

ABSTRACT
The identity of the platform supporting the initiation and formation of the nascent autophagosome, the phagophore, is not fully understood. Nucleation and expansion of the phagophore membrane requires a coordinated flux or activation of specific proteins and membrane lipids at the initiation site. The transmembrane protein ATG9A is essential for macroautophagy/autophagy and proposed to be an initiator of the phagophore by directing or facilitating the delivery of proteins and lipids to the initiation site. Upon amino acid starvation, ATG9A-containing vesicles are formed from the Golgi complex and endosomal compartments and translocate to the initiation site. Unravelling the complement of proteins and lipids brought by ATG9A vesicles to the forming autophagosome is essential to further understand the initiation of autophagy.
KEYWORDS: ARFIP2, ATG13, autophagosome initiation site, PI4KB, PI4KIIIβ
To establish the molecular function of ATG9A in autophagy, we used an immuno-isolation approach coupled with SILAC mass spectrometry to determine the composition of endogenous ATG9A-positive membranes during starvation. The analysis revealed an enrichment upon autophagy induction of known components of ATG9A vesicles such as the clathrin adaptor protein complexes AP1 and AP4, but also revealed a set of new proteins including BAR domain containing membrane-shaping proteins [1]. Golgi proteins are depleted from ATG9A-vesicles in starvation conditions. Interestingly, neither RAB11 nor SNX18 (sorting nexin 18), both implicated in delivery of ATG9A from the recycling endosome, are detected in the immuno-isolated vesicles, supporting the notion that the initiator vesicular population is derved from the Golgi complex, and ATG9A in the recycling endosome is part of a secondary wave of trafficking for phagophore expansion.
Figure 1.
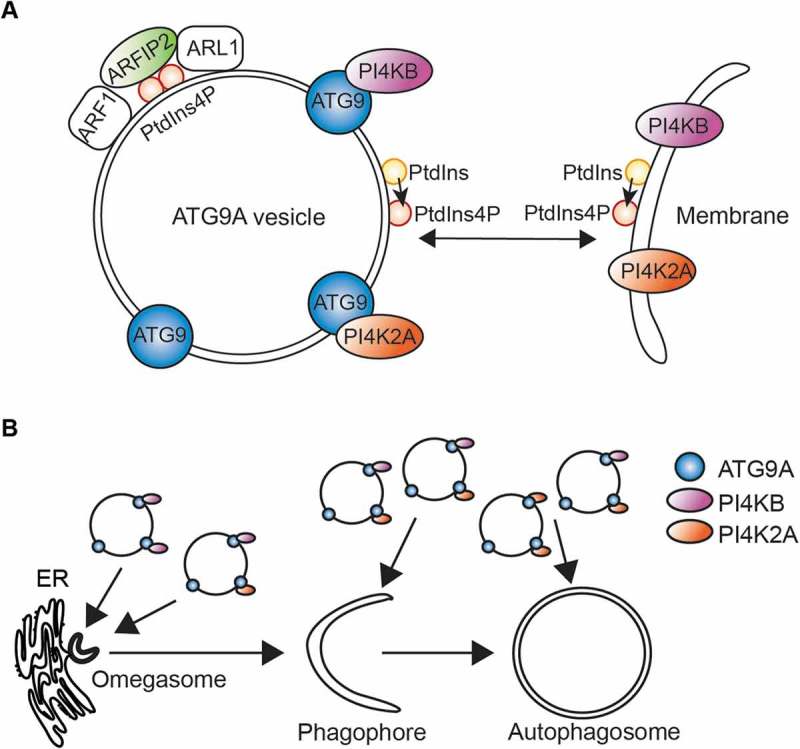
ATG9A vesicles deliver components to shape the phagophore membrane. (A) In association with ARF1 and ARL1, ARFIP2 may activate phosphoinositide (PI)-metabolizing enzymes contained on ATG9A vesicles, driving the production of PtdIns4P to generate a suitable environment for ATG9A function. ATG9A vesicles can then deliver PI4K2A/PI4KIIα and PI4KB/PI4KIIIβ to the phagophore initiation site or later to expanding phagophores to produce PtdIns4P. (B) ATG9A vesicles deliver PI4K2A/PI4KIIα and PI4KB/PI4KIIIβ to all stages of the autophagic membranes: omegasomes, phagophores and autophagosomes, participating in autophagy.
The BAR domain membrane-shaping proteins found in the ATG9A vesicles include ARFIP1/Arfaptin-1 (ADP ribosylation factor interacting protein 1), ARFIP2/Arfaptin-2 and SH3GLB1/BIF1. While SH3GLB1 was previously shown to regulate ATG9A trafficking from recycling endosomes, nothing was published implicating ARFIPs in autophagy. ARFIP1 and ARFIP2 bind small GTPases including ARF1 and ARL1 in their GTP-bound state, and function to regulate cargo exit from the Golgi. We showed that loss of ARFIP2 but not ARFIP1, leads to a defect in autophagy flux and in ULK1, WIPI2B and LC3B recruitment to the autophagosome formation site. ARFIP2 binds PtdIns4P through an amphipathic helix, and this binding is required for its role upon starvation even in the presence of a functional BAR domain.
ARFIP2 is required to maintain the steady-state distribution of ATG9A in normal conditions, and disruption of the normal distribution has an impact upon phagophore initation. Additional SILAC mass spectrometry experiments on ATG9A vesicles immuno-isolated from ARFIP2 CRISPR knockout cells in starvation revealed a loss of a broad range of proteins on ATG9A vesicles including phosphoinositide-metabolizing enzymes, PI4K2A/PI4KIIα and PI4KB/PI4KIIIβ, suggesting that the composition of these vesicles is dependent on ARFIP2. Further analysis showed that ATG9A interacts and localizes with both PI4K2A/PI4KIIα and PI4KB/PI4KIIIβ. In line with this, we detected PtdIns4P on ATG9A vesicles and observed by live-cell imaging that ATG9A-positive vesicles transiting during starvation contain PtdIns4P.
We then focused on the link between ATG9A, PtdIns4P and PI4KB/PI4KIIIβ, as PI4K2A/PI4KIIα was already described to be implicated in autophagosome maturation. We found that in starvation conditions the distribution of PtdIns4P and PI4KB/PI4KIIIβ follows ATG9A and is dependent on ARFIP2. Strikingly, their distribution depends also on ATG9A. PtdIns4P was found on ATG13-, ZFYVE1/DFCP-1- and WIPI2-positive phagophores and autophagosomes. We investigated the role of PI4KB/PI4KIIIβ in autophagy and revealed that loss of PI4KB/PI4KIIIβ alters the trafficking of ATG9A, and results in a defect in autophagosome formation suggesting that ATG9A and PI4KB/PI4KIIIβ must work hand in hand during the formation of autophagosomes (Figure 1).
ATG9A translocation to the autophagosome initiation site precedes the recruitment of the ULK1 complex subunit ATG13 suggesting that proteins and/or lipids delivered by ATG9A vesicles prepare the formation platform for delivery of the ULK1 complex, the autophagy initiator kinase complex. To prove that PtdIns4P and PI4KB/PI4KIIIβ are involved in priming of the early stage-autophagosome platform, we studied the composition of immuno-isolated PtdIns4P-positive membranes and found they contained ATG9A, and ULK1. Likewise, the ZFYVE1-positive omegasome and ATG13-positive phagophore contain PI4KB/PI4KIIIβ. Importantly, PI4KB/PI4KIIIβ binds ATG13 and contributes to ATG13-positive phagophore formation independently of PtdIns3P production. Strikingly, further experiments revealed that the activated form of PI4KB/PI4KIIIβ is enriched in ATG13-positive membranes. In line with this, it is known that ATG13 binds PtdIns3P, but also PtdIns4P, supporting our hypothesis that the delivery of PI4KB/PI4KIIIβ by ATG9A to the initiation site produces PtdIns4P, and thus facilitates local recruitment of ATG13, and thus the ULK1 complex.
Our data provide new insight into the molecular requirements of ATG9A in autophagy. ATG9A vesicles deliver lipid kinases necessary for the production of PtdIns4P on early stage autophagosomes, the omegasomes and phagophore. We speculate that ARFIP2 regulates ATG9A vesicle formation and generates a suitable environment for the function of ATG9A vesicles during autophagy. ARFIP2, by association with ARL1 and PRKD2 (protein kinase D2), known to regulate PI4KB/PI4KIIIβ, could activate the latter, increasing the lipid kinase activity and driving the production of PtdIns4P on ATG9A vesicles as well as on autophagosome initiation sites leading to the recruitment of the ULK1 complex. There are still many questions regarding the regulation of PI4KB/PI4KIIIβ activation by PRKD2 during amino acid starvation, including how, when and where this activation occurs. Our study opens up a whole new perspective on the lipids found in the initiation platform as it has been assumed that PtdIns3P, and its derivative PtdIns(3,5)P2, were the only phosphoinositides required at the early stage of autophagosome formation, and we speculate that PtdIns4P could be in fact the essential mono-phosphorylated phosphoinositide at the autophagosome initiation site.
Funding Statement
This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001187); the UK Medical Research Council (FC001187); and the Wellcome Trust (FC001187).
Acknowledgments
This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001187); the UK Medical Research Council (FC001187); and the Wellcome Trust (FC001187).
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Judith D, Jefferies HBJ, Boeing S, et al. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J Cell Biol. 2019;218:1634–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]