

ABSTRACT
PHLPP1 (PH domain and leucine rich repeat protein phosphatase 1) is a newly identified family of Ser/Thr phosphatases that catalyzes the dephosphorylation of a conserved regulatory motif of the AGC kinases resulting in a tumor suppressive function, while CDKN1B/p27 also acts as a tumor suppressor by regulating cell cycle, senescence, apoptosis, and cell motility. Our most recent studies reveal that CDKN1B is required for PHLPP1 abundance, which contributes to the inhibition of carcinogenic arsenite-induced cell malignant transformation through inhibition of RPS6-mediated Hif1a translation. However, nothing is known about the mechanisms underlying the crosstalk between these 2 key tumor suppressors in intact cells. Here, for the first time to the best of our knowledge, we show that CDKN1B is able to promote PHLPP1 protein translation by attenuating the abundance of Mir6981, which binds directly to the 5ʹuntranslated region (UTR) of Phlpp1 mRNA. Further studies indicate that the attenuation of Mir6981 expression is due to macroautophagy/autophagy-mediated degradation of Mir6981 in an SQSTM1/p62-dependent fashion. Moreover, we have determined that Sqstm1 is upregulated by CDKN1B at the level of transcription via enhancing SP1 protein stability in an HSP90-depdendent manner. Collectively, our studies prove that: 1) SQSTM1 is a CDKN1B downstream effector responsible for CDKN1B-mediated autophagy; 2) by promoting the autophagy-mediated degradation of Mir6981, CDKN1B exerts a positive regulatory effect on PHLPP1 translation; 3) Mir6981 suppresses PHLPP1 translation by binding directly to its mRNA 5ʹ-UTR, rather than classical binding to the 3ʹ-UTR. These findings provide significant insight into understanding the crosstalk between CDKN1B and PHLPP1.
Abbreviations: ATG: autophagy related; ACTB: actin beta; BAF: bafilomycin; BECN1: beclin 1; Cdkn1b/p27: cyclin-dependent kinase inhibitor 1B; CHX: cycloheximide; DMEM: dulbecco’s modified eagle medium; FBS: fetal bovine serum; GAPDH: glyceraldehyde −3-phosphate dehydrogenase; Hif1a: hypoxia inducible factor 1, alpha subunit; Hsp90: heat shock protein 90; JUN: Jun proto-oncogene, AP1 transcription factor subunit; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MG132: proteasome inhibitor; Mtor: mechanistic target of rapamycin kinase; Phlpp1: PH domain and leucine rich repeat protein phosphatase 1; Phlpp2: PH domain and leucine rich repeat protein phosphatase 2; Pp2c: protein phosphatase 2 C; RPS6: ribosomal protein S6; Sp1: trans-acting transcription factor 1; Sqstm1/p62: sequestosome 1; TUBA: alpha tubulin; 3ʹ-UTR; 3ʹ-untranslated region; 5ʹ-UTR: 5ʹ-untranslated region.
KEYWORDS: Autophagy, Cdkn1b, Mir6981, Phlpp1, Sqstm1
Introduction
The pleckstrin homology domain leucine-rich repeat protein phosphatases (PHLPPs) are novel members of the protein phosphatase 2C (PP2C) group in the family of Ser/Thr phosphatase [1]. Recent studies from various groups indicate that PHLPP functions as a tumor suppressor by regulating the signaling pathways of AKT, PKC, and JUN [2]. CDKN1B/KIP1 is also known to be a tumor suppressor by virtue of its action as a cyclin-dependent kinase inhibitor in the regulation of cell cycle, apoptosis, tumor progression, and invasion [3]. Unlike other classic tumor suppressors such as TP53 and RB, CDKN1B nor PHLPP1 are rarely mutated or deleted in human cancers [4]. The crosstalk between these 2 tumor suppressors has never been explored. Recently, our lab has shown that the N terminus of CDKN1B mediates PHLPP1 expression responsible for the inhibition of HIF1A translation following arsenite exposure [5]. Here we verify that CDKN1B can promote autophagy-mediated Mir6981 degradation, which subsequently decreases its binding to 5ʹ-UTR of Phlpp1 mRNA, in turn increasing PHLPP1 translation.
miRNAs are small non-coding RNAs that play critical roles in a wide variety of biologic processes through interaction with partially complementary target sites in mRNAs [6]. Similar to other Argonaute-bound small RNAs, miRNAs also target mRNAs based on approximately 7 nt complementary base-pairing, preferentially at nucleotides 2 to 8 from the 5ʹ end of a mature miRNA, consequently resulting in the degradation or translational suppression of their targeted mRNAs [7]. The expression level of a mature miRNA is determined by the rate of its transcription, biogenesis processing, and turnover [8]. Even though transcription regulation accounts for most of the alterations in miRNA expression, in spite of the elevation of their primary transcripts and precursors, a significant portion of mature miRNAs are downregulated [9]. The posttranscriptional regulation of miRNA is through either maturation or degradation. In our current studies, we have identified a novel mechanism of Mir6981 alteration through autophagy by binding with the autophagy receptor SQSTM1. This Mir6981 affects its targeted Phlpp1 mRNA translation by binding to the 5ʹ-untranslated region (UTR) rather than the classical 3ʹ-UTR of Phlpp1 mRNA.
Although it has been thought that miRNAs mainly target mRNA 3ʹ-UTR (3ʹ untranslated region) and result in gene silencing via translational repression and/or RNA degradation [10], a recent study provides new insight into the functional roles of the 5ʹ-UTR in mRNA repression mediated by miRNAs [11]. On the one hand, the 5ʹ-UTR contains several regulatory elements, such as binding sites for RNA binding proteins and upstream open reading frames, which have a significant impact on the regulation of protein translation. On the other hand, the 5ʹ-UTR has structured RNAs near the 5ʹ cap site, which is sufficient to block translation initiation [12]. Here, we have discovered that Mir6981 is able to bind to the 5ʹ-UTR of Phlpp1 mRNA and repress PHLPP1 protein translation.
Autophagy is an evolutionarily conserved cell survival mechanism used by stressed cells to degrade the unwanted cytoplasmic proteins or organelles [13]. Autophagy is activated by metabolic stresses (such as starvation), infective pathogens and other specific substrates, as well as mediated by a specific autophagy receptor to degrade targeted protein aggregates [14]. In mammalian cells, SQSTM1 has been identified as the first autophagy receptor and acts as a scaffold for the intracellular signaling that control various cell functions [15–18]. A specific region called LIR/LRS (LC3-interacting region/LC3-recognition sequence) of mouse Sqstm1 [19] enables the SQSTM1 targeted proteins to be recognized by the phagophore through LC3 [20]. In the current study, we reveal that CDKN1B is able to promote Sqstm1 transcription through substantially activating SP1, and increasing SQSTM1 binding to Mir6981, resulting in autophagy-dependent Mir6981 degradation, in turn attenuating Mir6981 inhibition of PHLPP1 translation. Our results, for the first time, to the best of our knowledge, highlight the novel crosstalk between 2 tumor suppressors, CDKN1B and PHLPP1.
Results
CDKN1B specifically promoted PHLPP1 protein translation in intact cells
CDKN1B is known to be a tumor suppressor as the result of its action as a cyclin-dependent kinase inhibitor for regulating the cell cycle and growth [21]. cdkn1b(Δ51) corresponds to MEFs that have been derived from an embryo with a disrupted Cdkn1b gene, encoding amino acids 52 to 198 of the CDKN1B protein, as described in Dr. Andrew Koff’s published paper [22]. The cdkn1b−/- MEFs harboring deletion of the entire coding region of mouse Cdkn1b were the generous gift of Dr. James Roberts [23]. cdkn1b(Δ51) and Cdkn1b+/+ cells were first subjected to cell cycle and cell proliferation assays to validate the behavior of the cell lines. As shown in Figure S1(a), the deletion of Cdkn1b in cdkn1b(Δ51) cells increased the proportion of cells in S phase and G2/M phase, and reduced the G0/G1 phase, in comparison to those in the Cdkn1b+/+ cells. Consistently, cell proliferation ability in cdkn1b(Δ51) cells was also elevated in comparison to Cdkn1b+/+cells (Figure S1(b)). We next explored the mechanisms of CDKN1B regulation of PHLPP1 by employing Cdkn1b+/+ and cdkn1b(Δ51) cells. As expected, knockout of Cdkn1b almost completely abolished PHLPP1 protein expression in comparison to Cdkn1b+/+ cells (Figure 1(a)), suggesting that CDKN1B positively regulates PHLPP1 protein expression. This notion was greatly supported by the data obtained from knockdown of Cdkn1b using Cdkn1b shRNA in both Cdkn1b+/+ cells (Figure 1(b)) and mouse epithelial Cl41 cells (Figure 1(c)). Consistently, reintroduction of GFP-CDKN1B into cdkn1b(Δ51) cells rescued the PHLPP1 protein level (Figure 1(d)). Given the results strongly indicating that CDKN1B is a positive regulator for PHLPP1 expression, the Phlpp1 mRNA levels were evaluated and compared between Cdkn1b+/+ and cdkn1b(Δ51) cells. The results showed that Phlpp1 mRNA levels are comparable in Cdkn1b+/+ vs. cdkn1b(Δ51) cells (Figure 1(e)), and Cdkn1b+/+(shCdkn1b) vs. Cdkn1b+/+(Nonsense) cells (Figure 1(f)). These results suggest that PHLPP1 might be regulated by CDKN1B through either protein degradation or protein translation. To explore this notion, MG132 and calpain inhibitors were used to test their effects on PHLPP1 protein accumulation in both Cdkn1b+/+ and cdkn1b(Δ51) cells. The results indicate that treatment of cells with either the calpain inhibitor or MG132 only cause PHLPP1 protein accumulation in Cdkn1b+/+ cells, but not in cdkn1b(Δ51) cells (Figure 1(g,h)), providing a strong indication that CDKN1B promotes PHLPP1 protein expression through a protein degradation-independent mechanism.
Figure 1.
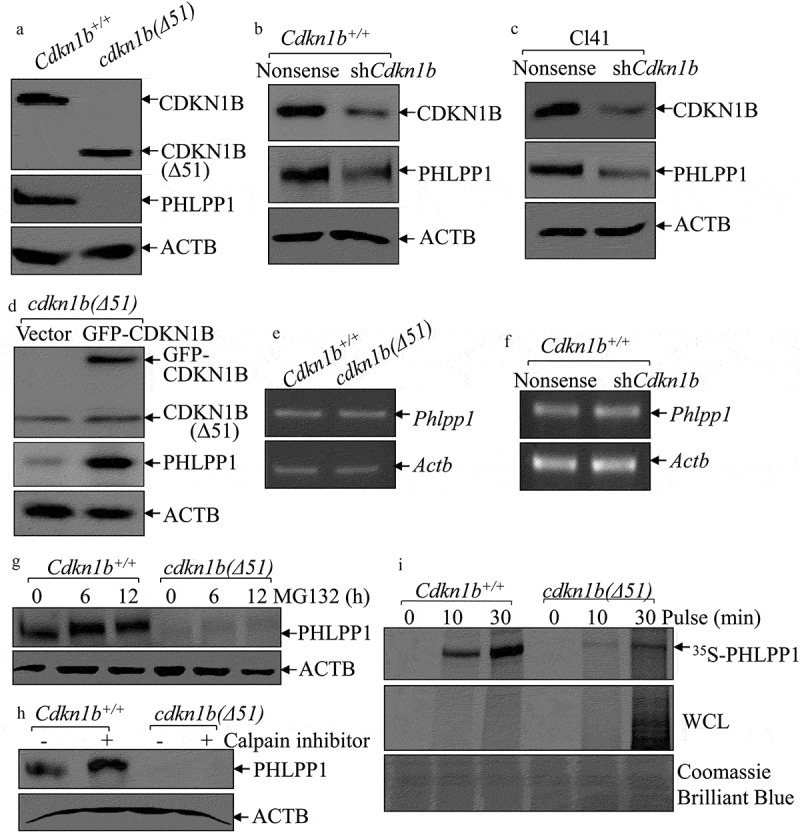
CDKN1B promoted PHLPP1 expression at protein translational level. (a–d) Western blots (WB) were used to determine the protein levels of PHLPP1 in Cdkn1b+/+ vs. cdkn1b(Δ51) cells (a), Cdkn1b+/+(Nonsense) vs. Cdkn1b+/+(shCdkn1b) (b), Cl41(Nonsense) vs. Cl41(shCdkn1b) (c), and cdkn1b(Δ51)(Vector) vs. cdkn1b−/-(Δ51)(GFP-CDKN1B) (d). ACTB was used as a protein loading control. (e,f) RT-PCR was applied to compare the Phlpp1 mRNA levels in Cdkn1b+/+ vs. cdkn1b(Δ51) (e) and Cdkn1b+/+(Nonsense) vs. Cdkn1b+/+(shCdkn1b) (f). Actb was used as an internal loading control. (g) Cdkn1b+/+ and cdkn1b(Δ51) cells were treated with MG132 for the indicated times. The cell extracts were then subjected to western blots to determine PHLPP1 protein accumulation. ACTB was used as a protein loading control. (h) Cdkn1b+/+ and cdkn1b(Δ51) cells were treated with Calpain inhibitor for 24 h. The cell extracts were then subjected to western blot analyses of the new PHLPP1 protein accumulation. ACTB was used as a protein loading control. (i) Newly synthesized PHLPP1 protein in Cdkn1b+/+ and cdkn1b(Δ51) cells was monitored by 35S-labeled methionine/cysteine pulse assay, as described in the section of ‘Materials and Methods’. WCL, whole cell lysate. Coomassie Brilliant Blue staining was used as a protein loading control.
To explore the potential effect of CDKN1B on PHLPP1 protein translation, an 35S-methionine/cysteine pulse labeling assay was performed to monitor PHLPP1 new protein translation in Cdkn1b+/+ and cdkn1b(Δ51) cells. As shown in Figure 1(i), the new PHLPP1 protein synthesizing rate in cdkn1b(Δ51) cells was remarkably impaired in comparison to Cdkn1b+/+ cells, although total new protein syntheses (WCL) were higher in cdkn1b(Δ51) cells than that in Cdkn1b+/+ cells. Our results reveal that CDKN1B is required for PHLPP1 protein translation.
Downregulation of the Mir6981 is responsible for CDKN1B promotion of PHLPP1 protein translation via reduction of its binding to 5ʹ-UTR, but not 3ʹUTR, of Phlpp1 mRNA.
The 3ʹ-UTR or 5ʹ-UTR of mRNA are major regulatory regions that are targeted by either miRNAs or mRNA binding proteins to regulate mRNA stability or protein translation [10,12]. To evaluate the potential contribution of Phlpp1 mRNA 3ʹ-UTR in CDKN1B’s promotion of PHLPP1 protein translation, we first constructed the Phlpp1 3ʹ-UTR-luciferase reporter then stably transfected it into Cdkn1b+/+ and cdkn1b(Δ51) cells. As shown in Figure 2(a), the Phlpp1 3ʹ-UTR activity was remarkably upregulated in cdkn1b(Δ51) cells (Figure 2(a)), revealing that downregulation of PHLPP1 protein translation in cdkn1b(Δ51) cells is not attributable to 3ʹ-UTR region of Phlpp1 mRNA. We next tested whether 5ʹ-UTR of Phlpp1 mRNA was involved in CDKN1B promotion of PHLPP1 protein translation. Following construction of the Phlpp1 mRNA 5ʹ-UTR-luciferase reporter, it was stably transfected into Cdkn1b+/+ and cdkn1b(Δ51) cells. As shown in Figure 2(b), the deletion of Cdkn1b resulted in a profound reduction of Phlpp1 mRNA 5ʹ-UTR activity in comparison to Cdkn1b+/+ cells, suggesting that the 5ʹ-UTR of Phlpp1 mRNA might be implicated in CDKN1B promotion of PHLPP1 protein translation. These results indicate that CDKN1B promotes PHLPP1 protein expression with upregulation of Phlpp1 mRNA 5ʹ-UTR activity as well as downregulation of Phlpp1 mRNA 3ʹ-UTR activity. To evaluate the effect of CDKN1B on the overall activity of Phlpp1 mRNA 3ʹ-UTR and 5ʹ-UTR, we constructed a Phlpp1 mRNA luciferase reporter that contains both 5ʹ-UTR and 3ʹ-UTR regulatory regions. The results indicate that CDKN1B does have a positive regulatory effect on Phlpp1 mRNA luciferase reporter under the control of both the 3ʹ-UTR and the 5ʹ-UTR regions (Figure 2(c)), indicating that CDKN1B positive regulation of Phlpp1 mRNA 5ʹ-UTR activity overcomes its negative modulation of Phlpp1 mRNA 3ʹ-UTR activity.
Figure 2.
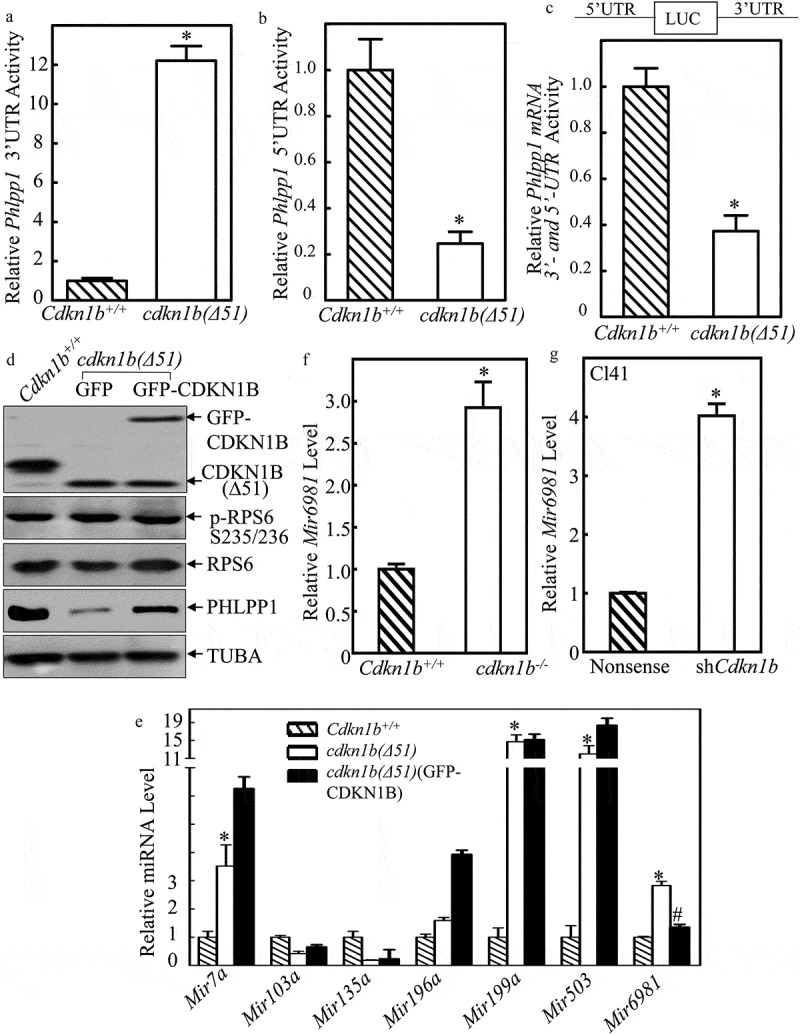
CDKN1B promoted Phlpp1 mRNA 5ʹ-UTR activity accompanied with inhibition of Mir6981 expression. (a–c) Wild-type Phlpp1 mRNA 3ʹ-UTR-driven luciferase reporters(a), 5ʹ-UTR-driven luciferase reporters (b), The luciferase reporter containing both the 5ʹ-UTR and 3ʹ-UTR regulatory regions (c), were cotransfected with pRL-TK into Cdkn1b+/+ and cdkn1b(Δ51) cells, respectively. Twenty-four h post transfection, the transfectants were extracted for evaluation of the luciferase activity. TK was used as an internal control. The results were presented as Phlpp1 3ʹ-UTR activity (a), 5ʹ-UTR activity (b), or Phlpp1 mRNA 3ʹ and 5ʹ-UTR activity (c) relative to Cdkn1b+/+ cells. Each bar indicates a mean±SD of triplicates. The symbol (*) indicates a significant difference in comparison to Cdkn1b+/+ cells (P < 0.05). (d) Cell extracts from Cdkn1b+/+, cdkn1b(Δ51)(GFP), and cdkn1b(Δ51)(GFP-CDKN1B) cells were subjected to western blot analysis for protein expression of CDKN1B, P-RPS6, RPS6, and PHLPP1. TUBA was used as a protein loading control. (e) For the cells indicated, the expression levels of Mir7a, Mir103a, Mir135a, Mir196a, Mir199a, Mir503, and Mir6981 were evaluated by real-time PCR. The results were normalized to Rnu6. The symbol (*) indicates a significant increase in comparison to Cdkn1b+/+ cells, while the symbol ‘#’ indicates significant inhibition in comparison to cdkn1b(Δ51) cells (P < 0.05). (f,g) The expression levels of Mir6981 were evaluated by real-time PCR in Cdkn1b+/+ vs. cdkn1b−/- cells (f), and Cl41(Nonsense) vs. Cl41(shCdkn1b) cells (g). The results were normalized to Rnu6. Each bar indicates a mean±SD of triplicates. The symbol (*) indicates a significant difference in comparison to Cdkn1b+/+ cells or Cl41(Nonsense) cells (P < 0.05).
It is well-characterized that RPS6 (ribosomal protein S6) activation is essential for initiation of canonical protein translation [24]. To test whether RPS6 is involved in CDKN1B regulation of PHLPP1 protein translation, the effects of CDKN1B on activation of RPS6 were evaluated in Cdkn1b+/+, cdkn1b(Δ51)(GFP) and cdkn1b(Δ51)(GFP-CDKN1B) cells. As shown in Figure 2(d), there was no observable alteration of RPS6 phosphorylation among the 3 cells, whereas Cdkn1b deletion resulted in a dramatic inhibition of PHLPP1 protein abundance. Moreover, the PHLPP1 protein defect can be rescued by constitutively expressing GFP-CDKN1B into cdkn1b(Δ51) cells. These results suggest that RPS6 activation might not be involved in CDKN1B promotion of PHLPP1 protein translation.
Therefore, we next sought to determine whether miRNA could be a CDKN1B downstream effector for its regulation of Phlpp1 mRNA 5ʹ-UTR activity. The results obtained from the bio-informatics analysis for the putative miRNA binding sites indicate that 5ʹ-UTR region of Phlpp1 mRNA contains the multiple miRNA binding sites, including Mir7a, Mir103a, Mir135a, Mir196a, Mir199a, Mir503a, and Mir6981 (Table 1). To define the specific miRNA that might be involved in CDKN1B promotion of Phlpp1 mRNA 5ʹ-UTR activity, the listed miRNA expression levels were determined in Cdkn1b+/+, cdkn1b(Δ51)(GFP), and cdkn1b(Δ51)(GFP-CDKN1B) cells which have recovered the CDKN1B expression. As shown in Figure 2(e), the expression levels of Mir7a, Mir199a, Mir503, and Mir6981, but not Mir103a, Mir135a, and Mir196a, were significantly elevated in cdkn1b(Δ51) cells in comparison to Cdkn1b+/+ cells. By ectopic expression of GFP-CDKN1B in cdkn1b(Δ51) cells, we discovered that only expression of Mir6981 could be inhibited by overexpression of GFP-CDKN1B in cdkn1b(Δ51) cells (Figure 2(e)), revealing that the inhibition of Mir6981 might contribute to CDKN1B promotion of PHLPP1 protein translation by targeting its mRNA 5ʹ-UTR region. This notion was further supported by the results obtained from utilization of Cdkn1b+/+ and cdkn1b−/- cells (Figure 2(f)) and knockdown of Cdkn1b in Cl41 cells (Figure 2(g)).
Table 1.
Sequence alignments of potential miRNAs with the Phlpp1 5ʹUTR.
| Phlpp1-5ʹUTR: 5ʹ…(44)CGCCGCCGCCGCCGCCGUCUCCCA (67)… |
| | | | | | | | |
| mmu-Mir7a: 3ʹ …UGUUGUUUUAGUGAUCAGAAGGU |
| Phlpp1-5ʹUTR: 5ʹ …(75)CUCACCUCCUCCUCCUAUGCUGC (97)… |
| | | | | | | | |
| mmu-Mir103a: 3ʹ AGUAUCGGGACAUGUUACGACGA |
| Phlpp1-5ʹUTR: 5ʹ …..(3)GAGCGGCCGCGCCGAACGCCAU (24)… |
| | | | | | | | |
| mmu-Mir135a: 3ʹ …..AGUGUAUCCUUAUUUUUCGGUAU |
| Phlpp1-5ʹUTR: 5ʹ …(79)CCUCCUCCUCCUCCACUACCU (99)… |
| | | | | | | | |
| mmu-Mir196a: 3ʹ ..GGGUUGUUGUACUUUGAUGGAU |
| Phlpp1-5ʹUTR: 5ʹ…(88) CCUCCGCUGCCUCCGGCACUGGG (110)… |
| | | | | | | | |
| mmu-Mir199a: 3ʹ CUUGUCCAUCAGACUUGUGACCC |
| Phlpp1-5ʹUTR: 5ʹ ….(111)GGAGCGACGUGAAGCCCTGCUA (132)… |
| | | | | | | | |
| mmu-Mir503: 3ʹ ….GACGUCUUGACAAGGGCGACGAU |
| Phlpp1-5ʹUTR: 5ʹ….(56) CGCCGUCUCCCACCUUCGCCUCAC (79)… |
| | | | | | | | |
| mmu-Mir6981: 3ʹ CGGAAGUCGGAGAAGGAAGAGGAGUG |
To define the role of Mir6981 in attenuating PHLPP1 translation in both Cdkn1b+/+ and cdkn1b(Δ51) cells, we stably transfected Mir6981 into Cdkn1b+/+ cells and Mir6981 inhibitor (Inh) into cdkn1b(Δ51) cells. The results indicated that the Mir6981 transfectant did exhibit overexpression, while Mir6981 Inh inhibited Mir6981 expression in comparison to their scrambled control vector transfectants (Figure 3(a)). Consistently, overexpression of Mir6981 in Cdkn1b+/+ cells profoundly inhibited Phlpp1 mRNA 5ʹ-UTR activity and attenuated PHLPP1 protein expression (Figure 3(b,c)), whereas ectopic expression of Mir6981 inhibitor significantly increased Phlpp1 mRNA 5ʹ-UTR activity, along with elevating PHLPP1 protein expression in cdkn1b(Δ51) cells (Figure 3(b,d)). Moreover, inhibition of Phlpp1 mRNA 5ʹ-UTR activity by Mir6981 in Cdkn1b+/+ cells was not observed in the Cdkn1b+/+ cells stably transfected with the Mir6981 binding site point-mutated Phlpp1 mRNA 5ʹ-UTR-luciferase reporter in comparison to that in Cdkn1b+/+ cells stably transfected with wild-type Phlpp1 mRNA 5ʹ-UTR-luciferase reporter (WT) (Figure 3(e,f)). Collectively, our results verify that upregulated Mir6981 in cdkn1b(Δ51) cells is able to inhibit PHLPP1 protein translation by directly targeting its binding site in Phlpp1 mRNA 5ʹUTR region, rather than targeting Phlpp1 mRNA 3ʹ-UTR region.
Figure 3.
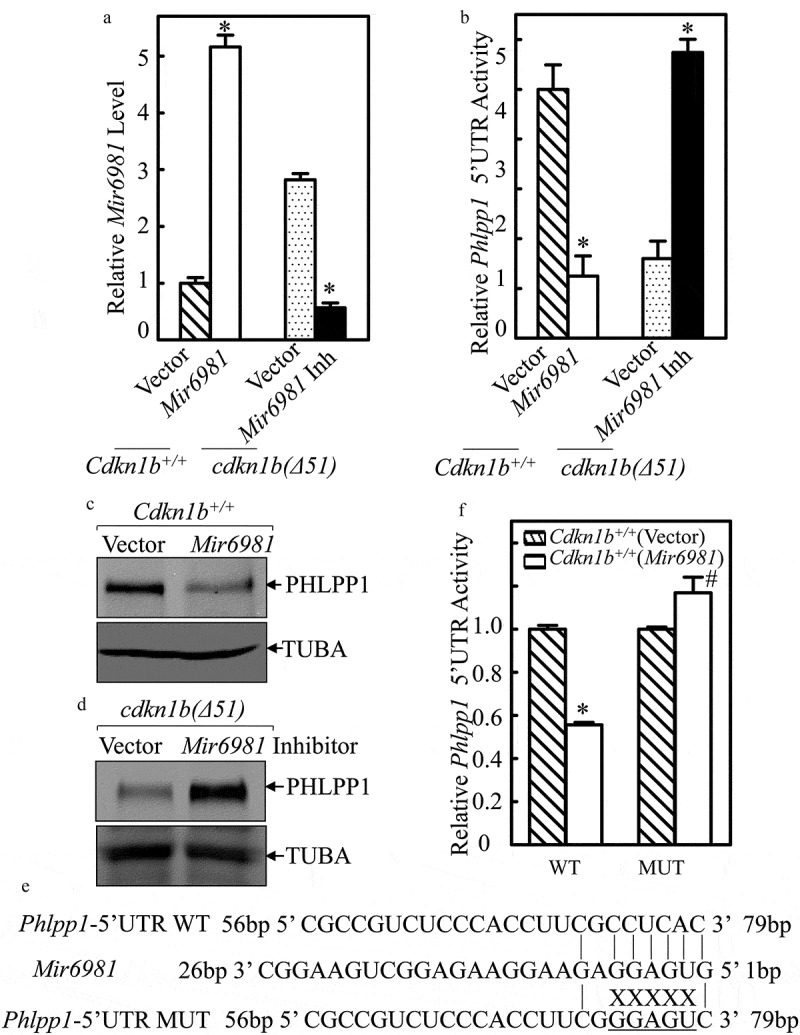
The Mir6981 binding site in 5ʹ-UTR of Phlpp1 mRNA was required for Mir6981 inhibition of Phlpp1 5ʹ-UTR activity and protein expression. (a) Real-time PCR was employed to identify the expression levels of Mir6981 in Cdkn1b+/+ cells with overexpressed Mir6981 and cdkn1b(Δ51) cells stably transfected with Mir6981 inhibitor. (b) Wild-type Phlpp1 mRNA 5ʹ-UTR-driven luciferase reporters were cotransfected with pRL-TK into Cdkn1b+/+ vs. Cdkn1b+/+(Mir6981) cells and cdkn1b(Δ51) vs. cdkn1b(Δ51) (Mir6981 inhibitor) cells, respectively. Twenty-four h post transfection, the transfectants were extracted to evaluate the luciferase activity. TK was used as an internal control. The results were presented as Phlpp1 5ʹ-UTR activity relative to scrambled control vector transfectants. The symbol (*) indicates a significant difference (P < 0.05). (c,d) The protein levels of PHLPP1 in Cdkn1b+/+ cells with overexpressed Mir6981 (c) and cdkn1b(Δ51) cells stably transfected with Mir6981 inhibitor (d) were determined by western blots with TUBA as a loading control. (e) The illustration of Mir6981 binding to the Phlpp1 mRNA 5ʹ-UTR region and Mir6981 binding site mutation. (f) Wild type and mutant of Phlpp1 mRNA 5ʹ-UTR-driven luciferase reporters were co-transfected with pRL-TK into Cdkn1b+/+ and Cdkn1b+/+(Mir6981) cells. Twenty-four h post transfection, the transfectants were extracted to determine the luciferase activity. TK was used as an internal control. The results are presented as Phlpp1 5ʹ-UTR activity relative to Cdkn1b+/+(vector) cells. The symbol (*) indicates a significant inhibition in comparison to Cdkn1b+/+(vector) cells (P < 0.05).
CDKN1B promoted Mir6981 degradation through upregulation of Sqstm1-mediated autophagy
The miRNA can be transcribed either along with its host gene under the control of its host gene promoter or it can be transcribed under control of its own promoter [9]. Using the NCBI gene database, Arap3 (ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 3) was identified as the host gene for Mir6981, as shown in Figure 4(a). To elucidate the mechanisms of CDKN1B modulation of Mir6981 expression, Arap3 mRNA expression was determined in Cdkn1b+/+ and cdkn1b(Δ51) cells. The result indicate that Arap3 mRNA levels in cdkn1b(Δ51) cells and cdkn1b−/- cells were significantly attenuated in comparison to Cdkn1b+/+ cells (Figure 4(b,c)). However, this is inconsistent with the elevation of Mir6981 expression in cdkn1b(Δ51) cells; thereby excluding the possibility that CDKN1B regulates Mir6981 transcription. This notion is further supported by the results showing that pre-Mir6981 was significantly impaired in both cdkn1b(Δ51) and cdkn1b−/- cells, in comparison to those observed in their corresponding Cdkn1b+/+ cells (Figure. 4(d,e)). Thus, we next tested the potential effect of CDKN1B on Mir6981 degradation. ACTD was used to block new miRNA transcription to evaluate Mir6981 degradation rates in both Cdkn1b+/+ and cdkn1b(Δ51) cells. The results indicate that Mir6981 degradation in cdkn1b(Δ51) cells is retarded in comparison to Cdkn1b+/+ cells (Figure 4(f)), providing significant evidence that CDKN1B promotes Mir6981 degradation.
Figure 4.
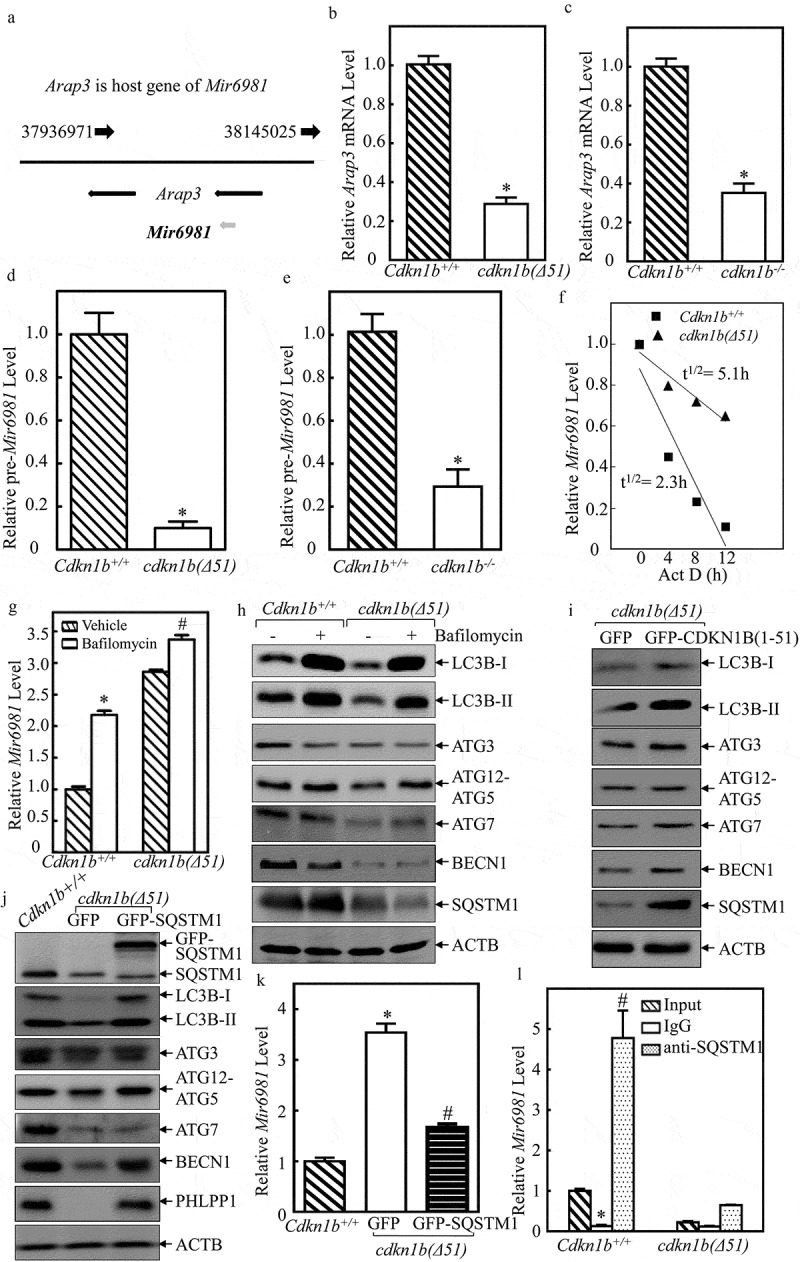
Mir6981 was degraded through a CDKN1B-mediated autophagic mechanism in SQSTM1-dependent manner. (a) The illustration of Arap3 as the host gene of Mir6981. (b,c) RT-PCR was employed to compare the mRNA levels of Arap3 in Cdkn1b+/+ vs. cdkn1b(Δ51) cells (b) and Cdkn1b+/+ vs. cdkn1b−/- cells (c), respectively. Actb was used as a loading control. (d,e) The expression levels pre-Mir6981 were evaluated by real-time PCR in Cdkn1b+/+ vs. cdkn1b(Δ51) cells (d) and Cdkn1b+/+ vs. cdkn1b−/- cells (e), respectively. The symbol (*) indicates significant inhibition (P < 0.05). (f) Mir6981 stabilities were determined by real-time PCR in the presence of actinomycin D in Cdkn1b+/+ and cdkn1b(Δ51) cells. (g) Real-time PCR was employed to evaluate the expression level of Mir6981 in Cdkn1b+/+ cells and cdkn1b(Δ51) cells in the presence or absence of BAF treatment. The symbol (*) indicates a significant increase in comparison to the vehicle control (P < 0.05). (h) Western blots were employed to evaluate the indicated protein expression in Cdkn1b+/+ cells and cdkn1b(Δ51) cells in the presence or absence of BAF treatment. (i,j) Western blots were employed to determine the indicated protein expressions in cdkn1b(Δ51) (Vector) vs. cdkn1b(Δ51)(GFP-CDKN1B) cells (i) or in Cdkn1b+/+ vs. cdkn1b(Δ51)(GFP) cells vs. cdkn1b(Δ51)(GFP-SQSTM1) cells (j). ACTB was used as a protein loading control. (k) The expression levels of Mir6981 in Cdkn1b+/+, cdkn1b(Δ51)(GFP) cells, and cdkn1b(Δ51)(GFP-SQSTM1) cells were determined by real-time PCR. (l) An RNA-IP assay was employed to evaluate the potential interaction between SQSTM1 protein and Mir6981 in Cdkn1b+/+ cells and cdkn1b(Δ51) cells. Real-time PCR was employed to determine Mir6981 levels in the immuno-complex affinity-isolation by anti-SQSTM1 antibody.
To the best of our knowledge, there have been very few reports about the mechanisms underlying the regulation of miRNA degradation by autophagy. To explore the potential involvement of autophagy in the degradation of Mir6981, we employed bafilomycin (BAF), an autophagy inhibitor. As shown in Figure 4(g,h), the treatment of cells with BAF led to a dramatic inhibition of autophagy, together with a remarkable elevation of Mir6981 expression in Cdkn1b+/+ cells, suggesting that autophagic response is involved in the regulation of Mir6981 stability. Consistently, cdkn1b(Δ51) cells showed a reduction of autophagy (Figure 4(h)) and an increase in Mir6981 levels in the absence of BAF treatment (Figure 4(g)). It was interesting to note that BAF treatment showed less effect on elevation of Mir6981 in cdkn1b−/-(Δ51) cells in comparison to Cdkn1b+/+ cells (Figure 4(g)). Possible reason that BAF shows less effect on autophagic activity in cdkn1b(Δ51) cells is because there already is a low basal level of autophagy in cdkn1b(Δ51) cells. Our results reveal that CDKN1B is critical for Mir6981 degradation through an autophagy-dependent mechanism. We next evaluated whether CDKN1B expression participated in autophagic response in intact cells. The results indicate that although Cdkn1b deletion exhibited a reduction of cell autophagy along with inhibition of protein expression of ATG3, ATG12–ATG5, ATG7, BECN1, and SQSTM1, ectopic restoration of GFP-CDKN1B in cdkn1b(Δ51) cells resulted in an increase in cell autophagy with a marked induction of only SQSTM1 and BECN1 (Figures 4(h,i) and S2), indicating that CDKN1B expression is positively related to expression of SQSTM1 and BECN1, which might contribute to the autophagic regulation of Mir6981 degradation. To test the potential contribution of SQSTM1 to CDKN1B-regulated Mir6981, GFP-SQSTM1 was stably transfected into cdkn1b(Δ51) cells. The results show that overexpression of SQSTM1 led to a profound increase in LC3B-II level and BECN1 expression, as well as PHLPP1 expression (Figure 4(j)), revealing that SQSTM1 is an upstream regulator for induction of BECN1 and autophagy. Our next step was to evaluate the possible regulatory effect of SQSTM1 on Mir6981 degradation in cdkn1b(Δ51) (GFP-SQSTM1) cells in comparison to cdkn1b(Δ51) (GFP) cells. As exhibited in Figure 4(k), ectopic expression of GFP-SQSTM1 in cdkn1b(Δ51) showed that there is a significant inhibition of Mir6981 expression. To assess the role of CDKN1B-regulated BECN1 and ATG7 in CDKN1B-mediated autophagy and PHLPP1 protein expression, we overexpressed GFP-ATG7 and GFP-BECN1, into cdkn1b(Δ51) cells. The results show that overexpression of ATG7 does not have any observable regulation of autophagy and PHLPP1 levels, whereas GFP-BECN1 overexpression has only a slight effect (Figure S3(a,b)). Moreover, the results obtained from RNA-IP assay indicate that SQSTM1 has a strong binding activity to Mir6981 in the immune-complex that was pulled down using anti-SQSTM1 antibodies (Figure 4(l)).
SP1 was a downstream transcription factor responsible for CDKN1B-regulated Sqstm1 transcription
To elucidate the mechanism of CDKN1B’s promotion of SQSTM1, Sqstm1 mRNA was first analyzed in Cdkn1b+/+ and cdkn1b(Δ51) cells. As shown in Figure 5(a), Sqstm1 mRNA was attenuated in cdkn1b(Δ51) cells. To determine whether CDKN1B modulates Sqstm1 at transcription level, the full length of the Sqstm1 promoter-driven luciferase reporters together with 3 deletions, as shown in Figure 5(b), were constructed and transfected into Cdkn1b+/+ cells and cdkn1b(Δ51) cells, respectively. The deletion of the Sqstm1 promoter region was analyzed for its potential transcription factor binding sites (listed in Figure 5(b)). As observed in Figure 5(c), both the full length and 3 deletions of Sqstm1 promoter-driven luciferase reporter showed significantly similar patterns of inhibition of promoter transcription activities in cdkn1b(Δ51) cells in comparison to the patterns that were seen in Cdkn1b+/+ cells. Based on this evidence, we anticipated that the transcription factor binding site responsible for CDKN1B promotion of Sqstm1 transcription might be in the −935 bp reporter. Our next step was to evaluate the effect of CDKN1B on the expression of the transcription factors that might potentially bind to this −935 bp region. We found that expression of E2F1, JUNB, p-JUN, JUN, and SP1 were impaired in cdkn1b(Δ51) cells, while ELK1 was comparable between the 2 cells (Figures 5(d) and S4(a)). Given that JUNB has been most commonly reported to inhibit its regulated gene transcription [25], JUN and SP1 were first analyzed for their potential effects on SQSTM1 expression. As shown in Figure 5(e,f) and S4(b,c), inhibition of JUN activation by ectopic expression of a dominant negative mutant of JUN (TAM67) slightly increased SQSTM1 expression, while knockdown of Sp1 led to a dramatic decrease in SQSTM1 expression in Cdkn1b+/+ cells, indicating that SP1, but not JUN, has a role in SQSTM1 expression in Cdkn1b+/+ cells. This notion is supported by the results showing that ectopic expression of SP1 increased SQSTM1 protein levels in cdkn1b(Δ51) cells (Figures 5(g) and S4(d)). Moreover, the results obtained from Sqstm1 promoter-driven luciferase reporter assay reveal that CDKN1B expression inhibits Sqstm1 promoter-driven luciferase reporter activity, whereas this inhibition by CDKN1B was completely abolished if the SP1 binding site in Sqstm1 promoter-driven luciferase reporter was point mutated (Figure 5(h,i)). In addition, the results from the CHIP assay reveal that SP1 can directly bind to the Sqstm1 promoter at the putative Sp1 binding site at −47 bp (Figure 5(j)). Taken together, these results provide strong evidence that SP1 is specifically regulated by CDKN1B, which in turn, activates Sqstm1 transcription and increases its protein expression.
Figure 5.
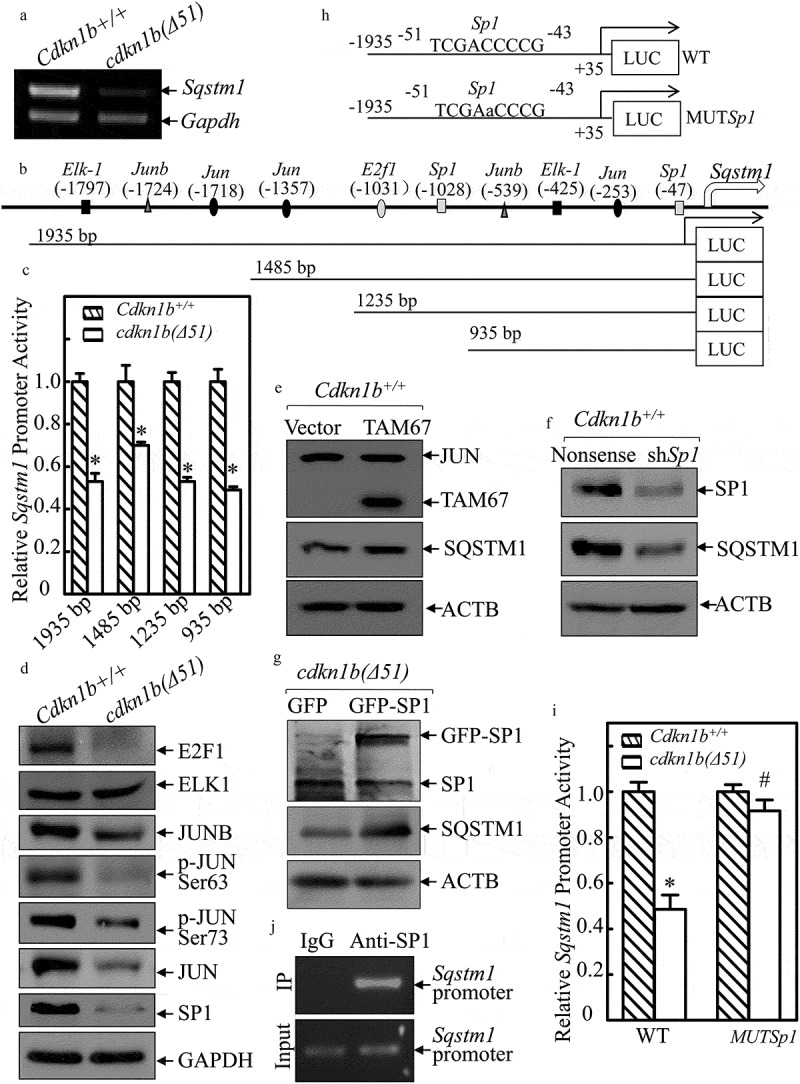
CDKN1B promoted Sqstm1 mRNA transcription specifically by elevating SP1 expression. (a) RT-PCR was applied to evaluate Sqstm1 mRNA levels in Cdkn1b+/+ and cdkn1b(Δ51) cells. (b) The various deletions of mouse Sqstm1 promoter-driven luciferase reporters were constructed and the putative transcription factor binding sites were analyzed using the TRANSFAC 8.3 engine online. (c) Various deletions of mouse Sqstm1 promoter luciferase reporters were cotransfected together with pRL-TK into Cdkn1b+/+ and cdkn1b(Δ51) cells. Twenty-four h post transfection, the transfectants were extracted to evaluate the luciferase activity. TK was used as an internal control. The results are presented as Sqstm1 promoter activity in cdkn1b(Δ51) cells relative to Cdkn1b+/+ cells. Each bar indicates a mean ± SD from 3 independent experiments. The symbol (*) indicates a significant reduction (P < 0.05). (d–g) The cell extracts from the indicated cells and their transfectants were subjected to western blots to evaluate protein expression using specific antibodies. Either GAPDH or ACTB were used as protein loading controls. (h) The diagram of the WT-Sqstm1 promoter-driven luciferase reporter and its mutation at the SP1 binding site at −47 bp. (i) Both WT- and the mutated forms of the Sqstm1 promoter luciferase reporters shown in Figure 5H were transiently cotransfected with pRL-TK into Cdkn1b+/+ and cdkn1b(Δ51) cells. Twenty-four h post-transfection, the transfectants were extracted to evaluate the luciferase activity. TK was used as an internal control. The results are presented as Sqstm1 promoter activity in cdkn1b(Δ51) cells relative to Cdkn1b+/+ cells. The symbol (*) indicates a significant decrease in comparison to Cdkn1b+/+ cells (P < 0.05). (j) ChIP assay was employed to evaluate SP1 binding to the Sqstm1 promoter region using anti-SP1 antibody.
Upregulated HSP90 mediated CDKN1B stabilization of SP1 protein
To explore the mechanism underlying SP1 regulation by CDKN1B, we evaluated Sp1 mRNA levels in Cdkn1b+/+ and cdkn1b(Δ51) cells and found that Sp1 mRNA expression was comparable between these 2 cells (Figure 6(a)), demonstrating that SP1 is regulated by CDKN1B, either through protein stability or protein translation. To test this, CHX was utilized to inhibit new protein synthesis to evaluate SP1 protein degradation rates between Cdkn1b+/+ and cdkn1b(Δ51) cells. As shown in Figure 6(b), SP1 degradation in cdkn1b(Δ51) cells was higher than that in Cdkn1b+/+ cells, indicating that CDKN1B might have a function in the stabilization of the SP1 protein. Because HSP90 can bind and stabilize SP1 protein [26], we analyzed HSP90 levels in Cdkn1b+/+ and cdkn1b(Δ51) cells and found that the protein abundance of HSP90 was significantly attenuated in cdkn1b(∆51) cells in comparison to the amount in Cdkn1b+/+ cells (Figure 6(c)). To further evaluate the contribution of HSP90 to the maintenance of SP1 stability, we introduced HSP90 and GFP-CDKN1B ectopic-expressing constructs into cdkn1b(∆51) cells. While GFP-CDKN1B construct was overexpressed in cdkn1b(∆51) to observe its effect on its downstream molecules and to determine whether GFP-CDKN1B restores HSP90 abundance. As shown in Figure 6(d), overexpression of GFP-CDKN1B in cdkn1b(∆51) cells restores HSP90 expression, while either ectopic expression of HSP90 or GFP-CDKN1B rescued protein expression of SP1, SQSTM1, LC3B, and PHLPP1 protein (Figure 6(d)), revealing that CDKN1B plays an essential role in maintaining these protein expressions. This is supported by the results obtained from the cdkn1b−/- and Cl41 (shCdkn1b) cells (Figure 6(e,f)). It is noted that although GFP-CDKN1B protein expression in cdkn1b(∆51)(GFP-CDKN1B) cells is relatively lower than that in Cdkn1b+/+ cells. However, this GFP-CDKN1B seems to be sufficient to rescue the levels of HSP90, SP1, SQSTM1, LC3B, and PHLPP1 proteins to levels similar to those observed in Cdkn1b+/+ cells (Figure 6(d)). It could be possible that the introduced GFP-CDKN1B is able to bring up the CDKN1B level in cdkn1b(∆51) cells to the threshold level that is necessary to activate the HSP90-SP1-SQSTM1 axis. Consistently, compared to its vector control transfectant, cdkn1b(∆51)(Vector), SP1 protein was more stable in cdkn1b(Δ51) (HSP90) cells with overexpressed HSP90 in cdkn1b(Δ51) cells (Figure 6(g)), suggesting that the increased SP1 protein degradation in Cdkn1b knockout cells could be stabilized by ectopic expression of HSP90. The results from coimmunoprecipitation assays also indicated that HSP90 interacts directly with SP1 protein in Cdkn1b+/+ cells (Figure 6(h)). Collectively, our results provide clear, direct evidence revealing that upregulated HSP90 is responsible for CDKN1B promotion of SP1 protein stability.
Figure 6.
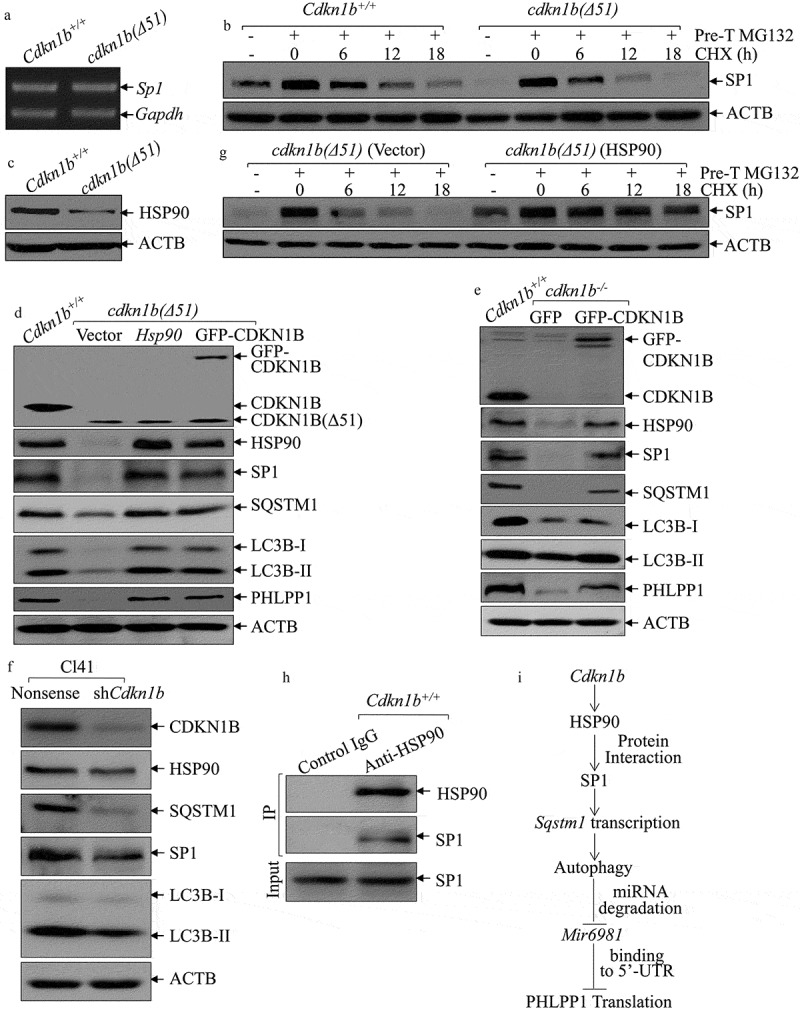
HSP90 acted as a CDKN1B downstream effector to stabilize the SP1 protein. (a) RT-PCR was employed to compare Sp1 RNA levels between Cdkn1b+/+ and cdkn1b(Δ51) cells. (b) Cdkn1b+/+ and cdkn1b(Δ51) cells were pretreated with MG132 for 12 h, and the cells were then incubated with CHX for the indicated times. The cell extracts were then subjected to western blots to analyze SP1 protein degradation rates. ACTB was used as a protein loading control. (c) The protein level of HSP90 in Cdkn1b+/+ vs. cdkn1b(Δ51) cells was determined by western blots and ACTB was used as a protein loading control. (d) Western blots were employed to compare protein expression between Cdkn1b+/+, cdkn1b(Δ51)(Vector), cdkn1b(Δ51)(HSP90) and cdkn1b(Δ51)(GFP-CDKN1B) cells, as indicated. ACTB was used as a protein loading control. (e,f) The cell extracts from the indicated cells and their transfectants were subjected to western blots to evaluate protein expression using specific antibodies. ACTB were used as protein loading controls. (g) cdkn1b(Δ51)(Vector) and cdkn1b(Δ51)(HSP90) cells were treated with MG132 for 12 h and the cells were then incubated with CHX for the indicated times. The cell extracts were subjected to western blots to analyze SP1 protein degradation rates. ACTB was used as a protein loading control. (h) An immunoprecipitation assay was employed to determine SP1 protein interaction with HSP90 using an anti-HSP90 antibody. (i) Schematic model for CDKN1B promotion of Mir6981 degradation and PHLPP1 protein translation.
Discussion
Although initially recognized as an inhibitor of cyclin E-CDK2 complexes, CDKN1B is also known to perform cyclin and CDK-independent functions such as, mediation of apoptosis, cytoskeleton rearrangements, and transcriptional regulation [27]. In the present study, we investigated the role of CDKN1B as an atypical tumor suppressor in regulating another key tumor suppressor, PHLPP1, which has never been explored before. Our results indicate that CDKN1B-regulated HSP90 could stabilize the SP1 protein, which initiates Sqstm1 transcription and expression, in turn promoting autophagy-mediated degradation of Mir6981, further reducing its binding to 5ʹ-UTR of Phlpp1 mRNA, and consequently increasing PHLPP1 protein translation. These novel findings provide new insight into the crosstalk between the 2 tumor suppressors, CDKN1B and PHLPP1 in modulation of normal cell function as schematic illustration in Figure 6(i).
PHLPP1 is known to be a tumor suppressor involved in regulation of various cellular functions. Although our recent studies show that PHLPP1 expression is regulated at the translational level by Mir190 in an NFKB1-dependent manner [28], its regulation remains largely unknown. In the current studies, we show that PHLPP1 is regulated by CDKN1B at the translational level through the newly identified Mir6981. Furthermore, we have discovered that the inhibition of PHLPP1 translation by Mir6981 is through its specific binding to the 5ʹ-UTR region of Phlpp1 mRNA, which is distinctly different from the mechanism where most miRNA binds to the 3ʹ- UTR of its targeted mRNA and affects mRNA stability and protein translation. Though the detailed mechanism between the mRNA 5ʹ-UTR and Mir6981 action is still unknown, the alteration of mRNA’s secondary structure following the interaction of the 5ʹ-UTR with miRNA [11,29] might be anticipated as an explanation for Mir6981-mediated PHLPP1 protein translation.
Mir6981 is a newly identified miRNA with unknown function, that has been reported in a study in mouse tissues [30]. Our current studies are not only the first to identify Mir6981 expression in MEF cell lines, we are also the first to show their function as directly binding to the 5ʹ-UTR of Phlpp1 mRNA, which attenuates tumor suppressor PHLPP1 protein translation, revealing a potential oncogenic function of Mir6981 in intact cells. The posttranscriptional regulation of mature miRNA has been barely elucidated in previous studies. To the best of our knowledge, there is only a 2014 report by Shenghui Lan et. al., which indicates that autophagy mediates the degradation of Mir224 in hepatocellular carcinoma [14]. In our current study, we have discovered that autophagy selectively degrades Mir6981, and that this degradation can be negatively regulated by CDKN1B. The inhibition of autophagy using the autophagy chemical inhibitor BAF significantly reverses Mir6981 expression. Moreover, our experiments show that Mir6981 is recruited to the autophagosome by virtue of its binding with the autophagy receptor SQSTM1, which is recognized by LC3. Therefore, the autophagy receptor SQSTM1 binds directly to Mir6981 and promotes the autophagy-dependent Mir6981 degradation initiated by CDKN1B in the intact cells.
It is well known that autophagy mediates SQSTM1 protein degradation, while the regulatory effects of SQSTM1 on autophagy are still controversial based on experimental systems [18]. SQSTM1 has been reported to promote MTORC1 activation, which negatively acts on initiation of autophagy in NIH-3T3 cells via modulation of MTORC1 translocation to the lysosomal surface [31], suggesting an inhibitory effect on autophagy [32], whereas in HEK293 and HeLa cells, SQSTM1 is able to liberate BECN1 by disrupting the association between BECN1 and BCL2, revealing that SQSTM1 positively induces autophagy [33]. Our current studies show that ectopic expression of SQSTM1 promotes autophagy in cells accompanied by the induction of BECN1 in Cdkn1b-deficient cells, revealing that BECN1 is a SQSTM1 downstream effector, responsible for the increased autophagic activity and degradation of SQSTM1-Mir6981 complex in Cdkn1b-deficient cells. Our results show the regulatory function of CDKN1B in the promotion of SQSTM1 expression and autophagy provides significant insight into understanding the nature of CDKN1B contribution to SQSTM1-induced cell autophagy in the intact cells.
SQSTM1 protein expression is regulated at multiple levels, including protein degradation and mRNA transcription [34,35]. Autophagy defects lead to accumulation of SQSTM1 protein [36]. Multiple transcription factors enhance tumor growth in some cancer tissues by upregulating SQSTM1 transcription, whereas SP1 has been reported to be a regulator of SQSTM1 promoter activity following H2O2 treatment in HEK293 cells [37]. Our recent studies indicate that environmental nickel exposure upregulates SQSTM1 protein expression through induction of mRNA and protein in human bronchial epithelial cells [38]. The exploration of the mechanisms underlying nickel regulation of SQSTM1 mRNA show that nickel upregulates SQSTM1 mRNA due to transcriptional activation of the SQSTM1 promoter via the NFKB-dependent cascade [38]. By tracking the upstream pathway involved in SQSTM1 alteration upon Cdkn1b deletion, our current studies show that CDKN1B can activate Sqstm1 transcription through an SP1-dependent manner, as supported by various deletions of Sqstm1 promoter-driven luciferase reporters and anti-SP1 antibody affinity-isolation ChIP assay that show SP1 binding to the SP1 binding site in the Sqstm1 promoter region. Our studies also provide evidence that HSP90 acts as a CDKN1B downstream mediator to stabilize SP1 protein via prevention of SP1 proteasome-dependent protein degradation in mouse normal MEFs and mouse epidermal Cl41 cells. Our most recent studies show that in human invasive bladder cancer cells, CDKN1B inhibits CAPN1 proteolysis cascade through attenuation of the JAK1 (Janus kinase 1)-STAT1 (signal transducer and activator of transcription 1) axis to stabilize HSP90 protein, which directly binds to PHLPP2 protein and protect it from degradation [23]. The elevated PHLPP2 promotes SQSTM1 transcription and in turn mediates autophagy to enhance MMP2 (matrix metallopeptidase 2) protein degradation, therefore exhibits an inhibition of bladder cancer cell invasion [23]. These results reveal that CDKN1B is able to promote Sqstm1 transcription and SQSTM1-mediated autophagy through elevating HSP90 expression in both normal MEFs and bladder cancer cells; however, the mechanisms underlying upregulation of Sqstm1 transcription by CDKN1B-HSP90 are different. Transcription factor SP1, but not JUN, acts as a direct downstream of HSP90 responsible for initiating transcription of Sqstm1 in normal MEFs (Figure 6), whereas HSP90 directly stabilizes PHLPP2 protein, and consequently activated JUN, but not SP1, leading to SQSTM1 transcription in human bladder cancer cells [39]. The differential mechanisms of CDKN1B-HSP90 axis in modulation of Sqstm1 transcription might be due to the differences between mouse normal cells and human cancer cells. It is noted that our current discovery of CDKN1B upregulation of HSP90 is distinctly different from our previous findings showing that CDKN1B exhibits a strong inhibitory effect on HSPB1/HSP27 (heat shock protein family B [small] member 1) and HSPA/HSP70 (heat shock protein family A [Hsp70]) expression at the transcriptional level through MAPK9-JUN- and MAPK9-HSF1-dependent pathways in cell response to arsenite exposure in the same cell lines [27]. Further elucidation of the mechanisms underlying CDKN1B promotion of HSP90 and attenuation of HSPB1 and HSPA will enable us to have a better understanding of the nature of CDKN1B in the regulation of biologic behaviors in the intact cells.
In summary, our studies define an inhibitory effect of CDKN1B on Mir6981 expression by promoting SQSTM1-dependent autophagic degradation of Mir6981. This results in a reduction of the direct binding of Mir6981 to the 5ʹ-UTR region of Phlpp1 mRNA, thereby leading to increased PHLPP1 protein translation. Our studies also show that CDKN1B mediates SQSTM1 protein induction through HSP90 stabilization of SP1 protein, which in turn binds to the Sqstm1 promoter SP1 binding site to initiate Sqstm1 transcription. Our current studies include the following discoveries: 1) SQSTM1 is a CDKN1B downstream effector and is responsible for CDKN1B-mediated autophagy; 2) by promoting autophagy-mediated degradation of Mir6981, CDKN1B exerts a positive regulatory effect on PHLPP1 protein translation; 3) Mir6981 suppresses PHLPP1 protein translation by binding directly to its mRNA 5ʹ-UTR, rather than to the classical 3ʹ-UTR of mRNA. These novel findings provide significant insight into understanding the crosstalk between 2 tumor suppressors, CDKN1B and PHLPP1, in the regulation of cell function.
Materials and methods
Reagents, antibodies and plasmids
Actinomycin D (sc-200906), CHX (sc-3508), MG132 (sc-201270), and bafilomycin A1 (BAF) (sc-201550) were purchased from Santa Cruz Biotechnology. Calpain inhibitor was bought from Cayman Chemical (14283). The Dual Luciferase Assay kit was purchased from Promega (E1960). TRIzol (15596026) reagent and SuperScript™ First-Strand Synthesis system (18080,51) were bought from Invitrogen. Antibodies specific against p-MTOR Ser2448, MTOR (9964S), LC3B, ATG3, ATG12–ATG5, ATG7, BECN1 (4445S, autophagy kit), SQSTM1 (88588), ELK1 (9182), GAPDH (5174), p-JUN Ser63 (2361), p-JUN Ser73 (9164), JUN (9165) and TAM67 (5000), were purchased from Cell Signaling Technology. Antibodies specific for CDKN1B (sc-528), JUNB (sc-73), SP1 (sc-14027), E2F1 (sc-193), ACTB (sc-58673) and TUBA (sc-53646) were from Santa Cruz Biotechnology. Specific antibody against PHLPP1 was bought from Bethyl Laboratories (A300661). The shRNAs that specifically target mouse Cdkn1b (RMM3981-97059450), Sp1 (RHS4430-200231665), and their nonsense control (RMM4534) were purchased from Open Biosystems (GE, Pittsburg, PA, USA). The mmu-Mir6981 expression plasmid (MmiR3993-MR03) and mmu-Mir6981 inhibitor plasmid (LPP-MMirAN4060-AM04p-100) was bought from GeneCopoeia. The plasmid GFP-Cdkn1b was described in our Previous Studies [5]. The plasmid GFP-Sqstm1 was a gift from Dr. Sung Ouk Kim (University of Western Ontario, London, Ontario, Canada). The plasmid for dominant negative JUN (TAM67) was described in our previous studies [27]. The plasmid containing luciferase reporter under control of mouse Sqstm1 gene promoter was kindly given by Dr. Alain Bruhat (University of Auvergne, France). The plasmid containing luciferase reporter under control of mouse WT Phlpp1 mRNA 3ʹUTR was constructed into pMIRreport vector using the primers: forward 5ʹ-CCC AAG CTT CCC AGC CTG AGT ACT GTT TTA -3ʹ, reverse 5ʹ- GGC AAG CTT TTA CGA CAT GTC CAT TTG G -3ʹ. The plasmid containing luciferase reporter under control of mouse WT Phlpp1 mRNA 5ʹUTR was constructed into PGL3-Control vector using the primers: forward 5ʹ- CCC AAG CTT AGC GGC CGC GCC GAA CGC CA -3ʹ, reverse 5ʹ- GGC AAG CTT CGT AGC GGG GCT TCA CGT CG -3ʹ. The primers for cloning the mutant of Phlpp1 mRNA 5ʹUTR luciferase reporter are: forward 5ʹ- CCA TCG GCG TCT CCG AAG AGT TCG CGC CGCCGC CG -3ʹ, reverse 5ʹ- CGG CGGCGG CGC GAA CTC TTC GGA GAC GCC GAT GG -3ʹ. The constructed Phlpp1 mRNA 5ʹ-UTR/3ʹ-UTR luciferase reporter which contains both 5ʹ-UTR and 3ʹ-UTR regulatory regions was constructed using the Phlpp1 mRNA 3ʹUTR luciferase reporter via the primers: forward 5ʹ- CGC GGA TCC TGG AGC GGC CGC G -3ʹ, reverse 5ʹ- CGC GGA TCC CGT AGC GGG GCT TCA CG -3ʹ. The Sqstm1 promoter-driven luciferase reporter with mutation of the Sp1 binding site was constructed using the primers: forward 5ʹ- CAG TGG GGT CGT CTC GAA CCC GTA CCT TTC AAG -3ʹ, reverse 5ʹ- CTT GAA AGG TAC GGG TTC GAG ACG ACC CCA CTG -3ʹ.
Cell lines and transfection
The 3T3 protocol-immortalized mouse embryonic fibroblasts (MEFs), wild-type Cdkn1b (Cdkn1b+/+) and Cdkn1b-deficient (cdkn1b(Δ51]), were established, as described in our previous studies [5] and cultured in DMEM with 10% FBS. Normal mouse epidermal Cl41 cells were cultured in MEM with 5% FBS [40,41]. Cell transfections were performed with PolyJetTM DNA in Vitro Transfection Reagent (SignaGen Laboratories, SL100468), according to the manufacturer’s instructions. For stable transfection, MEF cells were subjected to selection with hygromycin B (200 μg/mL) or puromycin (2.0 μg/mL) depending on the different antibiotic resistance of plasmids transfected. The cells surviving following the antibiotic selection were pooled as mass stable transfectants. Cl41(shCdkn1b) and Cl41(Nonsense) transfectants were selected by puromycin at 8.0 μg/mL (Alexis, BML-A260-0050).
Cell proliferation assay
Confluent monolayers of cells were trypsinized, and 1 × 103 viable cells suspended in 100 μl of medium were added to each well of 96-well plates. After adherence, cell synchronization was accomplished by replacing culture medium with 0.1% FBS medium for 24 h. Following this, the cells were cultured with 10% FBS medium for the indicated times. The proliferation of the cells was determined using the CellTiter-Glo luminescent cell viability assay kit (Promega, G7570) with a luminometer (Wallac 1420 Victor2 multipliable counter system, Madison, WI, USA).
Flow cytometry assay
The cell cycle distributions were determined by flow cytometry. The cells were cultured in 6-well plates until they were 60% confluent. The cell culture medium was replaced with 0.1% FBS medium for 24 h for cell synchronization, then replaced by 10% FBS medium for another 24 h. Following this, the cells were collected with ice-cold PBS and fixed with 5 ml of 75% ethanol at −20°C overnight. The fixed cells were stained in a buffer containing 0.1% Triton X-100 (Sigma, 9002-93-1), 0.2 mg/ml RNase A (Cell Signaling Technology, 9004S), and 50 μg/ml propidium iodide, then subjected to an EpicsXL flow cytometer (Beckman Coulter Inc., Miami, FL) for cell cycle analysis. The results were analyzed by histogram analysis software (Expo32 v. 1.2).
Western blot analysis
Whole cell extracts were prepared with the cell lysis buffer (10 mM Tris-HCl, pH 7.4, 1% SDS, 1 mM Na3VO4) [5] (Thermo Fisher Scientific, Inc. BP16650; Roche, 04693116001) with cocktail (Roche, 04693132001) added. Protein extracts were subjected to western blots with the primary antibodies indicated and probed with the AP-conjugated secondary antibody together with the enhanced chemifluorescence system, as described in a previous report [42]. The images were acquired by scanning with the Typhoon FLA 7000 laser scanner (GE, Pittsburg, PA). Some of the images were calculated by software ImageJ developed by NIH for statistical analysis.
Luciferase reporter assay
Phlpp1 mRNA 3ʹ-UTR luciferase reporter, Phlpp1 mRNA 5ʹ-UTR luciferase reporter, the Phlpp1 mRNA 5ʹ-UTR/3ʹ-UTR luciferase reporter and pRL-TK were transfected into Cdkn1b+/+, cdkn1b(∆51), and the related transfectants. Twenty-four h after transfection, luciferase activity was determined using the Dual Luciferase Assay system kit, as described in our previous studies [43]. The results were normalized by internal TK signal. The results are expressed as mean ± SD error from 3 independent experiments.
ChIP assay
The EZ-ChIP kit was used to carry out the ChIP assay according to the manufacturer’s instructions (Millipore Technologies, 17–371) [44]. Briefly, Cdkn1b+/+ cells were treated with 1% formaldehyde for 10 min at room temperature. Cells were then resuspended in lysis buffer and sonicated to generate 200 to 400 bp chromatin DNA fragments. After centrifugation (13,000 g for 10 min at 4°C), a 10-fold dilution of the supernatants was incubated with either an anti-SP1 antibody or the control rabbit IgG at 4°C overnight. The immune complex was captured with protein G–agarose beads (Millipore Technologies, 17–371) saturated with salmon sperm DNA (Millipore Technologies, 17–371), and then eluted with elution buffer. The reverse cross-linking of protein-DNA complexes to free DNA was conducted by incubating at 65°C overnight. The DNA was extracted and subjected to PCR analysis. To specifically amplify the region containing the SP1-binding sites in the human Sqstm1 promoter, PCR was performed with the following pair of primers: 5ʹ- GTC TCG ACC CCG TAC CTT TCA -3ʹ, and 5ʹ- GTC TAG GTA CGG ACG AAA CA -3ʹ. PCR products were separated on 2% agarose gels and stained with ethidium bromide. The images were scanned under UV light, as previously described [42].
RT-PCR
Total RNA was extracted using the TRIzol reagent as described in the manufacturer’s instructions and our previous studies [43]. Five µg of total RNA was used for first-strand cDNA synthesis with oligdT primer by SuperScriptTMIV First-Strand Synthesis System, as described in our previous studies [42]. Specific primers (Invitrogen) used for PCR amplification were: mouse Phlpp1, forward 5ʹ-ACA CCG TGA TTG CTC ACT CC −3ʹ, reverse 5ʹ-TTC CAG TCA GGT CTA GCT CC-3ʹ; mouse Arap3, forward 5ʹ-CCC TAC TGT TTG CAC CTA GTG TGT T-3ʹ, reverse 5ʹ-GCC TGG TCC GAG TCA ATA TCA A-3ʹ; mouse Sqstm1, forward 5ʹ-GAG AGT GTG GCA GCT GCC CT-3ʹ, reverse 5ʹ-GGC AGC TTC CTT CAG CCC TG-3ʹ; mouse Sp1, forward 5ʹ-ATT AAC CTC AGT GCA TTG GGT A-3ʹ, reverse 5ʹ-AGG GCA GGC AAA TTT CTT CTC-3ʹ; and mouse Actb, forward 5ʹ-GAC GAT ATT GCC GCA CT-3ʹ, reverse 5ʹ-GAT ACC ACG CTT GCT CTG AG-3ʹ.
Quantitative RT-PCR
The cells were extracted for total RNA using an miRNeasy Mini Kit (QIAGEN, 217,004). One μg of total RNA was used for reverse transcription. Analysis of Mir7a, Mir103a, Mir135a, Mir196a, Mir199a, Mir503, and Mir6981 expression were carried out using the miScript PCR system (QIAGEN,, 218073) by 7900HT Fast Real-time PCR system (Applied Biosystems, Waltham, MA, USA). The primers for real time PCR assay and internal control Rnu6 were purchased from Invitrogen. The initial activation was performed at 95°C for 15 min, followed by 40 cycles (denaturation at 95°C for 15 s, annealing at 55°C for 30 s and extension at 72°C for 30 s). The data were analyzed, as described in a previous publication [45].
RNA-IP assay
Cdkn1b+/+ cells were cultured in 10-cm dishes. When cell density reached 70 ~ 80%, the cells were extracted using the polysome lysis buffer [10 mM HEPES pH 7 (Sigma, H3375), 100 mM KCl (Mallinckrodt, 8648), 5 mM MgCl2 (EMD Bioscience, MX0045-1), 25 mM EDTA (EM science, 4005), 0.5% NP40 (Sigma, I-30201), 1 mM DTT (Fermentas, RO861), 100 units/ml RNAse OUT (Invitrogen, 10777-019), and complete proteinase inhibitor (Roche, 11697498001)]. The cell lysates were centrifuged at 14,000 × g for 10 min at 4°C. The anti-Sqstm1 antibody or IgG control (Cell Signaling Technology, 9004S) and A/G agarose beads (Cell Signaling Technology, 9004S) were added into the supernatant and rotated overnight at 4°C in NET2 buffer (50 mM Tris–HCl, pH 7.4, 150 mM sodium chloride, 1 mM magnesium chloride, 0.05% NP40). The beads were washed 3 times and resuspended in 100 µL NET2 and 100 μL SDS-TE (20 mM Tris-HCl, pH 7.5, 2 mM EDTA, 2% sodium dodecyl sulfate), then incubated at 55°C for 30 min, mixing occasionally. The total RNAs in the buffer of the beads were extracted by Trizol (Invitrogen, 15596-026) and real time-PCR was performed to identify the Mir6981 presented in the immune-complex.
Immunoprecipitation assay
The cells were cultured in 10-cm dishes until the cell concentration reached 70% to 80%. Culture medium was replaced by DMEM containing 0.1% FBS for 12 h. Cells were collected and lysed in 1 × Cell Lysis Buffer (Cell Signaling Technology, 9803) containing protease inhibitors (Roche, 04693132001) followed by brief sonication. Cell extracts were incubated with anti-Hsp90 or anti-mouse IgG (Cell Signaling Technology, 4875) with protein A/G agarose beads overnight at 4°C. Following a brief centrifuge, the beads were washed 3 times with 1 × Cell Lysis Buffer, and the bound proteins were eluted by 2 × SDS sample buffer (Cell Signaling Technology, 7722), then subjected to western blots.
[35S] methionine newly synthesized protein-labeling assays
Cdkn1b+/+ and cdkn1b(Δ51) cells were incubated with methionine–cysteine-free DMEM (Gibco-BRL, 21013024) containing 2% dialyzed fetal calf serum (Gibco-BRL, 10437036) in the presence of MG132 (50 µM) for 1 h. The cells were then incubated with 2% fetal bovine serum methionine–cysteine-free DMEM containing 35S-labeled methionine/cysteine (250 μCi per dish, 35S-label; Perkin Elmer, NEG009A001MC) for the time periods indicated. The cells were extracted with lysis buffer (Cell Signaling Technology, 9803) containing a complete protein inhibitor mixture and placed on ice for 10 min. Total lysate of 500 mg was incubated with anti-PHLPP1 antibody-conjugated agarose beads at 4°C overnight. The immunoprecipitated samples were washed with the cell lysis buffer 5 times, heated at 100°C for 5 min, and then subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis. The 35S-labeled PHLPP1 protein was imaged with the Typhoon FLA 7000 laser scanner.
Statistical analysis
The Student t test was used to determine the significance between treated and untreated groups and the LSD method was used to evaluate the significance among multiple groups. The results are expressed as mean±SD from at least 3 independent experiments. P < 0.05 was considered to be a significant difference between the compared groups.
Supplementary Material
Funding Statement
This work was supported by the NIH/NCI [CA165980];NIH/NIEHS [ES000260]; NIH/NCI [CA229234]; NIH/NCI [CA217923]; NIH/NCI [CA177665].
Acknowledgments
We would like to thank Dr. Alain Bruhat from University of Auvergne, France, for providing us with mouse Sqstm1 promoter-driven luciferase reporter, as well as Dr. James Roberts from Fred Hutchinson Cancer Research Center, Seattle, Washington 98104, USA, for the generous gifts of cdkn1b−/- MEF cells. We also thank Ms. Nedda Tichi for her critical reading of the manuscript. This work was partially supported by the grants of NIH/NCI CA165980, CA177665, CA217923 and CA229234, and NIH/NIEHS ES000260.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Brognard J, Newton AC.. PHLiPPing the switch on AKT and protein kinase C signaling. Trends Endocrinol Metab. 2008. August;19(6):223–230.PubMed PMID: 18511290; PubMed Central PMCID: PMCPMC2963565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhou C, Huang C, Wang J, et al. LncRNA MEG3 downregulation mediated by DNMT3b contributes to nickel malignant transformation of human bronchial epithelial cells via modulating PHLPP1 transcription and HIF-1alpha translation. Oncogene. 2017. July 06;36(27):3878–3889. PubMed PMID: 28263966; PubMed Central PMCID: PMC5525547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wander SA, Zhao D, Slingerland JM.. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res. 2011. January 1;17(1):12–18. PubMed PMID: 20966355; PubMed Central PMCID: PMCPMC3017239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sanseverino F, D’Andrilli G, Petraglia F, et al. Molecular pathology of ovarian cancer. Anal Quant Cytol Histol. 2005. June;27(3):121–124. PubMed PMID: 16121632. [PubMed] [Google Scholar]
- [5].Zhang D, Liu J, Mi X, et al. The N-terminal region of p27 inhibits HIF-1alpha protein translation in ribosomal protein S6-dependent manner by regulating PHLPP-Ras-ERK-p90RSK axis. Cell Death Dis. 2014;5:e1535 PubMed PMID: 25412313; PubMed Central PMCID: PMCPMC4260754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007. March;17(3):118–126. PubMed PMID: 17197185. [DOI] [PubMed] [Google Scholar]
- [7].Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013. July;14(7):447–459.PubMed PMID: 23732335. [DOI] [PubMed] [Google Scholar]
- [8].Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009. March;11(3):228–234. PubMed PMID: 19255566. [DOI] [PubMed] [Google Scholar]
- [9].Shen J, Hung MC. Signaling-mediated regulation of microRNA processing. Cancer Res. 2015. March 1;75(5):783–791. PubMed PMID: 25660948; PubMed Central PMCID: PMCPMC4348149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009. January 23;136(2):215–233. PubMed PMID: 19167326; PubMed Central PMCID: PMCPMC3794896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Meijer HA, Kong YW, Lu WT, et al. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science. 2013. April 5;340(6128):82–85. PubMed PMID: 23559250. [DOI] [PubMed] [Google Scholar]
- [12].Pickering BM, Willis AE. The implications of structured 5ʹ untranslated regions on translation and disease. Semin Cell Dev Biol. 2005. February;16(1):39–47.PubMed PMID: 15659338. [DOI] [PubMed] [Google Scholar]
- [13].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008. February 28;451(7182):1069–1075. PubMed PMID: 18305538; PubMed Central PMCID: PMCPMC2670399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lan SH, Wu SY, Zuchini R, et al. Autophagy-preferential degradation of MIR224 participates in hepatocellular carcinoma tumorigenesis. Autophagy. 2014. September;10(9):1687–1689. PubMed PMID: 25068270; PubMed Central PMCID: PMCPMC4206546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Duran A, Serrano M, Leitges M, et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004. February;6(2):303–309. PubMed PMID: 14960283. [DOI] [PubMed] [Google Scholar]
- [16].Rodriguez A, Duran A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006. March;3(3):211–222. PubMed PMID: 16517408. [DOI] [PubMed] [Google Scholar]
- [17].Sugimoto R, Warabi E, Katayanagi S, et al. Enhanced neointimal hyperplasia and carotid artery remodelling in sequestosome 1 deficient mice. J Cell Mol Med. 2010. June;14(6B):1546–1554. PubMed PMID: 19780870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lippai M, Low P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int. 2014;2014:832704 PubMed PMID: 25013806; PubMed Central PMCID: PMCPMC4075091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008. August 15;283(33):22847–22857. PubMed PMID: 18524774. [DOI] [PubMed] [Google Scholar]
- [20].Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009. June 12;137(6):1001–1004. PubMed PMID: 19524504; PubMed Central PMCID: PMCPMC3971861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008. April;8(4):253–267.PubMed PMID: 18354415. [DOI] [PubMed] [Google Scholar]
- [22].Kiyokawa H, Kineman RD, Manova-Todorova KO, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell. 1996. May 31;85(5):721–732. PubMed PMID: 8646780. [DOI] [PubMed] [Google Scholar]
- [23].Fero ML, Rivkin M, Tasch M, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996. May 31;85(5):733–744. PubMed PMID: 8646781. [DOI] [PubMed] [Google Scholar]
- [24].Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006. June;31(6):342–348. PubMed PMID: 16679021. [DOI] [PubMed] [Google Scholar]
- [25].Schmid DI, Schwertz H, Jiang H, et al. Translational control of JunB, an AP-1 transcription factor, in activated human endothelial cells. J Cell Biochem. 2013. July;114(7):1519–1528. PubMed PMID: 23297064; PubMed Central PMCID: PMC3999827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang SA, Chuang JY, Yeh SH, et al. Heat shock protein 90 is important for Sp1 stability during mitosis. J Mol Biol. 2009. April 17;387(5):1106–1119. PubMed PMID: 19245816. [DOI] [PubMed] [Google Scholar]
- [27].Liu J, Zhang D, Mi X, et al. p27 suppresses arsenite-induced Hsp27/Hsp70 expression through inhibiting JNK2/c-Jun- and HSF-1-dependent pathways. J Biol Chem. 2010. August 20;285(34):26058–26065. PubMed PMID: 20566634; PubMed Central PMCID: PMCPMC2924005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu Y, Zhang D, Huang H, et al. NF-kappaB1 p50 promotes p53 protein translation through miR-190 downregulation of PHLPP1. Oncogene. 2014. February 20;33(8):996–1005. PubMed PMID: 23396362; PubMed Central PMCID: PMCPMC3883870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gu W, Xu Y, Xie X, et al. The role of RNA structure at 5ʹ untranslated region in microRNA-mediated gene regulation. Rna. 2014. September;20(9):1369–1375. PubMed PMID: 25002673; PubMed Central PMCID: PMCPMC4138320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ladewig E, Okamura K, Flynt AS, et al. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res. 2012. September;22(9):1634–1645. PubMed PMID: 22955976; PubMed Central PMCID: PMCPMC3431481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell. 2011. November 11;147(4):724–727. PubMed PMID: 22078874; PubMed Central PMCID: PMCPMC3290994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011. October 7;44(1):134–146. PubMed PMID: 21981924; PubMed Central PMCID: PMCPMC3190169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou L, Wang HF, Ren HG, et al. Bcl-2-dependent upregulation of autophagy by sequestosome 1/p62 in vitro. Acta Pharmacol Sin. 2013. May;34(5):651–656. PubMed PMID: 23564079; PubMed Central PMCID: PMCPMC3647217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010. April 2;584(7):1374–1378. PubMed PMID: 20153326. [DOI] [PubMed] [Google Scholar]
- [35].Vadlamudi RK, Shin J. Genomic structure and promoter analysis of the p62 gene encoding a non-proteasomal multiubiquitin chain binding protein. FEBS Lett. 1998. September 18;435(2–3):138–142. PubMed PMID: 9762895. [DOI] [PubMed] [Google Scholar]
- [36].Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009. June 12;137(6):1062–1075. PubMed PMID: 19524509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Du Y, Wooten MC, Wooten MW. Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol Dis. 2009. August;35(2):302–310. PubMed PMID: 19481605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang H, Zhu J, Li Y, et al. Upregulation of SQSTM1/p62 contributes to nickel-induced malignant transformation of human bronchial epithelial cells. Autophagy. 2016. October 02;12(10):1687–1703. PubMed PMID: 27467530; PubMed Central PMCID: PMC5079680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peng M, Wang J, Zhang D, et al. PHLPP2 stabilization by p27 mediates its inhibition of bladder cancer invasion by promoting autophagic degradation of MMP2 protein. Oncogene. 2018. June 21 DOI: 10.1038/s41388-018-0374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhu J, Zhang J, Huang H, et al. Crucial role of c-Jun phosphorylation at Ser63/73 mediated by PHLPP protein degradation in the cheliensisin a inhibition of cell transformation. Cancer Prev Res (Phila). 2014. December;7(12):1270–1281. PubMed PMID: 25281487; PubMed Central PMCID: PMCPMC4256147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gao G, Chen L, Li J, et al. Isorhapontigenin (ISO) inhibited cell transformation by inducing G0/G1 phase arrest via increasing MKP-1 mRNA Stability. Oncotarget. 2014. May 15;5(9):2664–2677. PubMed PMID: 24797581; PubMed Central PMCID: PMCPMC4058035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jin H, Yu Y, Hu Y, et al. Divergent behaviors and underlying mechanisms of cell migration and invasion in non-metastatic T24 and its metastatic derivative T24T bladder cancer cell lines. Oncotarget. 2015. January 1;6(1):522–536. PubMed PMID: 25402510; PubMed Central PMCID: PMCPMC4381612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xu J, Wang Y, Hua X, et al. Inhibition of PHLPP2/cyclin D1 protein translation contributes to the tumor suppressive effect of NFkappaB2 (p100). Oncotarget. 2016. April 15 PubMed PMID: 27095572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fang Y, Cao Z, Hou Q, et al. Cyclin d1 downregulation contributes to anticancer effect of isorhapontigenin on human bladder cancer cells. Mol Cancer Ther. 2013. August;12(8):1492–1503. PubMed PMID: 23723126; PubMed Central PMCID: PMCPMC3744103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zeng X, Xu Z, Gu J, et al. Induction of miR-137 by isorhapontigenin (ISO) directly targets SP1 protein translation and mediates its anticancer activity both in vitro and in vivo. Mol Cancer Ther. 2016. March;15(3):512–522. PubMed PMID: 26832795; PubMed Central PMCID: PMCPMC4783212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.