

Abstract
Objective
We investigated genome-wide changes in gene expression and chromatin remodelling induced by tumour necrosis factor (TNF) in fibroblast-like synoviocytes (FLS) and macrophages to better understand the contribution of FLS to the pathogenesis of rheumatoid arthritis (RA).
Methods
FLS were purified from patients with RA and CD14+ human monocyte-derived macrophages were obtained from healthy donors. RNA-sequencing, histone 3 lysine 27 acetylation (H3K27ac), chromatin immunoprecipitation-sequencing (ChIP-seq) and assay for transposable accessible chromatin by high throughput sequencing (ATAC-seq) were performed in control and TNF-stimulated cells.
Results
We discovered 280 TNF-inducible arthritogenic genes which are transiently expressed and subsequently repressed in macrophages, but in RA, FLS are expressed with prolonged kinetics that parallel the unremitting kinetics of RA synovitis. 80 out of these 280 fibroblast-sustained genes (FSGs) that escape repression in FLS relative to macrophages were desensitised (tolerised) in macrophages. Epigenomic analysis revealed persistent H3K27 acetylation and increased chromatin accessibility in regulatory elements associated with FSGs in TNF-stimulated FLS. The accessible regulatory elements of FSGs were enriched in binding motifs for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), interferon-regulatory factors (IRFs) and activating protein-1 (AP-1). inhibition of bromodomain and extra-terminal motif (BET) proteins, which interact with histone acetylation, suppressed sustained induction of FSGs by TNF.
Conclusion
Our genome-wide analysis has identified the escape of genes from transcriptional repression in FLS as a novel mechanism potentially contributing to the chronic unremitting synovitis observed in RA. Our finding that TNF induces sustained chromatin activation in regulatory elements of the genes that escape repression in RA FLS suggests that altering or targeting chromatin states in FLS (eg, with inhibitors of BET proteins) is an attractive therapeutic strategy.
INTRODUCTION
Rheumatoid arthritis (RA) is characterised by chronic unremitting synovial inflammation, where sustained remission of synovitis is achieved only in a minority of patients, despite aggressive immuno-suppression with synthetic or biologic disease modifying antirheumatic drugs.1,2 The current model for RA pathogenesis is the so-called integrated model, where there is an interplay between environmental, genetic, hormonal and stochastic factors, resulting in a cross-talk between T and B lymphocytes, macrophages (Mφ) and fibroblast-like synoviocytes (FLS) within the synovium.3 This cross-talk between different cell types involves cell-to-cell interactions and soluble factors that drive the initiation and perpetuation of synovial inflammation.4,5 The pathologic hallmark of RA is pannus, an inflamed and hypercellular synovial lining that invades the adjacent bone and cartilage.6 Pannus consists of activated Mφ that secrete tumour necrosis factor (TNF), and numerous activated FLS that respond to paracrine TNF, establishing a Mφ–TNF–FLS axis.7 The effectiveness of biologics targeting TNF in RA suggests that the Mφ–TNF–FLS axis is a key driver of the chronic unremitting character of RA synovitis.8
The molecular mechanisms resulting in non-re-solving inflammation in the context of RA remain obscure.5,9,10 An inflammatory response typically follows a multistep evolution from induction to resolution aiming to finally restore the function and structure of the affected tissues.11,12 Along these lines, Mφ rapidly adjust the temporal order of their responses to inflammatory challenges (eg, exposure to TNF or toll-like receptor (TLR) ligands) by transitioning from an acute proinflammatory to a subsequent homeostatic phenotype that promotes tissue repair and the resolution of inflammation.13 This is a tightly regulated process mediated by: (1) feedback loops that limit inflammatory cytokine production (eg, the interleukin (IL)-10/signal transducer and activator of transcription (STAT) 3 axis),14 (2) signalling brakes (eg, A20, ABINs, SOCS) that restrain inflammatory signalling15,16 and (3) chromatin remodelling that represses expression of proinflammatory genes.17–19 The result of these homeostatic molecular events is that following exposure to inflammatory stimuli (eg, TNF) macrophages typically display: (1) robust but transient expression of proinflammatory transcripts (eg, TNF, IL-1β, CXCL8) and (2) desensitisation (tolerance) to subsequent inflammatory stimulation. Tolerance induction in macrophages is opposed by interferron (IFN)-γ and type I IFNs,19 and thus it is possible that the IFN signatures expressed in RA synovium or other unknown mechanisms prevent complete tolerisation and maintain synovial macrophage capacity to produce inflammatory cytokines.
Within the chronically inflamed RA synovium, FLS are exposed to long-term inflammatory stimulation and their gene expression transitions from an early to a late programme that shapes aspects of their aggressive phenotype.8 In previous studies, we have investigated the molecular mechanisms that shape this late gene expression programme and have identified fundamental differences between FLS and Mφ in terms of the kinetics, quality and quantity of their TNF-induced inflammatory program.20–22 Whereas macrophages display transient expression of inflammatory genes (eg, IL6 and CXCL8), one single pulse with TNF triggers in RA FLS prolonged activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), sustained chromatin accessibility in the promoters of key inflammatory genes (eg, IL6 and CXCL10), non-terminating transcription of IL6, continuous expression of cytokines, chemokines and tissue destructive enzymes, as well as mitogen-activated protein kinases (MAPK)-dependent messenger RNA (mRNA) stabilisation of arthritogenic transcripts (eg, IL6, CXCL1, CXCL3, CXCL8, CCL2 and PTGS2).20–22 In addition, on exposure to TNF, RA FLS acquire a ‘short-term inflammatory memory’ potentially resulting from the induction of transcription factors (eg, STAT1) and chromatin modifications that are not rapidly reversed or are turned over with slow kinetics.21 Overall, the results of our previous studies20–22 support a model where the aggressive FLS behaviour might be the result of sustained signalling and prolonged chromatin remodelling that cooperatively drive continuously active transcription together with mRNA stabilisation of arthritogenic transcripts.
To gain further insights into mechanisms that underlie the sustained inflammatory response of FLS to TNF, we undertook an integrated transcriptomic and epigenomic approach to analyse this response at the genome-wide level and to compare it to the transient inflammatory response induced by TNF in macrophages. A striking finding of this comparative analysis was that many TNF-inducible proinflammatory genes that are transiently expressed and/or tolerised in macrophages, are not repressed and are expressed with sustained kinetics in FLS. Aspects of this differential regulation are reflected by divergent baseline and TNF-induced chromatin states (epigenomic land-scapes) of these two cell types. These findings indicate that TNF reprograms the epigenomic landscape of FLS and suggest that such reprogramming contributes to sustained and prolonged TNF-induced inflammatory gene expression.
METHODS
Synovial tissues were obtained from patients with RA who underwent total knee replacement or elbow synovectomy. The diagnosis of RA was based on the 1987 American College of Rheumatology criteria.23 Detailed experimental procedures are described in the online supplementary methods, including cell purification and culture, sequencing (RNA-sequencing (RNA-seq), chromatin immunoprecipitation-sequencing (ChIP-seq), assay for transposable accessible chromatin by high throughput sequencing (ATAC-seq)), bioinformatics analysis, quantitative reverse transcription PCR (RT-qPCR) and statistical analysis.
RESULTS
TNF-induced gene set whose expression is transient in macrophages but sustained in FLS
We had previously found that expression of several inflammatory genes that is transient after TNF stimulation of macro-phages is instead sustained up to at least 24–72 hours in FLS.20–22 To better characterise this gene set that escapes downregulation in FLS, we analysed our previously generated RNA-seq data sets (dbGAP: phs001371.v1.p1 and online supplementary figure S1).19 Analysis of FLS RNA-seq data using an updated computational pipeline and more stringent statistical cut-off (adjusted p<0.05) (online supplementary methods and online supplementary figure S1) revealed six clusters of TNF-inducible genes (R1–R6), including gene clusters induced with sustained kinetics over a 72-hours time course (clusters R3, R4, R5 and R6; online supplementary figure S1A). A comparison of the TNF response in macrophages and FLS is presented in the online supplementary figure S2. We then identified a set of 280 genes induced transiently in macrophages, with peak expression at 1–6 hours, but with sustained kinetics in FLS, with peak expression at 24–72 hours (figure 1 and online supplementary figure S1D and S1E). This fibroblast-sustained gene set (FSGs, n=280) included key inflammatory genes such as IL6, CXCL8, CXCL10 and MMP19 (figure 1B) which is in accord with and confirms our previous work.20–22
Figure 1.
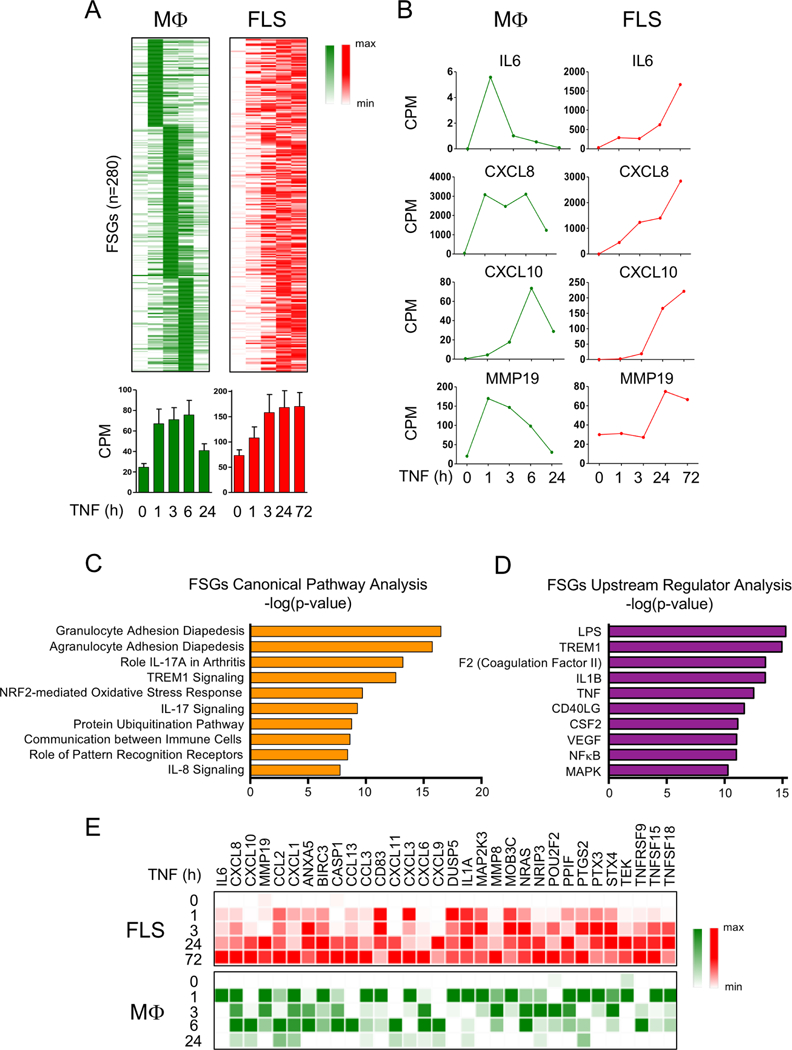
Genes that are transiently induced by TNF in macrophages exhibit sustained expression in FLS. (A) Kinetic analysis of TNF-induced mRNA transcripts whose expression is transient in macrophages but sustained in FLS. RNA-seq analysis was performed in a time course of TNF stimulation (10 ng/mL) using macrophages (three replicates from independent blood donors) and FLS (two replicates from independent patients with RA). Two hundred and eighty genes (FSGs) transiently expressed in macrophages but sustained in FLS are displayed on a heatmap (upper panels). Bar graphs (lower panels) represent CPM values for the FSGs. Error bars indicate SEM. (B) Representative genes from the gene set of the FSGs are presented in CPM values. (C and D) Ingenuity pathway analysis of the FSGs defined in (A). (E) Expression of a subset of gene transcripts from the FSGs defined in (A) is presented relative to the maximum expression. CPM, counts per million; FLS, fibroblast-like synoviocytes; FSG, fibroblast-sustained genes; IL, interleukin; LPS, lipopolysaccharide; Mφ, macrophages; mRNA, messenger RNA; MAPK, mitogen-activated protein kinases; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RA, rheumatoid arthritis; RNA-seq, RNA-sequencing; TNF, tumour necrosis factor.
To gain insight into the functions of these genes that escape downregulation in FLS, we performed ingenuity pathway analysis and found that these genes were significantly associated with inflammatory pathways implicated in RA pathogenesis (figure 1C). Analysis to identify upstream regulators of these genes likewise recovered inflammatory factors, including TNF and NF-κB (figure 1D). Expression of representative inflammatory genes contained within this gene set are shown in a heatmap in figure 1E and the entire gene list is provided in the online supplementary table 1. Overall, these genome-wide results support the notion that the expression of a substantial fraction of the TNF-induced inflammatory response is not effectively terminated in FLS.
Genes that are tolerised in macrophages escape repression in FLS
A subset of TNF-inducible genes transiently expressed in macro-phages are ‘tolerised’, a process whereby genes are repressed after a primary stimulation with inflammatory factors such as lipo-polysaccharide (LPS) or TNF and become resistant to induction by secondary challenge with prototypical inflammatory stimuli such as LPS.18,19 We analysed the patterns of expression of genes tolerised by initial stimulation of macrophages with TNF19 to test the notion that such genes would not be repressed in FLS. Interestingly, out of a well-defined set of 466 tolerised genes (T genes) in macrophages,19 80 exhibited sustained expression in FLS (figure 2A,B). These 80 genes were similarly associated with inflammatory pathways relevant for RA pathogenesis (figure 2C) and upstream regulators such as TNF and NF-κB (figure 2D); these genes are highlighted in red font in online supplementary table 1. These results reinforce the notion that mechanisms which suppress expression of a subset of TNF-inducible genes in macrophages are not operable in FLS and thus may enable an exaggerated inflammatory response.
Figure 2.
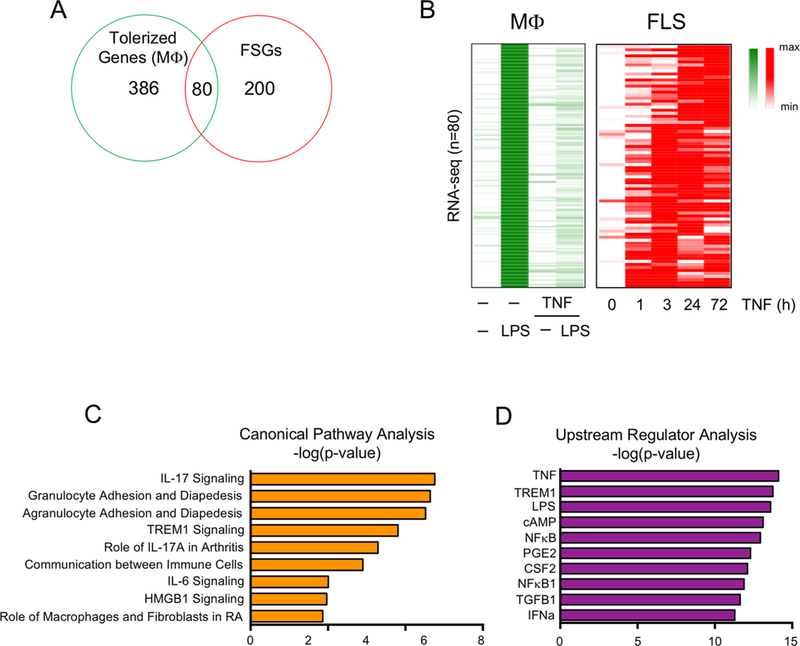
Tolerised genes in macrophage exhibit sustained expression in FLS after TNF exposure. (A) Venn diagram showing the overlap between 466 macrophage tolerised genes (robustly LPS-inducible genes that are minimally induced by secondary LPS in macrophages pretreated with TNF) and the FSGs identified in figure 1A. (B) Heatmap depicting expression of the 80 genes in the overlap region in (A) presented relative to the maximum expression. (C and D) Ingenuity pathway analysis of the 80 genes that were tolerised in macrophages but whose expression was sustained in RA FLS. FLS, fibroblast-like synoviocytes; FSG, fibroblast-sustained genes; IFN, interferron; IL, interleukin; IFN, interferron; LPS, lipopolysaccharide; Mφ, macrophages; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RA, rheumatoid arthritis; TGFB1, transforming growth factor beta 1; TNF, tumour necrosis factor.
Epigenomic analysis of TNF-stimulated FLS
Sustained TNF-induced transcription (as measured by primary/nascent RNA transcripts) and opening/activation of chromatin (as measured by formaldehyde-assisted isolation of regulatory elements (FAIRE) and ChlP-qPCR for histone acetylation) at promoters of select genes20,21 in FLS suggested that epigenetic chromatin-based mechanisms may contribute to sustained gene expression and thus escape from repression. We investigated this possibility by performing an epigenomic analysis combining ATAC-seq that measures open chromatin24 and ChlP-seq for histone 3 lysine 27 acetylation (H3K27ac), an established measure of regulatory element activity25,26 (online supplementary figure S3).
Similar to TNF-induced genes (online supplementary figure S1A: clusters R1–R6), TNF-induced H3K27ac peaks in FLS segregated into six clusters (figure 3A: clusters H1–H6), whose kinetic patterns of induction generally resembled the patterns of TNF-inducible gene expression (compare figure 3A to online supplementary figure S1A). Notably, both RNA-seq and ChIP-seq data revealed distinct clusters of genes expressed with transient versus sustained kinetics (clusters 1 and 2 vs 3–6). Gene ontology (GO) analysis of TNF-induced genes (online supplementary figure S1B) confirmed at a genome-wide level previous results with select genes21,27 that the TNF-induced inflammatory response in FLS expands to include IFN-STAT-inducible genes at later time points. In addition, genes expressed transiently only at 3 hours (online supplementary figure S1B, cluster R1) were related to transforming growth factor beta (TGFβ) and growth factor signalling, whereas late expressed genes were related to cytokine/chemokine production and cholesterol synthesis pathways (online supplementary figure S1B, cluster R6). Strikingly, genes associated with the various clusters of TNF-inducible H3K27ac peaks showed similar functional associations on GO analysis (figure 3B): H1 early transient peaks associated with TGFβ signalling, H2–H5 peaks with inflammatory and IFN-STAT pathway genes and H6 peaks associated with chemotaxis and lipid metabolic pathways. This analysis reveals that TNF induces sustained H3K27ac at a subset of regulatory elements and suggests that histone acetylation and thus activation of regulatory elements help drive the pattern of TNF-induced gene expression. Comparison of the relationship between gene sets R1–R6 and peak sets H1–H6 partially supported this notion by showing increased overlap of R2, R5 genes with H2, H5 peaks, R1 genes with H1 peaks and R6 genes with H6 peaks (figure 3C). Relatively weak gene–peak relationships observed in figure 3C may be related to regulation of several genes by individual regulatory elements or an individual gene by several regulatory elements, differential function of regulatory elements at different time points and the inherent uncertainty in knowing whether a given enhancer regulates the nearest genes. Furthermore, H3K27ac can act as a ‘bookmark’ that by itself does not recruit transcriptional machinery and thus drive transcription, but instead poises cells for robust responses to subsequent environmental challenges.
Figure 3.
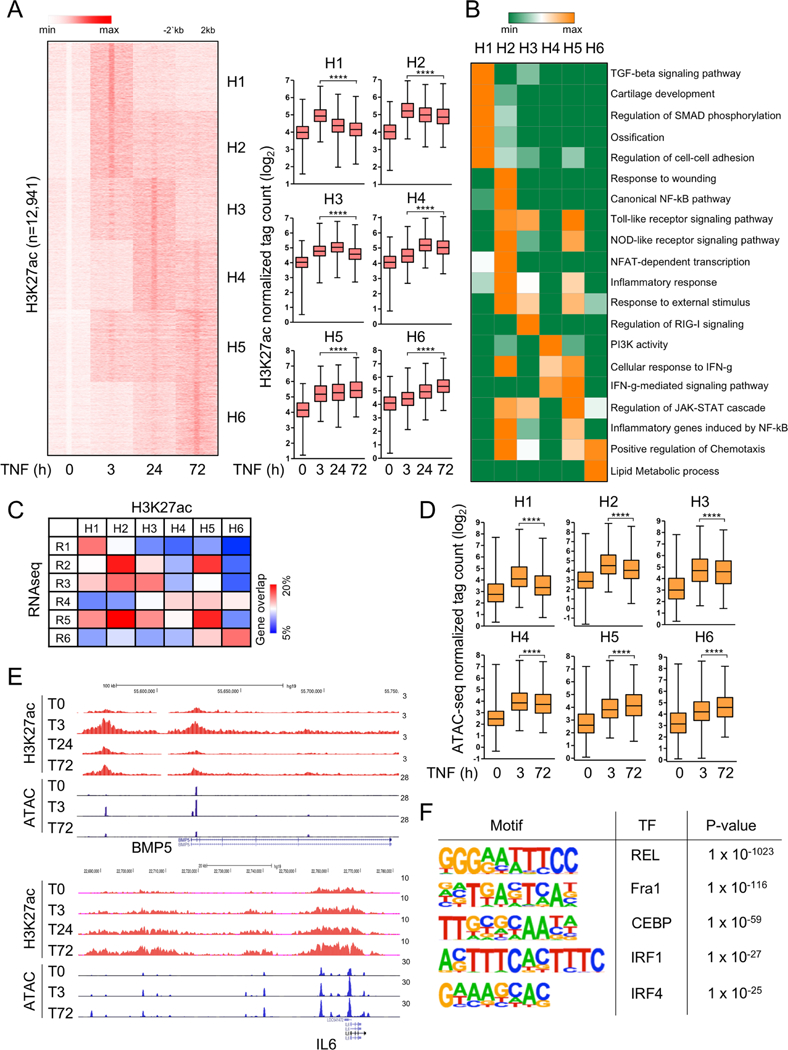
Epigenomic landscape changes in FLS after TNF exposure. (A) Genome-wide H3K27ac ChIP-seq analysis (two biological replicates from independent patients with RA) of differentially induced peaks at each TNF-stimulated time points (3, 24 and 72 hours). The left panel depicts K-means clustering of 12 941 peaks (one per row) that were induced more than fourfold by TNF (p<0.0001) into six clusters (H1–H6); expression is presented relative to the maximum (set as 1). Box graphs represent H3K27ac ChIP-seq normalised tag densities (log2) at enhancers of a given cluster under the indicated conditions (right). ****P<0.0001, Wilcoxon matched-pairs signed rank test. (B) GREAT gene ontology analysis using the peaks in each cluster defined in (A). (C) Heatmap presentation of the percentage of genes (percentage of gene overlap) in each gene expression clusters R1–R6 (online supplementary figure S1A) that overlap with genes associated with H3K27ac enhancer clusters H1–H6 identified in (A). (D) Box graphs represent ATAC-seq normalised tag densities (log2) at H3K27ac peaks defined in (A). This represents quantitation of the data shown as a heatmap in online supplementary figure S3D. ****P<0.0001, Wilcoxon matched-pairs signed rank test. (E) Representative University of California Santa Cruz Genome Browser tracks displaying normalised profiles for H3K27ac ChIP-seq and ATAC-seq signals at BMP5 (cluster 1) and IL6 (cluster 6) locus. (F) Motifs enriched under inducible ATAC-seq peaks at 72 hours of TNF stimulation. ChIP-seq and ATAC-seq analyses were performed with two FLS replicates from independent patients with RA that yielded similar results (online supplementary figure S3A). ATAC-seq, assay for transposable accessible chromatin by high throughput sequencing; ChIP-seq, chromatin immunoprecipitation-sequencing; FLS, fibroblast-like synoviocytes; FSG, fibroblast-sustained genes; H3K27ac, histone 3 lysine 27 acetylation; IFN, interferron; IL, interleukin; IRF, interferon regulatory factors; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; C/EBP, CCAAT/enhancer-binding protein; RA, rheumatoid arthritis; TGF, transforming growth factor; TNF, tumour necrosis factor.
To gain greater insight into regulation of chromatin accessibility during a TNF response in FLS, we performed ATACseq analysis (online supplementary figure S3D). Interestingly, chromatin accessibility (as defined by ATACseq normalised tag counts) at the regulatory regions H1–H6 defined in figure 3A paralleled the patterns of H3K27ac (compare figure 3D to right panel of figure 3A). Examples of chromatin regulation at a transiently expressed gene (BMP5) and a gene with sustained TNF-induced expression (IL6) are shown in the gene tracks in figure 3E. These results establish that TNF induces sustained acetylation and opening of chromatin in FLS.
ATACseq yields sharply delimited peaks centred around the region of greatest chromatin accessibility, which enables analysis to determine which DNA motifs are enriched under peaks and thus to infer which transcription factors (TFs) can bind and contribute to gene expression. De novo motif enrichment analysis under ATACseq peaks that were induced in a sustained manner by TNF revealed highly significant enrichment of binding motifs for canonical TNF-activated ‘inflammatory’ TFs of the NF-κB, AP-1, CCAAT/enhancer-binding protein (C/EBP) and interferon regulatory factor (IRF) families (figure 3F and online supplementary material 1 S3C); the IRF sequence also binds the STAT1/STAT2/IRF9-containing ISGF3 complex that is activated by type I IFNs. These results are in accord with previous work showing sustained TNF-induced inflammatory signalling in FLS20,21,28 and suggest that this late phase signal is delivered to chromatin to activate gene expression.
Persistent histone acetylation at genes that escape repression in FLS
The silencing of tolerized and transiently expressed genes in TNF-stimulated macrophages is characterised by a return to an inactive chromatin state, which can be refractory to inflammatory challenge.17–19 We therefore tested the notion that escape from transcriptional inactivation, and thus sustained expression, of genes in FLS may be maintained by persistent histone acetylation. To this end, we analysed the epigenomic landscape surrounding the FSGs that ‘escape repression’ as defined in figure 1. We compared H3K27ac peaks, which indicate active enhancers, surrounding these FSGs in FLS and macrophages. Strikingly, TNF induced H3K27ac at 783 regions associated with these genes in FLS, but H3K27ac induction was minimal if not absent at these regions in macrophages (figure 4A,B). In accord with the histone acetylation data, TNF substantially increased chromatin accessibility at these loci in FLS, but much less so in macrophages (figure 4C). Similar results were obtained when we analysed the 80 genes defined in figure 2 that escaped tolerance in FLS (online supplementary figure S4). These results demonstrate that the activation of enhancers and opening of chromatin by TNF in FLS is associated with sustained expression of genes that are not repressed in this cell type. When we extended the analysis to H3K27ac peaks associated with the FSGs in resting cells, we found that this segment of the epigenomic landscape was markedly different (online supplementary figure S4D): 358 macrophage-specific peaks (206 + 118 + 23 + 11; shaded in green), 1199 FLS-specific peaks (862 + 280 + 52 + 5; shaded in red) and 205 peaks common to both cell types at baseline (149 + 46 + 5 + 5; shaded). Thus, a different epigenomic landscape between the two cell types likely contributes to the distinct TNF-induced responses.
Figure 4.
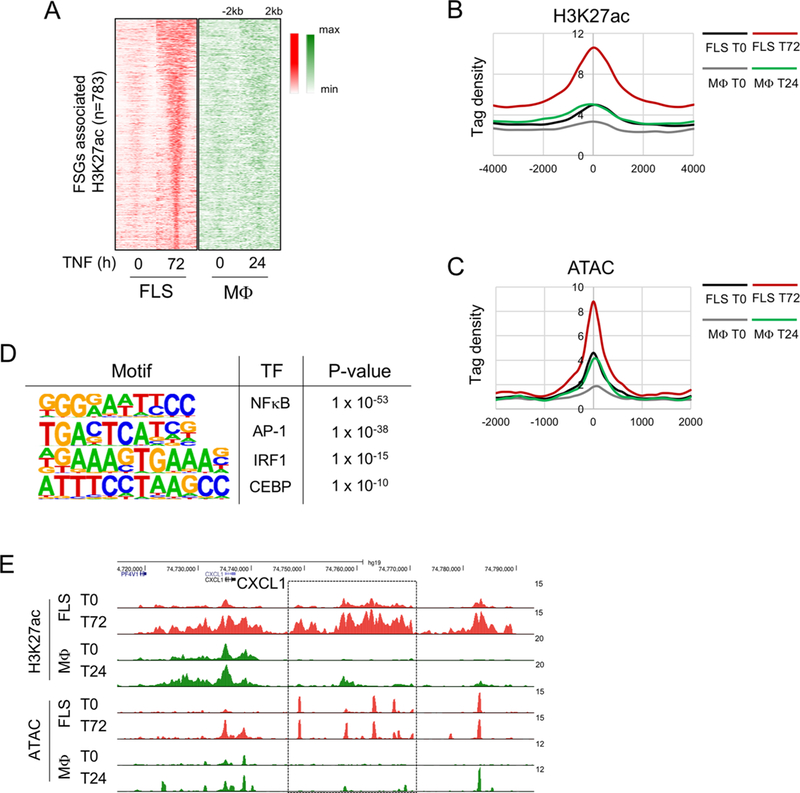
Persistent H3K27 acetylation is associated with sustained gene expression in FLS. (A) Heatmaps of normalised tag densities for H3K27ac ChIP-seq peaks associated with the FSGs defined in figure 1A in FLS and macrophages. (B) Histogram of H3K27ac-seq normalized tag densities for peaks defined in (A). (C) Histogram of ATAC-seq normalised tag densities at H3K27ac peaks defined in (A). (D) Motifs enriched under inducible ATAC-seq peaks associated with the FSGs at 72 hours of TNF stimulation. (E) Representative University of California Santa Cruz Genome Browser tracks displaying normalised profiles for H3K27ac ChIP-seq and ATAC-seq signals at the CXCL1 locus. Box enclose genomic regions that are differentially regulated across conditions. ATAC-seq, assay for transposable accessible chromatin by high throughput sequencing; ChIP-seq, chromatin immunoprecipitation-sequencing; FLS, fibroblast-like synoviocytes; FSG, fibroblast-sustained genes; H3K27ac, histone 3 lysine 27 acetylation; IRF, interferon regulatory factors; Mφ, macrophages; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RA, rheumatoid arthritis; TNF, tumour necrosis factor.
De novo motif enrichment analysis under TNF-induced ATACseq peaks in this gene set revealed highly significant enrichment of binding sequences for NF-κB, AP-1 and IRF family proteins (figure 4D). Representative gene tracks showing differential regulation of histone acetylation and chromatin accessibility in FLS and macrophages at the CXCL1 locus are depicted in figure 4E. These results are in accord with a model where TNF-induced NF-κB signalling is rapidly induced and terminated in macrophages,18 but a sustained TNF-induced NF-κB signalling in FLS20 maintains open chromatin and histone acetylation, which contributes to sustained transcription. The results also suggest a role for AP-1 and IRF proteins in sustaining transcription at this gene set, possibly in cooperation with NF-κB.
TNF-induced histone acetylation contributes to sustained gene expression in FLS
We reasoned that if sustained histone acetylation maintains expression of the ‘non-repressed’ genes in FLS, expression of these genes should be sensitive to inhibition of the function of acetylated histones. We therefore addressed the functional importance of TNF-induced histone acetylation for sustained gene expression using the inhibitor I-BET151 to suppress interaction of acetylated histones with bromodomain and extra-terminal (BET) proteins Brd2, Brd3 and Brd4.29 BET proteins ‘read’ histone acetylation and couple this positive histone mark to the transcriptional machinery, thereby promoting gene expression.29
Strikingly, I-BET significantly suppressed expression of 1697 genes in TNF-stimulated FLS (greater than twofold, p<0.05) (figure 5A). GO analysis of these genes showed IFN and chemo-kine-related pathways (figure 5B). Focusing on TNF-inducible genes, we found that among 911 genes which were significantly induced by TNF (greater than twofold, p<0.05), 617 genes (68%) were significantly suppressed (greater than twofold, p<0.05) by I-BET (figure 5C). Out of the 280 FSGs, 110 genes (39%) were significantly suppressed (>1.5-fold, p<0.05) by I-BET (figure 5D), including IL6, CXCL10, CXCL8, CCL2, MMP8, MMP19 (figure 5E and online supplementary figure S5A). These results support the notion that sustained TNF-induced histone acetylation contributes to gene expression that persists to late time points.
Figure 5.

Transcriptomic analysis of I-BET effects on TNF-induced inflammatory gene expression in FLS. (A) Volcano plot of transcriptomic changes in RA FLS stimulated with TNF (10 ng/mL) in the presence or absence of I-BET (10 M) for 24 hours; red dots correspond to genes with significant (p<0.05) and greater than twofold expression changes. RNA sequencing was performed in three independent biological replicates (FLS derived from three different patients with RA). (B) REACTOME pathway analysis of 1697 genes significantly suppressed by I-BET (greater than twofold and p<0.05) in TNF-stimulated FLS. (C) Heatmap depicting expression of genes that are TNF upregulated (greater than twofold and p<0.05) and I-BET suppressed (greater than twofold and p<0.05). Out of 911 TNF upregulated genes, 617 genes are suppressed by I-BET. (D) One hundred and ten genes out 280 FSGs were significantly suppressed by I-BET. (E) Representative TNF-inducible genes which were suppressed by I-BET (gene expression presented in CPM values). CPM, counts per million; FLS, fibroblast-like synoviocytes; FSG, fibroblast-sustained genes; I-BET, bromodomain and extra-terminal protein inhibitor; IL, interleukin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RA, rheumatoid arthritis; TNF, tumour necrosis factor.
DISCUSSION
Previous work from our team and others has shown that in macrophages, the expression of proinflammatory genes is tightly controlled: after an early acute induction, the expression of a large subset of these genes is repressed.17,18 Notably, a fraction of these repressed genes become desensitised (tolerised) to subsequent inflammatory stimulation. The current study reveals fundamental genome-wide differences between FLS and macro-phages in the transcriptional regulation of proinflammatory genes, with potential implications for FLS-mediated perpetuation of RA synovitis. We have discovered a set of TNF-inducible arthritogenic genes, termed FSGs, which are repressed and thus transiently expressed in macrophages, but in RA FLS, these genes escape from repression and are expressed with prolonged and unremitting kinetics that parallel the unremitting kinetics of RA synovitis. Our genome-wide analysis in FLS suggests that TNF induces sustained chromatin activation in regulatory elements of the genes that escape repression. Such lack of repression likely reflects a cell-type specific difference between FLS and macrophages, as it was also observed with osteoarthritis (OA) FLS and may represent a normal part of stromal cell physiology. Although these findings are not RA specific, they identify lack of transcriptional repression in FLS as a novel mechanism by which these cells may contribute to chronic unremitting synovitis in RA and other inflammatory diseases.
RA FLS, compared with FLS derived from OA, maintain ex vivo increased proliferative, migratory and invasive capacity.30,31 In two recent reports, we have also discovered that RA FLS display enhanced cytokine production, and on exposure to TNF they gain a short-term inflammatory memory.20,21 These phenotypic abnormalities have been explained on the basis of inflammation-induced chromatin modifications that are either permanent (chromatin imprinting)4 or reverse with slow kinetics.21 Most studies investigating the role of chromatin in regulating the aggressive behaviour of RA FLS have used gene-specific approaches20,21,32,33 and only recently genome-wide approaches have been incorporated.27,34–38 This prior work has provided insights into epigenomic mechanisms underlying gene expression that is sustained in RA FLS on ex vivo culture in the absence of TNF and other inflammatory factors characteristic of RA synovitis and has revealed stable differences between RA and OA FLS. Our study instead analyses for the first time at a genome-wide level the response of FLS to TNF as a model of inflammatory effects that may occur in vivo in patients and compares TNF responses in RA FLS and macrophages. These findings suggest mechanisms whereby an FLS-specific chromatin landscape can sustain unremitting expression of arthritogenic genes and thereby contribute to pathogenesis. Future work analysing freshly isolated RA, OA and normal FLS will reveal the effects of the in vivo inflammatory environment on the epigenomic landscape.
A key question that emerges from this study is how, during the course of an inflammatory response, a subset of arthritogenic genes repressed in macrophages escapes repression and tolerisation in RA FLS. Our previously reported work20–22 and the current study suggest in RA FLS a three-component model to explain the sustained unremitting expression of arthritogenic genes, mediated by inflammation triggered: (1) prolonged activation of NF-κB, (2) chromatin remodelling and (3) mRNA stabilisation (figure 6). According to this model, RA FLS display on exposure to TNF continuous upstream activation of NF-κB and MAPK signaling,20,22 potentially due to ineffective induction or function of signalling brakes.20 In parallel, RA FLS display at base line a permissive chromatin state that is further activated by TNF stimulation (TNF-induced histone acetylation and chromatin accessibility) allowing unopposed binding of transcription factors (such as p65, STAT1, IRFs) at promoters and enhancers of arthritogenic genes20,21 that maintain continuous transcription of these genes. In addition to the mechanisms that sustain chromatin accessibility and transcription, TNF induces in RA FLS a MAPK-dependent mRNA stabilisation of a subset of genes, allowing prolonged accumulation of transcripts which are translated to arthritogenic mediators.22
Figure 6.
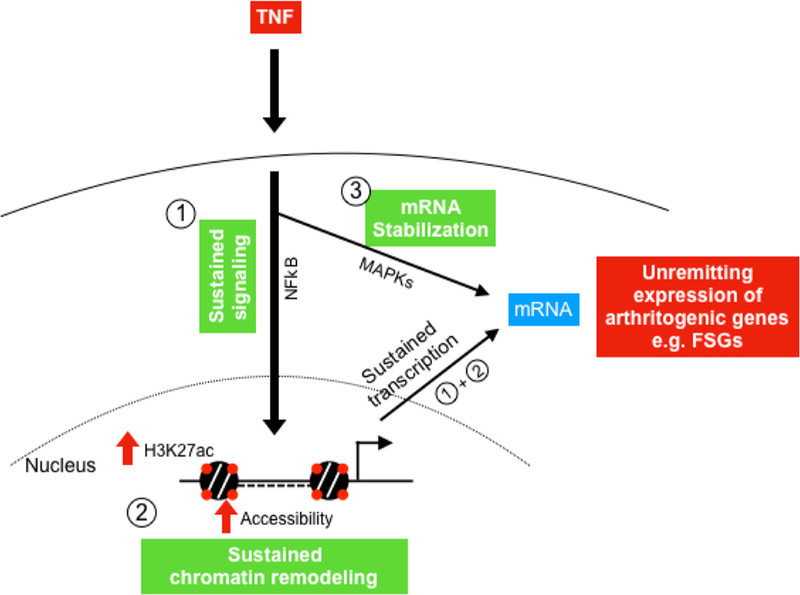
Three-component model that explains the sustained expression of arthritogenic genes (FSGs) in RA FLS. TNF triggers in RA FLS: (1) prolonged activation of NF-κB, (2) chromatin remodelling (increased H3K27ac and chromatin accessibility) and (3) MAPK-dependent mRNA stabilisation. Prolonged activation of NF-κB together with chromatin remodelling maintain continuous transcription of arthritogenic genes in RA FLS. Continuous transcription and mRNA stabilisation contribute to the sustained expression of these genes (FSGs) in RA FLS, ultimately perpetuating synovitis. FLS, fibroblast-like synoviocytes; FSG, fibroblast-sustained genes; H3K27ac, histone 3 lysine 27 acetylation; mRNA, messenger RNA; MAPK, mitogen-activated protein kinases; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RA, rheumatoid arthritis; TNF, tumour necrosis factor.
A striking finding of the current study is that 80 of the 280 FSGs that escape repression in FLS were desensitised (tolerised) in macrophages. A key mechanism that drives gene tolerisation in macrophages is chromatin remodelling from a permissive to a non-permissive state.19 What is the molecular mechanism preventing chromatin inactivation and tolerisation of these arthritogenic genes in RA FLS? One plausible explanation is that in contrast to macrophages, RA FLS lack the IL-10/STAT-3 homeostatic loop (online supplementary figure S2), potentially due to low IL-10 Ra expression,39 and instead a feed-forward IL-6/STAT-3 loop is in place that in the context of the FLS chromatin landscape operates as a proinflammatory signal.40 In addition, our team has recently shown a cooperative function on chromatin regulation of transcription factors of the IRFs and NF-κB families downstream of type I interferon and TNF signalling that prevents tolerisation of proinflammatory genes.19 Along these lines, we have found in this study that in FLS, the accessible chromatin in regulatory elements of the genes that escape tolerisation is enriched in binding motifs for NF-κB, IRFs and AP-1. This finding suggests that the cooperative binding of these TFs potentially maintains chromatin accessibility and prevents tolerisation of these genes. Overall, the results of the current study provide new insights about the role of the RA FLS-specific chromatin landscape in perpetuating synovitis and suggest that altering the chromatin states in RA FLS, for example, by using BET inhibitors, is a potentially attractive therapeutic strategy.
Supplementary Material
Key messages.
What is already known about this subject?
Fibroblast-like synoviocytes (FLS) contribute to perpetuation of synovitis in rheumatoid arthritis (RA), but the molecular mechanisms remain elusive.
What does this study add?
This study provides genome-wide insights into the mechanisms regulating tumour necrosis factor (TNF)-induced gene expression and chromatin landscape dynamics in RA FLS.
TNF-induced sustained chromatin activation in regulatory elements is a potential mechanism contributing to unremitting expression of arthritogenic genes in RA FLS.
How might this impact on clinical practice or future developments?
Altering or targeting chromatin states in FLS could be a potential therapeutic strategy in RA.
Acknowledgments
Funding This work was funded by grants from NIH AI046712, AR046713 and AR050401 (LBI) and the Feldstein Medical Foundation (GDK).
Footnotes
Competing interests None declared. GDK is at the time of publication of this study an employee at Regeneron Pharmaceuticals Inc. and declares no conflict of interest related to the content of this manuscript.
Patient consent for publication Not required.
Ethics approval Protocol approved by the Institutional Review Board of the Hospital for Special Surgery.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available in a public, open access repository.
Additional material is published online only. To view please visit the journal online (http://dx.doi.org/10.1136/annrheumdis-2018-214783).
REFERENCES
- 1.Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. The Lancet 2017;389:2338–48. [DOI] [PubMed] [Google Scholar]
- 2.Prince FHM, Bykerk VP, Shadick NA, et al. Sustained rheumatoid arthritis remission is uncommon in clinical practice. Arthritis Res Ther 2012;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mclnnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. The Lancet 2017;389:2328–37. [DOI] [PubMed] [Google Scholar]
- 4.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 2013;9:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donlin LT, Jayatilleke A, Giannopoulou EG, et al. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol 2014;193:2373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature 2003;423:356–61. [DOI] [PubMed] [Google Scholar]
- 7.Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev 2008;223:252–70. [DOI] [PubMed] [Google Scholar]
- 8.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol 2016;12:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haworth O, Buckley CD. Pathways involved in the resolution of inflammatory joint disease. Semin Immunol 2015;27:194–9. [DOI] [PubMed] [Google Scholar]
- 10.Alivernini S, Tolusso B, Ferraccioli G, et al. Driving chronicity in rheumatoid arthritis: perpetuating role of myeloid cells. Clin Exp Immunol 2018;193:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Origin Medzhitov R. and physiological roles of inflammation. Nature 2008;454:428–35. [DOI] [PubMed] [Google Scholar]
- 12.Nathan C, Ding A. Nonresolving inflammation. Cell 2010;140:871–82. [DOI] [PubMed] [Google Scholar]
- 13.Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol 2013;34:216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huynh L, Kusnadi A, Park SH, et al. Opposing regulation of the late phase TNF response by mT0RC1-IL-10 signaling and hypoxia in human macrophages. Sci Rep 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sokhi UK, Liber MP, Frye L, et al. Dissection and function of autoimmunity-associated TNFAIP3 (A20) gene enhancers in humanized mouse models. Nat Commun 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Afonina IS, Zhong Z, Karin M, et al. Limiting inflammation—the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol 2017;18:861–9. [DOI] [PubMed] [Google Scholar]
- 17.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007;447:972–8. [DOI] [PubMed] [Google Scholar]
- 18.Park SH, Park-Min K-H, Chen J, et al. Tumor necrosis factor induces GSK3 kinase–mediated cross-tolerance to endotoxin in macrophages. Nat Immunol 2011;12:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park SH, Kang K, Giannopoulou E, et al. Type i interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol 2017;18:1104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee A, Qiao Y, Grigoriev G, et al. Tumor necrosis factor α induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2013;65:928–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sohn C, Lee A, Qiao Y, et al. Prolonged tumor necrosis factor α primes fibroblast-like synoviocytes in a gene-specific manner by altering chromatin. Arthritis Rheumatol 2015;67:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loupasakis K, Kuo D, Sokhi UK, et al. Tumor necrosis factor dynamically regulates the mRNA stabilome in rheumatoid arthritis fibroblast-like synoviocytes. Plos One 2017;12:e0179762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnett FC, Edworthy SM, Bloch DA, et al. The American rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheumi 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 24.Buenrostro JD, Wu B, Chang HY, et al. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 2015;109:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 2010;107:21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long HK, Prescott SL, Wysocka J. Ever-Changing landscapes: transcriptional enhancers in development and evolution. Cell 2016;167:1170–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ai R, Laragione T, Hammaker D, et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. NatCommun 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosengren S, Corr M, Firestein GS, et al. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis 2012;71:440–7. [DOI] [PubMed] [Google Scholar]
- 29.Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010;468:1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller-Ladner U, Kriegsmann J, Franklin BN, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCiD mice. Am J Pathol 1996;149:1607–15. [PMC free article] [PubMed] [Google Scholar]
- 31.Lefèvre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med 2009;15:1414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maciejewska-Rodrigues H, Karouzakis E, Strietholt S, et al. Epigenetics and rheumatoid arthritis: the role of SENP1 in the regulation of MMP-1 expression. J Autoimmun 2010;35:15–22. [DOI] [PubMed] [Google Scholar]
- 33.Karouzakis E, Rengel Y, Jüngel A, et al. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun 2011;12:643–52. [DOI] [PubMed] [Google Scholar]
- 34.Karouzakis E, Raza K, Kolling C, et al. Analysis of early changes in DNA methylation in synovial fibroblasts of RA patients before diagnosis. Sci Rep 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank-Bertoncelj M, Trenkmann M, Klein K, et al. Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat Comms 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ai R, Hammaker D, Boyle DL, et al. Joint-specific DNA methylation and transcriptome signatures in rheumatoid arthritis identify distinct pathogenic processes. Nat Commun 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ai R, Whitaker JW, Boyle DL, et al. DNA methylome signature in synoviocytes from patients with early rheumatoid arthritis compared to synoviocytes from patients with longstanding rheumatoid arthritis. Arthritis Rheumatol 2015;67:1978–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol 2013;190:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neidhart M, Jüngel A, Ospelt C, et al. Deficient expression of interleukin-10 receptor α chain in rheumatoid arthritis synovium: limitation of animal models of inflammation. Arthritis Rheum 2005;52:3315–8. [DOI] [PubMed] [Google Scholar]
- 40.Williams LM, Sarma U, Willets K, et al. Expression of constitutively active STAT3 can replicate the cytokine-suppressive activity of interleukin-10 in human primary macrophages. J. Biol. Chem. 2007;282:6965–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.