

Abstract
Syndromic sensorineural hearing loss is multigenic and associated with malformations of the ear and other organ systems. Herein we describe a child admitted to the NIH Undiagnosed Diseases Program with global developmental delay, sensorineural hearing loss, gastrointestinal abnormalities, and absent salivation. Next generation sequencing revealed a uniparental isodisomy in chromosome 5, and a 22kb homozygous deletion in SLC12A2, which encodes for a sodium, potassium, and chloride transporter in the basolateral membrane of secretory epithelia. Functional studies using patient-derived fibroblasts showed truncated SLC12A2 transcripts and markedly reduced protein abundance when compared to control. Loss of Slc12a2 in mice has been shown to lead to deafness, abnormal neuronal growth and migration, severe gastrointestinal abnormalities, and absent salivation. Together with described phenotype of the Slc12a2 knockout mouse model, our results suggest that absence of functional SLC12A2 causes a new genetic syndrome and is crucial for the development of auditory, neurologic, and gastrointestinal tissues.
Keywords: NKCC1, cystic fibrosis, uniparental isodisomy, gut malrotation, absent salivation
Hearing loss is the most common neurosensory disorder in humans. Approximately 1 in 1000 newborns are diagnosed with bilateral sensorineural hearing loss (Paludetti, et al., 2012), and fifty to sixty percent are thought to be genetic in origin. Thirty percent of congenital, genetic hearing loss have accompanying clinical features and are therefore considered syndromic (Koffler, et al., 2015). More than 700 genetic syndromes include hearing impairment as a symptom, yet many causes of congenital hearing loss remain unknown. The development of whole genome sequencing, in combination with homozygosity mapping, has facilitated the discovery of new genes for syndromic and non-syndromic deafness (Parker and Bitner-Glindzicz, 2015).
The solute carrier 12 gene family includes nine genes, three of which are known to be associated with human diseases: SLC12A1 (MIM# 600839), SLC12A3 (MIM# 600968), and SLC12A6 (MIM# 604878), which cause Bartter, Gitelman, and Andermann syndromes, respectively. These well-described, autosomal recessive conditions are caused by defects in the movement and regulation of inorganic sodium (SLC12A1 & SLC12A3) and potassium (SLC12A6) cations against the movement of chloride anions (Gagnon and Delpire, 2013). The solute carrier 12, member 2 gene, SLC12A2 (MIM# 600840), encodes the Na-K-2Cl Cotransporter-1 (NKCC1). In mice, loss of Slc12a2 or NKCC1 lead to inner ear dysfunction, causing sensorineural deafness and balance problems that has been described as the “shaker/waltzer” phenotype (Gagnon and Delpire, 2013). Additional phenotyping has described decreased saliva production, intestinal transit problems, reduced neuron density, and many epithelial-related symptoms. Here, we describe a child with syndromic hearing loss due to a loss of function mutation in SLC12A2.
The proband was evaluated at the National Institutes of Health under the protocol 15-HG-0130, “Clinical and Genetic Evaluation of Patients with Undiagnosed Disorders Through the Undiagnosed Diseases Network,” approved by the National Human Genome Research Institute Institutional Review Board. Extensive phenotyping metrics and biospecimens were obtained following written informed consent from both parents. Please see Supplemental Data for description of methods.
The proband (Figure 1A, Supp. Figure S1) is a 5-year, 6-month-old male born at 32 weeks gestation to a non-consanguineous 26-year-old G1P0 mother and healthy 31-year-old father of mixed European ancestry. He was delivered by emergency cesarean section for pre-eclampsia. Birth weight was 1.7 kg. On newborn exam choanal atresia and bilateral syndactyly of the 2nd and 3rd toes, bilaterally, was noted. He failed his newborn hearing screen. Respiratory distress and difficulty feeding were apparent in the immediate postnatal period and he was discharged at eight weeks of life.
Figure 1. Clinical phenotype.
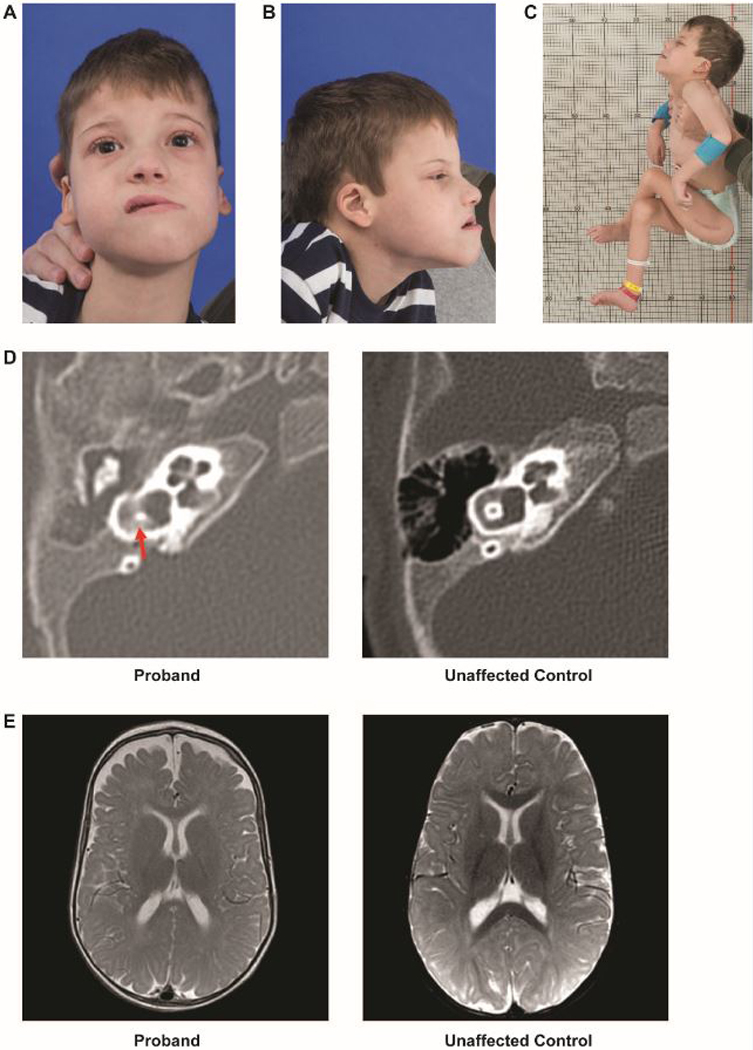
A. A photo of the proband depicting an oblong face with a long lower-third face, forward placed midface, and bitemporal and bicoronal narrowing.
B. Side profile of the proband showing long lower-third face, forward placed midface, and low-set ear with curling of the pinna
C. Typical posture and positioning of proband. Although hypotonic, he has significant contractures. Scar visible from osteotomy.
D. Computed tomography of the temporal bone showing vestibular dysplasia (left) as compared with an unaffected 5-month old.
E. Magnetic resonance imaging of the brain, with axial section, showing decreased cerebral volume and enlarged arachnoid spaces (left) as compared with an unaffected 9-month old.
An audiology examination at 2 months revealed bilateral auditory canal stenosis. Auditory- brainstem evoked potentials (ABR) demonstrated profound bilateral sensorineural hearing loss, Distortion Product Otoacoustic Emissions (DPOAE) were absent bilaterally, and he was fitted with hearing aids. Cochlear implants were placed although the parents not that he has functionally derived no benefit. The child had poor weight gain and chronic gastroesophageal reflux (GERD) was diagnosed at 11 months. A barium swallow at 13 months revealed midgut malrotation. A Ladd’s procedure, Nissen fundoplication, and appendectomy were performed. Pathology of the appendix showed focal cryptitis, crypt abscesses and mucosal glands with inspissated eosinophilic secretions suggestive of cystic fibrosis. Insufficient weight gain following surgery necessitated gastronomy tube placement.
At 10 months he was evaluated for concerns of laryngomalacia following episodes of thick oral secretions, coughing with feeds, and noisy breathing. Pulmonology evaluation at 12 months showed plastic bronchitis, suggesting primary ciliary dyskinesia. At 16 months, he developed acute airway obstruction due to mucous plugs requiring manual extraction following cardiopulmonary resuscitation.
At 5 years of age he was evaluated by the NIH UDP and was found to have dysmorphic features including an oblong face with bitemporal and bicoronal narrowing, low-set ears with curling of the pinna (Figure 1B), a fissured tongue, and severe xerostomia.
Growth parameters included weight 14.4 kilograms (<3%), length 108.5 centimeters (25–50%), and BMI 12.2 kilograms/meter2 (<3%, Z = −4.26). Systolic blood pressure was low (<7th centile). While his calorie, protein, and water intake exceed his estimated needs (>95% of required daily allowance), he was severely malnourished. The family reported periodic episodes of coffee-ground contents from the gastrostomy tube, though his hemoglobin was normal and stool for Hemoccult was negative. Morning cortisol was suboptimal (3.1 mcg/dl where normal is 5–25 mcg/dl) morning cortisol, though an ACTH stimulation test demonstrated normal cortisol production. Bone age was normal. There was normal muscle bulk but generalized hypotonia with hyporeflexia and lack of head control and the child was unable to sit independently (Figure 1C). Neuropsychological testing demonstrated profound delays in all developmental areas, with skills ranging from 1 to 6 months.
An ophthalmologic exam was normal, although a Schirmer test confirmed absence of tears (<1mm bilaterally). There was severe oral dryness and saliva was not able to be produced by massage of the salivary glands. Ultrasound showed normal salivary glands but underdeveloped parotid glands. No sympathetic postganglionic cholinergic function was observed in either the forearm or ankle by QSWEAT test, demonstrating sympathetic postganglionic sweat dysfunction
Awake and sleep electroencephalography showed focal slowing in the right temporal region with intermittent bursts of diffuse slowing. A muscle ultrasound, electrocardiogram and echocardiogram were normal.
Review of the computed tomography (CT) imaging of the temporal bones at 5 months demonstrated complete bilateral opacification of the middle ear spaces consistent with chronic otitis media, bilateral superior canal dehiscence (Figure 1D), and mild right vestibular dysplasia. Review of the brain MRI performed at 9 months showed decreased cerebral volume with sparing of the cerebellum involving both gray and white matter (Figure 1E).
Genetic testing, prior to the NIH evaluation, was non-diagnostic including testing for cystic fibrosis (97 common mutations) and spinal muscular atrophy. Sequence and deletion/duplication testing of the mitochondrial genome was normal. Exome sequencing identified four heterozygous variants of unknown significance (Supp. Table S1). A microarray revealed paternal, uniparental isodisomy in chromosome 5, which was confirmed during his NIH evaluation by whole genome microarray (Supp. Figure S2). Whole genome-sequencing identified a homozygous deletion of 22kb in SLC12A2. A second deletion was seen within the PCDHA (OMIM# 604966) gene cluster which is considered a normal variant and therefore unlikely related to our proband’s phenotype (Noonan, et al., 2003). Sanger sequencing of DNA from the proband and his parents confirmed the deletion was paternally inherited (Figure S1) and identified the breakpoints of the deletion from intron 1 to the beginning of exon 7 (chr5:127441491–127471419) — including an inversion of 34 base pairs — herein referred to as chr5:127441491–127471419delins34 (Figure 2A). Large deletions were not observed in the ExAC or gnomAD Browser and very few loss-of-function variants are reported; furthermore, no homozygote loss-of-function variants are seen in population databases.
Figure 2. Molecular analysis of SLC12A2 in proband.
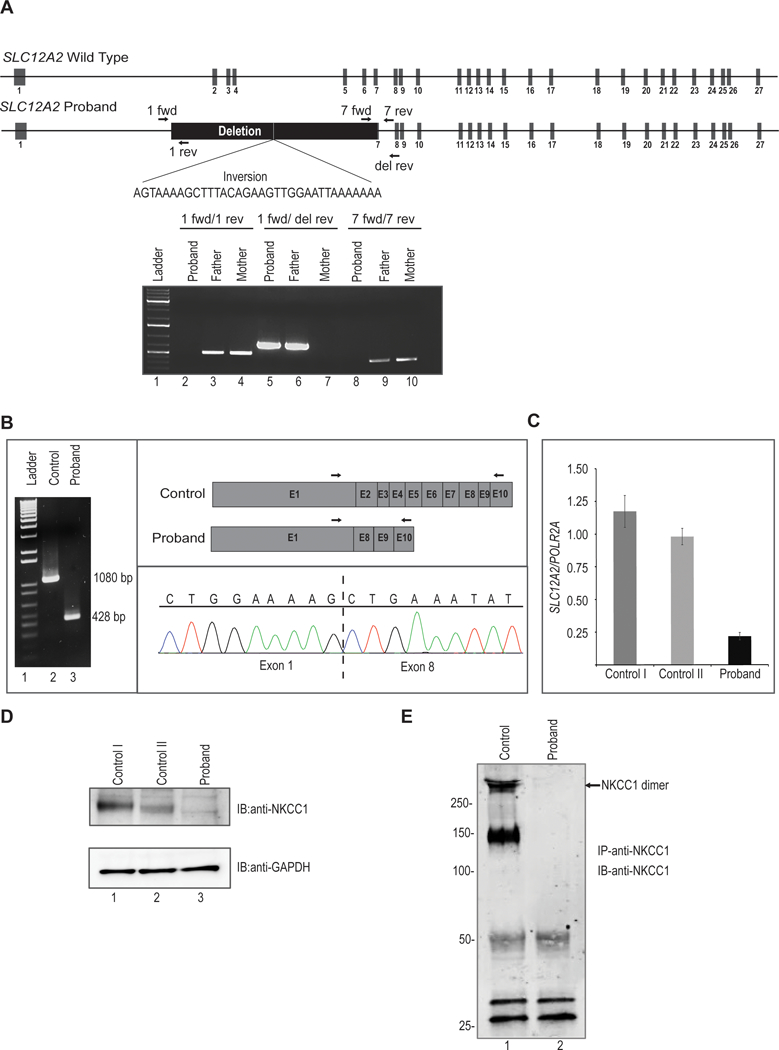
A. Upper, schematic representation of wild type SLC12A2 locus. Middle, structure of proband’s SLC12A2 locus showing homozygous deletion of 22kb and inversion of 34 nucleotides. Arrows indicate position of forward and reverse primers used for PCR amplification. Lower, agarose gel image showing amplification of genomic DNA demonstrates homozygous deletion in proband and heterozygous deletion in his father. The proband’s genomic DNA from blood amplifies only when primers are used outside of the deletion. The father’s DNA amplifies in all cases, showing he is a heterozygote. The mother’s genomic DNA does not amplify when primers straddle the breakpoint, as this region is presumably of a restrictive, wild type length of 22kb.
B. Analysis of cDNA synthesized from total RNA extracted from control and proband’s dermal fibroblasts. PCR amplification of cDNA results in truncated transcript in proband. Lane 1 is 1Kb molecular weight DNA ladder. Arrows indicate position of forward and reverse primers used for cDNA amplification. Cloning of PCR product followed by sequencing showed direct splicing of exon 1 to downstream exon 8 in proband.
C. Quantitative real time PCR analysis revealed significant reduced expression of SLC12A2 transcript normalized to POLR2A in proband fibroblasts compared to two independent fibroblast controls.
D. Western blot analysis of protein lysates from fibroblasts probed with polyclonal anti-SLC12A2 (NKCC1) antibody shows no detectable SLC12A2 protein in proband compared to two independent controls. The lower panel shows western blotting with anti-GAPDH to confirm equivalent sample loading.
E. Protein lysates from fibroblasts were immunoprecipitated with polyclonal anti-SLC12A2 (NKCC1) antibody conjugated to Protein A sepharose. Western blotting of immunoprecipitates with monoclonal anti-SLC12A2 (NKCC1) revealed the presence of SLC12A2 in both monomeric and dimeric forms in control while no SLC12A2 bands were detected in proband. Molecular weight in kilodaltons (kDa) is shown to the left of the panel.
By PCR we observed that the proband’s DNA was only amplified when primer pairs outside of the breakpoint were used, but not with a primer pair that straddled the breakpoints, thereby confirming the deletion. In contrast, the father’s DNA amplified with both sets of primers, demonstrating heterozygosity. To confirm that this deletion indeed disrupts SLC12A2 expression, we extracted mRNA from fibroblasts of the proband and two normal controls. Amplification of the proband’s cDNA showed a smaller product (428 bp) than control (1080 bp) (Figure 2B), and cloning the PCR product revealed the deletion of exons 2–7 and the splicing of exon 1 directly to exon 8 (Figure 2B, right panel). This splicing effect introduced a premature termination codon in exon 9 which is predicted to eliminate aberrant SLC12A2 by nonsense mediated decay. Indeed, quantitative real time PCR demonstrated that SLC12A2 mRNA expression was reduced to about 80% compared to controls (Figure 2C), suggesting that the aberrant transcripts were not translated and likely subjected to nonsense mediated decay. To confirm the disruption at the protein level, we performed western blot analysis of dermal fibroblasts, and documented an absent SLC12A2 (NKCC1) protein in the proband compared to controls (Figure 2D). Immunoprecipitation of fibroblasts lysates with NKCC1 antibody further revealed the presence of NKCC1 protein, (presumed to be glycosylated forms) only in control fibroblasts while no detectable protein was found in proband (Figure 2E). Taken together, these data suggest that the homozygous deletion found in the proband is pathogenic, and the NKCC1 protein non-functional.
Disruption of genes in the SLC12 gene family account for three human diseases: Bartter’s disease (SLC12A1), Gitelmann’s disease (SLC12A3), and Andermann’s syndrome (SLC12A6). The patient described in this report has overlapping features with these disease phenotypes: failure to thrive and sensorineural deafness in Bartter’s, hypotension and muscle weakness in Gitelmann’s, and 2–3 syndactyly and hypotonia in Andermann’s. This new syndrome is associated with a loss of function mutation in SLC12A2; we now refer to this novel disease as Kilquist syndrome, which is additionally characterized by the absence of saliva, tears, and sweat, mucus plugging and respiratory problems reminiscent of cystic fibrosis, and severe gastrointestinal problems.
Like other SLC12 gene family disorders, Kilquist syndrome is an autosomal recessive condition caused by the loss of function of SLC12A2. Our proband’s homozygous deletion of SLC12A2 was the result of uniparental paternal isodisomy. As a carrier, his father shows no symptoms of Kilquist syndrome. The gnomAD database shows only 26 loss of function variants in SLC12A2 and no homozygotes, further supporting loss of function due to homozygous deletion of SLC12A2 as the mechanism of disease.
The patient has a strikingly similar phenotype to the Slc12a2 knockout mouse and, like the mouse model, is also markedly deficient in NKCC1 mRNA and protein (Supp. Table S2). The mice have multisystem disease with significant overlap to our proband’s phenotype (Figure 1). This significant overlap with the published mouse phenotype and the loss of NKCC1 expression in vitro suggest that homozygous mutations in SLC12A2 are responsible for his symptoms. Our case is the first known human presentation of complete loss of NKCC1 and demonstrates the role of SLC12A2 in human secretory systems and disease.
The hallmark feature of the NKCC1 knockout mouse is a “shaker/waltzer” phenotype characteristic of inner ear dysfunction, histologically seen as a complete collapse of the cochlear duct (Flagella, et al., 1999). In addition, these mice show a lack of endolymphatic fluid creating the division between the vestibular chamber and the cochlear chamber (Delpire, et al., 1999). Further, Reissner’s membrane was completely collapsed towards the vestibular chamber in all turns of the cochlea. Delpire’s work shows that this structural damage is caused by a decrease in endolymph secretion due to lack of the functional co-transporter, NKCC1. The mice do not respond to a hand-clap and ABRs show no wave form for any sound stimuli (Flagella, et al., 1999). Our patient similarly has profound sensorineural hearing loss.
We were unable to completely assess the inner ear structures due to the presence of cochlear implants. However, review of an earlier MRI study shows bilateral superior canal dehiscence consistent with endolymph secretion abnormalities. In addition to sensorineural hearing loss, there are further phenotypic similarities between the mouse model and our patient. In mice, evidence of bleeding in the intestine and blockage of the colon were documented on autopsy (Flagella, et al., 1999). Our patient has a history of significant gastrointestinal problems including malrotation, constipation, and concern for blood in the GI tract based on periodic coffee-ground G-tube aspirations. In the mouse model, NKCC1 is necessary for maintaining normal neuronal migration, and is implicated in dendritic growth and increasing neuron density through its regulation of GABAergic signaling (Young, et al., 2012). Since the placement of cochlear implants, our proband did not have updated imaging and therefore the concern regarding a progressive versus static encephalopathy remains. Nonetheless, based on NKCC1’s role in early murine brain development, together with the absence of progressive neurodegeneration, we hypothesize that NKCC1is likely important in human brain development.
The only secretory deficiency reported in the mouse model is in salivation. NKCC1 is responsible for controlling the rate of salivation by regulating chloride entry to the acinar cells. NKCC1 knockout mice show a significant reduction in saliva production due to the inability of the parotid gland to conserve NaCl (Evans, et al., 2000). When presented with a muscarinic agonist, NKCC1 mice show > 60% reduction in saliva production, as compared to wild type animals. Likewise, our patient does not produce detectable saliva even with massage of the salivary glands.
Our patient shares features in common with cystic fibrosis patients, specifically respiratory mucous plugging. Due to his respiratory and GI symptoms he was evaluated multiple times for cystic fibrosis both by sweat chloride analysis and CFTR (MIM# 602421) mutation analysis. NKCC1 is critical for maintaining CFTR-mediated Cl- secretion since NKCC1 transports chloride from the blood into the epithelial cells where it is secreted by CFTR (Gagnon and Delpire, 2013). mRNA encoding CFTR has been shown to be slightly reduced in the colon of knockout mice, suggesting a regulatory relationship between NKCC1 and CFTR (Grubb, et al., 2001). The fact that sweat chloride testing was not able to be successfully performed in our patient is consistent with such a relationship. It is possible that children who have insufficient sweat for analysis and negative CFTR mutation testing may, in fact, have mutations in the SLC12A2 gene, particularly if they also have hearing loss.
Delpire (Delpire, et al., 2016) described a 13-year-old female with multisystem dysfunction and a truncating, heterozygous mutation in SLC12A2. They hypothesize that disruption in NKCC1 may affect sensory and smooth muscle function resulting in her phenotype of ketotic hypoglycemia with illness, decreased energy and fatigue, episodes of vomiting and dehydration, and dilated cardiomyopathy. Of note, the patient had normal hearing and cognition with no ataxia or inner ear deficits, similar to the mouse that was recently generated to model the patient’s mutation (Koumangoye, et al., 2018). It is possible that the de novo truncated protein is disrupting SLC12A2 function by mistargeting NKCC1 localization to the apical pole of epithelial cells, as the authors suggested (Koumangoye, et al., 2018), thus the variant could be a dominant negative mutation specifically in epithelial cells, which is different from the loss of function mutation that we see in our patient.
In summary, we present a novel cause of syndromic hearing loss associated with global developmental delay, failure to thrive, respiratory problems, absent salivation and sweat production, and gastrointestinal abnormalities. Additional descriptions of individuals with biallelic mutations in, or deletions of, SLC12A2 will be needed to better understand the phenotypic variability and possible genotype-phenotype correlations.
Supplementary Material
Acknowledgement:
This work was supported by the NIH NHGRI Intramural Research Program and the NIH Common Fund, Office of the Director. J.M.H. was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U01HG007943. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Delpire E, Lu J, England R, Dull C, Thorne T. 1999. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 22(2):192–5. [DOI] [PubMed] [Google Scholar]
- Delpire E, Wolfe L, Flores B, Koumangoye R, Schornak CC, Omer S, Pusey B, Lau C, Markello T, Adams DR (2016). A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb Mol Case Stud, 2(6), a001289 10.1101/mcs.a001289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. 2000. Severe impairment of salivation in Na+/K+/2Cl- cotransporter (NKCC1)-deficient mice. J Biol Chem 275(35):26720–6. [DOI] [PubMed] [Google Scholar]
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T and others. 1999. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274(38):26946–55. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, Delpire E. 2013. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am J Physiol Cell Physiol 304(8):C693–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BR, Pace AJ, Lee E, Koller BH, Boucher RC. 2001. Alterations in airway ion transport in NKCC1-deficient mice. American Journal of Physiology-Cell Physiology 281(2):C615–C623. [DOI] [PubMed] [Google Scholar]
- Koffler T, Ushakov K, Avraham KB. 2015. Genetics of Hearing Loss: Syndromic. Otolaryngol Clin North Am 48(6):1041–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumangoye R, Omer S, Delpire E. 2018. Mistargeting of a truncated Na-K-2Cl cotransporter in epithelial cells. Am J Physiol Cell Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan JP, Li J, Nguyen L, Caoile C, Dickson M, Grimwood J, Schmutz J, Feldman MW, Myers RM. 2003. Extensive linkage disequilibrium, a common 16.7-kilobase deletion, and evidence of balancing selection in the human protocadherin alpha cluster. Am J Hum Genet 72(3):621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludetti G, Conti G, W DIN, E DEC, Rolesi R, Picciotti PM, Fetoni AR. 2012. Infant hearing loss: from diagnosis to therapy Official Report of XXI Conference of Italian Society of Pediatric Otorhinolaryngology. Acta Otorhinolaryngol Ital 32(6):347–70. [PMC free article] [PubMed] [Google Scholar]
- Parker M, Bitner-Glindzicz M. 2015. Genetic investigations in childhood deafness. Arch Dis Child 100(3):271–8. [DOI] [PubMed] [Google Scholar]
- Young SZ, Taylor MM, Wu S, Ikeda-Matsuo Y, Kubera C, Bordey A. 2012. NKCC1 knockdown decreases neuron production through GABA(A)-regulated neural progenitor proliferation and delays dendrite development. J Neurosci 32(39):13630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.