

Abstract
Objective
To identify molecular correlates of primary angiitis of the CNS (PACNS) through proteomic analysis of CSF from a biopsy-proven patient cohort.
Methods
Using mass spectrometry, we quantitatively compared the CSF proteome of patients with biopsy-proven PACNS (n = 8) to CSF from individuals with noninflammatory conditions (n = 11). Significantly enriched molecular pathways were identified with a gene ontology workflow, and high confidence hits within enriched pathways (fold change >1.5 and concordant Benjamini-Hochberg–adjusted p < 0.05 on DeSeq and t test) were identified as differentially regulated proteins.
Results
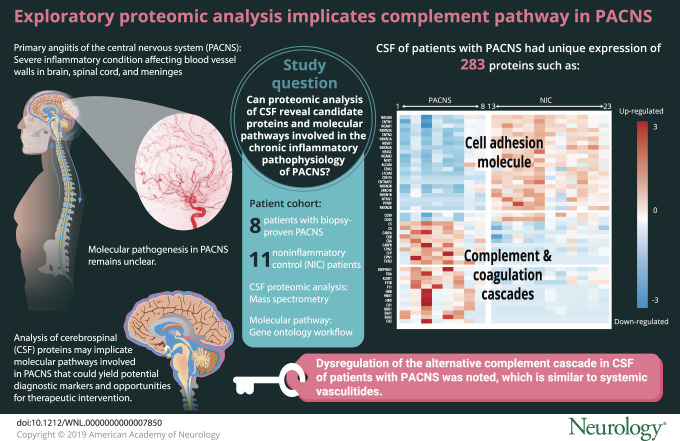
Compared to noninflammatory controls, 283 proteins were differentially expressed in the CSF of patients with PACNS, with significant enrichment of the complement cascade pathway (C4-binding protein, CD55, CD59, properdin, complement C5, complement C8, and complement C9) and neural cell adhesion molecules. A subset of clinically relevant findings were validated by Western blot and commercial ELISA.
Conclusions
In this exploratory study, we found evidence of deregulation of the alternative complement cascade in CSF from biopsy-proven PACNS compared to noninflammatory controls. More specifically, several regulators of the C3 and C5 convertases and components of the terminal cascade were significantly altered. These preliminary findings shed light on a previously unappreciated similarity between PACNS and systemic vasculitides, especially anti-neutrophil cytoplasmic antibody–associated vasculitis. The therapeutic implications of this common biology and the diagnostic and therapeutic utility of individual proteomic findings warrant validation in larger cohorts.
Primary angiitis of the CNS (PACNS) is a severe inflammatory disease affecting the blood vessel walls in the brain, spinal cord, and meninges.1 Without treatment, PACNS is frequently progressive.2,3 Although broadly acting immunosuppressants prevent mortality in ≈80% of patients, these medications have adverse side effects, and nearly half of patients relapse with debilitating neurologic symptoms.4 The basis for the variable response to therapy is unknown and cannot be reconciled with clinical data alone.3 The lack of molecular tools to aid in the clinical investigation of PACNS is an obstacle to improving patient outcomes.
Because of the low prevalence of PACNS and lack of available mouse models, the molecular pathogenesis in PACNS is poorly understood.5 To elucidate molecular correlates, a proteomic survey of patient CSF with mass spectrometry is a compelling approach. CSF is an accessible biological fluid that circulates throughout the meninges and parameningeal structures of the brain and spinal cord. Molecular analysis of CSF can provide diagnostic information on disease pathologies occurring within these regions.1,6 CSF abnormalities, including increased protein content, are observed in ≈90% of patients with PACNS, and reversal of CSF abnormalities correlates with improved patient outcomes.7 Previous attempts to characterize molecular abnormalities in PACNS CSF have been made but without the benefit of biopsy-proven disease.8
Here, we perform an unbiased proteomic analysis comparing the CSF profiles of patients with biopsy-proven PACNS to those of noninflammatory controls (NICs) and controls with reversible cerebral vasoconstriction syndrome (RCVS). Our goals were to identify candidate proteins and molecular pathways involved in the chronic inflammatory pathophysiology of PACNS and to highlight molecular targets for future therapeutic and diagnostic studies.
Methods
Full details on molecular protocols, including mass spectrometry and orthogonal validations, can be found in appendix e-1 available from Dryad (doi.org/10.5061/dryad.k52h33d).
Patient recruitment and study protocol
Patients with PACNS and NICs were recruited as part of a larger study analyzing biological samples from patients with suspected neuroinflammatory disease at the University of California, San Francisco (UCSF). The UCSF Institutional Review Board approved the study protocol, and participants or their surrogates provided written informed consent. RCVS controls were recruited as part of a larger study analyzing biological samples from patients with CNS vascular disorders at Cleveland Clinic. The Cleveland Clinic Institutional Review Board approved the study protocol, and participants or their surrogates provided written informed consent. Patients with PACNS and RCVS were diagnosed according to standard clinical diagnostic criteria, including neuropathology evaluation in all of the patients diagnosed with PACNS.
PACNS clinical vignettes
Patient 1
A previously healthy 50-year-old woman developed mild headaches with episodic, migrainous features, including visual auras together with worsening mental fogging independent of the headaches, all of which worsened over 4 years. At that time, she was discovered to have a thoracic myelopathy on examination and inflammatory CSF of unclear etiology. On MRI, she was found to have a thoracic myelitis with nodular leptomeningeal enhancement throughout the spine and the brain. A magnetic resonance (MR) angiogram of the head and neck was unremarkable. A brain biopsy revealed evidence for a small vessel vasculitis and chronic meningitis. An extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing. The CSF profile from a sample from later in the patient's clinical course was used for this study and showed a white blood cell (WBC) count of 2 cells/μL (66% lymphocytes, 34% monocytes) (0–5 cells/μL), red blood cell (RBC) count of 0 cells/μL (0–5 cells/μL), glucose of 45 mg/dL (45–80 mg/dL), total protein of 192 mg/dL (15–45 mg/dL), immunoglobulin (Ig) G index of 1.8 (<0.6), and <5 unique oligoclonal bands (OCBs) (≤1 band).
Patient 2
A 39-year-old woman with a history of ulcerative colitis well controlled on mesalamine and oral budesonide developed increasing fatigue and increasingly painful, new left-sided headaches and left facial paresthesias over 6 weeks to the point that they prompted hospitalization. A brain MRI revealed T2 hyperintensities in a gyriform pattern over the left parietal and temporal lobes with associated leptomeningeal enhancement. A CSF examination revealed a WBC count of 15 cells/μL (54% lymphocytes, 39% granulocytes, 7% other), an RBC count of 0 cells/μL, glucose of 65 mg/dL, total protein of 50 mg/dL, and 0 OCBs. A CT angiogram of the head and neck was unremarkable except for the suggestion of mild smooth narrowing of the left carotid artery terminus and left M1 segment of the middle cerebral artery. Over the next few days, the patient developed aphasia and apraxia, prompting a brain biopsy that revealed a small vessel vasculitis. Extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing.
Patient 3
A 56-year-old man was hospitalized for rapid cognitive decline and was found to have bilateral papilledema and a left abducens nerve palsy on examination. He had extensive confluent white matter T2 hyperintensities and multiple small areas of restricted diffusion consistent with acute infarcts on brain MRI. An MR angiogram of the head and neck was unremarkable. A CSF examination showed a WBC count of 26 cells/μL (53% neutrophils, 39% lymphocytes, 8% monocytes), RBC count of 3,275 cells/μL, glucose 60 mg/dL, total protein 141 mg/dL and an IgG index of 0.9. An extraventricular drain was placed for elevated intracranial pressure, and a brain biopsy revealed a small vessel vasculitis. Extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing.
Patient 4
A 55-year-old woman with a history of non–insulin-dependent diabetes mellitus had progressive difficulty walking over 6 months before she acutely lost sensation in her right leg and developed severe urinary retention. She was found to have a longitudinally extensive transverse myelitis on MRI, and a CSF examination revealed a WBC count of 8 cells/μL, an RBC count of 1 cell/μL, glucose of 92 mg/dL, total protein of 99 mg/dL, an IgG index of 0.57, and 1 unique OCB. Despite initial attempts at immunosuppression with glucocorticoids, the patient developed new weakness in her left leg and urinary and fecal incontinence. Serial imaging revealed new inflammatory lesions in the cerebellum and overlying leptomeninges. All vascular imaging, including a cerebral and spinal angiogram, was unremarkable. A brain biopsy revealed a small vessel vasculitis. Extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing.
Patient 5
A 36-year-old man with a history of possible relapsing polychondritis, with 1 episode of ear chondritis, presented with a few weeks of new-onset daily headaches and fatigue followed by visual hallucinations that prompted a neurologic evaluation. A CSF examination revealed a WBC count of 6 cells/μL (60% lymphocytes, 22% monocytes, 18% neutrophils), an RBC count of 3 cells/μL, glucose of 51 mg/dL, total protein of 27 mg/dL, and an IgG index of 1.62. A brain MRI revealed patchy leptomeningeal enhancement, multiple areas of T2 hyperintensity with patchy gadolinium enhancement, and multifocal areas of restricted diffusion consistent with acute infarcts. An MR angiogram of the head was normal, but a cerebral angiogram showed diffuse vasculopathy bilaterally in the distal vasculature (i.e., M3, M4, ophthalmic arteries, P3 and P4 vessels). A brain biopsy showed small vessel vasculitis. Extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing.
Patient 6
A previously healthy 57-year-old woman presented with 2 months of new-onset headaches with visual aura, 10 days of dizziness and vertigo, and an isolated episode of hemi-body sensory symptoms and was found to have a subarachnoid T2 hyperintensity on brain MRI and faint leptomeningeal enhancement. A CT angiogram of the head and neck was normal. CSF examination revealed a WBC count of 31 cells/μL (90% lymphocytes, 6% monocytes, 2% neutrophils, 1% eosinophils, and 1% unidentified), an RBC count of 102 cells/μL, glucose of 54 mg/dL, total protein of 75 mg/dL, an IgG index of 2.02, and >2 unique OCBs. A brain biopsy revealed a small vessel vasculitis. Extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing.
Patient 7
A previously healthy 13-year-old girl presented with fever, headache, altered mental status, and seizure and was found to have unihemispheric, subcortical T2 hyperintense lesions on brain MRI, many of which enhanced with gadolinium. An MR angiogram of the head and neck was unremarkable. A CSF examination revealed a WBC count of 11 cells/μL (84% lymphocytes, 16% monocytes), an RBC count of 6 cells/μL, glucose of 49 mg/dL, total protein of 37 mg/dL, and >5 unique OCBs. Brain biopsy revealed a small vessel vasculitis. Extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing.
Patient 8
A 51-year-old man with a history of atrial fibrillation, hypertension, and seizure presented with bony aches and migratory joint pains that went away and were followed months later by bilateral episcleritis, fever, numbness in his feet, and confusion. A brain MRI showed leptomeningeal enhancement and overlying multifocal areas of swollen and T2-hyperintense cortex and possible subcortical U-fiber enhancement. An MR angiogram of the head and neck was unremarkable. CSF examination revealed a WBC count of 3 cells/μL (82% lymphocytes, 12% monocytes, 6% neutrophils), an RBC count of 1 cell/μL, glucose of 59 mg/dL, total protein of 42 mg/dL, an IgG index 0.7, and 5 unique OCBs. The clinical impression by the treating neurologist was that the systemic symptoms were unrelated to the patient's neuroinflammatory disease. A brain biopsy revealed a small vessel vasculitis. Extensive workup for neoplastic, infectious, and other autoimmune etiologies was unrevealing.
RCVS clinical vignettes
Patient 9
A 54-year-old woman with a history of hepatitis C virus infection and epilepsy presented with altered mental status, dysarthria, expressive aphasia, and left- greater than right-sided weakness. An MR angiogram of the head revealed irregularities of the right anterior cerebral artery and left internal carotid artery. A cerebral angiogram showed narrowing and beading in multiple vessels, including the basilar artery, bilateral posterior cerebral arteries, and left M1 and A1 segments. There was also focal beading in the distal left anterior and middle cerebral arteries and an aneurysm at the origin of the right temporal artery. Her CSF profile revealed a WBC count of 11 cells/μL (93% lymphocytes, 3% monocytes, 4% other), an RBC count of 340 cells/μL, glucose of 57 mg/dL, total protein of 22 mg/dL, and no OCBs. She started on calcium channel blockers, and repeated MR angiogram of intracranial vessel 10 days later showed marked improvement in the intracranial vessel abnormalities.
Patient 10
A 33-year-old woman presented with a sudden-onset, thunderclap headache that was clearly different from her typical migraine headaches. A CT angiogram of the head and a noncontrast head CT revealed diffuse beading of the vasculature throughout the anterior and posterior circulation, and a parietal subarachnoid hemorrhage, respectively. A cerebral angiogram similarly found segmental irregularities of the intracranial vessels of the distal left anterior circulation. Her CSF profile revealed a WBC count of 1 cell/μL (82% lymphocytes, 8% monocytes, 10% other), an RBC count of 1,150 cells/μL, glucose of 65 mg/dL, total protein of 115 mg/dL, and an IgG index of 1.0. The patient was started on calcium channel blockers and had rapid resolution of her symptoms and no disease recurrence.
Patient 11
A 57-year-old woman with a history of depression treated with citalopram and bupropion hydrochloride presented with 4 days of dizziness, lightheadedness, left greater than right leg weakness, and falls followed by a rapid decline in mental status. She was found to have large areas of restricted diffusion in the bilateral parietal lobes on brain MRI. She became unresponsive and was intubated and transferred to the intensive care unit. A CT angiogram of the head revealed irregularities in the distal portions of the anterior cerebral arteries, and a cerebral angiogram was similarly consistent with vasospasm. A CSF examination revealed a WBC count of 1 cell/μL, an RBC count of 29 cells/μL, glucose of 95 mg/dL, and total protein of 22 mg/dL. The patient improved clinically and radiologically after administration of intra-arterial nicardipine and verapamil. She was started on a calcium channel blocker, and citalopram and bupropion hydrochloride were discontinued.
Patient 12
A previously healthy 30-year-old woman was admitted with new left-sided weakness and severe hypothermia after experiencing new-onset, recurrent thunderclap headaches for 2 weeks. A CT angiogram of the head and neck showed multiple foci of intracranial vascular narrowing in the bilateral anterior cerebral arteries, middle cerebral arteries, and posterior cerebral arteries, which was corroborated by a cerebral angiogram. A brain MRI showed acute bilateral subcortical infarcts. A high-resolution brain MRI found no evidence of abnormal vessel wall enhancement. Her CSF examination revealed a WBC count of 0 cells/μL, an RBC count of 133 cells/μL, glucose of 78 mg/dL, and total protein of 33 mg/dL. She was started on calcium channel blockers and improved clinically and radiographically.
Mass spectrometry
Total protein concentration in patient CSF was determined to be 0.1 to 0.6 mg/mL by Bradford assay (Sigma, B6916). A total of 5 μg protein was used from each patient’s CSF sample for liquid chromatography–dual mass spectrometry analysis. Appendix e-1 available from Dryad (doi.org/10.5061/dryad.k52h33d) provides full details on sample processing and data acquisition.
Statistical analyses
The following statistical analyses were performed in R version 3.4.1. For comparative analyses of the individual CSF proteomes, spectral counts were aggregated by protein (i.e., protein abundance) for each sample. The protein abundances for the unique 1,043 proteins identified in CSF were compared between the PACNS (n = 8) and NIC (n = 11) cohorts with 2 statistical approaches commonly used for mass spectrometry datasets, DESeq2 version 3.7 and the t test.9,10 DESeq2 uses a method based on the negative binomial distribution to assess differential expression in count data. Spectral counts for all 1,043 unique proteins were used as the input for this package, which was then run with default settings. For the t test, spectral count values of zero were first replaced with counts of 0.16, a value empirically determined to best approximate normal distributions for each protein within the NIC samples. Spectral counts were then divided by the sum of spectral counts for each sample and multiplied by 10,000, generating normalized spectral counts. Only proteins at sufficient abundance were considered in the t test, defined as having a sum across all NIC and PACNS samples of ≥10 normalized spectral counts and being observed in at least 5 of the combined group of NIC and PACNS samples. Normalized spectral counts for these 713 abundant proteins were then transformed by natural logarithm to avoid large differences in variances for different proteins and then analyzed with 2-sided t tests assuming unequal variance to compare abundances between the PACNS and NIC cohorts for each protein. The resulting p values were then adjusted for multiple comparisons with the Benjamini-Hochberg method. Fold changes were calculated as the ratio between the mean of PACNS and NIC samples for each protein. In our conservative approach, only proteins that were significantly different (fold change >1.5 and Benjamini-Hochberg–adjusted p < 0.05) between PACNS and NIC in both tests were considered differentially regulated proteins in PACNS. Because of the low sample number in the RCVS cohort (n = 4), this comparative statistical analysis was restricted to the PACNS and NIC cohorts only.
Unbiased hierarchical clustering was performed with the numpy (version 1.15.0) and seaborn (version 0.9.0) packages in python (version 3.7.0). For this analysis, abundant proteins across PACNS, NIC, and RCVS samples were grouped to combine counts for isoforms of the same protein with shared peptides (peptides mapping to multiple isoforms). Proteins and samples were then clustered by applying an unweighted pair group method with arithmetic mean to the correlation distance matrix.
Molecular pathway enrichment analysis
Pathway enrichment analysis was performed with the Database for Annotation, Visualization and Integrated Discovery Bioinformatics resource (david.ncifcrf.gov/). For enrichment analysis, the 222 downregulated proteins and 61 upregulated proteins in PACNS were analyzed against the CSF background of all 1,043 observed proteins. For 50 of the proteins most highly downregulated in PACNS, annotations for transmembrane and topologic domains were retrieved from UniProt. The locations of these domains and the locations of peptides observed in the NIC samples by mass spectrometry were then mapped onto the protein chains.
Data availability
Raw mass spectrometry data files and peak list files have been deposited at ProteoSAFE (massive.ucsd.edu) with accession number MSV000082129.
Results
Summary of clinical characteristics
In addition to the individual patient vignettes above, an aggregated summary of patient demographics, clinical features, imaging abnormalities, and clinical CSF parameters for the PACNS, NIC, and RCVS cohorts is provided for comparison in the table. The majority of our patients with biopsy-proven PACNS were refractory to initial treatment strategies or required continual immunosuppression to achieve remission. Notably, all patients with PACNS had a chronic disease course, compared to the monophasic nature of RCVS.
Table.
Demographics and clinical features of the PACNS, RCVS, and NIC cohorts

All 8 patients with PACNS had a brain biopsy with documented transmural inflammation and vessel wall damage of small to medium-sized vessels in the brain parenchyma or meninges (figure 1). A single patient's biopsy was described as having granulomatous vasculitis; the remainder were lymphocytic. Biopsies were not performed in individuals with RCVS or NIC. No patient with PACNS had evidence of amyloid angiitis or systemic vasculitis. All patients tested negative for anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitis.
Figure 1. Histopathologic features of a diagnostic biopsy for PACNS patient cohort.

Biopsies from all patients in our primary angiitis of the CNS (PACNS) cohort (n = 8) showed inflammation of small to medium-sized CNS blood vessels. (A) Representative images from a patient biopsy (patient 3) demonstrating several hallmark features of PACNS histopathology, including (I) perivascular inflammation, (II) intramural inflammation with thickening of blood vessel wall, (III) leptomeningeal inflammation, and (IV) rupture of blood vessel wall. (B) Additional section of meningeal vessels (brain parenchyma, bottom left), showing transmural inflammation. (C) Immunohistochemistry with anti-CD3 shows enrichment of T cells among immune cell infiltrates corresponding to panel A. (D) High magnification of perivascular and intramural inflammation. Arrowheads in the inset indicate mononuclear lymphocytes. Tissue sections were stained with hematoxylin and eosin unless otherwise noted. Scale bar, 100 μm.
Unbiased discovery of a putative molecular phenotype in PACNS
CSF samples from individuals with PACNS (n = 8), RCVS (n = 4), and NIC (n = 11) were analyzed by mass spectrometry. A total of 1,043 proteins were identified across all cohorts (appendix e-2 available from Dryad, doi.org/10.5061/dryad.k52h33d). Unbiased clustering of individuals based on the normalized protein counts showed that patients with PACNS were more similar to each other than to individuals in the RCVS and NIC cohorts (figure 2).
Figure 2. Unbiased discovery of a molecular phenotype in patients with PACNS.

Dendrogram (left) depicts the distance between samples (sample identifiers shown at branch tips). Two major clusters emerge from an unbiased clustering analysis: top, containing mostly primary angiitis of the CNS (PACNS) samples (purple); and bottom, containing noninflammatory control (light green) and reversible cerebral vasoconstriction (dark green) samples. Heat map displays levels of 651 proteins for every sample as a by-protein z score. Hierarchical clustering was performed by applying the unweighted pair group method with arithmetic mean method to the correlation distance matrix. Length of horizontal lines on the dendrogram represents the distance between samples.
Defining differentially expressed proteins and molecular pathways in PACNS
To identify specific proteins that distinguish PACNS from NIC, we compared protein abundances in PACNS vs NIC. We identified 283 proteins that had statistically significant differential regulation in PACNS, with 61 upregulated and 222 downregulated proteins compared to NIC (appendix e-3 available from Dryad, doi.org/10.5061/dryad.k52h33d). Pathway enrichment analysis showed significant enrichment for the KEGG pathways complement and coagulation cascades (adjusted p < 0.05) and cell adhesion molecules (adjusted p < 0.01). Differentially expressed proteins in PACNS relative to NIC are shown in figure 3. Although statistical analyses were not performed on the RCVS cohort due to the small number of patients, the relative protein abundances are displayed for qualitative comparison. Note that patients with RCVS have elevated levels of serum-derived proteins (hemoglobin, serum amyloid 1, serum amyloid 2, carbonic anhydrase), similar to PACNS, but show minimal evidence of complement protein dysregulation.
Figure 3. Differentially expressed proteins and molecular pathways in PACNS.
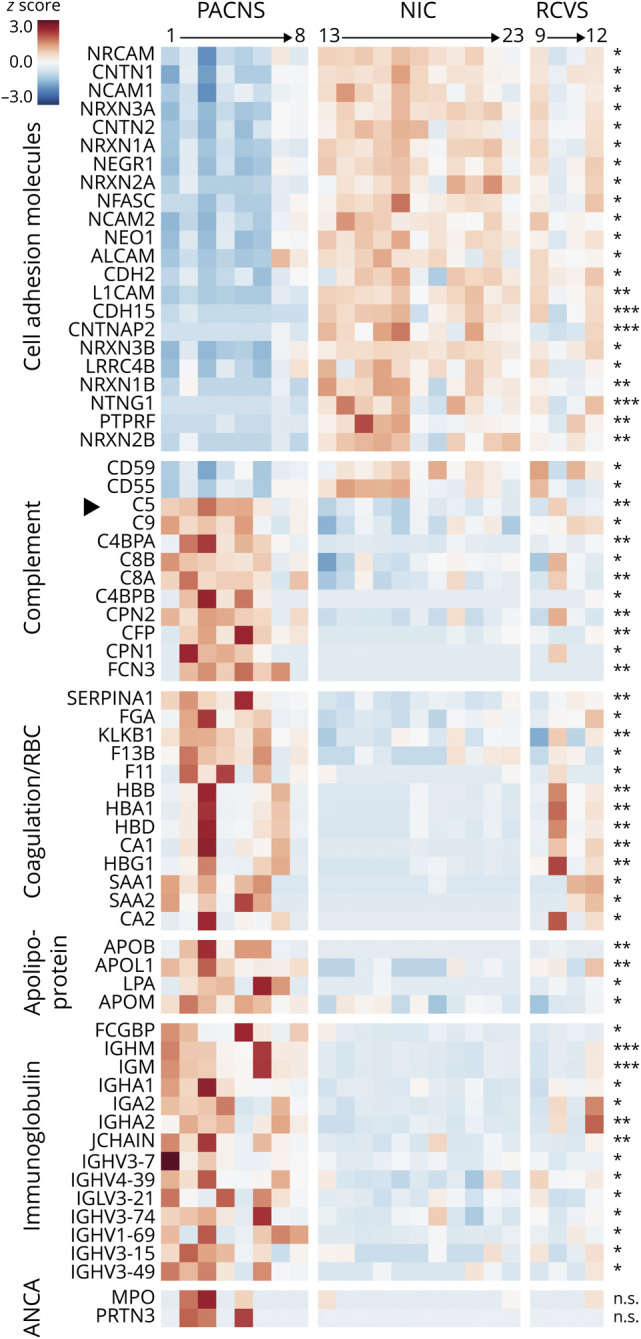
Relative protein abundances for a subset of differentially regulated proteins in primary angiitis of the CNS (PACNS) vs noninflammatory control (NIC) are reported. Proteins representing the statistically significantly enriched pathways cell adhesion molecules and complement and coagulation cascades are included, as well as a subset of proteins manually curated to reflect findings of significant clinical interest, with functional classifiers informed by Database for Annotation, Visualization and Integrated Discovery and KEGG annotations. Heat map displays relative protein abundance across individual samples (PACNS n = 8, NIC n = 11, and reversible cerebral vasoconstriction [RCVS], n = 4), plotted as the z score of the normalized spectral counts. Differential expression was evaluated with DESeq2 and the t test (see Methods), with significance defined as having fold change >1.5 and Benjamini-Hochberg–adjusted p < 0.05 for both tests (*p < 0.05, **p ≤ 0.01, ***p ≤ 0.001). n.s. = not significant.
There are many significantly altered proteins within the complement pathway in PACNS, including decay accelerating factor (CD55), membrane attack complex inhibitory protein (CD59), properdin (CFP), complement C4 binding protein A (C4BPA), complement C4 binding protein B (C4BPB), ficolin-3 (FCN3), carboxypeptidase N catalytic chain (CPN1), carboxypeptidase N subunit 2 (CPN2), and complement C5, C8, and C9.
The enrichment for cell adhesion molecules was driven by 22 proteins downregulated in PACNS compared to NIC. More specifically, these proteins are transmembrane cell adhesion molecules expressed in neural tissue. Manual inspection of the remaining 200 downregulated proteins revealed many more proteins that have roles in neural cell adhesion and contain transmembrane domains, including β-amyloid (APP), despite the absence of these proteins in the core set of the KEGG cell adhesion molecules pathway. We localized the recovered peptides from mass spectrometry according to protein domain annotations assigned by UniProt and found that NIC CSF contained peptides specifically from the extracellular domains of transmembrane proteins that are downregulated in PACNS CSF (figures 4 and 5).
Figure 4. Downregulated peptides in PACNS CSF map to extracellular domains of transmembrane cell adhesion proteins.

A subset of proteins that are strongly downregulated in primary angiitis of the CNS (PACNS) compared to noninflammatory control (NIC) are shown with UniProt annotations for transmembrane and topologic domains. Locations of peptides from mass spectrometry of NIC samples are plotted. Note the absence of peptides from cytoplasmic regions of proteins with transmembrane domains.
Figure 5. Proposed model of the complement phenotype of PACNS.

Molecular phenotype in primary angiitis of the CNS (PACNS) CSF is informed by proteomic comparison of CSF between PACNS and noninflammatory control (NIC) cohorts. Molecular data are overlaid onto the complement cascade pathway (adapted from KEGG, hsa04610). Fold change in protein abundance between PACNS and NIC was evaluated for all proteins annotated in the pathway. Proteins are reported as elevated (red, >1.5 fold significant increase in PACNS), unchanged (pink, not significantly changed in PACNS), decreased (blue, >1.5 fold significant reduction in PACNS), or not observed (white, no abundance in NIC or PACNS). The prediction for C3 and C5 convertase activity (activated, yellow; inhibited, gray) is informed specifically by molecular changes observed in the complement regulatory proteins CFP, complement C4 binding protein (C4BP), CD59, and CD55. The proposed model predicts a shift toward activation of the alternative pathway (CFP, CD55), active inhibition of classical pathway (C4BPA and C4BPB), and elevated signaling from the terminal cascade (CD59).
In addition, we noted several significant findings that do not correspond to a specific pathway but pique clinical interest. These findings include elevated levels of immunoglobulins (IgM and IgA) and apolipoproteins [including apolipoprotein B100 (ApoB100) and lipoprotein(a)] in PACNS.
Orthogonal confirmation
To validate our technical approach, we reproduced a subset of findings by Western blot and commercial ELISA. Because of the potential clinical and therapeutic implications of identifying a role for the alternative complement cascade in PACNS, specifically complement C5, we validated elevated C5 levels in PACNS CSF through Western blotting with commercial antibody and commercial ELISA (figures e-1 and e-2 available from Dryad, doi.org/10.5061/dryad.k52h33d). Notably, the relative C5 levels by mass spectrometry analysis and by ELISA are correlated, suggesting that the variation in these data across patients is reproducible across technical approaches. The substantial changes in IgM and IgA levels identified by mass spectrometry were also reproduced orthogonally through Western blotting.
Discussion
In this exploratory study, a mass spectrometry–based approach was used to characterize the CSF proteome associated with ongoing PACNS pathology relative to noninflammatory disease, with the intention to discover new diagnostic or therapeutic candidates. Currently, the diagnosis of PACNS remains challenging.3 The diagnostic criteria for PACNS, including CSF cytology, imaging abnormalities, and neurologic manifestations, are largely nonspecific, and long-term outcomes are variable.1,11 Several of these features, including elevated protein and imaging abnormalities, are also observed in individuals diagnosed with an early mimic, RCVS, as detailed in the clinical summary of our patient cohorts (table) and individualized clinical vignettes (Methods) for patients with PACNS and RCVS.12,13 We restricted our analyses to patients with biopsy-proven PACNS to enhance the rigor of this exploratory study and to ensure that our molecular data were most closely correlated to the key features that are associated with PACNS pathology.14
Our findings highlight the complement cascade as a significant feature of PACNS CSF. We found up to 12 significantly dysregulated proteins in PACNS CSF that function within the complement cascade pathway, and these changes are highly reproducible across the PACNS patient cohort. In addition, the proteomic changes within the complement pathway are specific, affecting the alternative and terminal cascade only, and include changes in transient, fluid-phase regulators, the presence and alteration of which are suggestive of an ongoing pathologic process.15 Lastly, the robust changes in complement are not observed in patients with RCVS, which, early in the disease course, can mimic PACNS clinically and radiologically but does not manifest with chronic inflammation.12,16 Thus, changes in the complement pathway in PACNS are unlikely to be due to secondary processes from acute vascular injury alone. Taken together, these data suggest that differential expression of the complement pathway is a potential correlate of the ongoing inflammatory processes in PACNS.
The specific proteomic findings within the complement cascade have predictable consequences on complement pathway function in PACNS. The complement cascade is one of the primary effector systems of an immunologically induced inflammatory reaction that can be triggered via 2 separate proteolytic pathways, the classical pathway (homologous to the lectin pathway) and the alternative pathway.15,17 Signaling events from the classical pathway and alternative pathway converge onto a common effector pathway, known as the terminal cascade, which enables lysis and phagocytosis of foreign/inflammatory material. The alternative pathway and classical pathway are molecularly unique at the level of the complement C3 and C5 convertases, which drive cleavage of C3 and C5, respectively. In PACNS CSF, elevated levels of the 2 major regulators of the C3 and C5 complement cascade convertases, CFP and C4BPA/C4BPB, were observed. CFP is the only known positive regulator of the alternative pathway, stabilizing the alternative pathway convertases, while C4BPA and C4BPB are well-studied inactivators of the C3 convertases specific to the classical pathway.18,19 These 2 features are consistent with an alternative pathway activation state. Downstream, among the terminal complement components, an increase in C5 and members of the membrane attack complex (C8A, C8B, C9) and a reduction in CD59, an inhibitor of the membrane attack complex, were also observed.15 Thus, we speculate that abnormal signaling of the terminal cascade is occurring in PACNS CSF and that this is due to sustained activation of the alternative pathway.
While the role of a dysregulated alternative complement cascade with respect to the pathogenesis of PACNS remains unclear, these findings implicate a critical pathway that has been exploited for therapeutic intervention in similar diseases.15,20,21 Specifically, activation of the alternative complement pathway has been implicated in ANCA-associated vasculitis. Blockade of the alternative pathway reduced disease activity in an ANCA-mediated mouse model of peripheral vasculitis, and clinical trials have demonstrated the effectiveness of anti-C5a receptor therapies in ANCA-associated vasculitis in humans.20–22 Our results reveal a previously unappreciated overlap in molecular targets between PACNS and ANCA-associated vasculitis, suggesting that similar therapeutic interventions should be considered for future PACNS trials, assuming that these findings extend to larger cohorts of patients with PACNS and controls.
In addition, several observations in this cohort, beyond complement pathway components, warrant further investigation, either as diagnostic biomarkers or as therapeutic candidates themselves. These include elevated IgM, IgA, and ApoB100. While an elevated CSF IgG index is commonly found in PACNS, our finding of elevated IgA and IgM in CSF from patients with PACNS has not been previously described.3,11,14 While the functional significance of elevated IgA and IgM in the context of PACNS is unclear, elevated IgA and IgM levels may be explored as a separate diagnostic differentiator.
In contrast, several anecdotal pieces of evidence implicate ApoB100 with a subset of PACNS pathological features. For one, an immune response to ApoB100, mediated by T cells and IgM antibodies, is commonly observed in the progressive development of atherosclerotic lesions.23,24 Furthermore, elevated low-density lipoprotein and antibodies to ApoB100 have been identified in alternative non-PACNS vasculitides, including ANCA-associated vasculitis. In the case of both MPO-ANCA and PR3-ANCA, elevated levels of anti-apoB100 antibodies are thought to be an indirect result of the chronic inflammatory disease.25 Given the robust elevation of ApoB100 in this PACNS cohort, further investigation into the direct or indirect role of ApoB100 in PACNS pathology is warranted.
Finally, an unexpected finding of this study was the loss of neural cell adhesion molecules in PACNS CSF. For noninflammatory CSF, we observe peptides almost exclusively from the extracellular domains (ectodomains) of transmembrane proteins. Changes in these proteins in PACNS may be the result of transcriptional, translational, or posttranslational regulatory differences.26–28 The last may include abnormal regulation of the normal process of ectodomain shedding.26,29,30 The clear absence of ectodomain peptides in PACNS CSF suggests a loss of these proteins or a loss of proteolytic homeostasis associated with shedding. While the roles of ectodomain shedding are diverse, the mechanism and impact of dysregulated shedding in CSF are unknown.
Overall, these exploratory findings suggest potential new biomarkers of PACNS, subject to validation in larger cohorts. These results also underline the importance of future mechanistic studies around the role of complement pathways in PACNS disease pathobiology, with the ultimate goal of creating targeted therapeutic interventions for this devastating and poorly understood disease.
Glossary
- ANCA
anti-neutrophil cytoplasmic antibody
- ApoB100
apolipoprotein B100
- C4BPA
complement C4 binding protein A
- C4BPB
complement C4 binding protein B
- Ig
immunoglobulin
- MR
magnetic resonance
- NIC
noninflammatory control
- OCB
oligoclonal band
- PACNS
primary angiitis of the CNS
- RBC
red blood cell
- RCVS
reversible cerebral vasoconstriction syndrome
- UCSF
University of California, San Francisco
- WBC
white blood cell
Appendix. Authors
Study funding
Funded by the NIH National Institutes for Neurological Disorders and Stroke (award K08NS096117); the UCSF Center for Next-Gen Precision Diagnostics supported by the Sandler Foundation and William K. Bowes, Jr. Foundation; UCSF Medical Scientist Training Program; the Rachleff Foundation; and Chan Zuckerberg Biohub.
Disclosure
C. Mandel-Brehm, H. Retallack, G. Knudsen, A. Yamana, R. Hajj-Ali, L. Calabrese, T. Tihan, H. Sample, K. Zorn, M. Gorman, J. Madan Cohen, A. Sreih, J. Marcus, S. Josephson, and V. Douglas report no disclosures relevant to the manuscript. J. Gelfand reports personal compensation for consulting for Biogen and Alexion, research support to UCSF from Genentech, service contract support from MedDay, and personal compensation for medical legal consulting/expert witness. M. Wilson reports no disclosures relevant to the manuscript. J. DeRisi is a scientific consultant for Allen & Company. Go to Neurology.org/N for full disclosures.
References
- 1.Hajj-Ali RA, Calabrese LH. Primary angiitis of the central nervous system. Autoimmun Rev 2013;12:463–466. [DOI] [PubMed] [Google Scholar]
- 2.Salvarani C, Brown RD, Christianson TJH, et al. Adult primary central nervous system vasculitis treatment and course: analysis of one hundred sixty-three patients. Arthritis Rheumatol 2015;67:1637–1645. [DOI] [PubMed] [Google Scholar]
- 3.Byram K, Hajj-Ali RA, Calabrese L. CNS vasculitis: an approach to differential diagnosis and management. Curr Rheumatol Rep 2018;20:37. [DOI] [PubMed] [Google Scholar]
- 4.Hutchinson C, Elbers J, Halliday W, et al. Treatment of small vessel primary CNS vasculitis in children: an open-label cohort study. Lancet Neurol 2010;9:1078–1084. [DOI] [PubMed] [Google Scholar]
- 5.Alba MA, Espigol-Frigole G, Prieto-Gonzalez S, et al. Central nervous system vasculitis: still more questions than answers. Curr Neuropharmacol 2011;9:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bastos P, Ferreira R, Manadas B, Moreira PI, Vitorino R. Insights into the human brain proteome: disclosing the biological meaning of protein networks in cerebrospinal fluid. Crit Rev Clin Lab Sci 2017;54:185–204. [DOI] [PubMed] [Google Scholar]
- 7.Oliveira V, Póvoa P, Costa A, Ducla-Soares J. Cerebrospinal fluid and therapy of isolated angiitis of the central nervous system. Stroke 1994;25:1693–1695. [DOI] [PubMed] [Google Scholar]
- 8.Ruland T, Wolbert J, Gottschalk MG, et al. Cerebrospinal fluid concentrations of neuronal proteins are reduced in primary angiitis of the central nervous system. Front Neurol 2018;9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langley SR, Mayr M. Comparative analysis of statistical methods used for detecting differential expression in label-free mass spectrometry proteomics. J Proteomics 2015;129:83–92. [DOI] [PubMed] [Google Scholar]
- 10.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res 2006;5:2339–2347. [DOI] [PubMed] [Google Scholar]
- 11.Salvarani C, Brown RD, Calamia KT, et al. Primary central nervous system vasculitis: analysis of 101 patients. Ann Neurol 2007;62:442–451. [DOI] [PubMed] [Google Scholar]
- 12.Ducros A, Boukobza M, Porcher R, Sarov M, Valade D, Bousser MG. The clinical and radiological spectrum of reversible cerebral vasoconstriction syndrome: a prospective series of 67 patients. Brain 2007;130:3091–3101. [DOI] [PubMed] [Google Scholar]
- 13.Rocha EA, Topcuoglu MA, Silva GS, Singhal AB. RCVS2 score and diagnostic approach for reversible cerebral vasoconstriction syndrome. Neurology 2019;92:e639–e647. [DOI] [PubMed] [Google Scholar]
- 14.Miller DV, Salvarani C, Hunder GG, Brown JE, Christianson TJ, Giannini C. Biopsy findings in primary angiitis of the central nervous system. Am J Surg Pathol 2009;33:35–43. [DOI] [PubMed] [Google Scholar]
- 15.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol 2006;176:1305–1310. [DOI] [PubMed] [Google Scholar]
- 16.de Boysson H, Parienti J-J, Mawet J, et al. Primary angiitis of the CNS and reversible cerebral vasoconstriction syndrome: a comparative study. Neurology 2018;91:e1468–e1478. [DOI] [PubMed] [Google Scholar]
- 17.Gigli I. Immunochemistry and immunobiology of the complement system. J Invest Dermatol 1976;67:346–353. [DOI] [PubMed] [Google Scholar]
- 18.Blatt AZ, Pathan S, Ferreira VP. Properdin: a tightly regulated critical inflammatory modulator. Immunol Rev 2016;274:172–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gigli I, Fujita T, Nussenzweig V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Am J Physiol 1979;273:C883–C892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol 2007;170:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol 2017;28:2756–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bekker P, Dairaghi D, Seitz L, et al. Characterization of pharmacologic and pharmacokinetic properties of CCX168, a potent and selective orally administered complement 5a receptor inhibitor, based on preclinical evaluation and randomized phase 1 clinical study. PLoS One 2016;11:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nilsson J, Björkbacka H, Fredrikson GN. Apolipoprotein B100 autoimmunity and atherosclerosis-disease mechanisms and therapeutic potential. Curr Opin Lipidol 2012;23:422–428. [DOI] [PubMed] [Google Scholar]
- 24.Nilsson J, Hansson GK. Autoimmunity in atherosclerosis: a protective response losing control? J Intern Med 2008;263:464–478. [DOI] [PubMed] [Google Scholar]
- 25.Slot MC, Theunissen R, van Paassen P, Damoiseaux JG, Tervaert JW. Anti-oxidized low-density lipoprotein antibodies in myeloperoxidase-positive vasculitis patients preferentially recognize hypochlorite-modified low density lipoproteins. Clin Exp Immunol 2007;149:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsumagari K, Shirakabe K, Ogura M, Sato F, Ishihama Y, Sehara-Fujisawa A. Secretome analysis to elucidate metalloprotease-dependent ectodomain shedding of glycoproteins during neuronal differentiation. Genes Cells 2017;22:237–244. [DOI] [PubMed] [Google Scholar]
- 27.Shirakabe K, Omura T, Shibagaki Y, et al. Mechanistic insights into ectodomain shedding: susceptibility of CADM1 adhesion molecule is determined by alternative splicing and O-glycosylation. Sci Rep 2017;7:46174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lichtenthaler SF, Lemberg MK, Fluhrer R. Proteolytic ectodomain shedding of membrane proteins in mammals: hardware, concepts, and recent developments. EMBO J 2018;37:e99456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waldera-Lupa DM, Etemad-Parishanzadeh O, Brocksieper M, et al. Proteomic changes in cerebrospinal fluid from primary central nervous system lymphoma patients are associated with protein ectodomain shedding. Oncotarget 2017;8:110118–110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller MA, Sullivan RJ, Lauffenburger DA. Molecular pathways: receptor ectodomain shedding in treatment, resistance, and monitoring of cancer. Clin Cancer Res 2017;23:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw mass spectrometry data files and peak list files have been deposited at ProteoSAFE (massive.ucsd.edu) with accession number MSV000082129.