasts into myofibroblasts (Rønnov-Jessen and Petersen, 1993), thereby promoting tumor growth and invasion (Casey et al., 2008; Guido et al., 2012; Zhang et al., 2015). Furthermore, TGFβ is essential for the differentiation of regulatory T-cells (Fantini et al., 2004) and contributes to their intratumoral accumulation (Liu et al., 2007); as a result, TGFβ production is associated with an inferior response to immunotherapy (Mariathasan et al., 2018; Tauriello et al., 2018). Recently, it has been observed that TGFβ may impose these effects on non-malignant cells via effects on cellular metabolism. For example, TGFβ-dependent activation of fibroblasts requires an increase in glutaminolysis (Bernard et al., 2018), and TGFβ-dependent induction of FoxP3 promotes the ability of regulatory T-cells to survive under glucose-limiting conditions by enhancing their capacity to utilize lactate as a bioenergetic substrate (Angelin et al., 2017).
A second potent example of the metabolic effects of receptor-ligand interactions within the tumor microenvironment is PD-1/PD-L1. PD-L1 expression limits excess inflammation and tissue damage by suppressing T-cell function following activation (Frebel et al., 2012), in part by shifting T-cell differentiation towards “exhaustion”, rendering T-cells non-responsive to antigen (Barber et al., 2006). There is now evidence to support the idea that alterations in cellular metabolism support the effect of PD-1/PD-L1 interactions. PD-L1/PD-1 interactions can enhance glucose uptake in tumor cells (Chang et al., 2015) while diminishing glucose uptake in T-cells (Parry et al., 2005), both of which support the ability of the tumor microenvironment to suppress effector T-cell function. Inhibition of PD-1/PD-L1 interactions enhances glucose uptake in effector T-cells (Chang et al., 2015), suggesting that enhancement of anti-tumor immune responses by checkpoint inhibitors may be in part due to metabolic reprogramming of immune cells within the tumor environment.
Lastly, the role of exosomes in mediating communication and metabolic interaction between cancer cells and stromal cells merits further exploration. These small membrane-derived vesicles transport a variety of cargo between cells including macromolecules and biologically active substances (Maia et al., 2018). For example, exosomal transport of TGFβ from cancer cells to fibroblasts induces CAF differentiation (Ringuette Goulet et al., 2018), and CAF-derived exosomes containing TGFβ promote epithelial-mesenchymal transition in cancer cells (Li et al., 2017). CAF-secreted exosomes rewire cancer cell metabolism by promoting a shift from oxidative metabolism to aerobic glycolysis and reductive carboxylation of glutamine (Zhao et al., 2016). In addition, exosomes secreted by CAFs can sustain cancer cell viability under amino acid deprivation by providing a source of free metabolites that supply up to one third of TCA cycle fluxes (Achreja et al., 2017; Zhao et al., 2016). Whether cancer cell-mediated metabolic changes in the tumor microenvironment can promote exosome secretion by stromal cells is yet to be determined.
Conclusion and perspectives
The study of cancer metabolism has revealed the remarkable plasticity of cancer cells to acquire nutrients for growth and survival under even the harshest conditions. In contrast, non-malignant cells remain exquisitely sensitive to nutrient depletion and metabolic waste accumulation. Establishment and maintenance of the metabolic microenvironment by tumor cells therefore contributes strongly to stromal cell identity. Many of the phenotypes stromal cells acquire in the microenvironment of a tumor resemble those induced during wound healing. As summarized above, tumor cell-mediated depletion of glucose, oxygen and amino acids as well as accumulation of lactate and amino acid catabolites, contribute to a stromal microenvironment that suppresses inflammation and supports regeneration.
Despite the highly overlapping features of stromal cells in the tumor stroma and a healing wound, differences in cell phenotypes should be noted. For instance, although tumor-associated macrophages share many similarities with M2 macrophages, their gene expression program is not identical. Furthermore, both M1- and M2-like macrophage subtypes can be found within tumors. While macrophage polarization can be regulated by their environment, as evidenced by differential prevalence of macrophage subtypes based on oxygen tension (Movahedi et al., 2010), other factors undoubtedly contribute to their accumulation in tumors. More work will be needed to delineate how multiple metabolic inputs converge to establish the diversity of stromal cell subtypes within the tumor microenvironment.
It is important to note the myriad of additional nutrients, metabolic intermediates, and metabolic end-products that is present within the extracellular environment of tumors, which may alter the fate of stromal cells. Recently, there has been a growing interest in additional waste products of tumor metabolism, including formaldehyde and ammonia (Burgos-Barragan et al., 2017; Meiser et al., 2018; Spinelli et al., 2017). Ammonia derived from tumor cell glutaminolysis has been shown to trigger autophagy in a paracrine manner (Eng et al., 2010). As autophagy is an important mediator of the metabolic exchange between stromal and cancer cells (Katheder et al., 2017; Sousa et al., 2016), it is possible that ammonia production by tumor cells could be an additional trigger of stromal catabolic processes that facilitate support of tumor cell growth. The role of lipid metabolism in tumor-stroma crosstalk is also unclear. Recently, it has been demonstrated that tumor cells can scavenge lipids from extracellular sources to support proliferation (Kamphorst et al., 2013). The source of extracellular lipids is not clear, but stromal adipocytes appear to be likely candidates (Huang et al., 2018; Ladanyi et al., 2018; Nieman et al., 2011). Whether nutrient depletion reprograms stromal adipocytes to provide tumor cells with a source of lipids remains to be determined.
Finally, the increasing evidence of bidirectional metabolic crosstalk between tumor and stromal cells underscores the need to directly interrogate the metabolic behavior of distinct cell types in vivo. Recent work has highlighted that the metabolic behavior of tumors in vivo is distinct from the same tumors grown in vitro, including differential metabolism of glucose (Courtney et al., 2018) as well as variable utilization of alternative nutrients such as branched-chain amino acids based on the tissue of residence (Mayers et al., 2016). The technical challenges of interrogating metabolic fluxes have largely limited evaluation of the metabolic interaction of cancer and stromal cells to in vitro systems. Indeed, these systems have revealed how the metabolic behavior of cells changes under defined nutrient conditions as detailed in the numerous studies referenced above, including how stromal fibroblasts can sustain viability of cancer cells by acting as a nutrient source, or that depletion of some and enrichment of other metabolites creates an inhospitable milieu for some immune cell types, while others can thrive. However, when considered in the context of the growing list of potential metabolic fuels and waste products present within the tumor microenvironment as well as the distinct metabolic phenotypes emerging from in vivo systems, an integrated assessment of tumor metabolism that includes direct interrogation of the metabolic behavior of multiple interacting cell types in vivo is an area of growing need. Further advances in the utilization of stable isotope tracers, imaging-based assays of metabolic activity, and improvements in the sensitivity of mass spectrometry-based metabolite evaluation permitting the isolation and interrogation of multiple cellular subsets will allow us to validate previous findings and establish novel metabolic vulnerabilities in an in vivo setting with multiple stromal cell types interacting in parallel. Creating such an understanding of tumor metabolism-driven stromal cell fate changes will finally help to overcome many stroma-mediated therapeutic barriers.
Acknowledgement
We thank the members of the Thompson laboratory for critical discussions during manuscript preparation. S.S. is supported by postdoctoral fellowships from the Human Frontier Science Program and the European Molecular Biology Organization. S.A.V. is supported by a Fellow Award from the Parker Institute of Cancer Immunotherapy. This work was supported by grants from the NCI (C.B.T.) and the MSKCC Cancer Center Support Grant P30 CA008748.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
C.B.T. is a founder of Agios Pharmaceuticals and a member of its scientific advisory board. He is a former member of the Board of Directors and a stockholder of both Merck and Charles River Laboratories. He is a named inventor on patents related to cellular metabolism.
References
- Achreja A, Zhao H, Yang L, Yun TH, Marini J, and Nagrath D (2017). ExoMFA – A 13C metabolic flux analysis framework to dissect tumor microenvironmentsecreted exosome contributions towards cancer cell metabolism. Metab. Eng 43, 156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler M, Mayo A, Zhou X, Franklin RA, Jacox JB, Medzhitov R, and Alon U (2018). Endocytosis as a stabilizing mechanism for tissue homeostasis. Proc. Natl. Acad. Sci 115, E1926–E1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammirante M, Shalapour S, Kang Y, Jamieson CAM, and Karin M (2014). Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl. Acad. Sci 111, 14776–14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianifahanana M, Hernandez DM, Yin X, Kang JH, Jung MY, Wang Y, Yi ES, Roden AC, Limper AH, and Leof EB (2016). Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J 30, 3733–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L, et al. (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25, 1282–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, Clarke F, Sitia R, and Rubartelli A (2002). Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc. Natl. Acad. Sci. U. S. A 99, 1491–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardawi MS (1988). Skeletal muscle glutamine production in thermally injured rats. Clin. Sci. (Lond) 74, 165–172. [DOI] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, and Chazaud B (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek GH, Tse YF, Hu Z, Cox D, Buboltz N, McCue P, Yeo CJ, White MA, DeBerardinis RJ, Knudsen ES, et al. (2014). MCT4 Defines a Glycolytic Subtype of Pancreatic Cancer with Poor Prognosis and Unique Metabolic Dependencies. Cell Rep 9, 2233–2249. [DOI] [PubMed] [Google Scholar]
- Baker SK, McCullagh KJA, and Bonen A (1998). Training intensity-dependent and tissue-specific increases in lactate uptake and MCT-1 in heart and muscle. J. Appl. Physiol 84, 987–994. [DOI] [PubMed] [Google Scholar]
- Balkwill F (2004). Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550. [DOI] [PubMed] [Google Scholar]
- Balkwill F, and Mantovani A (2001). Inflammation and cancer: Back to Virchow? Lancet 357, 539–545. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, and Ahmed R (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. [DOI] [PubMed] [Google Scholar]
- Bergman BC, Wolfel EE, Butterfield GE, Lopaschuk GD, Casazza GA, Horning MA, and Brooks GA (1999). Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol 87, 1684–1696. [DOI] [PubMed] [Google Scholar]
- Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, Rangarajan S, Zmijewski JW, Mitra K, Liu G, Darley-Usmar VM, et al. (2015). Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem 290, 25427–25438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard K, Logsdon NJ, Benavides GA, Sanders Y, Zhang J, Darley-Usmar VM, and Thannickal VJ (2018). Glutaminolysis is required for transforming growth factor-β1-induced myofibroblast differentiation and activation. J. Biol. Chem 293, 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi R, Adami C, Giambanco I, and Donato R (2007). S100B binding to RAGE in microglia stimulates COX-2 expression. J. Leukoc. Biol 81, 108–118. [DOI] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. (2013). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663. [DOI] [PubMed] [Google Scholar]
- Bonen A (2001). The expression of lactate transporters (MCT1 and MCT4) in heart and muscle. Eur. J. Appl. Physiol 86, 6–11. [DOI] [PubMed] [Google Scholar]
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. (2016). LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 24, 657–671. [DOI] [PubMed] [Google Scholar]
- Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, Alansary D, Sonner JK, Green E, Deumelandt K, et al. (2018). Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med 24, 1192–1203. [DOI] [PubMed] [Google Scholar]
- Burgos-Barragan G, Wit N, Meiser J, Dingler FA, Pietzke M, Mulderrig L, Pontel LB, Rosado IV, Brewer TF, Cordell RL, et al. (2017). Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 548, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, et al. (2013). A Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell 155, 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell MD (1989). Local glutamine metabolism in wounds and inflammation. Metabolism 38, 34–39. [DOI] [PubMed] [Google Scholar]
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, et al. (2013). Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol 15, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps JL, Chang SM, Hsu TC, Freeman MR, Hong SJ, Zhau HE, von Eschenbach a C., and Chung LW (1990). Fibroblast-mediated acceleration of human epithelial tumor growth in vivo. Proc. Natl. Acad. Sci. U. S. A 87, 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen LA, et al. (2016). Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 30, 968–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, and Xavier JB (2017). Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci 114, 2934–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, and Frauwirth KA (2010). Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J. Immunol 185, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, and Mazzone M (2014). Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene 33, 1743–1754. [DOI] [PubMed] [Google Scholar]
- Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al. (2018). Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab 27, 977–987.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey TM, Eneman J, Crocker A, White J, Tessitore J, Stanley M, Harlow S, Bunn JY, Weaver D, Muss H, et al. (2008). Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-β1) increase invasion rate of tumor cells: a population study. Breast Cancer Res. Treat 110, 39–49. [DOI] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, Huang SCC, Van Der Windt GJW, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, Van Der Windt GJW, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, Van De Rijn M, Botstein D, and Brown PO (2004). Gene expression signature of fibroblast serum response predicts human cancer progression: Similarities between tumors and wounds. PLoS Biol 2, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Nuyten DSA, Sneddon JB, Hastie T, Tibshirani R, Sørlie T, Dai H, He YD, van’t Veer LJ, Bartelink H, et al. (2005). Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc. Natl. Acad. Sci. U. S. A 102, 3738–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, and Sato H (2012). The oxidative stress-inducible cystine/glutamate antiporter, system xc-: Cystine supplier and beyond. Amino Acids 42, 231–246. [DOI] [PubMed] [Google Scholar]
- Constant JS, Feng JJ, Zabel DD, Yuan H, Suh DY, Scheuenstuhl H, Hunt TK, and Hussain MZ (2000). Lactate elicits vascular endothelial growth factor from macrophages: a possible alternative to hypoxia. Wound Repair Regen 8, 353–360. [DOI] [PubMed] [Google Scholar]
- Cori CF (1981). The glucose-lactic acid cycle and gluconeogenesis. Curr. Top. Cell. Regul 18, 377–387. [PubMed] [Google Scholar]
- Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, Cho H-I, Celis E, Quiceno DG, Padhya T, et al. (2010). HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med 207, 2439–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P, Lotan Y, et al. (2018). Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab 28, 793–800.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, and Werb Z (2002). Inflammation and Cancer. Nature 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. (2003). HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran T-A, Jalili RB, Farrokhi A, and Ghahary A (2014). IDO expressing fibroblasts promote the expansion of antigen specific regulatory T cells. Immunobiology 219, 17–24. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo D, Gaetano C, and Capogrossi MC (2002). Acidification prevents endothelial cell apoptosis by Axl activation. Circ. Res 91, e4–12. [DOI] [PubMed] [Google Scholar]
- Dai X-M, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, and Stanley ER (2002). Targeted disruption of the mouse colonystimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 99, 111–120. [DOI] [PubMed] [Google Scholar]
- Deberardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, and Thompson CB (2007). Beyond aerobic glycolysis : Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci 104, 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmoulière A, and Gabbiani G (1988). The Role of the Myofibroblast in Wound Healing and Fibrocontractive Diseases. In The Molecular and Cellular Biology of Wound Repair, (Boston, MA: Springer US; ), pp. 391–423. [Google Scholar]
- Desmoulière A, Geinoz A, Gabbiani F, and Gabbiani G (1993). Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol 122, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst MW, Secomb TW, Ong ET, Hsu R, and Gross JF (1994). Determination of local oxygen consumption rates in tumors. Cancer Res 54, 3333–3336. [PubMed] [Google Scholar]
- Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, Hellerbrand C, Kastenberger M, Kunz-Schughart LA, Oefner PJ, et al. (2010). Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes. J. Immunol 184, 1200–1209. [DOI] [PubMed] [Google Scholar]
- Dimmer KS, Friedrich B, Lang F, Deitmer JW, and Bröer S (2000). The lowaffinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J 350 Pt 1, 219–227. [PMC free article] [PubMed] [Google Scholar]
- Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, and Goldrath AW (2013). Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol 14, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Li Z, Leffler NR, Asch AS, Chi J-T, and Yang LV (2013). Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS One 8, e61991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubouchaud H, Butterfield GE, Wolfel EE, Bergman BC, and Brooks GA (2000). Endurance training, expression, and physiology of LDH, MCT1, and MCT4 in human skeletal muscle. Am. J. Physiol. Metab 278, E571–E579. [DOI] [PubMed] [Google Scholar]
- Dunnill C, Patton T, Brennan J, Barrett J, Dryden M, Cooke J, Leaper D, and Georgopoulos NT (2017). Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int. Wound J 14, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF (1986). Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- Dvorak HF (2015). Tumors: Wounds That Do Not Heal--Redux. Cancer Immunol. Res 3, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H (1955a). The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J. Exp. Med 102, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H (1955b). Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514. [DOI] [PubMed] [Google Scholar]
- Edinger AL, and Thompson CB (2002). Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 13, 2276–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Nakasone ES, and Werb Z (2010). Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 18, 884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. (2004). Akt stimultes aerobic glycolysis in cancer cells. Cancer Res 64, 3892–3899. [DOI] [PubMed] [Google Scholar]
- Eming SA, Martin P, and Tomic-Canic M (2014). Wound repair and regeneration: mechanisms, signaling, and translation. Sci. Transl. Med 6, 265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CH, Yu K, Lucas J, White E, Abraham RT, Eng CH, Yu K, Lucas J, White E, and Abraham RT (2010). Ammonia Derived from Glutaminolysis Is a Diffusible Regulator of Autophagy. Sci. Signal 3, 1–11. [DOI] [PubMed] [Google Scholar]
- Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, and Rabinowitz JD (2014). Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, and Neurath MF (2004). Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol 172, 5149–5153. [DOI] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, and Chiarugi P (2012). Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 72, 5130–5140. [DOI] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. (2015). Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood 109, 3812–3820. [DOI] [PubMed] [Google Scholar]
- Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, Sanchez-Pino D, Hernez C, Wyczechowska DD, Ochoa AC, et al. (2015). LArginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res 75, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, and Thompson CB (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777. [DOI] [PubMed] [Google Scholar]
- Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, Wagner CA, Loffing-Cueni D, Kurrer M, Ludewig B, et al. (2012). Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med 209, 2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, and Makowski L (2014). Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem 289, 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zheng S, Lin J, Ryerse J, and Chen A (2008). Curcumin Protects the Rat Liver from CCl 4 -Caused Injury and Fibrogenesis by Attenuating Oxidative Stress and Suppressing Inflammation 73, 399–409. [DOI] [PubMed] [Google Scholar]
- Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, and Utsumi S (1996). Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect. Immun 64, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G, Ryan GB, and Majne G (1971). Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27, 549–550. [DOI] [PubMed] [Google Scholar]
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al. (2016). L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167, 829–842.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladden LB (2004). Lactate metabolism: A new paradigm for the third millennium. J. Physiol 558, 5–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleave M, Hsieh J, Gao C, Cleave M, Eschenbach A.C. Von, and Chung LWK (1991). Acceleration of Human Prostate Cancer Growth in Vivo by Factors Produced by Prostate and Bone Fibroblasts. Sci. Rep 3753–3761. [PubMed]
- Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, and MuellerKlieser W (2011). Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol 39, 453–463. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, and Giaccia AJ (1996). Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379, 88–91. [DOI] [PubMed] [Google Scholar]
- Gualdoni GA, Mayer KA, Göschl L, Boucheron N, Ellmeier W, and Zlabinger GJ (2016). The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB J 30, 3800–3809. [DOI] [PubMed] [Google Scholar]
- Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Ando S, Martinez-Outschoorn U, et al. (2012). Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg- like” cancer metabolism and L-lactate production. Cell Cycle 11, 3019–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullino PM, Clark SH, and Grantham FH (1964). The Interstitial Fluid of Solid Tumors. Cancer Res 24, 780–794. [PubMed] [Google Scholar]
- Guo D, Murdoch CE, Xu H, Shi H, Duan DD, Ahmed A, and Gu Y (2017). Vascular endothelial growth factor signaling requires glycine to promote angiogenesis. Sci. Rep 7, 14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, and Longaker MT (2008). Wound repair and regeneration. Nature 453, 314–321. [DOI] [PubMed] [Google Scholar]
- Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, et al. (2015). Lactate regulates metabolic and proinflammatory circuits in control of T cell migration and effector functions. PLoS Biol 13, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, and Balkwill FR (2008). “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med 205, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, and Price NT (1999). The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J 343 Pt 2, 281–299. [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, and Wilson MC (2012). The monocarboxylate transporter familyRole and regulation. IUBMB Life 64, 109–119. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Coussens LM (2012). Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 21, 309–322. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al. (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab 15, 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger G, Yuan F, Dellian M, and Jain RK (1997). Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat. Med 3, 177–182. [DOI] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Celetta G, Tomasek JJ, Gabbiani G, and Chaponnier C (2001). AlphaSmooth Muscle Actin Expression Upregulates Fibroblast Contractile Activity. Mol. Biol. Cell 12, 2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, MacIntyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR, and Vander Heiden MG (2016). Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 36, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y, Hung J, Chiang S, Jian S, Wu C, Lin Y-S, Tsai Y-M, Chou S-H, Tsai M-J, and Kuo P-L (2016). Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 7, 27584–27598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Vandekeere S, Kalucka J, Bierhansl L, Zecchin A, Brüning U, Visnagri A, Yuldasheva N, Goveia J, Cruys B, et al. (2017). Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J 36, e201695518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Duran A, Reina-Campos M, Valencia T, Castilla EA, Müller TD, Tschöp MH, Moscat J, and Diaz-Meco MT (2018). Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell 33, 770–784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, et al. (2017a). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Zhan Le, Yanxiang Guo J, et al. (2017b). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulikova A, Black N, Hsia L-T, Wilding J, Bodmer WF, and Swietach P (2016). Stromal uptake and transmission of acid is a pathway for venting cancer cellgenerated acid. Proc. Natl. Acad. Sci 113, E5344–E5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt TK, Aslam RS, Beckert S, Wagner S, Ghani QP, Hussain MZ, Roy S, and Sen CK (2007). Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid. Redox Signal 9, 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Byrns RE, Buga GM, Wood KS, and Chaudhuri G (1988). Pharmacological evidence that endothelium-derived relaxing factor is nitric oxide: use of pyrogallol and superoxide dismutase to study endothelium-dependent and nitric oxide-elicited vascular smooth muscle relaxation. J. Pharmacol. Exp. Ther 244, 181–189. [PubMed] [Google Scholar]
- Im MJC, and Hoopes JE (1970). Energy metabolism in healing skin wounds. J. Surg. Res 10, 459–464. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, Salah S, Gunner MR, Chodera JD, Cross JR, et al. (2017). L-2Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol 13, 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, Giannoni E, Schrader T, Puhr M, Chiarugi P, et al. (2016). Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 7, 61890–61904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, Kohn LD, and Klinman DM (2001). Genomic DNA released by dying cells induces the maturation of APCs. J. Immunol 167, 2602–2607. [DOI] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, and Rathmell JC (2008). Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol 180, 4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, FeghaliBostwick C, Mutlu GM, Budinger GRS, and Chandel NS (2013). Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem 288, 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Auwerx J, ClémentSchatlo V, Radovanovic I, Rheinbay E, Provero P, and Stamenkovic I (2012). Imp2 controls oxidative phosphorylation and is crucial for preservin glioblastoma cancer stem cells. Genes Dev 26, 1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. (2017). Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355, 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, and Rabinowitz JD (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhász G, et al. (2017). Microenvironmental autophagy promotes tumour growth. Nature 541, 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keibler MA, Wasylenko TM, Kelleher JK, Iliopoulos O, Vander Heiden MG, and Stephanopoulos G (2016). Metabolic requirements for cancer cell proliferation. Cancer Metab 4, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Li J, Jang C, and Arany Z (2017). Glutamine fuels proliferation but not migration of endothelial cells. EMBO J 36, 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, et al. (2015). Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal 8, ra97. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Balaji U, Freinkman E, McCue P, and Witkiewicz AK (2016). Unique metabolic features of pancreatic cancer stroma: relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget 7, 78396–78411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, and Mathis D (2016). Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 44, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, et al. (2018). Adipocyteinduced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37, 2285–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, and Sotgia F (2014). Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT½ inhibition. Oncotarget 5, 11029–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, and Harris AL (1996). Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res 56, 4625–4629. [PubMed] [Google Scholar]
- Li W, Zhang X, Wang J, Li M, Cao C, Tan J, Ma D, and Gao Q (2017). TGFβ1 in fibroblasts-derived exosomes promotes epithelial-mesenchymal transition of ovarian cancer cells. Oncotarget 8, 96035–96047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM, and Diaz-Meco MT (2017). ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab 26, 817–829.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, Zhang Q, Lonning S, Teicher BA, and Lee C (2007). Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumorderived TGF-beta. J. Immunol 178, 2883–2892. [DOI] [PubMed] [Google Scholar]
- Liu Y-C, Li F, Handler J, Huang CRL, Xiang Y, Neretti N, Sedivy JM, Zeller KI, and Dang CV (2008). Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3, e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage UK, Moore TT, Joo H-G, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, et al. (2002). Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol 169, 2756–2761. [DOI] [PubMed] [Google Scholar]
- Loo JM, Scherl A, Nguyen A, Man FY, Weinberg E, Zeng Z, Saltz L, Paty PB, and Tavazoie SF (2015). Extracellular metabolic energetics can promote cancer progression. Cell 160, 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev D, Klebanov B, Kojima H, Grinberg A, Ohta A, Berenfeld L, Wenger RH, Ohta A, and Sitkovsky M (2006). Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J. Immunol 177, 4962–4965. [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, and Thompson CB (2007). The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev 21, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, et al. (2017). Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 25, 345–357. [DOI] [PubMed] [Google Scholar]
- Maddocks ODK, Labuschagne CF, Adams PD, and Vousden KH (2016). Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 61, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen CD, Pedersen JT, Venning FA, Singh LB, Moeendarbary E, Charras G, Cox TR, Sahai E, and Erler JT (2015). Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep 16, 1394–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia J, Caja S, Strano Moraes MC, Couto N, and Costa-Silva B (2018). Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol 6, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, et al. (2010a). Oxidative stress in cancer associated fibroblasts drives tumor-stroma coevolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 9, 3256–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, et al. (2010b). Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 9, 3515–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P (2002). The danger model: a renewed sense of self. Science 296, 301–305. [DOI] [PubMed] [Google Scholar]
- Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM, et al. (2016). Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiser J, Schuster A, Pietzke M, Vande Voorde J, Athineos D, Oizel K, Burgos-Barragan G, Wit N, Dhayade S, Morton JP, et al. (2018). Increased formate overflow is a hallmark of oxidative cancer. Nat. Commun 9, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modarressi A, Pietramaggiori G, Godbout C, Vigato E, Pittet B, and Hinz B (2010). Hypoxia impairs skin myofibroblast differentiation and function. J. Invest. Dermatol 130, 2818–2827. [DOI] [PubMed] [Google Scholar]
- Moncada S, Gryglewski R, Bunting S, and Vane JR (1976). An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 263, 663–665. [DOI] [PubMed] [Google Scholar]
- Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, Sanz N, Ferry A, Sakamoto K, Foretz M, et al. (2013). AMPK alpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab 18, 251–264. [DOI] [PubMed] [Google Scholar]
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van Bossche J. Den, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, et al. (2010). Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 70, 5728–5739. [DOI] [PubMed] [Google Scholar]
- Munn DH (2006). Indoleamine 2, 3-dioxygenase, tumor-induced tolerance and counter-regulation. Curr. Opin. Immunol 18, 220–225. [DOI] [PubMed] [Google Scholar]
- Muranen T, Iwanicki MP, Curry NL, Hwang J, DuBois CD, Coloff JL, Hitchcock DS, Clish CB, Brugge JS, and Kalaany NY (2017). Starved epithelial cells uptake extracellular matrix for survival. Nat. Commun 8, 13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Giannoudis A, and Lewis CE (2004). Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 104, 2224–2234 [DOI] [PubMed] [Google Scholar]
- Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, Bundick RV, Cook ID, Craggs RI, Edwards S, et al. (2005). Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol 1, 371–376. [DOI] [PubMed] [Google Scholar]
- Nabeyama A, Kurita A, Asano K, Miyake Y, Yasuda T, Miura I, Nishitai G, Arakawa S, Shimizu S, Wakana S, et al. (2010). xCT deficiency accelerates chemically induced tumorigenesis. Proc. Natl. Acad. Sci. U. S. A 107, 6436–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, Libby P, Weissleder R, and Pittet MJ (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med 204, 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, Blonska M, Lin X, and Sun S-C (2014). Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 40, 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional Transport of Amino Acids Regulates mTOR and Autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, et al. (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med 17, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman JT, Clark IM, and Garcia PL (2000). Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int 58, 2351–2366. [DOI] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, and Olefsky JM (2010). GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-inflammatory and Insulin-Sensitizing Effects. Cell 142, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, and Medzhitov R (2016). Tissue biology perspective on macrophages. Nat. Immunol 17, 9–17. [DOI] [PubMed] [Google Scholar]
- Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut M-N, Berthezène P, et al. (2017). Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun 8, 16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, and Cunha GR (1999). Carcinoma-associated Fibroblasts Direct Tumor Progression of Initiated HumanProstatic Epithelium.pdf. Cancer Res 5002–5011. [DOI] [PMC free article] [PubMed]
- Ono A, Utsugi M, Masubuchi K, Ishizuka T, Kawata T, Shimizu Y, Hisada T, Hamuro J, Mori M, and Dobashi K (2009). Glutathione redox regulates TGF- b induced fibrogenic effects through Smad3 activation 583, 357–362. [DOI] [PubMed] [Google Scholar]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, et al. (2011). An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203. [DOI] [PubMed] [Google Scholar]
- Orphanides C, Fine LG, and Norman JT (1997). Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int 52, 637–647. [DOI] [PubMed] [Google Scholar]
- De Palma M, and Lewis CE (2013). Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23, 277–286. [DOI] [PubMed] [Google Scholar]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, and Riley JL (2005). CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol 25, 9543–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastò A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, Rasola A, Frasson C, Nardo G, Zulato E, et al. (2014). Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 5, 4305–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, and Magistretti PJ (1998). Evidence supporting the existence of an activitydependent astrocyte-neuron lactate shuttle. Dev. Neurosci 20, 291–299. [DOI] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardí M, Caelles C, Serrano AL, and Muñoz-Cánoves P (2011). p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol 195, 307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Payen VL, De Saedeleer CJ, Préat V, Thissen JP, Feron O, and Sonveaux P (2012). Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis 15, 581–592. [DOI] [PubMed] [Google Scholar]
- Raff MC (1996). Size Control: The Regulation Minireview of Cell Numbers in Animal Development. Cell 86, 173–175. [DOI] [PubMed] [Google Scholar]
- Rattigan YI, Patel BB, Ackerstaff E, Sukenick G, Koutcher JA, Glod JW, and Banerjee D (2012). Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res 318, 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringuette Goulet C, Bernard G, Tremblay S, Chabaud S, Bolduc S, and Pouliot F (2018). Exosomes Induce Fibroblast Differentiation into CancerAssociated Fibroblasts through TGFβ Signaling. Mol. Cancer Res 16, 1196–1204. [DOI] [PubMed] [Google Scholar]
- Robson H, Anderson E, James RD, and Schofield PF (1996). Transforming growth factor beta 1 expression in human colorectal tumours: an independent prognostic marker in a subgroup of poor prognosis patients. Br. J. Cancer 74, 753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Prados J-C, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, and Bosca L (2010). Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol 185, 605–614. [DOI] [PubMed] [Google Scholar]
- Rodriguez PC, Quiceno DG, and Ochoa AC (2007). L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood J 109, 1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rønnov-Jessen L, and Petersen OW (1993). Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Invest 68, 696–707. [PubMed] [Google Scholar]
- Saito H, Tsujitani S, Oka S, Kondo A, Ikeguchi M, Maeta M, and Kaibara N (1999). The expression of transforming growth factor-beta1 is significantly correlated with the expression of vascular endothelial growth factor and poor prognosis of patients with advanced gastric carcinoma. Cancer 86, 1455–1462. [DOI] [PubMed] [Google Scholar]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Graña O, et al. (2015). MYC/PGC1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab 22, 590–605. [DOI] [PubMed] [Google Scholar]
- Savolainen ER, Leo MA, Timpl R, and Lieber CS (1984). Acetaldehyde and lactate stimulate collagen synthesis of cultured baboon liver myofibroblasts. Gastroenterology 87, 777–787. [PubMed] [Google Scholar]
- Schornack PA, and Gillies RJ (2003). Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia 5, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Evans JE, and Rock KL (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Zhu H, Yvanka De Soysa T, Shinoda G, Seligson MT, Tsanov KM, Nguyen L, Asara JM, Cantley LC, and Daley GQ (2013). Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell 155, 778–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer AJ, and Clark RA (1999). CUTANEOUS WOUND HEALING. N. Engl. J. Med 341, 738–746. [DOI] [PubMed] [Google Scholar]
- Solinas G, Germano G, Mantovani A, and Allavena P (2009). Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol 86, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, Saedeleer C.J. De, Kennedy KM, Diepart C, Jordan BF, et al. (2008). Targeting lactate-fueled respiration selectivelt kills hypoxic tumor cells in mice. J. Clin. Invest 118, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, and Haigis MC (2017). Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, et al. (2010). Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med 362, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganami T, Nishida J, and Ogawa Y (2005). A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol 25, 2062–2068. [DOI] [PubMed] [Google Scholar]
- Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, and Liu G (2015). Pyruvate Dehydrogenase Kinase 1 Participates in Macrophage Polarization via Regulating Glucose Metabolism. J. Immunol 194, 6082–6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, BadiaRamentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, et al. (2018). TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543. [DOI] [PubMed] [Google Scholar]
- Taylor G, and Odili JL (1970). Tumour specific T-like antigen of human breast carcinoma. Br. J. Cancer 24, 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, Richardson M, Rigaill G, Parrini MC, Lucchesi C, et al. (2010). Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med 2, 211–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabold O, Wagner S, Wicke C, Scheuenstuhl H, Hussain MZ, Rosen N, Seremetiev A, Becker HD, and Hunt TK (2003). Lactate and oxygen constitute a fundamental regulatory mechanism in wound healing. Wound Repair Regen 11, 504–509. [DOI] [PubMed] [Google Scholar]
- Turley SJ, Cremasco V, and Astarita JL (2015). Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol 15, 669–682. [DOI] [PubMed] [Google Scholar]
- Ullah MS, Davies AJ, and Halestrap AP (2006). The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1αdependent mechanism. J. Biol. Chem 281, 9030–9037. [DOI] [PubMed] [Google Scholar]
- Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, Duran A, Castilla EA, Metallo CM, Diaz-Meco MT, et al. (2014). Metabolic Reprogramming of Stromal Fibroblasts through p62-mTORC1 Signaling Promotes Inflammation and Tumorigenesis. Cancer Cell 26, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekeere S, Dubois C, Kalucka J, Sullivan MR, García-Caballero M, Goveia J, Chen R, Diehl FF, Bar-Lev L, Souffreau J, et al. (2018). Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab 1–15. [DOI] [PubMed]
- Vaupel P, Kallinowski F, and Okunieff P (1989). Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 49, 6449–6465. [PubMed] [Google Scholar]
- Végran F, Boidot R, Michiels C, Sonveaux P, and Feron O (2011). Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 71, 2550–2560. [DOI] [PubMed] [Google Scholar]
- Venter G, Oerlemans FTJJ, Wijers M, Willemse M, Fransen JAM, and Wieringa B (2014). Glucose controls morphodynamics of LPS-stimulated macrophages. PLoS One 9, e96786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. (2014). Oncogene ablationresistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, et al. (2011). Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci 108, 16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, and Mueller-Klieser W (2000). High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res 60, 916–921. [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, Mccormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, and Negelein E (1927). THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol 8, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, Birbe RC, Howell A, Pavlides S, Gandara R, et al. (2011). Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 10, 1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci 105, 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, Sneddon S, Martinez-Outschoorn UE, Sotgia F, and Lisanti MP (2012). Using the “reverse Warburg effect” to identify high-risk breast cancer patients: Stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle 11, 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Wolf AM, Rumpold H, Fiegl H, Zeimet AG, Muller-Holzner E, Deibl M, Gastl G, Gunsilius E, and Marth C (2005). The Expression of the Regulatory T Cell-Specific Forkhead Box Transcription Factor FoxP3 Is Associated with Poor Prognosis in Ovarian Cancer. Clin. Cancer Res 11, 8326–8331. [DOI] [PubMed] [Google Scholar]
- Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R, et al. (2016). Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab 24, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, Zhang J, Zhao S, Zhou BP, and Mi J (2015). Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep 10, 1335–1348. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, et al. (2017). Enhancing CD8+T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 32, 377–391.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, et al. (2016). Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 5, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]