

Abstract
Perfluorochemicals produce hepatotoxic effects via activation of peroxisome proliferator-activated receptor alpha (PPARα) and constitutive androstane receptor (CAR) nuclear receptors in animals. Bile formation is one major liver function. But it remains unknown whether perfluorochemicals alter metabolism of bile acids (BAs) in liver. The present study was designed to determine the impact of perfluorononanoic acid (PFNA) on BA and cholesterol homeostasis in mice. A single dose of PFNA (0.1 mmol/kg) was intraperitoneally administered to adult male wild-type (WT), PPARα-null, and CAR-null mice. PFNA caused cholestasis in the WT mice, indicated by increased serum alanine aminotransferase, hyperbilirubinemia, elevated BA concentrations in mouse serum, and appearance of bile plugs in mouse liver. In addition, PFNA decreased total and some individual BAs in mouse liver. PFNA increased the concentrations of total and taurine-conjugated, as well as some individual BAs in the serum of WT and CAR-null mice but not in PPARα-null mice, indicating a PPARα-dependent mechanism. PFNA decreased mRNA expression of most BA-related transporters (sodium-taurocholate cotransporting polypeptide, organic anion transporting polypeptide [Oatp]1a1, Oatp1b2, and bile salt export pump) and BA biosynthetic enzymes (Cyp7a1, 7b1, 8b1, and 27a1) in mouse liver, but increased mRNA expression of some efflux transporters (breast cancer resistance protein, multidrug resistance transporter 2, multidrug resistance-associated protein [Mrp] 2, Mrp3, and Mrp4), primarily via a PPARα-dependent mechanism. Moreover, PFNA increased free and total cholesterol in mouse liver but not in mouse serum. Furthermore, PFNA increased mRNA expression of sterol transporters, namely Abca1, g1, g5/g8, and steroidogenic acute regulatory protein via PPARα. In conclusion, PFNA produced cholestasis in mouse liver, and the activation of PPARα plays a central role in regulating BA and cholesterol metabolism and transport in mouse serum and liver.
Keywords: bile acids, cholesterol, PFNA, PPARα, CAR
Perfluorochemicals have been widely used in many industrial and consumer products. Due to their environmental persistence and toxicities in laboratory animals, the Environmental Protection Agency (EPA) in 2006 initiated a global program called “EPA 2010/15 PFOA Stewardship Program” to phase out perfluorooctanoic acid (PFOA) and PFOA related higher homologue chemicals (Calafat et al., 2007; Kudo and Kawashima, 2003; Lau et al., 2007; Mitro et al., 2016; Olsen and Zobel, 2007). So far, as shown in the EPA progress report (https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/2007-2014-company-progress-reports-summary-tables-pfoa; last accessed June 15, 2017) global manufacturing emissions of perfluorooctanoate and perfluorooctanesulfonate have been markedly reduced (Calafat et al., 2007; Prevedouros et al., 2006). However, the manufacture of perfluorononanoic acid (PFNA) and its ammonium salt, which are primarily used in facilitating the manufacture of fluoropolymers, are not officially restricted (Prevedouros et al., 2006). In addition, through environmental degradation, fluorotelomer alcohols can also be transformed into PFNA (Ellis et al., 2004). As a consequence, PFNA concentrations in serum of the U.S. population in 2003–2004 were approximately twice the levels in 1999–2000 (Calafat et al., 2007). However, the mean PFNA concentrations appeared to decline in 2011–2012 and 2013–2014 survey periods (CDC, 2017; Kato et al., 2011). Similar to other perfluorochemicals, PFNA causes wasting syndrome, immunotoxicities, developmental toxicity, hepatomegaly, hepatosteatosis, peroxisome proliferation, increases in hepatic lipid content, and hepatic necrosis in laboratory animals (Brewster and Birnbaum, 1989; Das et al., 2015, 2017; Fang et al., 2008, 2009; Harris et al., 1989; Kudo and Kawashima, 1997; Rockwell et al., 2013, 2017; Son et al., 2008; Van Rafelghem et al., 1987; Wolf et al., 2008; Yamamoto and Kawashima, 1997).
Bile formation is a major function of liver. More than 90% of total cholesterol in the body is excreted after conversion into bile acids (BAs). In addition, BAs are critical for absorption of lipids and fat-soluble vitamins from the intestine. However, little is known about the impact of perfluorochemicals on BA homeostasis and disposition.
We, together with others, have previously reported that perfluorochemicals, such as perfluorooctanoic acid (PFOA) and perfluorodecanoic acid (PFDA), activate xenobiotic nuclear receptors, such as peroxisome proliferator-activated receptor alpha (PPARα) and constitutive androstane receptor (CAR) (Cheng and Klaassen, 2008a,b; Maher et al., 2008; Rosen et al., 2008, 2017; Yang et al., 2002). We have reported that PFDA PPARα-dependently decreased the expression of hepatic uptake transporters, such as sodium-taurocholate cotransporting polypeptide (Ntcp) and organic anion transporting polypeptide (Oatp)1b2 in mouse liver (Cheng and Klaassen, 2008a).
The present study was designed to determine the impact of PFNA, the one-carbon lower homologue of PFDA, on the BA and cholesterol metabolism and transport in mice, and to determine the roles of activation of PPARα and CAR in the PFNA-produced alterations.
MATERIALS AND METHODS
Materials
Sodium bicarbonate, sodium dodecyl sulfate, sodium chloride, lithium lauryl sulfate, ethylenediaminetetraacetic acid, and d-(+)-glucose were purchased from Sigma-Aldrich (St. Louis, Missouri). PFNA (free acid form; 97% purity; M.W. 464 g/mol) was also purchased from Sigma-Aldrich Co. All other chemicals, unless otherwise indicated, were purchased from ThermoFisher Scientific (Fairlawn, New Jersey).
Animals and chemical exposure
Eight-week-old adult male C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, Maine), and housed according to the Institutional Animal Care and Use Committee guidance of University of Kansas Medical Center (Kansas City, Kansas). The PPARα-null mice were originally engineered by the laboratory of Dr. Frank J. Gonzalez (Lee et al., 1995), then back-crossed to a C57BL/6 background (Akiyama et al., 2001), and kindly provided by Dr. Jeffrey M. Peters (Pennsylvania State University, University Park, Pennsylvania). Breeding pairs of CAR-null mice in the C57BL/6 background were obtained from Dr. Ivan Rusyn (University of North Carolina, Chapel Hill, North Carolina), which were engineered by Tularik Inc (South San Francisco, California) as described previously (Ueda et al., 2002).
We have previously reported that a single i.p. administration of PFDA (0.1 mmol/kg) decreased expression of hepatic uptake transporters, such as Ntcp and Oatps in the livers of C57BL/6 wild-type (WT) mice, but not in PPARα-null mice (Cheng and Klaassen, 2008a). In addition, we have reported that a single i.p. administration of PFNA (0.1 mmol/kg) produced acute immunotoxic effects in C57BL/6 WT mice (Rockwell et al., 2013, 2017). To be consistent, in the present study, adult male C57BL/6 WT mice, and age-matched PPARα-null mice and CAR-null mice (n = 5/genotype/exposure) were administered a single i.p. dose of PFNA (0.1 mmol/kg) in a volume of 5 ml/kg of body weight. Control mice were administered i.p. with the vehicle propylene glycol: water (1:1, v/v). After 1 week, livers were collected and snap-frozen in liquid nitrogen and stored at −80 °C. A piece of freshly collected liver from each mouse was fixed in 10% zinc formalin for histological examination. In addition, blood was collected and allowed to coagulate, and then centrifuged at 10, 000 x g for 15 min. The resulting supernatant (serum) was collected for analysis.
In addition, adult male C57BL/6 mice (n = 6/exposure) were administered a single i.p. dose of PFNA (0.1 mmol/kg). Control mice were exposed i.p. with the vehicle propylene glycol: water (1:1, v/v). These mice were sacrificed after 14 or 90 days. Then, the mouse livers were collected and snap-frozen in liquid nitrogen and stored at −80 °C. A piece of freshly collected liver from individual mice was fixed in 10% zinc formalin for histological examination.
Histopathology
Liver samples, which were fixed in 10% zinc formalin, underwent routine processing and paraffin embedding. Liver sections (5 µm in thickness) were stained with hematoxylin and eosin and evaluated for histology examination.
Alanine aminotransferase activity in mouse serum
Serum alanine aminotransferase (ALT) activity was quantified as a biochemical indicator of hepatocellular injury using Liquid ALT Reagent Set (Pointe Scientific, Inc, Canton, Michigan), according to the manufacturer’s protocol.
Quantification of conjugated bilirubin in mouse serum
Serum conjugated bilirubin concentrations were determined according to the manufacturer’s protocol (Direct Bilirubin Reagent Set Kit, Pointe Scientific, Inc).
RNA extraction and quantification
Total RNAs were extracted using RNA-Bee reagent (Fisher Scientific Inc, Waltham, Massachusetts) per the manufacturer’s instructions. Total RNA concentrations were quantified at 260 nm with a spectrophotometer (Eppendorf Biospectrometer, Hauppauge, New York). RNA samples with an A260/A280 ratio between 1.8 and 2.0 were used for further analysis.
Quantitative real time-PCR assay
Total RNA was reverse transcribed into cDNA using SuperScript II reverse transcriptase (Life Technologies, Carlsbad, California) following the manufacturer’s instructions. Quantitative PCR was performed using SYBR Select Master Mix (Life Technologies) in an AriaMx Real-Time PCR system (Agilent Technologies, Santa Clara, California). Data were calculated according to the comparative delta-delta CT method and presented as relative fold of the control. Primers used in quantitative real time-PCR were designed with Primer3 software (version 4), synthesized by Integrated DNA Technologies (Coralville, Iowa), and are listed in Supplementary Table 1.
Quantification of individual BAs in mouse serum and liver
Total BAs were extracted from mouse serum and liver using previously described methods (Alnouti et al., 2008). Individual BAs (cholic acid [CA], α-murocholic acid [MCA], β-MCA, chenodeoxycholic acid [CDCA], ursodeoxycholic acid [UDCA], and deoxycholic acid [DCA] and their glycine- and taurine-conjugates) were quantified by an Ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method (Alnouti et al., 2008). Various concentrations of individual pure BAs (external standards) were analyzed among the extracted samples. In addition, internal standards, including the 2H4-G-CDCA and 2H4-CDCA, were used to determine extraction efficiency (Alnouti et al., 2008).
Quantification of free and total cholesterol in mouse serum and liver
Liver lipid content including cholesterol was extracted as described previously (Tanaka et al., 2008). Serum and liver lipid samples were analyzed spectrophotometrically at 505 nm for free and total cholesterol in accordance with the manufacturer’s protocols (Wako Chemicals USA, Inc, Richmond, Virginia).
Statistical analysis
Data are expressed as Mean ± SEM. Data of 3 or more groups were analyzed by one-way analysis of variance, followed by Duncan’s post hoc test using Statistical software (StatSoft Inc, Tulsa, Oklahoma). Data of 2-sample comparisons were analyzed by Student’s t-test. Statistical significance was considered at p < .05.
RESULTS
Histological Examination of Livers of WT, PPARα-Null, and CAR-Null Mice After PFNA Exposure
A single i.p. administration of PFNA to WT and CAR-null mice induced hypertrophy of hepatocytes around the central vein, and increased the number of denser eosinophilic granules in the cytoplasm (Figure 1A). In PPARα-null mice, PFNA administration resulted in swollen hepatocytes, and hepatocytes contained small fat droplet vacuoles, indicating prominent microvesicular steatosis (Figure 1A), similar as recently reported (Das et al., 2017). In addition, histology examination by Dr. Fan Fang (Department of Pathology & Laboratory Medicine, KU Medical Center, Kansas), a certified pathologist, revealed that PFNA produced cholestasis in WT mouse liver, which became apparent 2 weeks and 3 months after PFNA exposure (Figure 1B), as indicated by the appearance of yellow bile droplets (accumulated bile) and bile plugs in the liver sections (arrows).
Figure 1.
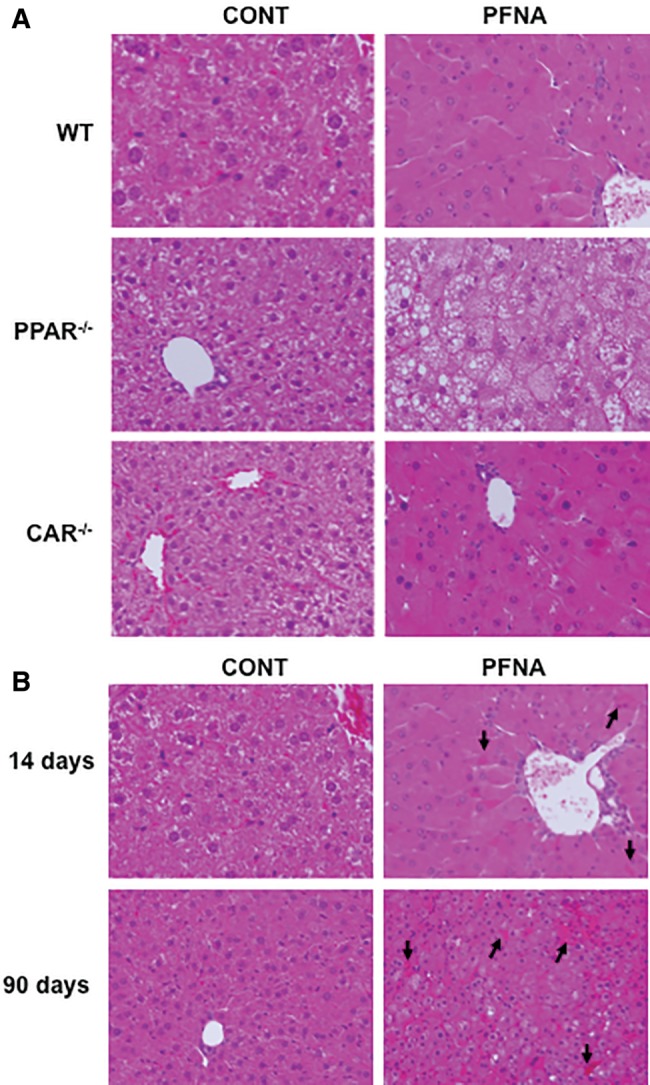
Liver histology of PFNA-exposed WT, PPARα-null, and CAR-null mice. The mouse livers were collected 1 week (A) or 2 weeks or 3 months after PFNA exposure (B) after a single i.p. administration of PFNA (0.1 mmol/kg of body weight) (n = 5/genotype/exposure). The freshly collected livers were fixed in 10% zinc formalin, and then processed for hematoxylin and eosin (H/E) staining. Representative images are shown in x200 magnification. The arrows point to yellow bile droplets/bile plugs formed in the liver section.
Alteration of Liver-to-Body Weight Ratios by PFNA in WT, PPARα-Null, and CAR-Null Mice
PFNA progressively decreased body weight similarly in WT and CAR-null mice. Specifically, a single PFNA administration decreased body weights of WT and CAR-null mice 7–8 g after 14 days. In contrast, PFNA did not apparently decrease body weight in PPARα-null mice. PFNA increased the ratios of liver-to-body weight 2.4-, 2-, and 1.6-fold, respectively, in WT, PPARα-null, and CAR-null mice (Figure 2).
Figure 2.
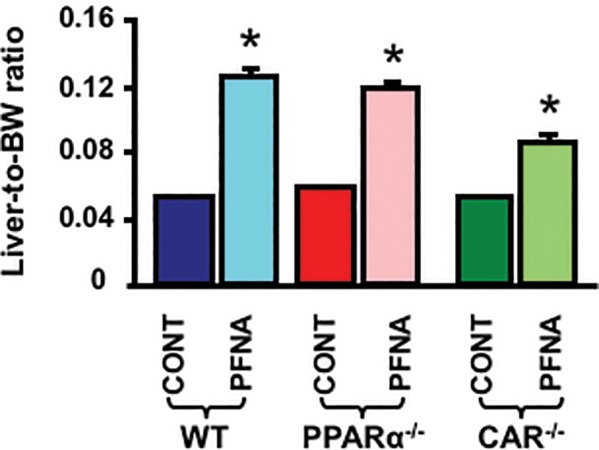
Alteration of liver-to-body weight ratios by PFNA. The body weight and liver weight of adult male C57BL/6 WT mice and age-matched male PPARα-null and CAR-null mice (n = 5/genotype/exposure) mice following a single i.p. administration of PFNA (0.1 mmol/kg) were determined 1 week later. The ratios of liver-to-body weight were calculated. The data are presented as mean ± SEM. Asterisks (*) represent a statistical difference (p < .05) between control and PFNA exposure.
Alterations of ALT and Conjugated Bilirubin by PFNA in WT, PPARα-Null, and CAR-Null Mouse Serum
As shown in Figure 3A, PFNA increased serum ALT approximately 15-fold in WT mice. In CAR-null mice, PFNA increased serum ALT about 3.5-fold. In contrast, PFNA did not increase serum ALT in PPARα-null mice.
Figure 3.
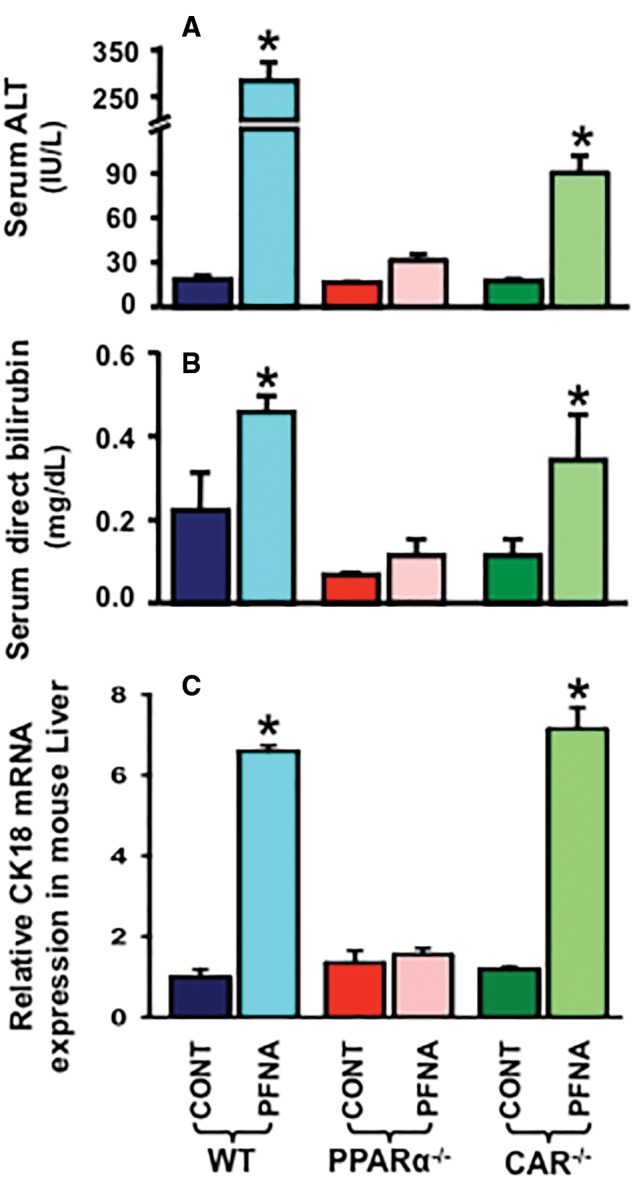
Effect of PFNA on serum ALT activity (A), serum conjugated bilirubin levels (B), and hepatic CK18 mRNA expression (C) in WT, PPARα-null, and CAR-null mice. Data are reported as mean ± SEM (n = 5/group). Asterisks (*) indicate statistically significant differences between control and PFNA exposure (p < .05).
Liver injury, including cholestasis, is often characterized by increased serum levels of conjugated bilirubin. As shown in Figure 3B, PFNA increased serum conjugated bilirubin 2.1-fold in WT mice, but not in PPARα-null mice. In contrast, PFNA increased conjugated bilirubin about 3-fold in serum of CAR-null mice.
Regulation of CK18 by PFNA in WT, PPARα-Null, and CAR-Null Mouse Liver
Cytokeratin 18 (CK18), a major intermediate filament protein expressed by hepatocytes, is a newly characterized biomarker of liver injury (Weiler et al., 2015). As shown in Figure 3C, PFNA increased CK18 mRNA expression in WT (↑ 6.6-fold) and CAR-null (↑ 6-fold) mouse livers, but not in PPARα-null mouse livers.
Impacts of PFNA on BA Concentrations in WT, PPARα-Null, and CAR-Null Mouse Serum
Disruption of PPARα or CAR function, as observed in PPARα-null or CAR-null mice, did not alter the constitutive concentrations of individual BAs (Figure 4A), unconjugated BAs, glycine-conjugated BAs, taurine-conjugated BAs, or total BAs (Figure 4B) in mouse serum.
Figure 4.
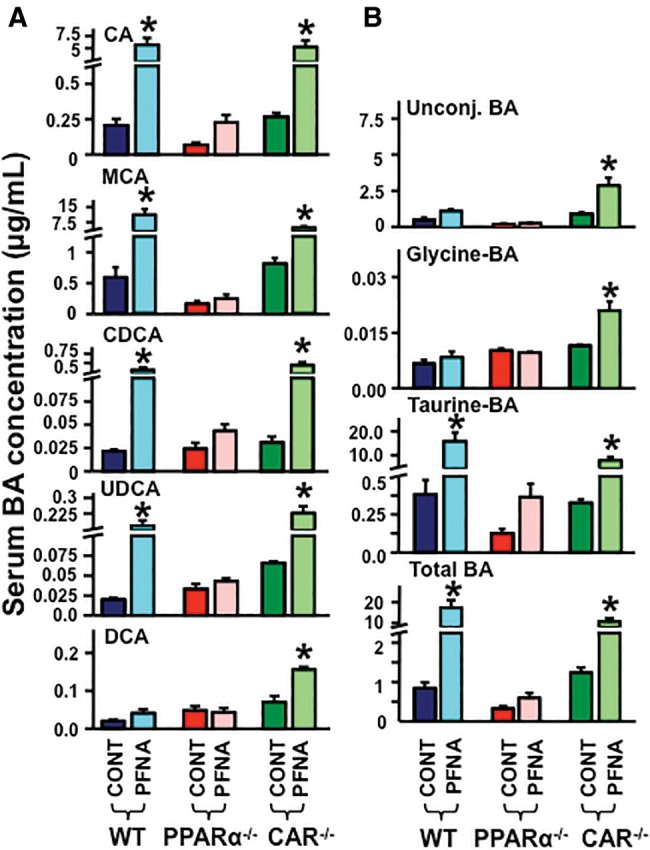
Alteration of BA concentrations by PFNA in serum of WT, PPARα-null, and CAR-null mice. The serum concentrations of (A) CA, MCA, CDCA, UDCA, and DCA, as well as (B) individual unconjugated BAs, glycine-conjugated BAs, taurine-conjugated BAs, and total serum BAs were quantified by an UPLC-MS/MS method. The data are presented as mean ± SEM. Asterisks (*) represent a statistical difference (p < .05) between control and PFNA exposure.
As shown in Figure 4A, PFNA increased serum concentrations of CA (↑ 21-fold), MCA (↑ 25-fold), CDCA (↑ 20-fold), UDCA (↑ 8-fold), but not DCA. PFNA did not increase BA concentrations in PPARα-null mice, but increased BAs in CAR-null mice.
As observed in Figure 4B, PFNA did not alter the serum concentrations of unconjugated and glycine-conjugated BAs. In contrast, PFNA increased taurine-conjugated (↑ 40-fold) and total serum BAs (↑ 19-fold) in WT mice, but not in PPARα-null mice. In CAR-null mice, PFNA increased serum concentrations of unconjugated (↑ 3-fold), glycine-conjugated (↑ 2.2-fold), taurine-conjugated (↑ 30-fold), and total serum BAs (↑ 9.8-fold).
Effects of PFNA on BA Concentrations in Livers of WT, PPARα-Null, and CAR-Null Mice
Disruption of PPARα or CAR function decreased the constitutive concentrations of individual BAs, except for DCA, in mouse livers (Figure 5A). Disruption of PPARα function, as observed in the PPARα-null mice, resulted in decreased constitutive concentrations of unconjugated, taurine-conjugated, and total BAs, but not glycine-conjugated BAs in mouse livers. Disruption of CAR function, as observed in the CAR-null mice, decreased constitutive concentrations of total and taurine-conjugated BAs, but not unconjugated and glycine-conjugated BAs in mouse livers (Figure 5B).
Figure 5.
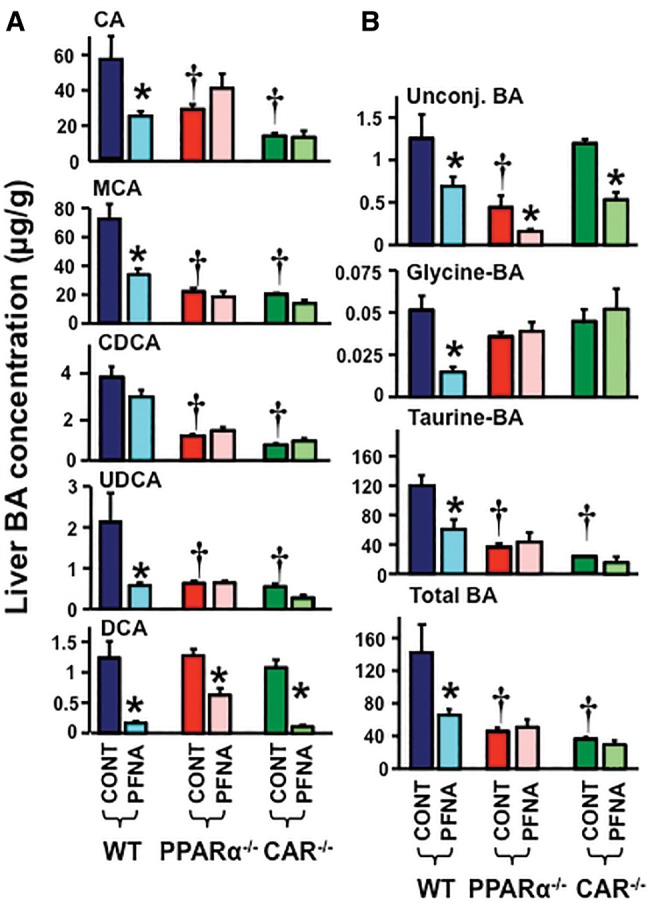
Alteration of BA concentrations by PFNA in liver of WT, PPARα-null, and CAR-null mice. The liver concentrations of (A) CA, MCA, CDCA, UDCA, and DCA, as well as (B) individual unconjugated BAs, glycine-conjugated BAs, taurine-conjugated BAs, and total liver BAs were quantified by an UPLC-MS/MS method. The data are presented as mean ± SEM. Asterisks (*) represent a statistical difference (p < .05) between control and PFNA exposure. Single daggers (†) represent a statistical difference (p < .05) between WT and PPARα-null/CAR-null mice.
In livers of WT mice, PFNA decreased the concentrations of CA (↓ 60%), MCA (↓ 55%), UDCA (↓ 65%), and DCA (↓ 88%), but not CDCA. In contrast, in PPARα-null and CAR-null mice, PFNA decreased the concentrations of DCA in livers, but did not decrease the concentrations of CA, MCA, CDCA, or UDCA (Figure 5A).
In WT mice, PFNA decreased concentrations of unconjugated (↓ 50%), glycine-conjugated (↓ 70%), taurine-conjugated (↓ 50%), and total BAs (↓ 60%) in mouse livers. In PPARα-null mice, PFNA did not decrease BA concentrations in liver, except for unconjugated BA concentrations. In CAR-null mice, PFNA decreased concentrations of unconjugated BAs (↓ 60%) in liver, but did not decrease liver concentrations of glycine-conjugated, taurine-conjugated, and total liver BAs (Figure 5B).
Effects of PFNA on Serum and Liver Cholesterol in WT, PPARα-Null, and CAR-Null Mice
Cholesterol is the precursor for the de novo biosynthesis of BAs. More than 90% of total cholesterol in the body is excreted after conversion into BAs. Therefore, the impact of PFNA on cholesterol homeostasis in mouse serum and liver was determined.
Disruption of PPARα or CAR function as observed in PPARα-null and CAR-null mice, respectively, did not alter free and total cholesterol levels in mouse serum (Figure 6A). In contrast, disruption of PPARα and CAR function reduced free and total cholesterol levels >50% in mouse liver (Figure 6B).
Figure 6.
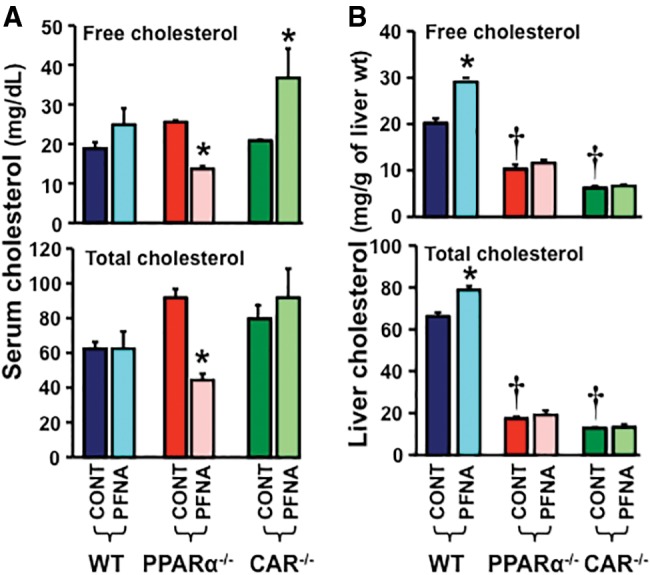
Alteration of free and total cholesterol concentrations by PFNA in sera and livers of WT, PPARα-null, and CAR-null mice. The concentrations of free and total cholesterol in mouse (A) serum and (B) liver were analyzed spectrophotometrically at 505 nm by using a commercially available kit. The data are presented as mean ± SEM (n = 5/exposure). Asterisks (*) represent a statistical difference (p < .05) between control and PFNA exposure. Single daggers (†) represent a statistical difference (p < .05) between WT and PPARα-null/CAR-null mice.
In mouse serum, PFNA did not alter free or total cholesterol concentrations in the serum of WT mice, but decreased them in the serum of PPARα-null mice. PFNA increased free cholesterol but not total cholesterol in the serum of CAR-null mice (Figure 6A). In mouse liver, PFNA increased free and total cholesterol concentrations 150% and 125%, respectively, in WT mouse livers, but not in PPARα-null or CAR-null mouse livers (Figure 6B).
Effect of PFNA on mRNA Expression of Drug and BA Transporters in WT, PPARα-Null, and CAR-Null Mouse Liver
Disruption of PPARα or CAR function increased Oatp1a4 (↑ 4- and 4.8-fold, respectively) (Figure 7A). In addition, disruption of CAR function increased bile salt export pump (Bsep) (↑ 2-fold), multidrug resistance transporter 2 (Mdr2) (↑ 2.3-fold), and multidrug resistance-associated protein 2 (Mrp2) (↑ 2.3-fold) mRNA expression in mouse livers (Figure 7A).
Figure 7.
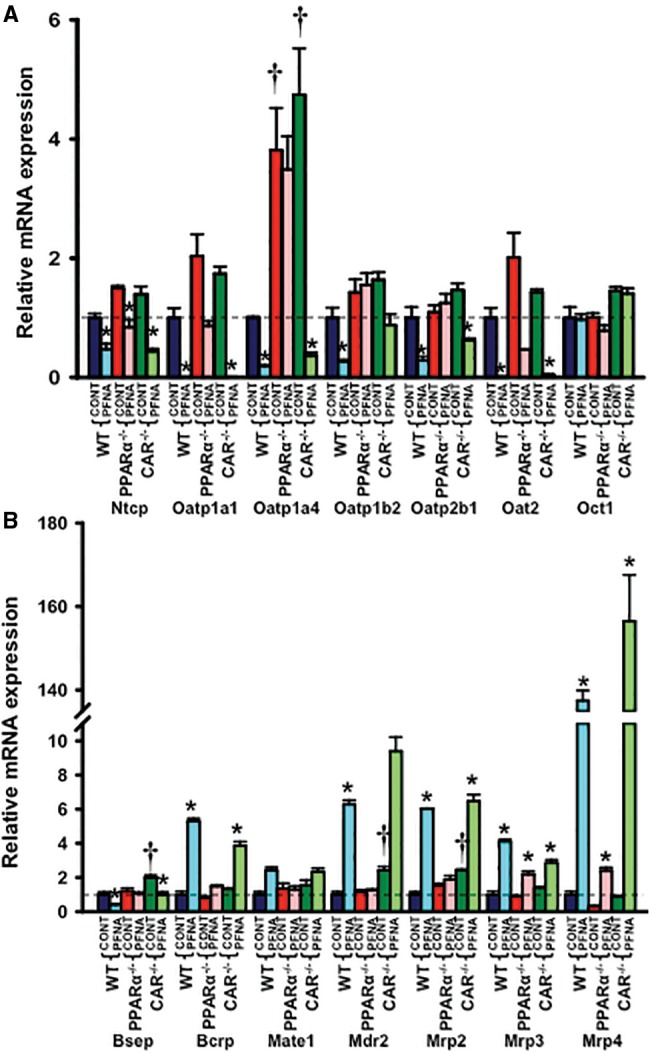
Messenger RNA expression of (A) uptake and (B) efflux transporters in WT, PPARα-null, and CAR-null mouse livers after PFNA exposure. The data are presented as mean ± SEM (n = 5/exposure). Asterisks (*) represent a statistical difference (p < .05) between control and PFNA exposure in mice of the same genotype. Single daggers (†) represent a statistical difference (p < .05) between WT and PPARα-null/CAR-null mice.
PFNA administration decreased most uptake transporters (Ntcp [↓ 55%], Oatp1a1 [↓ 99%], 1a4 [↓ 80%], 1b2 [↓ 75%], 2b1 [↓ 75%], and Oat2 [↓ 99%]) in WT mouse livers, also in CAR-null mouse livers (Ntcp [↓ 70%], Oatp1a1 [↓ 99%], 1a4 [↓ 92%], 1b2 [↓ 47%], 2b1 [↓ 58%], and Oat2 [↓ 97%]), but generally not in PPARα-null mouse livers (Figure 7A).
PFNA decreased the major BA efflux transporter Bsep (↓ approximately 50%), but increased some other efflux transporters such as breast cancer resistance protein (Bcrp) (↑ >4-fold), Mdr2 (↑ >6.4-fold), and Mrp2 (↑ >6-fold) in both WT and CAR-null mouse livers, but not in PPARα-null mouse livers. In contrast, PFNA induced Mrp3 and 4 mRNA expression in WT, PPARα-null, and CAR-null mouse livers, indicating that PFNA increased these 2 Mrps independent of PPARα or CAR activation (Figure 7B).
Effect of PFNA on mRNA Expression of BA Biosynthesis Enzymes in WT, PPARα-Null, and CAR-Null Mouse Liver
Disruption of CAR function increased Cyp7a1 mRNA (↑ 4-fold) in mouse livers (Figure 8A).
Figure 8.
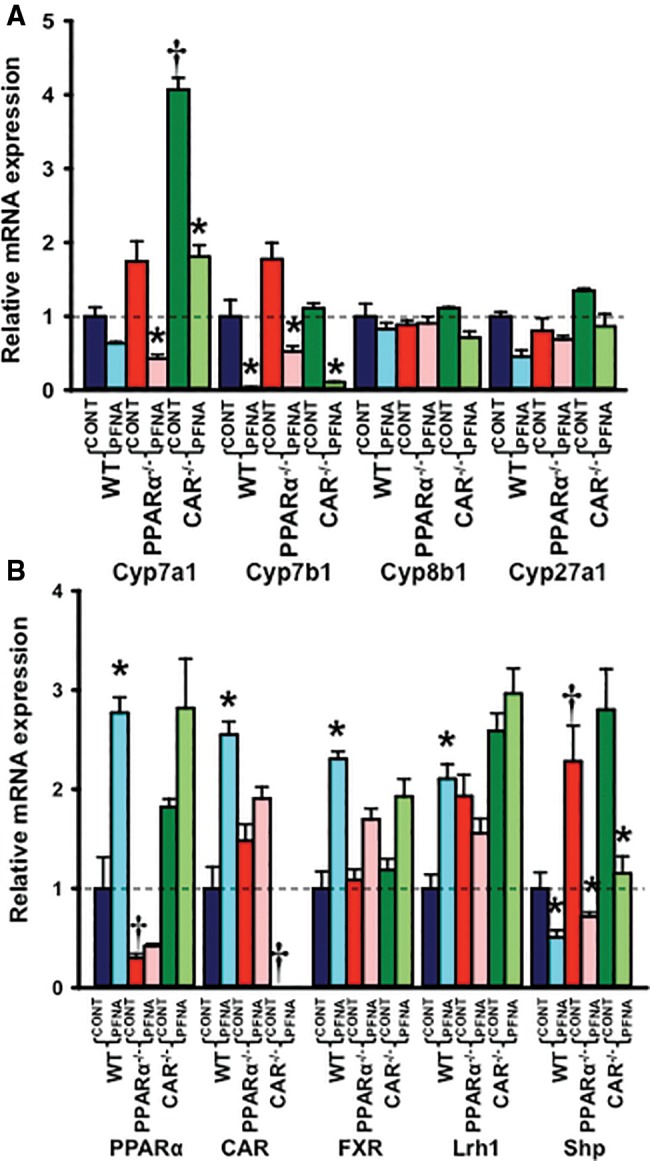
Messenger RNA expression of (A) BA biosynthesis enzymes and (B) some transcription factors in WT, PPARα-null, and CAR-null mouse livers after PFNA exposure. The data are presented as mean ± SEM (n = 5/exposure). Asterisks (*) represent a statistical difference (p < .05) between control and PFNA exposure in mice of the same genotype. Single daggers (†) represent a statistical difference (p < .05) between WT and PPARα-null/CAR-null mice.
PFNA tended to decrease Cyp7a1 mRNA expression in WT mouse livers (↓ 40%), but decreased it 75% and 55%, respectively, in PPARα-null and CAR-null mouse livers (Figure 8A). Cyp7b1 mRNA expression was decreased by PFNA in WT (↓ 96%), PPARα-null (↓ 70%), and CAR-null (↓ 90%) mouse livers. In contrast, PFNA did not alter Cyp8b1 or Cyp27a1 mRNA expression in WT, PPARα-null, or CAR-null mice (Figure 8A).
Effect of PFNA on mRNA Expression of Some Transcription Factors in WT, PPARα-Null, and CAR-Null Mouse Liver
PFNA increased mRNA expression of CAR, PPARα, and farnesoid X receptor >2-fold in WT mouse livers, but not in PPARα-null or CAR-null mouse livers (Figure 8B). PFNA increased mRNA expression of liver receptor homolog 1 (Lrh1) 2-fold in WT, but not in PPARα-null or CAR-null mouse livers. In addition, PFNA decreased small heterodimer partner (Shp) mRNA expression in WT (↓ 50%), PPARα-null (↓ 70%), and CAR-null (↓ 60%) mouse livers (Figure 8B).
Effect of PFNA on mRNA Expressions of Some Sterol-Associated Enzymes and Transporters in WT, PPARα-Null, and CAR-Null Mouse Liver
PFNA increased mRNA expression of sterol efflux transporters Abca1 (↑ 2.5-fold), Abcg1 (↑ 6-fold), Abcg5 (↑ 6.5-fold)/g8 (↑ 4.7-fold), and mitochondrial cholesterol transporter (steroidogenic acute regulatory protein, StAR) at least 2-fold in WT and CAR-null mouse livers, but not in PPARα-null mouse livers, indicating a PPARα-dependent mechanism (Figure 9). In addition, PFNA increased the expression of Scavenger receptor class B member 1 (SR-B1) (↑ 2-fold), which facilitates the uptake of cholesterol into liver, and 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase (↑ 1.7-fold), the rate-limiting enzyme of de novo cholesterol biosynthesis, in WT but not in CAR-null mouse livers. In contrast, PFNA tended to decrease SR-B1 and HMG-CoA reductase mRNA expression in PPARα-null mouse livers (Figure 9).
Figure 9.
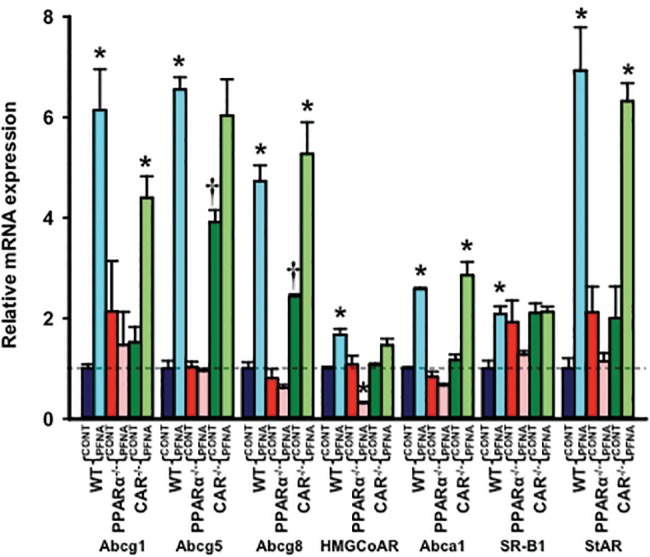
Messenger RNA expression of sterol-associated enzymes and transporters in WT, PPARα-null, and CAR-null mouse livers after PFNA exposure. The data are presented as mean ± SEM (n = 5/exposure). Asterisks (*) represent a statistical difference (p < .05) between control and PFNA exposure in mice of the same genotype. Single daggers (†) represent a statistical difference (p < .05) between WT and PPARα-null/CAR-null mice.
DISCUSSION
We previously reported that PFDA increased total BA concentrations in mouse serum, and decreased Ntcp and Oatp1b2 mRNA expression in a PPARα-dependent manner in mouse liver (Cheng and Klaassen, 2008a). In the present study, we showed that similar to PFDA effects, PFNA increased total BA concentrations in mouse serum and decreased Ntcp and Oatp1b2 mRNA expression in mouse liver. Furthermore, PFNA also decreased total BA concentrations in mouse liver, and produced cholestasis in mice (Figs. 4 and 5).
It has been reported that after absorption, PFNA is accumulated in mouse liver (Tatum-Gibbs et al., 2011). Therefore, the liver is the logical target organ of PFNA exposure. We reported herein that PFNA causes apparent liver injury in WT mice, indicated by increased serum levels of ALT activity (Figure 3A). In addition, PFNA produced liver injury in a PPARα-dependent but CAR-independent manner, as indicated by attenuated serum ALT levels in PPARα-null mice but elevated serum ALT levels in CAR-null mice (Figure 3A). CK18 is an emerging biomarker of liver injury. CK18 can bind bilirubin and is a membrane reservoir for the transport and secretion of bile pigments. Recent studies demonstrated that CK18 is up-regulated by toxic BAs, particularly in cholestatic conditions. Increased CK18 expression has been suggested to indicate a stress response against the toxic effects of BAs (Fickert et al., 2002). The data indicate that PFNA markedly increased CK18 mRNA expression in WT mouse livers, which further indicates that PFNA produces liver injury and alters BA homeostasis (Figure 3C).
In addition, PFNA similarly produced mouse hepatomegaly in WT, PPARα-null, and CAR-null mice (Figure 2). This indicated that activation of PPARα alone or CAR alone is not sufficient for PFNA-induced hepatomegaly. By engineering PPARα/CAR double-null mouse model, our unpublished data showed that activation of both PPARα and CAR are responsible for PFNA-induced hepatomegaly.
Homeostasis of BAs in liver is maintained through sinusoidal BA extraction from blood, de novo BA biosynthesis from cholesterol in the liver, as well as sinusoidal and canalicular BA excretion. PFNA decreased Ntcp and Oatp1b2 expression (Figure 7A). Ntcp and Oatp1b2 play important roles in the hepatic uptake of BAs from sinusoidal blood. Ntcp transports conjugated BAs, especially taurine-conjugated BAs. Oatps, including Oatp1b2, primarily transport unconjugated BAs and also some drugs. Therefore, PFNA administration may lead to less extraction of BAs from the sinusoidal blood. Bsep and Mrp2 are two important canalicular efflux transporters, with Bsep transporting BAs, whereas Mrp2 transporting conjugated bilirubin and drugs. PFNA decreased Bsep mRNA, but increased Mrp2 mRNA (Figure 7B). PFNA also increased Mrp3 and 4 mRNA expression, which may enhance sinusoidal BA excretion from mouse liver into blood (Figure 7B). Thus, less sinusoidal BA extraction from blood and enhanced sinusoidal BA excretion by PFNA may account for the increased total BAs in serum and decreased total BAs in liver. In addition, PFNA markedly increased cholesterol efflux transporters Abcg1, Abcg5/g8, and Abca1, and phospholipid efflux transporter Mdr2 (Figs. 7B and 9). Therefore, except for Bsep, PFNA increased a majority of efflux transporters in mouse liver.
De novo BA biosynthesis from cholesterol by the liver can lead to an increase in total BA concentrations in the liver. The present data indicate that PFNA did not alter mRNA expression of Cyp7a1, the rate-limiting enzyme in the classic BA biosynthesis pathway (Figure 8A). In addition, PFNA decreased mRNA expression of Cyp7b1, a key enzyme in the alternative BA biosynthesis pathway. Therefore, although PFNA increases free and total cholesterol levels in the liver, it apparently decreases de novo BA biosynthesis.
Cyp2B10 and Cyp4A14 are classical target genes of CAR and PPARα activation in mouse liver. Similar to the effects of PFDA, a single i.p. administration of 0.1 mmol/kg PFNA markedly induced mRNA expression of Cyp2B10 (21-fold) and Cyp4A14 (49-fold) in mouse liver (data not shown), indicating that PFNA activates PPARα and CAR. Activation of both PPARα and CAR nuclear receptors play important roles in the regulation of drug processing genes and carcinogenesis. Therefore, PFNA may regulate the expression of drug processing genes and promote hepatocarcinogenesis via activation of PPARα and CAR nuclear receptors. In addition, it is well known that perfluorochemicals activate PPARα. However, more and more evidence shows that in addition to PPARα activation, perfluorochemicals activate other signaling pathways to produce its effects, such as hepatomegaly and reproductive toxicity (Cheng and Klaassen, 2008a,b; Rosen et al., 2017). In the present study, we showed that activation of both PPARα and CAR signaling are involved in the regulation of BA and cholesterol metabolism by PFNA. In addition, PPARα activation accounted for most of the alterations of BA and cholesterol homeostasis produced by PFNA, similar to the effects of PFDA (Cheng and Klaassen, 2008a; Maher et al., 2008). For instance, PFNA altered serum ALT, bilirubin, serum, and liver BA concentrations and the expression of most of the BA transporters, such as Bsep, Oatp1a1, 2b1, Bcrp, and Mrp2 in a PPARα-dependent manner (Figs. 7A and B). In addition, PFNA PPARα-dependently increased mRNA expression of Abcg1, Abcg5/g8 and Abca1, and StAR, which are all involved in cholesterol efflux or elimination from hepatocytes (Figure 9). In contrast, in a CAR-dependent manner, PFNA decreased total and individual BA concentrations, increased free and total cholesterol, as well as induced Lrh1 and SR-B1 mRNA expression in mouse liver (Figs. 5, 6B, 8B, and 9).
Furthermore, PFNA did not alter serum free and total cholesterol levels in WT mice, but decreased them in PPARα-null mice (Figure 6A). It was recently reported that gastric gavage of 5 mg/kg PFNA once daily for 14 days decreased total serum cholesterol levels in male BALB/C mice. However, this increase in total serum cholesterol was not observed in mice exposed to 0.2 and 1 mg/kg/day PFNA (Wang et al., 2015). Therefore, our data are consistent with the BALB/C mouse serum data dosed with 0.2 and 1 mg/kg/day PFNA, but somewhat inconsistent with that dosed with 5 mg/kg/day PFNA (Wang et al., 2015). To interpret the discrepancy in two studies, one should consider different mouse models (BALB/C mice vs C57BL/6 mice), different dosage and routes, and different exposure time (2 weeks vs 1 week) in the two studies. Similar as previously reported (Wang et al., 2015), PFNA increased free and total cholesterol levels in WT mouse liver, but in neither PPARα-null nor CAR-null mouse liver (Figure 6). In addition, PFNA induced mRNA expression of SR-B1 (liver uptake transporter of cholesterol) and HMG-CoA reductase (the rate-limiting enzyme of de novo cholesterol biosynthesis) in WT mouse liver, but in neither PPARα-null nor CAR-null mouse liver (Figure 9). This indicates that PFNA-activated PPARα and CAR may synergistically impact on a common regulatory mechanism to alter cholesterol metabolism in the mouse liver.
In Figure 9, we reported that the expression of cholesterol efflux transporters (Abcg5/g8) is up-regulated by PFNA in WT mouse liver. However, PFNA exposure increase free and total cholesterol levels in WT mouse liver (Figure 6B). To address this, we need to realize that the overall cholesterol levels in mouse liver are determined by multiple factors, such as hepatic uptake of cholesterol from sinusoidal blood, de novo cholesterol biosynthesis in the liver, conversion of cholesterol into BAs, and efflux from hepatocytes. Therefore, increased cholesterol in WT mouse liver may be due to decreased BA biosynthesis via Cyp7a1 and Cyp7b1 (down-regulated by PFNA), increased cholesterol biosynthesis via HMG-CoA reductase (up-regulated by PFNA), and increased liver uptake via SR-B1 (up-regulated by PFNA) (Figs. 8A and 9).
Previous studies have demonstrated that in addition to PPARα activation, Nrf2 activation plays important roles in the up-regulation of Mrp3 and 4 efflux transporters by PFDA (Maher et al., 2008), which may provide an explanation of how PFNA increased Mrp3 and 4 mRNA expression in WT, PPARα-null, and CAR-null mouse liver (Figure 7B).
Taken together, PFNA produced cholestasis in mouse liver, as well as increased total and individual BA concentrations in mouse serum, and decreased them in mouse liver. PPARα plays a central role in regulating BA and cholesterol homeostasis by PFNA in serum and liver of mice. Although PFNA was reported to decline from 2011 to 2014 in United States, it is still a global health problem with high concentrations in tap water in Japan and China (Mak et al., 2009). In addition, some fluorotelomers, which can be biotransformed to PFNA by biological systems (Butt et al., 2010; Dauchy et al., 2012), are still widely used. Therefore, exposure to PFNA can still pose a human health risk. In addition, because it is currently unknown whether perfluorochemicals impact bile formation, the present study provides important insight into the physiopathology of perfluorochemical-induced hepatotoxicity and the underlying mechanisms.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
This work was supported by internal research funds including new faculty start fund and Seed grant to X.C. from St. John’s University; and National Institutes of Health (R01 ES09649 to C.D.K.).
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr Fang Fan (Cytopathology program, Department of Pathology & Laboratory Medicine, KU Medical Center, Kansas) for microscopic evaluation of liver histology. The authors declared that there is no conflict of interest.
AUTHORSHIP CONTRIBUTIONS
Participated in research design: Klaassen and Cheng. Conducted experiments: Zhang, Zhang, and Cheng. Performed data analysis: Zhang, Zhang, and Cheng. Wrote or contributed to the writing of the manuscript: Zhang, Zhang, Klaassen, and Cheng.
REFERENCES
- Akiyama T. E., Nicol C. J., Fievet C., Staels B., Ward J. M., Auwerx J., Lee S. S., Gonzalez F. J., Peters J. M. (2001). Peroxisome proliferator-activated receptor-alpha regulates lipid homeostasis, but is not associated with obesity: Studies with congenic mouse lines. J. Biol. Chem. 276, 39088–39093. [DOI] [PubMed] [Google Scholar]
- Alnouti Y., Csanaky I. L., Klaassen C. D. (2008). Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 873, 209–217. 10.1016/j.jchromb.2008.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster D. W., Birnbaum L. S. (1989). The biochemical toxicity of perfluorodecanoic acid in the mouse is different from that of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 99, 544–554. 10.1016/0041-008X(89)90161-0 [DOI] [PubMed] [Google Scholar]
- Butt C. M., Muir D. C., Mabury S. A. (2010). Biotransformation of the 8:2 fluorotelomer acrylate in rainbow trout. 1. In vivo dietary exposure. Environ. Toxicol. Chem. 29, 2726–2735. 10.1002/etc.349 [DOI] [PubMed] [Google Scholar]
- Calafat A. M., Wong L. Y., Kuklenyik Z., Reidy J. A., Needham L. L. (2007). Polyfluoroalkyl chemicals in the U.S. population: Data from the National Health and Nutrition Examination Survey (NHANES) 2003-2004 and comparisons with NHANES 1999-2000. Environ. Health Perspect. 115, 1596–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. (2017). The Fourth National Report on Human Exposure to Environmental Chemicals. Centers for Disease Control and Prevention (CDC.gov), 347–350.
- Cheng X., Klaassen C. D. (2008a). Critical role of PPAR-alpha in perfluorooctanoic acid- and perfluorodecanoic acid-induced downregulation of Oatp uptake transporters in mouse livers. Toxicol. Sci. 106, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Klaassen C. D. (2008b). Perfluorocarboxylic acids induce cytochrome P450 enzymes in mouse liver through activation of PPAR-alpha and CAR transcription factors. Toxicol. Sci. 106, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K. P., Grey B. E., Rosen M. B., Wood C. R., Tatum-Gibbs K. R., Zehr R. D., Strynar M. J., Lindstrom A. B., Lau C. (2015). Developmental toxicity of perfluorononanoic acid in mice. Reprod. Toxicol. 51, 133–144. [DOI] [PubMed] [Google Scholar]
- Das K. P., Wood C. R., Lin M. T., Starkov A. A., Lau C., Wallace K. B., Corton J. C., Abbott B. D. (2017). Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology 378, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauchy X., Boiteux V., Rosin C., Munoz J. F. (2012). Relationship between industrial discharges and contamination of raw water resources by perfluorinated compounds. Part I: Case study of a fluoropolymer manufacturing plant. Bull. Environ. Contam. Toxicol. 89, 525–530. 10.1007/s00128-012-0704-x [DOI] [PubMed] [Google Scholar]
- Ellis D. A., Martin J. W., De Silva A. O., Mabury S. A., Hurley M. D., Sulbaek Andersen M. P., Wallington T. J. (2004). Degradation of fluorotelomer alcohols: A likely atmospheric source of perfluorinated carboxylic acids. Environ. Sci. Technol. 38, 3316–3321. [DOI] [PubMed] [Google Scholar]
- Fang X., Feng Y., Shi Z., Dai J. (2009). Alterations of cytokines and MAPK signaling pathways are related to the immunotoxic effect of perfluorononanoic acid. Toxicol. Sci. 108, 367–376. 10.1093/toxsci/kfp019 [DOI] [PubMed] [Google Scholar]
- Fang X., Zhang L., Feng Y., Zhao Y., Dai J. (2008). Immunotoxic effects of perfluorononanoic acid on BALB/c mice. Toxicol. Sci. 105, 312–321. 10.1093/toxsci/kfn127 [DOI] [PubMed] [Google Scholar]
- Fickert P., Trauner M., Fuchsbichler A., Stumptner C., Zatloukal K., Denk H. (2002). Cytokeratins as targets for bile acid-induced toxicity. Am. J. Pathol. 160, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. W., Uraih L. C., Birnbaum L. S. (1989). Acute toxicity of perfluorodecanoic acid in C57BL/6 mice differs from 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Fundam. Appl. Toxicol. 13, 723–736. 10.1016/0272-0590(89)90330-8 [DOI] [PubMed] [Google Scholar]
- Kato K., Wong L. Y., Jia L. T., Kuklenyik Z., Calafat A. M. (2011). Trends in exposure to polyfluoroalkyl chemicals in the U.S. Population: 1999-2008. Environ. Sci. Technol. 45, 8037–8045. [DOI] [PubMed] [Google Scholar]
- Kudo N., Kawashima Y. (1997). Fish oil-feeding prevents perfluorooctanoic acid-induced fatty liver in mice. Toxicol. Appl. Pharmacol. 145, 285–293. 10.1006/taap.1997.8186 [DOI] [PubMed] [Google Scholar]
- Kudo N., Kawashima Y. (2003). Toxicity and toxicokinetics of perfluorooctanoic acid in humans and animals. J. Toxicol. Sci. 28, 49–57. 10.2131/jts.28.49 [DOI] [PubMed] [Google Scholar]
- Lau C., Anitole K., Hodes C., Lai D., Pfahles-Hutchens A., Seed J. (2007). Perfluoroalkyl acids: A review of monitoring and toxicological findings. Toxicol. Sci. 99, 366–394. [DOI] [PubMed] [Google Scholar]
- Lee S. S., Pineau T., Drago J., Lee E. J., Owens J. W., Kroetz D. L., Fernandez-Salguero P. M., Westphal H., Gonzalez F. J. (1995). Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 15, 3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher J. M., Aleksunes L. M., Dieter M. Z., Tanaka Y., Peters J. M., Manautou J. E., Klaassen C. D. (2008). Nrf2- and PPAR alpha-mediated regulation of hepatic Mrp transporters after exposure to perfluorooctanoic acid and perfluorodecanoic acid. Toxicol. Sci. 106, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak Y. L., Taniyasu S., Yeung L. W., Lu G., Jin L., Yang Y., Lam P. K., Kannan K., Yamashita N. (2009). Perfluorinated compounds in tap water from China and several other countries. Environ. Sci. Technol. 43, 4824–4829. [DOI] [PubMed] [Google Scholar]
- Mitro S. D., Dodson R. E., Singla V., Adamkiewicz G., Elmi A. F., Tilly M. K., Zota A. R. (2016). Consumer product chemicals in indoor dust: A quantitative meta-analysis of U.S. studies. Environ. Sci. Technol. 50, 10661–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen G. W., Zobel L. R. (2007). Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. Int. Arch. Occup. Environ. Health 81, 231–246. 10.1007/s00420-007-0213-0 [DOI] [PubMed] [Google Scholar]
- Prevedouros K., Cousins I. T., Buck R. C., Korzeniowski S. H. (2006). Sources, fate and transport of perfluorocarboxylates. Environ. Sci. Technol. 40, 32–44. [DOI] [PubMed] [Google Scholar]
- Rockwell C. E., Turley A. E., Cheng X., Fields P. E., Klaassen C. D. (2013). Acute Immunotoxic Effects of Perfluorononanoic Acid (PFNA) in C57BL/6 Mice. Clin. Exp. Pharmacol Suppl 4, S4–002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell C. E., Turley A. E., Cheng X., Fields P. E., Klaassen C. D. (2017). Persistent alterations in immune cell populations and function from a single dose of perfluorononanoic acid (PFNA) in C57Bl/6 mice. Food Chem. Toxicol. 100, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen M. B., Das K. P., Rooney J., Abbott B., Lau C., Corton J. C. (2017). PPARalpha-independent transcriptional targets of perfluoroalkyl acids revealed by transcript profiling. Toxicology 387, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen M. B., Lee J. S., Ren H., Vallanat B., Liu J., Waalkes M. P., Abbott B. D., Lau C., Corton J. C. (2008). Toxicogenomic dissection of the perfluorooctanoic acid transcript profile in mouse liver: Evidence for the involvement of nuclear receptors PPAR alpha and CAR. Toxicol. Sci. 103, 46–56. [DOI] [PubMed] [Google Scholar]
- Son H. Y., Kim S. H., Shin H. I., Bae H. I., Yang J. H. (2008). Perfluorooctanoic acid-induced hepatic toxicity following 21-day oral exposure in mice. Arch. Toxicol. 82, 239–246. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Aleksunes L. M., Yeager R. L., Gyamfi M. A., Esterly N., Guo G. L., Klaassen C. D. (2008). NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 325, 655–664. [DOI] [PubMed] [Google Scholar]
- Tatum-Gibbs K., Wambaugh J. F., Das K. P., Zehr R. D., Strynar M. J., Lindstrom A. B., Delinsky A., Lau C. (2011). Comparative pharmacokinetics of perfluorononanoic acid in rat and mouse. Toxicology 281, 48–55. [DOI] [PubMed] [Google Scholar]
- Ueda A., Hamadeh H. K., Webb H. K., Yamamoto Y., Sueyoshi T., Afshari C. A., Lehmann J. M., Negishi M. (2002). Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol. Pharmacol. 61, 1–6. [DOI] [PubMed] [Google Scholar]
- Van Rafelghem M. J., Mattie D. R., Bruner R. H., Andersen M. E. (1987). Pathological and hepatic ultrastructural effects of a single dose of perfluoro-n-decanoic acid in the rat, hamster, mouse, and guinea pig. Fundam. Appl. Toxicol. 9, 522–540. [PubMed] [Google Scholar]
- Wang J., Yan S., Zhang W., Zhang H., Dai J. (2015). Integrated proteomic and miRNA transcriptional analysis reveals the hepatotoxicity mechanism of PFNA exposure in mice. J. Proteome Res. 14, 330–341. 10.1021/pr500641b [DOI] [PubMed] [Google Scholar]
- Weiler S., Merz M., Kullak-Ublick G. A. (2015). Drug-induced liver injury: the dawn of biomarkers? F1000prime Rep. 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D. C., Moore T., Abbott B. D., Rosen M. B., Das K. P., Zehr R. D., Lindstrom A. B., Strynar M. J., Lau C. (2008). Comparative hepatic effects of perfluorooctanoic acid and WY 14, 643 in PPAR-{alpha} knockout and wild-type mice. Toxicol. Pathol. 36, 632–639. [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Kawashima Y. (1997). Perfluorodecanoic acid enhances the formation of oleic acid in rat liver. Biochem. J. 325, 429–434. 10.1042/bj3250429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Xie Y., Alexson S. E., Nelson B. D., DePierre J. W. (2002). Involvement of the peroxisome proliferator-activated receptor alpha in the immunomodulation caused by peroxisome proliferators in mice. Biochem. Pharmacol. 63, 1893–1900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.