

ABSTRACT
Coding or non-coding mutations in FUS (fused in sarcoma) cause amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). In addition to familial ALS, abnormal aggregates of FUS are present in a portion of FTD and other neurodegenerative diseases independent of their mutations. Broad expression within the central nervous system of either wild-type or two ALS-linked human FUS mutants produces progressive motor phenotypes accompanied by characteristic ALS-like pathology. FUS levels are autoregulated to maintain an optimal steady-state level. Increasing FUS expression by saturating its autoregulatory mechanism results in rapidly progressive neurological phenotypes and dose-dependent lethality. Genome-wide expression analysis reveals genetic mis-regulations distinct from those via FUS reduction. Among these are increased expression of lysosomal proteins, suggestive of disruption in protein homeostasis as a potential gain-of-toxicity mechanism. Indeed, increased expression of wild-type FUS or ALS-linked mutant forms of FUS inhibit macroautophagy/autophagy. Collectively, our results demonstrate that: (1) mice expressing FUS develop progressive motor deficits, (2) increased FUS expression by overriding its autoregulatory mechanism accelerates neurodegeneration, providing a basis for FUS involvement without mutation, and (3) disruption in both protein homeostasis and RNA processing contribute to FUS-mediated toxicity.
KEYWORDS: Amyotrophic lateral sclerosis (ALS), autophagy, frontotemporal dementia (FTD), FUS, lysosome, p62, SQSTM1/p62
Amyotrophic lateral sclerosis (ALS, also known as motor neuron disease) and frontotemporal dementia (FTD) was first described by Jean-Martin Charcot (1874) and Arnold Pick (1892), respectively. The names ALS and FTD highlight the defining features of the diseases: muscle wasting progressing to paralysis with degeneration of motor neurons and their axonal tracks in ALS, and pre-senile dementia, aphasia and lobar atrophy for FTD. Although seemingly different, FTD and ALS symptoms can occur simultaneously in the same patient, and either ALS or FTD or both can present in familial cases, suggesting genetic and clinical overlaps of these diseases. Indeed, identification of their common genetic causes and pathological hallmarks for FTD and ALS indicate that FTD and ALS are one disease continuum. Regardless of the cause, pathological inclusions containing TARDBP/TDP-43 and FUS are found in >95% and 55% of ALS and FTD, respectively. Furthermore, dominant FUS mutations cause 5% of inherited ALS and <1% of FTD cases, establishing a gene-pathology relationship. Disease-causing mutations in genes that encode pathological hallmark proteins are commonly seen in most adult-onset neurodegenerative diseases, including Alzheimer disease (AD) and Parkinson disease (PD). Therefore, it is crucial to elucidate how FUS causes ALS and FTD.
We have established and characterized transgenic mice expressing wild-type (WT) human FUS and two disease-linked mutants (FUSR514 and FUSR21C) to identify the pathogenic roles of FUS in neurodegeneration [1]. The murine Prnp (prion protein) promoter was used to broadly drive FUS cDNA expression in the central nervous system (CNS). We chose transgenic lines that mimic the endogenous FUS expression patterns with matching expression levels of the transgene. Both WT FUS and FUSR514G mice develop ALS-like phenotypes and symptoms in age-dependent and mutant-enhanced manners.
Regardless of numbers of transgene copies, the total FUS protein level remained unchanged, likely due to autoregulatory mechanisms, where FUS binds to its own mRNA. Because of its autoregulation, the transgene is roughly 1:1 to endogenous protein and total FUS protein is similar to that in non-transgenic animals. Thus, these mice genetically mimic the human disease condition. Furthermore, overriding the auto-regulatory mechanisms resulted in a 2-fold increase of FUS expression, generating severe neurological deficits and early lethality by 40 days. The neurological deficits are likely caused by motor neurons/axon degeneration and loss of neuromuscular junctions (NMJs). Elevated gene expressions encoding the pathological hallmarks are not without precedent, such as duplication of APP (causal for AD), and triplication of SNCA/α-synuclein (causal for PD). Furthermore, ALS-causing mutations in the FUS 3ʹ-UTR increase FUS levels, indicating disruption of FUS homeostasis within the CNS could drive ALS pathogenesis.
FUS binds to DNA and RNA, and participates in RNA metabolism. We performed RNA-seq analysis to identify how increased FUS levels affect RNA maturation/stability. Unbiased gene ontology (GO) analyses of differentially expressed genes (DEGs) showed distinct up- and downregulated genes. When these DEGs are classified with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, the upregulated genes are found to be involved in lysosomes, cytokine-cytokine receptor interactions, and the MAPK or TP53/p53 signaling pathways, whereas the downregulated genes are involved in steroid and terpenoid backbone biosynthesis.
Upregulation of lysosomal genes in the homozygous WT FUS transgenic mice suggests changes in protein homeostasis. Although ubiquitin-positive aggregates are not observed, SQSTM1/p62 (sequestosome 1) accumulation is found in the motor neurons of homozygous WT FUS mice, but not in non-transgenic, hemizygous WT FUS mice and neighboring non-motor neurons. This suggests that an autophagy deficit occurs in these motor neurons. Overexpressing WT FUS and ALS-linked mutants (FUSR514G and FUS521C) in vitro inhibits autophagy induction and flux using both LC3 and SQSTM1 as markers (Figure 1). Furthermore, most FUS proteins remain in the nuclei of the spinal motor neurons and do not colocalize with SQSTM1. Thus, protein homeostasis is perturbed and motor neurons may become susceptible to death. Because various ALS-FTD genes, including SQSTM1, OPTN, and TBK1, are involved in autophagy, autophagy dysfunction is pivotal in ALS-FTD pathogenesis.
Figure 1.
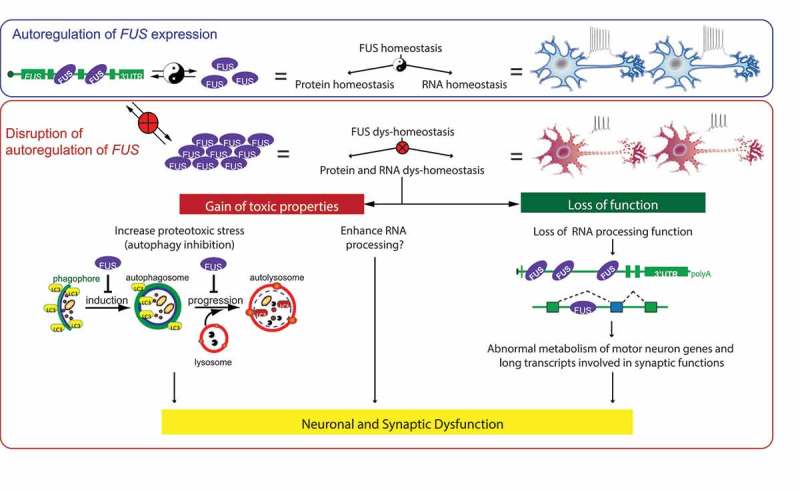
Proposed model of FUS-mediated neurodegeneration. FUS homeostasis is essential for maintaining both protein and RNA homeostasis. FUS protein binds directly to FUS mRNA. The FUS level is possibly maintained through nonsense-mediated decay and/or miRNA-mediated mechanisms. An elevated FUS level produces both gain-of-toxicity properties (by either increasing proteotoxic stress through autophagy inhibition or expression of stress genes), and loss of RNA-processing function affecting genes involved with long transcripts and synaptic regulation. Together these changes cause neuronal and synaptic dysfunction and eventual neuronal death.
The WT FUS transgene rescues the Fus-null lethality, suggesting that human FUS can functionally replace the mouse gene. Furthermore, the strong phenotype from overexpression of wild-type FUS does not correlate with FUS aggregation, where it mostly remains in the nucleus. These results suggest that the toxic cascade is initiated at the nucleus with dysregulation of RNA processing being one of the potential mechanisms. The counter-intuitive loss of RNA processing functions includes downregulation of genes with long introns (>100 kb per average intron length) that encode proteins essential for neuronal function and integrity (Figure 1). Genome-scale comparison of these transcriptomic alterations driven by FUS depletion or FUS overexpression are different. Therefore, it is likely that human FUS overexpression, but not the endogenous FUS reduction, is the core driver for the observed phenotypes. This suggests that FUS-mediated toxicity is primarily driven by a toxic gain of function.
In conclusion, protein expression in our mouse models resemble the FUS endogenous expression pattern and levels, and these mice develop an age-dependent and mutant-enhanced phenotype. Elevated FUS levels by overriding its autoregulation produce gain-of-toxicity properties that disrupt protein and RNA homeostasis. These disruptions are, at least in part, due to autophagy inhibition to cause proteotoxic stress and loss of normal RNA processing functions. Together they cause neuronal and synaptic dysfunction and eventual neuronal death (Figure 1). Thus, an optimal FUS level to maintain protein and RNA homeostasis is essential in neuronal function, and increasing FUS level exerts gain-of-toxicity effects.
Funding Statement
This work was supported by the Ministry of Education, Singapore [MOE2016-T2-1-024]; National Medical Research Council, Singapore [NMRC/OFIRG/0042/2017]; National Medical Research Council, Singapore [NMRC/OFIRG/0001/2016].
Acknowledgments
We thank Daniel Klionsky and Eddy Leman for editing the manuscript. This work was supported in part by grants to S.-C. L. from the National Medical Research Council (NMRC/OFIRG/0001/2016 and NMRC/OFIRG/0042/2017) and Ministry of Education (MOE2016-T2-1-024), Singapore.
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Ling S-C, Dastidar SG, Tokunaga S, et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. Elife. 2019;8:602. [DOI] [PMC free article] [PubMed] [Google Scholar]