

Abstract
Alterations of the cellular proteome over time due to spontaneous or toxin-mediated enzymatic deamidation of glutamine (Gln) and asparagine (Asn) residues contribute to bacterial infection and might represent a source of aging-related diseases. Here, we put into perspective what is known about the mode of action of the CNF1 toxin from pathogenic Escherichia coli, a paradigm of bacterial deamidases that activate Rho GTPases, to illustrate the importance of determining whether exposure to these factors are risk factors in the etiology age-related diseases, such as cancer. In particular, through in silico analysis of the distribution of the CNF1-like deamidase active site Gly-Cys-(Xaa)n-His sequence motif in bacterial genomes, we unveil the wide distribution of the super-family of CNF-like toxins and CNF-like deamidase domains among members of the Enterobacteriacae and in association with a large variety of toxin delivery systems. We extent our discussion with recent findings concerning cellular systems that control activated Rac1 GTPase stability and provide protection against cancer. These findings point to the urgency for developing holistic approaches toward personalized medicine that include monitoring for asymptomatic carriage of pathogenic toxin-producing bacteria and that ultimately might lead to improved public health and increased lifespans.
Keywords: deamidation, glutamine, cancer, dermonecrotic, toxins, gut microbiota
CNF-like deamidase modules show high prevalence in the arsenal of a wide spectrum of pathogenic bacterial species therefore representing a risk factor in etiology of aging-related diseases.
INTRODUCTION
The field of microbiology and notably the study of toxins in host–pathogen interactions has been a remarkable source of knowledge regarding the consequences of proteome alterations in human diseases. Pathogens are indeed endowed with the remarkable ability to deliver toxic enzymes into host cells that are able to catalyze post-translational modifications of critical cell regulators that in turn promote potent pathophysiological outcomes (Alouf, Ladant and Popoff 2015). Although we now have a better appreciation of the contributing role of these toxin-mediated processes during infection, little is known about the long-term impact of these environmental alterations of the proteome on the onset and/or progression of aging-related diseases. This contrasts with the study of late onset diseases, such as cancer, that have been most advanced with regard to processes involving alterations of the genome. Colorectal cancer (CRC) is a progressive disease of the large intestine that develops over extended periods of time, typically 10–40 years, as a result of accumulating mutations (Sears and Garrett 2014). Since the large intestine is an organ populated by a unique array of bacteria, we are confident that study of the impact of bacterial toxins produced by the microbiota on the host proteome will provide valuable insights regarding environmental factors contributing to human diseases that develop with age.
To illustrate our perspective for the need to define environmental risk factors that alter the proteome and thereby promote late-onset disease, we review recent advances made about the connection between potential cancer risk and exposure to the cytotoxic necrotizing factor 1 (CNF1) toxin from several pathogenic strains of Escherichia coli. CNF1 is a paradigm of bacterial deamidase toxins, as evidenced below by the unveiling of the unexpectedly broad distribution of CNF1-related deamidase domains in bacterial genomes. Extensive information on the mode of action and function of CNF1 has been collected since the discovery of this bacterial toxin with deamidase activity (Flatau et al.1997; Schmidt et al.1997). Remarkably, by catalyzing the deamidation of a specific glutamine (Gln) residue of host Rho GTPases, CNF1 confers these regulatory proteins with a gain-of-function by switching on the signaling pathways that they control. Rho GTPases, like other small G proteins of the Ras superfamily, are active in their GTP-bound form, associating with and activating a panel of effectors, many of which work at the membrane interface, but then hydrolyze the GTP into GDP to convert them into an inactive GDP-bound form (Hodge and Ridley 2016). Small GTPases are proven drivers of inflammation and tumorigenesis and serve as critical elements supporting tumor development (Kazanietz and Caloca 2017). Activation of Rho GTPases by bacterial toxins confers to pathogens the capacity to alter the architecture of host cells and tissues, thereby promoting their ability to evade host defenses, spread within and among hosts and form intracellular reservoirs of persistent bacteria (Lemichez and Aktories 2013; Popoff 2014). Due to the role played by Rho GTPases as critical drivers of inflammation and cancer progression, it is of importance to determine whether chronic dysregulation of Rho GTPases mediated by exposure to bacterially produced deamidases plays a role in the etiology of cancer. In agreement with this idea, epidemiological and functional links in mouse chronic infection models have started to establish casual links between the presence of pathogenic strains of E. coli producing CNF1 and progression of certain forms of cancer (Gagnière et al.2016; Guo et al.2017).
Spontaneous non-enzymatic deamidation of glutamine (Gln) and asparagine (Asn) residues is a common occurrence and is generally considered a molecular clock of protein aging (Robinson and Robinson 2001). The deamidation rate of an amide group within a protein is dependent on solvent accessibility, physicochemical conditions and the amino acid sequence surrounding the amide group. How long-living organisms cope with these alterations of the proteome and how these changes link to late onset diseases largely remain to be defined. Specific enzymatic deamidase activities are not restricted to infection with bacterial or viral pathogens (Washington, Banfield and Dangl 2013; Zhao et al.2016). In eukaryotes, specific protein deamidation controls the level of the anti-apoptotic factor Bcl-XL with important consequences for treatment response to DNA-damaging anti-neoplastic agents (Dho et al.2013). Thus, the study of the environmental effects conferred by CNF1 and other deamidase toxins could inform us about the influence of host deamidation reactions. Notably, we also consider the importance of studying CNF1 in the discovery of the regulation of activated forms of Rho GTPases by the host ubiquitin proteasome system. This system seems to play a crucial role in maintaining the equilibrium of Rho GTPase activation levels. A number of key enzymes that catalyze the ubiquitination of Rho GTPases for their subsequent targeting to proteasomal degradation are tumor suppressors. This raises the question of a possible relationship between efficiency of quality control systems of the proteome, chronic infection and late-onset diseases such as cancer.
The CNF1 toxin from E. coli and its prevalence
The name ‘cytotoxic necrotizing factor’ for this bacterial protein toxin produced by necrotoxigenic E. coli was coined to refer to the characteristic necrotic lesions that develop upon subcutaneous injection of the toxin. CNF1 is a 114 kDa, AB-like toxin that binds host cell surface receptors, p67/LR and Lu/BCAM, and enters endosomes, where upon acidification the activity domain is translocated across the vesicle membrane and delivered into the cytosol (Boquet and Lemichez 2003; Kim, Chung and Kim 2005; Piteau et al.2014) (Fig. 1). The internalization and intracellular trafficking of CNF1 in target cells proceeds via a mechanism that does not involve detergent-resistant membrane microdomains (filipin-insensitive), the brefeldin A-sensitive trans-Golgi network to endoplasmic reticulum pathway or the clathrin-dependent uptake pathway (Contamin et al.2000). CNF1 contains two hydrophobic alpha-helices that insert into membranes at acidic pH to promote the translocation of the catalytic domain into the cytosol (Pei, Doye and Boquet 2001). During the membrane insertion and translocation process, an endo-proteolytic cleavage of CNF1 occurs between amino-acids 532 and 544 (Knust et al.2009), and a C-terminal fragment of 55 kDa is released into the cytosol. Residues 720–1014 of this fragment constitute the enzymatic domain that catalyzes the irreversible deamidation of a specific Gln residue of Rho GTPases (63 in RhoA and 61 in Rac1/Cdc42) into a glutamic acid (Glu) (Flatau et al.1997; Schmidt et al.1997). This modification abrogates the GTPase activity of Rho proteins to promote their permanent activation.
Figure 1.

Schematic representation of CNF1 and its mode of action. CNF1 is a protein toxin of 1014 amino acids, comprised of several functional regions for (i) binding to the p67-LR (laminin receptor) and Lu/BCAM (Lutheran adhesion glycoprotein/basal cell adhesion molecule) receptors; (ii) two hydrophobic helices involved in the insertion of the toxin into membranes at acidic pH; (iii) a 55-kDa cargo delivered into the cytosol after proteolytic cleavage and comprising the deamidase domain (amino-acids 720–1014). The various stages of the intoxication process depicted in the scheme include: (1) toxin binding to the receptor complex; (2) internalization of the toxin receptor complex; (3) delivery of the catalytic cargo upon acidification; (4) deamidation of Rho GTPases; and (5) subsequent cellular responses to Rho GTPase activation.
The solved crystal structure of the catalytic domain of CNF1 provides some molecular insights about the specificity of action of CNF1 (Buetow et al.2001). This domain contains the invariant histidine (His) and cysteine (Cys) catalytic residues at the active site. The catalytic domain consists of two beta-sheets surrounded by alpha-helices and a crown of loops at the entrance of the catalytic site, which creates a deep, narrow cavity that restricts the accessibility of the catalytic site to specific substrates (Flatau et al.2000; Buetow and Ghosh 2003). The specificity of recognition of the substrate is provided by residues within certain loops, in particular loop-8, as well as loop-3 and 9 (Hoffmann, Aktories and Schmidt 2007; Buetow and Ghosh 2003). The distance between the loops and the catalytic site allows accommodation of the side chain of a Gln, but not the shorter side chain of Asn. Molecular determinants in the region surrounding the targeted Gln, and in particular arginine-68 and leucine-72 in RhoA, contribute to specific recognition of members of the Rho GTPase family among small GTPases (Lerm et al.1999; Buetow and Ghosh 2003). The tight specificity in the interactions between CNF1 and Rho GTPases accounts for differences in the panel of Rho GTPases modified by members of the CNF family. In E. coli for example, whereas CNF1 acts on RhoA, Rac1 and Cdc42 isoforms (Doye et al.2002), CNF2 acts on RhoA and Rac1, but not Cdc42 (Sugai et al.1999) and CNF3, like CNF1, acts on all three substrates, but more strongly on RhoA than CNF1 does (Stoll et al.2009). Moreover, CNFy from Yersinia pseudotuberculosis shows preference for RhoA over Rac1 and Cdc42 isoforms (Hoffmann et al.2004; Schweer et al.2013).
An association of E. coli strains containing the gene encoding CNF1 with acute intestinal and extra-intestinal infections has been established (Hashemizadeh, Kalantar-Neyestanaki and Mansouri 2017). CNF1 was first isolated and characterized from pathogenic strains of E. coli isolated from young children suffering from diarrhea (Caprioli et al.1983; Falbo et al.1993; Fabbri et al.2013). In humans, commensal and pathogenic strains of E. coli fall into four main phylogroups A, B1, B2 and D, which make up about 108 colony forming units per gram of feces (Tenaillon et al.2010). It is now established that E. coli bearing the cnf1 gene belong to the pathogenic phylogroups B2 and D and have a prevalence ranging from 8% to 30% in the human gut microbiota (Smati et al.2015). The prevalence of cnf1-positive strains is significantly higher in E. coli isolated from urinary tract infections, than in strains recovered from fecal samples, with a prevalence of up to 50% in the case of hemorrhagic urinary tract infections (Real et al.2007; Hashemizadeh, Kalantar-Neyestanaki and Mansouri 2017). The cnf1 gene displays its highest prevalence in E. coli isolates responsible for prostatitis with 64% (Mitsumori et al.1999). The gene for CNF1 is also present in some virulent strains of the intercontinental clonal group of E. coli sequence type 131 (ST131) that displays resistance to fluoroquinolone and expression of extended-spectrum β-lactamase (ESBL) (Nicolas-Chanoine, Bertrand and Madec 2014; Li et al.2017).
Breaching of cell homeostasis by CNF1
CNF1 has been classified as a cyclomodulin, a toxin that regulates cell-cycle progression (Nougayrede et al.2005). Specifically, CNF1 promotes accumulation of cells in the G2/M phase of the cell cycle and multi-nucleation in some cell lines. CNF1 achieves this by blocking apoptosis and inducing mitotic catastrophe, thereby promoting aneuploidy and a dysregulation of cytokinesis (Malorni and Fiorentini 2006). CNF1 also promotes cell-cycle progression via induction of cyclin D1 expression due to Rac1-mediated reactive oxygen species (ROS) production (Daugaard et al.2013). Moreover, by inducing activation of Rho GTPases and downstream actin cytoskeleton dynamics, CNF1 directly promotes major actin cytoskeletal remodeling, thereby disrupting uroepithelial cell junctions and inducing epithelial cell motility (Doye et al.2002). CNF1 also confers remarkable macropinocytic abilities to epithelial cells (Falzano et al.1993), and by stimulating Rac1 signaling CNF1 enhances cellular entry of E. coli expressing Type-I pili via the hijacking of beta-1 integrins (Eto et al.2007; Visvikis et al.2011). The internalized bacteria form reservoirs of persister bacterial cells that likely contribute to recurrent urinary tract infections (Mulvey, Schilling and Hultgren 2001).
Direct activation of Rho GTPases and their signaling pathways by CNF1 induces relatively minor changes in the global transcriptome landscape, except for promoting the expression of a specific array of inflammatory and anti-apoptotic factors associated with engagement of NF-κB signaling (Munro et al.2004). Similarly, production of CNFy by Y. pseudotuberculosis exacerbates inflammation and tissue damage during infection, even though this strain contains a plasmid encoding a type-3 secretion system (T3SS) that injects a plethora of Rho GTPase-inactivating factors (Schweer et al.2013). Critical components relaying signals from Rac1 and Cdc42 to the NF-κB pathway include the nucleotide-binding oligomerization domain-containing protein 1 (NOD1) and the receptor-interacting protein kinase 2 (RIPK2) (Keestra et al.2013; Stuart and Boyer 2013). Moreover, CNF1-mediated Rac activation stimulates the ASC/caspase-1 inflammasome to promote IL-1β processing and secretion (Diabate et al.2015), while RhoA activation antagonizes pyrin inflammasome activation (Xu et al.2014; Chung et al.2016). Induction of inflammasome in response to perturbations of Rho GTPase activities likely serves as a means for cells to sense the attack by pathogens expressing virulence factors targeting Rho GTPases (Stuart, Paquette and Boyer 2013; Xu et al.2014). Remarkably, this modulation of host immune responses makes CNF1 an immunostimulator of anti-microbial responses for prophylactic and therapeutic vaccine applications (Munro et al.2005; Munro, Flatau and Lemichez 2007; Michel et al.2016).
CNF1, in cooperation with the genetically linked alpha-hemolysin, is positively correlated with bloodstream infections of E. coli (Kadhum et al.2008; Diabate et al.2015). The inflammatory-based damaging impact of both CNF1 and alpha-hemolysin on host mucosa, as suggested by epidemiological studies mentioned above, is supported by experimental studies using small animals (Rippere-Lampe et al.2001; Garcia et al.2013). Pioneering epidemiological studies aimed at characterizing the gut microbiota associated with CRC has pinpointed E. coli of phylogroups B2 and D as common colonizers of colorectal polyps (Buc et al.2013; Jobin 2013; Raisch et al.2014). These bacteria produce a panel of different toxins that disrupt the host cell cycle and DNA integrity (Nougayrede et al.2005). Notably, about one fourth of these pathogenic strains of E. coli produce CNF1. Thus, chronic infection-associated inflammation coupled with dysregulation of cell fate, migration, and division by exposure to CNF1 represents a potential risk factor for cancer onset and progression. In support of this, a recent animal study has unveiled a role for CNF1 in promoting prostatic cancer progression and metastasis (Guo et al.2017). In this case, CNF1-mediated Cdc42-PAK1 signaling appears to mediate prostate cancer progression.
The extended family of CNF1 like toxins among pathogenic bacteria
There are at least nine CNF toxins that share strong homology with CNF1 throughout the entire sequence length (Fig. 2). CNF2 and CNF3 share 85% and 71% amino-acid sequence identity with CNF1, respectively, and are associated with diarrheagenic strains of E. coli in animals, while CNF1 is more prevalent in human isolates (Caprioli et al.1983; Falbo et al.1993; Mitsumori et al.1999; Real et al.2007; Kadhum et al.2008; Fabbri et al.2013; Diabate et al.2015), CNF3 and the plasmid-encoded CNF2 were identified in pathogenic strains of E. coli isolated from sheep, cows and goats (Oswald et al.1989; Orden et al.2007). CNF3 has also been identified in an isolate of Shigella boydii. Although a functional, full-length CNFy (65.1% shared identity with CNF1) is expressed in a few isolates of Y. pseudotuberculosis that belong to serogroup III, other strains of this group and the Yersinia pestis strain CO92 contain partially deleted cnfy genes (Lockman et al.2002). No full-length cnfy homologs have yet been detected in Yersinia enterocolitica, Y. pestis and non-pathogenic Yersinia species (Lockman et al.2002). Additional full-length CNF homologs have been discovered in the genomes of Moritella viscosa (CNFm) (Tunsjø et al.2011; Karlsen et al.2014) and Photobacterium damselae strain CIP 102761 (CNFp), each with 62%–65% protein sequence identity with the other CNF toxins. A CNF3-related homologue has also been identified in the meningitis causative agent Salmonella enterica serovar Loma Linda (CNFs), sharing 67% and 86% identity with CNF1 and CNF3, respectively. A more distant CNF homolog has been identified in E. coli strain GN02091 (CNFx) that has only 50%–55% identity with all other CNF homologs. The exchangeability of the deamidase domains among members of the CNF family has recently been explored and results suggest that these domains can be swapped and evolved for efficient cytosolic cargo delivery and intracellular activity (Haywood, Ho and Wilson 2018).
Figure 2.

Molecular phylogenetic relationships among the full-length members of the CNF family. Nine representative full-length CNF protein sequences were used to construct a phylogenetic tree, in which the evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model (Jones, Taylor and Thornton 1992). The percentage of trees in which the associated taxa clustered together is shown next to the branches (blue). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site (below the branches). Evolutionary analyses were conducted in MEGA7 (Kumar, Stecher and Tamura 2016).
CNF1-like deamidase modules in the arsenal of virulent bacteria
Functional CNF1-like domains have started to be described in the armament of several pathogenic bacteria (Table 1). The dermonecrotic toxin (DNT) from Bordetella spp. was the first AB-like toxin outside of the CNF family known to share homology (30% identity) within the CNF1 catalytic domain (Lemichez et al.1997). The CNF1-like domain of DNT is located at the C-terminus of the 1464-residue protein. However, the receptor-binding and translocation domains of the toxins are not related, and in contrast to the CNF toxins that exhibit low transglutaminase activity, DNT catalyzes preferentially transglutamination of the same Gln residue targeted by the CNF toxins leading to Rac1 activation (Schmidt et al.1998; Masuda et al.2000; Munro et al.2005). Interestingly, CNF1-like activity domains have also been found as small effector proteins of specialized secretion systems. The T3SS effector VopC from Vibrio parahaemolyticus acts on Rac1 and Cdc42 GTPases to mediate bacterial invasion of non-phagocytic cells (Zhang et al.2012). VopC shares limited identity (25%) with the CNF1 catalytic domain, including the active-site Cys and His residues. A CNF1-like domain is found in a MIX-effector Vpr01570 of the type 6 secretion system (T6SS) from Vibrio proteolyticus, which has been shown to induce actin cytoskeletal rearrangements upon exogenous expression in cells (Ray et al.2017).
Table 1.
Selected bacterial protein deamidases.
| Toxin | Bacterial source | Cellular activity | Target(s) | Refs |
|---|---|---|---|---|
| CNF | E. coli, Y. pseudotuberculosis, Shigella boydii | Rearrangement of actin cytoskeleton, modulation of inflammatory and apoptotic responses | Rho GTPases | (Flatau et al.1997; Schmidt et al.1997; Doye et al.2002) |
| DNT | Bordetella spp. | Rearrangement of actin cytoskeleton, modulation of inflammatory and apoptotic responses | Rho GTPases | (Masuda et al.2000) |
| VopC | Vibrio parahemolyticus | Rearrangement of actin cytoskeleton, modulation of inflammatory and apoptotic responses | Rho GTPases | (Zhang et al.2012) |
| MIX-CNF | Vibrio proteolyticus | Rearrangement of actin cytoskeleton, modulation of inflammatory and apoptotic responses | Rho GTPases | (Ray et al.2017) |
| PMT | Pasteurella multocida | Activation of mitogenic and calcium signaling pathways, actin rearrangements, protein synthesis, and cell proliferation | Heterotrimeric G proteins: Gαq/11, Gα12/13, Gαi | (Orth et al.2008) |
| SseI | Salmonella enterica serovar Typhi | Increased cell migration | Binds IQ motif-containing GTPase-activating protein 1 (IQGAP1) | (Bhaskaran and Stebbins 2012) |
| BLF1 | Burkholderia pseudomallei | Inhibition of protein synthesis | eIF4a | (Cruz-Migoni et al.2011) |
| Cifs | E. coli (EPEC, EHEC), Burkholderia pseudomallei, Photorhabdus luminescens, other bacterial species | Inhibition of Cullin-RING ligases, ubiquintination | NEDD8 and Ubiquitin | (Cui et al.2010) |
| OspI | Shigella flexneri | Disruption of TRAF6-mediated responses | Ubc13 | (Sanada et al.2012) |
| TecA | Burkholderia cenocepacia | Rearrangement of actin cytoskeleton, modulation of inflammatory and apoptotic responses | Rho GTPases | (Aubert et al.2016) |
| PaTox | Photorhabdus aymbiotica | Rearrangement of actin cytoskeleton, activation of mitogenic signaling pathways | Heterotrimeric G proteins: Gαq/11, Gαi | (Jank et al.2013) |
We set out to depict the landscape of CNF1 related deamidase domain containing virulence factors among pathogenic bacteria. All known sequences of CNF1-like domains contain conserved active-site Cys and His residues (Fig. 3A), and the crystal structure of CNF1 (PDB 1HQ0) (Buetow et al.2001) shows that these two residues are located at the ends of two anti-parallel β-strands, forming a Cys-beta-turn-beta-His motif (Fig. 3B). Among CNF1-like deamidase domains, the effector protein HopBJ1 from Pseudomonas syringae also contains a similar sequence motif that can be modeled to show a structure resembling that found in CNF1, and its presence has been correlated with disease in plants (Hockett et al.2014), including dependency on the active-site Cys and His residues for its intracellular activity in plants. Burkholderia pseudomallei lethal factor 1 (BLF1) kills mice upon injection and inhibits protein synthesis in macrophages through Gln deamidation of the eukaryotic initiation factor 4A (eIF4A) (Cruz-Migoni et al.2011). Although the target is not a Rho GTPase and BLF1 has no discernable amino acid sequence homology with that of CNF1 (Fig. 3A), BLF1 (PDB 3TU8) does show similar topology surrounding the active-site Cys-beta-turn-beta-His motif. We propose that a CNF1-like domain can be determined on the basis of proven Gln deamidase activity and a common active-site motif. This motif consists of the sequence Gly-Cys-(Xaa)n-His, where n is the number of residues that permit formation of a beta-sheet loop that would bring the Cys and His residues into close proximity for catalysis to occur (Fig. 3B).
Figure 3.

CNF1 signature active-site motif. (A) Shown are PyMOL renderings of the crystal structures of the C-terminal CNF1 catalytic domain (1HQ0) and the Burkholderia lethal factor 1 (3BT8) (right panels). The bottom portion of the structure (green) contains the beta-turn-beta motif (cyan) with the active site His and Cys residues shown in red. The portion of the structure above the active site beta sheet is shown in grey. Shown to the right are the corresponding zoomed-in views of the beta-turn-beta motifs with Gly, Cys and His residues shown as stick structures. (B) Alignment of sequences in the region near the Gly-Cys-beta-turn-beta-His motif in proteins known to have CNF1-like Gln-deamidase activity.
Using this Gly-Cys-(Xaa)n-His sequence motif, where n = 10–18, additional proteins with possible CNF-like deamidase domains can be identified from the collection of BLASTp hits, obtained by using the above-defined CNF-like domains as query in the search. Fig. 4 shows a phylogenetic relationship tree of 167 CNF-like domains containing proteins obtained from such a search based on the 150-residue regions flanking the Gly-Cys-(Xaa)n-His motif (Fig. 4). Besides the cluster of full-length CNF toxin homologs, other CNF1-like proteins with known deamidase activities, notably DNT, VopC, MIX-CNF, BLF1 and HopBJ1, fall into distinct clusters. From this analysis, there are a large number of additional bacterial proteins with CNF1-like motifs, but without confirmed Gln deamidase activities or with uncharacterized roles in virulence, encoded in bacterial genomes.
Figure 4.

Molecular phylogenetic relationships among the extended members of the CNF-domain-containing family. 167 representative sequences were collected from NCBI BLASTp search using VopC, DNT, CNF1, MIX-CNF, BLF1 and HopBJ1 as query, followed by screening for Gly-Cys-(Xaa)n-His motif, where n = 10–18, and trimming to within 150-residues flanking the motif. Sequences were aligned with MUSCLE (Edgar 2004), and then refined with Clustal W (Thompson, Higgins and Gibson 1994), before generating a Neighbor-Joining tree using MEGA7 (Kumar, Stecher and Tamura 2016). In some cases, Jalview (Waterhouse et al.2009) was used for editing the alignments. Those with known CNF1-like deamidase activity and a few select ones of interest (PNF, VgrG-CNF and YenC1) are indicated with solid symbols. Open red circles and squares are untested CNF or DNT sequences, respectively. Otherwise, open circles denote suspected T3SS effectors; triangles denoted suspected T6SS effectors; squares denote suspected large, multi-modular cytotoxins; and diamonds denote suspected Rhs-containing cytotoxins. Colors correspond to the indicated genera.
Additional homologs containing CNF1-like motifs can be found in other Gram-negative bacteria, including Morganella, Pseudomonas, Shewanella, Burkholderia and Photorhabdus species. From comparison of these homologs, it is evident that the CNF1-like domain can be delivered into the host cell cytosol through multiple systems (Fig. 5). For example, the N-terminal receptor-binding and translocation domains of the full-length CNF homologs share sequence homology with the N-terminus of the mitogenic toxin from Pasteurella multocida (PMT-N), but their respective catalytic domains share no homology in their sequences (Lemichez et al.1997) or structures (Wilson and Ho 2015). The existing sequences of other proteins containing CNF1-like domains indicate that these fall into several types, including a number of T3SS effectors like VopC and Pnf, T6SS effectors like VgrG-CNF and MIX-CNF, the RHS-containing cytotoxins such as YenC1 and the large modular cytotoxins (ranging from 2000 to 5000 amino acids in length), some of which contain a TcdA_TcdB pore-forming domain with the CNF-like domain being located at various positions in the protein.
Figure 5.
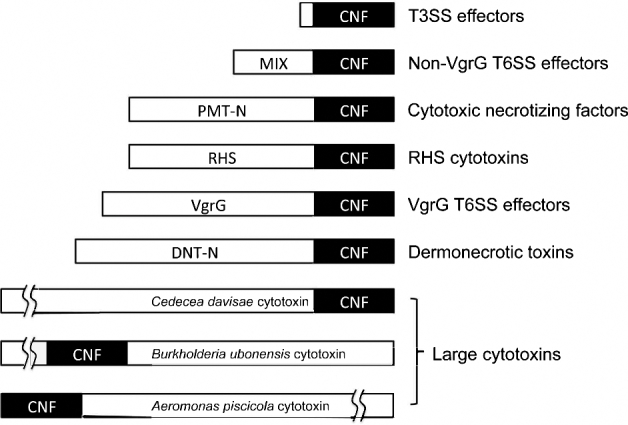
Schematic diagram of the CNF-like superfamily of modular protein toxins. Shown is the location of the CNF1-like domain within each toxin or effector protein. MIX, non-VgrG T6SS effector motif. PMT-N, N terminus of CNF that shares homology with N terminus of PMT. RHS, N-terminal rearrangement hotspot-repeat containing domain. VgrG, T6SS effector motif. DNT-N, N terminus unique to DNT.
Other host cell-modulating deamidase modules in virulent bacteria
In addition to the CNF-like deamidases containing Cys and His active site residues, there are additional Gln deamidases, such as PMT, with Cys-His-Asp catalytic triad at the active site (Table 1). However, the catalytic domains of CNF1 (PDB 1HQ0) and PMT (PDB 2EBF) belong to two very different structural folds (Wilson and Ho 2010). PMT deamidates and activates a subset of heterotrimeric G-protein α subunits (Orth et al.2008; Orth et al.2013) and thereby stimulating critical calcium and mitogenic signaling pathways and notably indirectly activating RhoA GTPase signaling. Homologs of the PMT catalytic domain are found in a wide array of protein toxins with diverse intracellular delivery machinery (Wilson and Ho 2015). For example, the T3SS effector protein SseI from S. enterica serovar Typhimurium involved in host cell migration has a PMT-like catalytic domain (PDB 4G29) (Bhaskaran and Stebbins 2012). The T3SS effector OspI from Shigella flexneri has a PMT-like active site arrangement (PDB 3B12) and acts on Ubc13 to disrupt K63-ubiquitin conjugation that is essential to relay signals of pathogen perception (Sanada et al.2012). The cycle inhibiting factors (Cifs) are translocated effector proteins that catalyze Gln deamidation of ubiquitin and the related protein NEDD8 (Crow et al.2012). The active-site structure of the cyclomodulin Cifs from enteropathogenic and enterohemorrhagic E. coli (PDB 3EFY), B. pseudomallei (PDB 3GQM) and Photorhabdus luminescens (PDB 3GQJ) shows that their active sites are PMT-like (Hsu et al.2008; Crow et al.2009), but have a Cys-His-Gln motif instead of a Cys-His-Asp motif. The non-VgrG T6SS effector TecA from Burkholderia cenocepacia also has a PMT-like active site, but it deamidates Rho GTPases at a conserved Asn instead of a Gln (Aubert et al.2016). This modification disrupts the actin cytoskeleton and triggers caspase-1-mediated inflammation and cell death. As was observed for CNF1-like domains, PMT/SseI-like domains can also be found in large modular toxins produced by a number of emerging human pathogens. For example, a large modular toxin produced by the entomopathogenic Photorhabdus asymbiotica (PaTox) contains a tyrosine-glycosyltransferase domain that inactivates Rho GTPases and a PMT/SseI-like domain that deamidates the active-site Gln of Gαq/11 and Gαi proteins (Jank et al.2013). Another large modular toxin (WP_046975705) can be found in the genome of Photorhabdus temperata strain DSM 15199. In addition to having a VIP2-like ADP-ribosylating domain, this toxin contains both a CNF1-like domain and a PMT/SseI-like domain.
Host protective responses against Rho GTPase activation by CNF1
Once Rho GTPases are activated by either CNF1, by point mutations, or by guanine nucleotide exchange factors (GEFs), they undergo ubiquitin-modification for degradation by the proteasome (Doye et al.2002). In light of the inherent occurrence of spontaneous deamidation reactions of proteins in living cells, this raises the general question of whether long-lived organisms might have evolved protective systems against the alteration of these key Gln residues in G proteins or as a mechanism of defense against pathogen attack. This question is of critical importance considering the panel of bacteria carrying genes encoding deamidase-containing factors homologous to CNF1, described above. Indeed, regulation of the flux of active Rho GTPases is under the scrutiny of the ubiquitination and proteasome machineries (UPS) (Mettouchi and Lemichez 2012). Ubiquitination is a post-translational modification of proteins that results in covalent attachment of ubiquitin, a small polypeptide of 76 amino-acids displaying 7 lysine residues on its surface, to residues on the target protein or on previously crosslinked ubiquitin molecules. Target proteins are thus tagged with different combinations of mono-, multi-, or poly-ubiquitin chains (Swatek and Komander 2016). The ubiquitination reaction involves successively three major classes of enzymes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3), where the E3 ligase bridges E2-linked ubiquitin with the substrate to ensure the direct or indirect transfer of ubiquitin to the target. By interacting with ubiquitin-binding proteins having different chain specificities, this allows for assembly of signal transduction complexes, modulation of protein trafficking to particular cellular locations, alteration of protein–protein interactions, or control of protein fate by targeting substrates to proteasome degradation. Owing to the control they operate on cell signaling and their critical role in detoxifying processes, E3 ligases not only play a role in cell signaling, but also function as gatekeepers of late-onset diseases (Scheffner and Kumar 2014).
Progress has been made in deciphering the E3 ligases responsible for ubiquitination of active Rho GTPases (Ding, Yin and Wang 2011; Hodge and Ridley 2016). Recently, the HECT domain and ankyrin repeats-containing E3 ubiquitin protein ligase 1 (HACE1) was identified by several groups as the critical enzyme catalyzing the ubiquitin-mediated proteasomal degradation of active Rac1 (Torrino et al.2011; Castillo-Lluva et al.2013). Depletion of HACE1 impairs degradation of CNF1-modified Rac1 (Torrino et al.2011; Castillo-Lluva et al.2013). The ankyrin repeats and middle region of HACE1 cooperate to recognize selectively the GTP-bound, active form of Rac1 and facilitate its poly-ubiquitination (Andrio et al.2017). Such tight control of Rac1 activity by HACE1, in addition to GEF/GAP regulators, is likely essential considering the pleiotropic role played by Rac1 in the control of cell division, transformation, migration, cohesion of the epithelium, phagocytosis, production of ROS and expression of anti-apoptotic and inflammatory factors (Hodge and Ridley 2016). In line with this, HACE1-mediated regulation of the flux of active Rac1 controls cell division and migration, as well as the Rac1-dependent NADPH oxidase production of ROS (Castillo-Lluva et al.2013; Goka and Lippman 2015; Daugaard et al.2013). By controlling cell fate and inflammation via TNFR1-RIP3 kinase necroptotic signaling, Hace1 provides an important protective function during Listeria monocytogenes infection and development of CRC caused by chemical-induced colitis (Tortola et al.2016). HACE1 appears to play another critical role in the control of cell homeostasis through its action on autophagic flux (Liu et al.2014; Zhang et al.2014). Hace1 is a tumor suppressor of broad function that is commonly inactivated in cancer due to genetic alterations, such as chromosome translocation (Zhang et al.2007), aberrant methylation of hace1 promoter (Hibi et al.2008), and discrete cancer-associated mutations (Andrio et al.2017). Now that a link between HACE1 and the control of the flux of active Rac1 has been established, it will be important to determine whether HACE1 tampers certain cytotoxic effects triggered by CNF1 and also operates as a control on gain-of-function alterations of Rac1 found in cancers (Jordan et al.1999; Hodis et al.2012; Krauthammer et al.2012; Kawazu et al.2013). Considering the general role played by E3 ligase dysfunction in human aging-related diseases, the study linking these factors to the regulation of GTPases in pathogens and disease will undoubtedly be a major source of findings relevant to human health.
Deep sequencing studies of the mutational landscape in melanoma have established the high occurrence of mutants in Rho GTPases and heterotrimeric G-protein subunits, notably Rac1 and Gαq, which are also targets of CNF1 and PMT toxins, respectively (Hodis et al.2012; Krauthammer et al.2012; O’Hayre et al.2013). In the case of Rac1, a hot spot of cancer-associated mutations is observed in position 29 (P29S and P29L). Melanoma-associated mutations at P29 carry ultraviolet alteration signatures. Although the type of alterations of these host proteins with pro-oncogenic activities do not bear the same signature between proteome and genome alterations, they all result in gain-of-function phenotypes with activation of downstream signaling pathways. For Rho GTPases, it will be interesting in the future to define whether these differences reflect particular constraints linked to the proteasomal degradation system that acts on activated GTPase forms, as discussed below, or reflect differences in the mechanism of action of mutagenic agents.
CONCLUSIONS
Defining bacterial risk factors in the etiology of late-onset diseases, such as cancer, are of critical importance so as to intervene with appropriate therapies. As we have outlined here, proteins containing CNF1-like activity domains and other host cell-modulating deamidase domains, such as that from PMT, can be found in a wide range of bacterial genomes and are associated with a diverse number of cytosolic delivery systems. The scope and prevalence of these diverse proteins point to extensive horizontal gene transfer and recombination events involving these modules, as well as strong selective pressures to maintain them or inactivate them in some specific contexts, as observed in Y. pseudotuberculosis. This also brings to light an unrealized potential health hazard associated with exposure to these cell-modulating, deamidating proteins during infections or colonization with the bacteria that produce them.
Much remains to be learned about how host organisms cope with these proteome alterations, as for the case of deamidated Rho GTPases, and about what the long-term consequences of these modifications are. Indeed, the host genetic background clearly plays a significant role in mitigating bacterially promoted late-onset diseases by limiting the extent of infection and the extent of the cytotoxic effects of bacterial virulence factors. The action of CNF1 and the ability of host cells to eliminate permanently activated forms of Rho GTPases represent a remarkable example of this interplay.
While the connection between cancer and other late-onset diseases has been established for only a limited number of cases, the rapidly growing list of bacteria and/or their toxins that target pro-oncogenic signaling pathways, promote chronic inflammation, or cause DNA damage, places an urgency for better understanding the risks posed by exposure to these toxin-producing bacteria. Coupled with this knowledge, individualized microbial profiling made possible through current advanced sequencing technologies could be applied towards surveillance of these risk factors and the design of appropriate intervention strategies.
The application of targeted pre- or probiotics, vaccines, and antibiotic regimens that are tailored toward an individual's microbiome and life history could have a major impact on the occurrence of late-onset, recurrent, or chronic diseases, similar to that which occurred a century ago upon implementation of widespread, standardized hygiene measures. Ultimately, these holistic approaches in personalized medicine will lead to significant public health savings and increased lifespan.
Acknowledgements
We gratefully acknowledge the many important contributions of colleagues whose work could not be cited here due to space limitations. The authors declare no conflict of interest.
FUNDING
This work was supported by INSERM with funding from Institut Pasteur Paris and the Ligue Nationale Contre le Cancer (LNCC, équipe labellisée) (to E.L.) and from the Research Board of the University of Illinois at Urbana-Champaign and the National Institutes of Health award AI038396 (to B.A.W).
Conflict of interest. None declared.
REFERENCES
- Alouf JE, Ladant D, Popoff MR. The Comprehensive Sourcebook of Bacterial Protein Toxins. 4th edn., (Elsevier: Oxford, UK; ), 2015. [Google Scholar]
- Andrio E, Lotte R, Hamaoui D et al. Identification of cancer-associated missense mutations in Hace1 that impair cell growth control and Rac1 ubiquitylation. Sci Rep 2017;7:44779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert DF, Xu H, Yang J et al. A Burkholderia type VI effector deamidates Rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe 2016;19:664–74. [DOI] [PubMed] [Google Scholar]
- Bhaskaran SS, Stebbins CE. Structure of the catalytic domain of the Salmonella virulence factor SseI. Acta Crystallogr D Biol Crystallogr 2012;68:1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boquet P, Lemichez E. Bacterial virulence factors targeting Rho GTPases: parasitism or symbiosis? Trends Cell Biol 2003;13:238–46. [DOI] [PubMed] [Google Scholar]
- Buc E, Dubois D, Sauvanet P et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 2013;8:e56964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow L, Flatau G, Chiu K et al. Structure of the Rho-activating domain of Escherichia coli cytotoxic necrotizing factor 1. Nat Struct Biol 2001;8:584–8. [DOI] [PubMed] [Google Scholar]
- Buetow L, Ghosh P. Structural elements required for deamidation of RhoA by cytotoxic necrotizing factor 1. Biochemistry 2003;42:12784–91. [DOI] [PubMed] [Google Scholar]
- Caprioli A, Falbo V, Roda LG et al. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect Immun 1983;39:1300–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Lluva S, Tan CT, Daugaard M et al. The tumour suppressor HACE1 controls cell migration by regulating Rac1 degradation. Oncogene 2013;32:1735–42. [DOI] [PubMed] [Google Scholar]
- Chung LK, Park YH, Zheng Y et al. The Yersinia virulence factor YopM hijacks host kinases to inhibit type III effector-triggered activation of the pyrin inflammasome. Cell Host Microbe 2016;20:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contamin S, Galmiche A, Doye A et al. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol Biol Cell 2000;11:1775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow A, Hughes RK, Taieb F et al. The molecular basis of ubiquitin-like protein NEDD8 deamidation by the bacterial effector protein Cif. Proc Natl Acad Sci 2012;109:E1830–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow A, Race PR, Jubelin G et al. Crystal structures of Cif from bacterial pathogens Photorhabdus luminescens and Burkholderia pseudomallei. PLoS One 2009;4:e5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Migoni A, Hautbergue GM, Artymiuk PJ et al. A Burkholderia pseudomallei toxin inhibits helicase activity of translation factor eIF4A. Science 2011;334:821–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Yao Q, Li S et al. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science 2010;329:1215–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugaard M, Nitsch R, Razaghi B et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat Commun 2013;4:2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dho SH, Deverman BE, Lapid C et al. Control of cellular Bcl-xL levels by deamidation-regulated degradation. PLoS Biol 2013;11:e1001588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabate M, Munro P, Garcia E et al. Escherichia coli alpha-hemolysin counteracts the anti-virulence innate immune response triggered by the Rho GTPase activating toxin CNF1 during bacteremia. PLoS Pathog 2015;11:e1004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Yin Z, Wang HR. Ubiquitination in Rho signaling. Curr Top Med Chem 2011;11:2879–87. [DOI] [PubMed] [Google Scholar]
- Doye A, Mettouchi A, Bossis G et al. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002;111:553–64. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004;32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto DS, Jones TA, Sundsbak JL et al. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog 2007;3:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri A, Travaglione S, Ballan G et al. The cytotoxic necrotizing factor 1 from E. coli: a janus toxin playing with cancer regulators. Toxins 2013;5:1462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falbo V, Pace T, Picci L et al. Isolation and nucleotide sequence of the gene encoding cytotoxic necrotizing factor 1 of Escherichia coli. Infect Immun 1993;61:4909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzano L, Fiorentini C, Donelli G et al. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type1. Mol Microbiol 1993;9:1247–54. [DOI] [PubMed] [Google Scholar]
- Flatau G, Landraud L, Boquet P et al. Deamidation of RhoA glutamine 63 by the Escherichia coli CNF1 toxin requires a short sequence of the GTPase switch 2 domain. Biochem Biophys Res Commun 2000;267:588–92. [DOI] [PubMed] [Google Scholar]
- Flatau G, Lemichez E, Gauthier M et al. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997;387:729–33. [DOI] [PubMed] [Google Scholar]
- Gagnière J, Raisch J, Veziant J et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 2016;22:501–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia TA, Ventura CL, Smith MA et al. Cytotoxic necrotizing factor 1 and hemolysin from uropathogenic Escherichia coli elicit different host responses in the murine bladder. Infect Immun 2013;81:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goka ET, Lippman ME. Loss of the E3 ubiquitin ligase HACE1 results in enhanced Rac1 signaling contributing to breast cancer progression. Oncogene 2015;34:5395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zhang Z, Wei H et al. Cytotoxic necrotizing factor 1 promotes prostate cancer progression through activating the Cdc42-PAK1 axis. J Pathol 2017;243:208–19. [DOI] [PubMed] [Google Scholar]
- Hashemizadeh Z, Kalantar-Neyestanaki D, Mansouri S. Association between virulence profile, biofilm formation and phylogenetic groups of Escherichia coli causing urinary tract infection and the commensal gut microbiota: a comparative analysis. Microb Pathog 2017;110:540–5. [DOI] [PubMed] [Google Scholar]
- Haywood EE, Ho M, Wilson BA. Modular domain swapping among the bacterial cytotoxic necrotizing factor (CNF) family for efficient cargo delivery into mammalian cells. J Biol Chem 2018;293:3860–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi K, Sakata M, Sakuraba K et al. Aberrant methylation of the HACE1 gene is frequently detected in advanced colorectal cancer. Anticancer Res 2008;28:1581–4. [PubMed] [Google Scholar]
- Hockett KL, Nishimura MT, Karlsrud E et al. Pseudomonas syringae CC1557: a highly virulent strain with an unusually small type III effector repertoire that includes a novel effector. Mol Plant Microbe Interact 2014;27:923–32. [DOI] [PubMed] [Google Scholar]
- Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol 2016;17:496–510. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Aktories K, Schmidt G. Change in substrate specificity of cytotoxic necrotizing factor (CNF) unmasks proteasome-independent down-regulation of constitutively active RhoA. J Biol Chem 2007;282:10826–32. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Pop M, Leemhuis J et al. The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J Biol Chem 2004;279:16026–32. [DOI] [PubMed] [Google Scholar]
- Hsu Y, Jubelin G, Taieb F et al. Structure of the cyclomodulin Cif from pathogenic Escherichia coli. J Mol Biol 2008;384:465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jank T, Bogdanović X, Wirth C et al. A bacterial toxin catalyzing tyrosine glycosylation of Rho and deamidation of Gq and Gi proteins. Nat Struct Mol Biol 2013;20:1273–80. [DOI] [PubMed] [Google Scholar]
- Jobin C. Colorectal cancer: looking for answers in the microbiota. Cancer Discov 2013;3:384–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 1992;8:275–82. [DOI] [PubMed] [Google Scholar]
- Jordan P, Brazåo R, Boavida MG et al. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999;18:6835–9. [DOI] [PubMed] [Google Scholar]
- Kadhum HJ, Finlay D, Rowe MT et al. Occurrence and characteristics of cytotoxic necrotizing factors, cytolethal distending toxins and other virulence factors in Escherichia coli from human blood and faecal samples. Epidemiol Infect 2008;136:752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsen C, Ellingsen AB, Wiik-Nielsen C et al. Host specificity and clade dependent distribution of putative virulence genes in Moritella viscosa. Microb Pathog 2014;77:53–65. [DOI] [PubMed] [Google Scholar]
- Kawazu M, Ueno T, Kontani K et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc Natl Acad Sci 2013;110:3029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanietz MG, Caloca MJ. The Rac GTPase in cancer: from old concepts to new paradigms. Cancer Res 2017;77:5445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keestra AM, Winter MG, Auburger JJ et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 2013;496:233–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Chung JW, Kim KS. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J Biol Chem 2005;280:1360–8. [DOI] [PubMed] [Google Scholar]
- Knust Z, Blumenthal B, Aktories K et al. Cleavage of Escherichia coli cytotoxic necrotizing factor 1 is required for full biologic activity. Infect Immun 2009;77:1835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 2016;33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemichez E, Aktories K. Hijacking of Rho GTPases during bacterial infection. Exp Cell Res 2013;319:2329–36. [DOI] [PubMed] [Google Scholar]
- Lemichez E, Flatau G, Bruzzone M et al. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol Microbiol 1997;24:1061–70. [DOI] [PubMed] [Google Scholar]
- Lerm M, Schmidt G, Goehring UM et al. Identification of the region of rho involved in substrate recognition by Escherichia coli cytotoxic necrotizing factor 1 (CNF1). J Biol Chem 1999;274:28999–9004. [DOI] [PubMed] [Google Scholar]
- Li B, Lu Y, Lan F, He Q, Li C, Cao Y. Prevalence and characteristics of ST131 clone among unselected clinical Escherichia coli in a Chinese university hospital Antimicrob Resist Infect Control 2017;6:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen P, Gao H et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell 2014;26:106–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman HA, Gillespie RA, Baker BD et al. Yersinia pseudotuberculosis produces a cytotoxic necrotizing factor. Infect Immun 2002;70:2708–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malorni W, Fiorentini C. Is the Rac GTPase-activating toxin CNF1 a smart hijacker of host cell fate? FASEB J 2006;20:606–9. [DOI] [PubMed] [Google Scholar]
- Masuda M, Betancourt L, Matsuzawa T et al. Activation of rho through a cross-link with polyamines catalyzed by Bordetella dermonecrotizing toxin. EMBO J 2000;19:521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettouchi A, Lemichez E. Ubiquitylation of active Rac1 by the E3 ubiquitin-ligase HACE1. Small GTPases 2012;3:102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel G, Ferrua B, Munro P et al. Immunoadjuvant properties of the Rho activating factor CNF1 in prophylactic and curative vaccination against Leishmania infantum. PLoS One 2016;11:e0156363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsumori K, Terai A, Yamamoto S et al. Virulence characteristics of Escherichia coli in acute bacterial prostatitis. J Infect Dis 1999;180:1378–81. [DOI] [PubMed] [Google Scholar]
- Mulvey MA, Schilling JD, Hultgren SJ. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun 2001;69:4572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro P, Flatau G, Anjuère F et al. The Rho GTPase activators CNF1 and DNT bacterial toxins have mucosal adjuvant properties. Vaccine 2005;23:2551–6. [DOI] [PubMed] [Google Scholar]
- Munro P, Flatau G, Doye A et al. Activation and proteasomal degradation of rho GTPases by cytotoxic necrotizing factor-1 elicit a controlled inflammatory response. J Biol Chem 2004;279:35849–57. [DOI] [PubMed] [Google Scholar]
- Munro P, Flatau G, Lemichez E. Intranasal immunization with tetanus toxoid and CNF1 as a new mucosal adjuvant protects BALB/c mice against lethal challenge. Vaccine 2007;25:8702–6. [DOI] [PubMed] [Google Scholar]
- Nicolas-Chanoine MH, Bertrand X, Madec JY. Escherichia coli ST131, an intriguing clonal group. Clin Microbiol Rev 2014;27:543–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nougayrede JP, Taieb F, De Rycke J et al. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol 2005;13:103–10. [DOI] [PubMed] [Google Scholar]
- O’Hayre M, Vázquez-Prado J, Kufareva I et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer 2013;13:412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orden JA, Domínguez-Bernal G, Martínez-Pulgarín S et al. Necrotoxigenic Escherichia coli from sheep and goats produce a new type of cytotoxic necrotizing factor (CNF3) associated with the eae and ehxA genes. Int Microbiol 2007;10:47–55. [PubMed] [Google Scholar]
- Orth JH, Fester I, Preuss I et al. Activation of Galpha (i) and subsequent uncoupling of receptor-Galpha(i) signaling by Pasteurella multocida toxin. J Biol Chem 2008;283:23288–94. [DOI] [PubMed] [Google Scholar]
- Orth JH, Fester I, Siegert P et al. Substrate specificity of Pasteurella multocida toxin for α subunits of heterotrimeric G proteins. FASEB J 2013;27:832–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald E, De Rycke J, Guillot JF et al. Cytotoxic effect of multinucleation in HeLa cell cultures associated with the presence of Vir plasmid in Escherichia coli strains. FEMS Microbiol Lett 1989;49:95–99. [DOI] [PubMed] [Google Scholar]
- Pei S, Doye A, Boquet P. Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol Microbiol 2001;41:1237–47. [DOI] [PubMed] [Google Scholar]
- Piteau M, Papatheodorou P, Schwan C et al. Lu/BCAM adhesion glycoprotein is a receptor for Escherichia coli cytotoxic necrotizing factor 1 (CNF1). PLoS Pathog 2014;10:e1003884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff MR. Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases 2014;5:e28209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisch J, Buc E, Bonnet M et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol 2014;20:6560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Schwartz N, de Souza Santos M et al. Type VI secretion system MIX-effectors carry both antibacterial and anti-eukaryotic activities. EMBO Rep 2017;18:1978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Real JM, Munro P, Buisson-Touati C et al. Specificity of immunomodulator secretion in urinary samples in response to infection by alpha-hemolysin and CNF1 bearing uropathogenic Escherichia coli. Cytokine 2007;37:22–25. [DOI] [PubMed] [Google Scholar]
- Rippere-Lampe KE, Lang M, Ceri H et al. Cytotoxic necrotizing factor type 1-positive Escherichia coli causes increased inflammation and tissue damage to the prostate in a rat prostatitis model. Infect Immun 2001;69:6515–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NE, Robinson AB. Molecular clocks. Proc Natl Acad Sci 2001;98:944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada T, Kim M, Mimuro H et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 2012;483:623–6. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta 2014;1843:61–74. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Sehr P, Wilm M et al. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997;387:725–9. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Selzer J, Lerm M et al. The Rho-deamidating cytotoxic necrotizing factor 1 from Escherichia coli possesses transglutaminase activity. Cysteine 866 and histidine 881 are essential for enzyme activity. J Biol Chem 1998;273:13669–74. [DOI] [PubMed] [Google Scholar]
- Schweer J, Kulkarni D, Kochut A et al. The cytotoxic necrotizing factor of Yersinia pseudotuberculosis (CNFY) enhances inflammation and Yop delivery during infection by activation of Rho GTPases. PLoS Pathog 2013;9:e1003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smati M, Clermont O, Bleibtreu A et al. Quantitative analysis of commensal Escherichia coli populations reveals host-specific enterotypes at the intra-species level. MicrobiologyOpen 2015;4:604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll T, Markwirth G, Reipschläger S et al. A new member of a growing toxin family–Escherichia coli cytotoxic necrotizing factor 3 (CNF3). Toxicon 2009;54:745–53. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Boyer L. RhoGTPases - NODes for effector-triggered immunity in animals. Cell Res 2013;23:980–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart LM, Paquette N, Boyer L. Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat Rev Immunol 2013;13:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugai M, Hatazaki1 K, Mogami A et al. Cytotoxic necrotizing factor type 2 produced by pathogenic Escherichia coli deamidates a Gln residue in the conserved G-3 domain of the Rho family and preferentially inhibits the GTPase activity of RhoA and Rac1. Infect Immun 1999;67:6550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Komander D. Ubiquitin modifications. Cell Res 2016;26:399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Skurnik D, Picard B et al. The population genetics of commensal Escherichia coli. Nat Rev Microbiol 2010;8:207–17. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrino S, Visvikis O, Doye A et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell 2011;21:959–65. [DOI] [PubMed] [Google Scholar]
- Tortola L, Nitsch R, Bertrand MJM et al. The tumor suppressor Hace1 is a critical regulator of TNFR1-mediated cell fate. Cell Rep 2016;15:1481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunsjø HS, Wiik-Nielsen CR, Grove S et al. Putative virulence genes in Moritella viscosa: activity during in vitro inoculation and in vivo infection. Microb Pathog 2011;50:286–92. [DOI] [PubMed] [Google Scholar]
- Visvikis O, Boyer L, Torrino S et al. Escherichia coli producing CNF1 toxin hijacks tollip to trigger Rac1-dependent cell invasion. Traffic 2011;12:579–90. [DOI] [PubMed] [Google Scholar]
- Washington EJ, Banfield MJ, Dangl JL. What a difference a Dalton makes: bacterial virulence factors modulate eukaryotic host cell signaling systems via deamidation. Microbiol Mol Biol Rev 2013;77:527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM et al. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009;25:1189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Ho M. Evolutionary aspects of toxin-producing bacteria. In: The Comprehensive Sourcebook of Bacterial Protein Toxins. 4th edn., (Elsevier: Oxford, UK: ) 2015,3–39. [Google Scholar]
- Wilson BA, Ho M. Recent insights into Pasteurella multocida toxin and other G-protein-modulating bacterial toxins. Future Microbiol 2010;5:1185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang J, Gao W et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014;513:237–41. [DOI] [PubMed] [Google Scholar]
- Zhang L, Anglesio MS, O'Sullivan M et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med 2007;13:1060–9. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen X, Sharma P et al. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat Commun 2014;5:3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Krachler AM, Broberg CA et al. Type III effector VopC mediates invasion for Vibrio species. Cell Rep 2012;1:453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Li J, Xu S et al. Emerging roles of protein deamidation in innate immune signaling. J Virol 2016;90:4262–8. [DOI] [PMC free article] [PubMed] [Google Scholar]